
Abstract
There is considerable evidence that signaling through cannabinoid 1 receptors (CB1Rs) contributes to the effects of stress on the brain as well as stress-adaptation. Similarly, serotonin, norepinephrine, and dopamine are important modulators and effectors of stress. The purpose of this review is to present and discuss the results of studies that have investigated the interactions between endocannabinoid-CB1 receptor signaling and each of the biogenic amines in the context of stress.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Cannabinoid
- Cannabis
- Marijuana
- Delta-9-tetrahydrocannabinol
- N-arachidonylethanolamine
- 2-arachidonoylglycerol
- Dopamine
- Norepinephrine
- Dopamine
9.1 Introduction
Chronic exposure to psychological stress is a fact of life in the twenty-first century society. Chronic stress exposure contributes to neuropsychiatric diseases and disorders; including depression, anxiety, substance abuse, and schizophrenia . In addition, stress is a risk factor for the development of obesity, cardiovascular disease, gastrointestinal disorders, and functional pain disorders. Drugs that target monoamine signaling have efficacy in the treatment of several stress-related diseases, particularly depression and schizophrenia, but not all affected individuals are responsive and adverse drug effects can interfere with efficacy. In addition, pharmacotherapies targeting the monoamines can treat disease symptoms but do not reduce the impact of stress. In fact, there are very few approaches available to reduce the consequences of stress and thereby reduce the risk of developing stress-related disorders.
It could be argued that Chinese and Indian cultures discovered a stress-reducing therapeutic, Cannabis sativa, thousands of years ago. Cannabis extracts were used as medicinals by these cultures to reduce anxiety, pain, seizures, mania, and muscle spasms; and to stimulate appetite. Modern research confirms some of these benefits; for example, recreational use of Cannabis can be associated with elevation of mood, and euphoria along with reduced feelings of stress and anxiety (Tournier et al. 2003 ). The constituent of Cannabis that is likely responsible for the stress-reducing effects is ∆9-tetrahydrocannabinol (THC), though another abundant phytocannabinoid, cannabidiol, is also an effective antianxiety agent (Bergamaschi et al. 2011 ). THC is a partial agonist of two G-protein coupled cannabinoid receptors (CBRs), CB1R (Matsuda et al. 1990 ) and CB2R (Munro et al. 1993 ). The effects of THC on stress, anxiety, and mood are mediated by these receptors, particularly CB1R, which will be the focus of this review (Pertwee et al. 2010 ).
Considerable evidence indicates that CB1R are located on presynaptic, axonal terminals (Herkenham et al. 1991a ) and couple to inhibition of calcium influx (Mackie and Hille 1992 ), thereby inhibiting neurotransmitter release. CB1R are present on glutamatergic and GABAergic terminals throughout the central nervous system (CNS) and, via regulation of release of these primary excitatory and inhibitory transmitters, exert a profound effect on postsynaptic neuronal activity (Freund et al. 2003 ). Through this mechanism, CB1R activity regulates activational drive on principal, outflow neurons in many brain regions. CB1R found on axon terminals of noradrenergic (Oropeza et al. 2007 ; Scavone et al. 2010 ), and serotonergic (Hermann et al. 2002 ) neurons can inhibit the release of the biogenic amines under some circumstances.
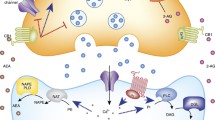
The endogenous ligands for CB1R (endocannabinoids (eCBs)) are two arachidonic acid derivatives: N-arachidonylethanolamine (AEA) (Devane et al. 1992 ) and 2-arachidonoylglycerol (2-AG) (Mechoulam et al. 1995 ; Sugiura et al. 1995 ). Considerable evidence indicates that the eCBs are synthesized and released from neurons postsynaptic the CB1R-containing axon terminals (Freund et al. 2003 ). Multiple stimuli can induce eCB mobilization, including depolarization (Wilson and Nicoll 2001 ) and activation of phospholipase C (PLC) by G protein coupled receptors (GPCRs) that couple to Gq family heterotrimeric G proteins (Kim et al. 2002 ; Varma et al. 2001 ). As discussed further below, glucocorticoids also mobilize eCBs. Although there are likely other functions of endocannabinoid-CB1R signaling (ECS), it is a primary regulator of synaptic plasticity via changes in presynaptic release, specifically subserving short-term, activity-driven changes in synaptic strength as well as other forms of presynaptic plasticity (Patel and Hillard 2009a ).
9.2 ECS and Stress: Endocrine Aspects
Considerable evidence has accumulated to support the hypothesis that ECS is altered by stress exposure and modulates stress responses through effects on synaptic activity. Stress activates the hypothalamic-pituitary-adrenal (HPA) axis and induces the release of glucocorticoid hormones, which exert widespread effects on the body, including the brain (McEwen 2008 ). Exogenous administration of glucocorticoids to rats results in a rapid (i.e. within 10 min) increase in eCB contents in several limbic structures (Hill et al. 2010a ). Multiple studies indicate that ECS is an effector of glucocorticoids in the brain (Hill and McEwen 2009 ). In the hypothalamus , glucocorticoids act through a membrane receptor to rapidly mobilize eCBs that, through CB1R on glutamatergic axons, inhibit excitatory drive onto corticotropes in the paraventricular nucleus (PVN) (Di et al. 2003 ; Di et al. 2005). Glucocorticoid infusion into the PVN rapidly inhibits HPA axis activation, an effect that is blocked by the CB1R antagonist, AM-251 (Evanson et al. 2010 ). Collectively, these data demonstrate that glucocorticoid-mediated fast feedback inhibition of the HPA axis requires mobilization of ECS in the PVN.
Both the hippocampus (Sapolsky et al. 1985 ) and medial prefrontal cortex (mPFC) (Diorio et al. 1993 ) are responsive to glucocorticoids and participate in long-loop feedback regulation of the HPA axis. Recent evidence indicates that this function of glucocorticoids also requires ECS. In the hippocampus, exposure to acute stress, in vivo glucocorticoid treatment, and direct application of glucocorticoids to slices all act through GR to increase eCB-mediated inhibition of gamma-aminobutyric acid (GABA) release (Wang et al. 2012b ). In the PFC, glucocorticoid treatment of slices produces eCB-mediated inhibition of GABA release and dysinhibition of mPFC pyramidal neurons (Hill et al. 2011a ). Increased pyramidal neuronal activity contributes to termination of the HPA axis response through activation of inhibitory projections from the anterior bed nucleus of the stria terminalis to the PVN (Radley and Sawchenko 2011 ).
ECS signaling is also involved in the behavioral effects of glucocorticoids . In these functions, ECS is required for the positive effects of glucocorticoids and therefore can be considered as a contributor to the stress response. For example, glucocorticoid-mediated enhancement of memory consolidation (Campolongo et al. 2009 ) and stress-induced reinstatement of cocaine seeking behavior in mice (Vaughn et al. 2012 ) are both inhibited by antagonism of CB1R.
In sum, these data support a critical role of ECS as a “second messenger” for glucocorticoids in the brain. Our understanding of the mechanisms by which glucocorticoids mobilize ECS is incomplete; however, it is likely that multiple mechanisms are at play with different time courses that are well matched to the function of the glucocorticoids in the specific brain region.
In spite of the evidence discussed above that acute stress and glucocorticoid treatment increase eCB-mediated signaling in hypothalamus , hippocampus, and PFC, acute stress exposure in rodents results in decreased amygdalar and PFC AEA concentrations (Hill et al. 2009b ; McLaughlin et al. 2012 ; Patel et al. 2005 ; Rademacher et al. 2008 ). The reduction in AEA content is accompanied by an increase in the activity of fatty acid amide hydrolase (FAAH, (Hill et al. 2009b ; McLaughlin et al. 2012 )), the primary catabolic enzyme for AEA in brain (Cravatt et al. 1996 ). If the decline in amygdalar AEA is prevented, HPA axis activation by stress is reduced (Hill et al. 2009b ); evidence that tonic CB1R signaling in the amygdala opposes stress-induced HPA axis activation and must be inhibited in order for the HPA stress response to occur. In other words, AEA functions as a gatekeeper for the stress response (Patel et al. 2004 ). The mechanism by which stress increases the activity of FAAH is unknown, but is likely a very early event in stress response cascade. Although stress decreases AEA concentrations in the amygdala, glucocorticoid treatment increases amygdalar AEA concentrations (Hill et al. 2010a ). Thus, the sensory perception of stress decreases AEA, reduces CB1R tone and allows for HPA axis activation while subsequent elevation of glucocorticoids re-establishes the CB1R “gate” by elevating AEA. This, glucocorticoid-induced elevation in amygdalar AEA could be another example of ECS involvement in feedback regulation of HPA axis activity.
These data have important implications for therapeutic treatment of disorders in which hyperactive HPA axis activity contributes to disease. Preclinical models demonstrate that FAAH inhibition inhibits stress-induced increases in circulating glucocorticoids (Patel et al. 2004 ), reduces anxiety in adverse environments (Patel and Hillard 2006 ), and decreases immobility in rats in the forced swim assay (Gobbi et al. 2005 ; McLaughlin et al. 2007 ). Several authors have suggested that therapeutic agents that increase AEA (e.g., FAAH inhibitors) should be examined in humans for treatment of anxiety and depressive disorders that are characterized by excessive or prolonged HPA axis activation (Hill and Gorzalka 2009 ; Hill et al. 2009a; Parolaro et al. 2010 ).
9.3 ECS and Stress: Neural Aspects
Acute stress exposure evokes characteristic physiological and behavioral changes that are mediated by activation of the neuronal defense pathway. Among the changes evoked are sympathoexcitation, including an increase in arterial blood pressure. ECS has been shown to regulate the sympathetic response to stress at multiple sites in the CNS. First, activation of ECS in the dorsal periaqueductal gray (PAG) enhances sympathetic nerve activity through inhibition of GABA release (Dean 2011 ). As ECS is increased in this region by stress (Hohmann et al. 2005 ), it could contribute to the effects of stress to enhance sympathetic outflow via this mechanism. Second, administration of corticotropin releasing factor (CRF) into the cerebral ventricle (i.c.v.) activates the sympathetic nervous system in anesthetized rats (Shimizu et al. 2010 ). This response is inhibited by CB1R direct and indirect agonists and is potentiated by CB1R antagonists administered i.c.v. (Shimizu et al. 2010 ). The site of this suppressive CB1R mechanism is unknown. Third, CB1R signaling in the nucleus tractus solitarius (NTS) enhances baroreceptor sympathoactivation (Seagard et al. 2004 ) through inhibition of GABA release (Chen et al. 2010 ). These brain-regional effects of ECS on regulation of sympathetic outflow illustrate an important point regarding the ECS: that it is a local modulatory system. As a result, it is not unusual to find that ECS exerts non-congruent changes at individual sites within a circuit.
CB1R are expressed at the terminals of sympathetic axons innervating blood vessels and activation of presynaptic CB1R reduces the release of NE in anesthetized (Ishac et al. 1996 ) and pithed (Pfitzer et al. 2005 ) rats. While there is evidence that these receptors are not endogenously active in healthy animals (Pfitzer et al. 2005 ), they could contribute to blood pressure regulation during inflammation. For example, treatment of conscious rats with lipopolysaccharide (LPS) induces vasodilation and hypotension that is reversed by both blockade of CB1R and ß-adrenergic signaling (Gardiner et al. 2005 ). LPS releases eCBs from circulating platelets and macrophages (Varga et al. 1998 ), suggesting that inflammation brings ECS “on-line” to reduce blood pressure and contribute to inflammation-induced shock. Indeed, human studies support a role for ECS in hypotension that occurs during endotoxic shock (Sakamoto et al. 2008 ; Wang et al. 2001 ).
It is hypothesized that chronic inflammation contributes to major depressive illness in humans (Haroon et al. 2012 ). It is interesting in this regard that women with depression exhibit a positive correlation between circulating eCBs and systolic and diastolic blood pressure while there is no correlation among eCBs and blood pressure measurements in healthy women (Ho et al. 2012 ). One explanation of these findings is that chronic inflammation leads to increased eCBs, depression, and hypertension in a coordinated manner.
9.4 ECS and Chronic Stress
Like most other signaling pathways that utilize GPCRs, CB1R density and coupling to downstream effectors exhibit considerable plasticity in response to increased agonist availability. In particular, exogenous agonist treatment and excessive amounts of 2-AG cause desensitization and down-regulation of CB1R signaling (Schlosburg et al. 2010 ; Sim et al. 1996 ). Chronic, non-habituating stress decreases CB1R density in the hippocampus (Hill et al. 2005 ) and CB1R function in the ventral striatum (Wang et al. 2010 ); possibly as a result of sustained increases in 2-AG. On the other hand, repeated exposure of rodents to the same stress, which is accompanied by habituation, enhances ECS at the level of the eCB ligand (Patel and Hillard 2008 ; Patel et al. 2009 ). The increase in ECS is required for the dampened responsivity to stress and fear that occurs during habituation (Hill et al. 2010b ; Kamprath et al. 2011 ; Patel et al. 2005 ).
An important point illustrated by these studies is that ECS can be considered a component of the stress response that provides plasticity and a molecular memory of prior stress exposures. These features, along with the function of ECS to regulate neurotransmitter release throughout the neural axis, places ECS in a vital position to regulate the impact of stress on the body through modulation of neural circuits.
9.5 Monoamine Pathways and Stress
There is considerable evidence that serotonergic, noradrenergic, and dopaminergic networks are stress-responsive and contribute to the behavioral responses to stress. In light of the role of ECS to regulate synaptic activity, and the sensitivity of ECS to stress, it is logical to ask to what extent do ECS and monoamine signaling overlap and/or act in series to regulate behavioral and endocrine responses to stress? The purpose of this review is to examine the evidence regarding the relationships between ECS and each of the three, CNS active monoamines : serotonin (5-HT), norepinephrine (NE) and dopamine (DA) to address this question.
9.6 Interactions Between ECS and 5-HT Signaling: Mechanisms
Physiological and behavioral studies demonstrate significant interactions between cannabinoid and serotonergic signaling . In some cases, ECS and serotonergic signaling produce similar effects and act in series to affect physiological or behavioral change. For example, activation of either 5-HT1A or CB1R profoundly reduces body temperature (Malone and Taylor 1998 ) and pharmacological data suggest that the hypothermic effects of CB1R activation require increased 5-HT release in the medial raphe nucleus (Malone and Taylor 2001 ). Similarly, both CB1R (Patel and Hillard 2006 ) and 5-HT receptors (Griebel et al. 1997 ) regulate anxiety in rodents. Pharmacological blockade of 5-HT1A receptors inhibits both the anxiolytic (Braida et al. 2007) and anxiogenic (Marco et al. 2004 ) effects of the cannabinoids. On the other hand, CBR agonists can produce pharmacological effects via suppression of 5-HT signaling. For example, THC-induced impairment of spatial memory is accompanied by a reduction in 5-HT release in the ventral hippocampus and is attenuated by pharmacological treatments that maintain 5-HT signaling (Egashira et al. 2002 ).
Molecular evidence supports the pharmacological data that ECS and serotonergic systems interact functionally in both positive and negative ways. In the forebrain, 5-HT1B and CB1R are coexpressed in principal neurons, while 5-HT3 and CB1R are colocalized on GABA interneurons (Hermann et al. 2002 ). CB1R are also present on serotonergic neuron cell bodies in the dorsal raphe nucleus (DRN) and on axon terminals of serotonergic neurons in projection areas, including the hippocampus and amygdala (Haring et al. 2007 ; Lau and Schloss 2008 ). CB1R on serotonergic axon terminals inhibit 5-HT release in cortical slice preparations (Nakazi et al. 2000 ) and in vivo (Egashira et al. 2002 ; Merroun et al. 2009 ; Moranta et al. 2004; 2009 ). Blockade of CB1R increases basal extracellular concentrations of 5-HT in mPFC (Darmani et al. 2003 ; Tzavara et al. 2003 ), indicating ECS exerts tonic inhibition of 5-HT release. Sustained changes in CB1R activity alter the expression and/or functional coupling of 5-HT receptors in hippocampus and PFC (Aso et al. 2009 ; Mato et al. 2007 ; Moranta et al. 2009 ). Since 5-HT receptors are readily up- and down-regulated by changes in 5-HT availability, these findings are consistent with the hypothesis that ECS is a physiologically important regulator of 5-HT release, and therefore, of 5-HT signaling in the terminal regions of serotonergic projection neurons.
CB1R activity also affects serotonergic signaling through changes in the firing of serotonergic neurons. In vitro studies of synaptic activity within the DRN reveal that CB1R agonists inhibit glutamatergic activation of DRN serotonergic neurons through presynaptic inhibition of glutamate release (Haj-Dahmane and Shen 2005; 2009 ). Extracellular recordings provide evidence that ECS also inhibits GABA release in the DRN (Mendiguren and Pineda 2009 ). Therefore, the net effect of ECS on serotonergic neuronal activity will depend on synaptic strength, endogenous eCB tone, and CB1R density on terminals impinging on DRN serotonergic neurons (Haj-Dahmane and Shen 2011 ).
Previous work has demonstrated that DRN serotonergic neuron activity is regulated by an afferent projection from the mPFC to inhibitory interneurons of the DRN (Celada et al. 2001 ). Activation of this important input results in reduced activity of DRN serotonergic neurons while its inhibition is associated with decreased anxiety and depressive behaviors (Celada et al. 2001 ). CBIR agonist administration into the ventromedial PFC results in increased firing of serotonergic neurons in the DRN; an effect abolished by transection of the medial but not lateral PFC (Bambico et al. 2007 ). Since ECS also regulates the activity of serotonergic neurons projecting to the frontal cortex (Haj-Dahmane and Shen 2011 ), it has been suggested that ECS and 5-HT participate in a reciprocal loop; increased ECS in the PFC leads to increased 5-HT neuronal activity in the DRN, resulting in increased 5-HT release in the PFC (McLaughlin et al. 2012 ).
The cellular localization of CB1Rs in a brain region is one critical determinant of the effect of changes in ECS on the serotonergic circuit. Another important determinant involves the mechanisms that trigger eCB mobilization. Haj-Dahmane and Shen have demonstrated that serotonergic neurons in the DRN synthesize and release eCBs (Haj-Dahmane and Shen 2005, 2009 ). As has been demonstrated at other synapses, eCB mobilization can be initiated by membrane depolarization and increased intracellular calcium in serotonergic neurons (Haj-Dahmane and Shen 2009 ). Mobilization of eCBs in DRN neurons is also triggered by activation of GPCRs that couple to Gq proteins, including orexin receptors (Haj-Dahmane and Shen 2005 ). Orexin receptor activation results in recruitment of ECS, likely via increased synthesis of 2-AG since it is dependent upon PLC and diacylglycerol lipase (DGL) activities (Haj-Dahmane and Shen 2005 ). These studies indicate that orexin-mediated regulation of serotonergic signaling is likely indirect, through changes in ECS. The result of eCB synthesis and release from serotonergic cell bodies in the DRN can be decreased glutamate or GABA release, thus either decreased or increased excitatory drive, respectively, onto the serotonergic projections.
5-HT , acting through 5-HT2 receptors which are known to couple to Gq family proteins (Barnes and Sharp 1999 ), activates ECS in the inferior olive, a brain region that receives extensive serotonergic input (Best and Regehr 2008 ). As a result, serotonin inhibits glutamate release through ECS and thereby regulates both pre- and post-synaptic function in this brain region. The authors of this study speculate that this mechanism could contribute responses to other 5-HT2 receptor effects, including food intake, sexual behavior, and sleep. Studies in rat cerebellar membranes suggest that, when CB1R and 5-HT2 receptors are colocalized, activation of the 5-HT2 receptor increases the high affinity binding site for at least one synthetic agonist (Devlin and Christopoulos 2002 ), another mechanism by which 5-HT regulates ECS.
Thus, currently available evidence indicates that ECS regulates the serotonergic system via multiple mechanisms, including regulation of DRN neuronal firing and of 5-HT release from axon terminals. Current data suggest that ECS regulation of 5-HT signaling occurs in many brain regions and is the primary mechanism of interaction of these two systems in the healthy, unstressed brain. However, 5-HT can reciprocally regulate ECS through 5-HT2 receptors, and, as is discussed further below, studies in rodent models of depression suggest that 5-HT regulation of ECS could contribute to behaviors in these models.
9.7 Interactions Between ECS and 5-HT Signaling: Stress Context
9.7.1 HPA Axis Regulation
It is well-documented that stress increases 5-HT turnover and serotonergic signaling plays an important and complex role in the regulation of autonomic and endocrine responses to stress (Lanfumey et al. 2008 ; Lowry 2002 ). In particular, exposure of rodents to inescapable stressors increases c fos expression in DRN serotonergic neurons (Greenwood et al. 2003 ) and increases 5-HT release in projection areas, such as the PFC (Bland et al. 2003 ). Serotonergic inputs contribute to regulation of HPA axis activity in the PVN, primarily in a positive manner (Lanfumey et al. 2008 ). For example, activation of either 5-HT1A (Osei-Owusu et al. 2005 ) or 5-HT2C (Klaassen et al. 2002 ) receptors in the PVN increase ACTH secretion. Glucocorticoids inhibit DRN neuronal activity, resulting in feedback inhibition on the serotonergic drive to increase HPA axis activity (Lanfumey et al. 2008 ). Recent data demonstrate that glucocorticoids regulate DRN neuronal activity via recruitment of ECS in the DRN (Wang et al. 2012a ). In particular, glucocorticoids act via activation of a membrane-localized, GPCR to increase ECS which results in decreased glutamatergic drive to serotonergic neurons in the DRN (Wang et al. 2012a ). ECS regulation of activity in the DRN is consistent with a general function of the ECS to inhibit or dampen HPA axis activity through effects in multiple brain regions, including hypothalamus (Di et al. 2003 ), PFC (Hill et al. 2011a ), hippocampus (Wang et al. 2012b ), and amygdala (Hill et al. 2009b ).
9.7.2 Behavior in the Forced Swim (FST) and Tail Suspension Tests
A large body of evidence supports the hypothesis that 5-HT1A receptors play a pivotal but complex role in the mechanism of action of anti-depressant drugs (Lanfumey et al. 2008 ). A commonly employed rodent assay for efficacy of anti-depressant drugs involves the examination of behavioral responses to an inescapable and stressful situation, usually either a period of forced swim or suspension by the tail. The read-out of these assays is a comparison of the time spent using active behaviors that enhance chances to escape, such as climbing or swimming, and passive behaviors, such as immobility, which conserve energy. Chronic stress and other manipulations that are considered pro-depressive increase time spent immobile while treatment with antidepressants increase active behavioral repertoires (Cryan et al. 2005 ).
There are multiple reports of antidepressant-like effects in the FST following treatment of rats with CB1R agonists and indirect agonists (Bambico et al. 2007 ; Gobbi et al. 2005 ; Hill and Gorzalka 2005 ; McLaughlin et al. 2007 ) and emerging evidence indicates that the cannabinoids produce these effects via increased 5-HT release. In a seminal study, Bambico et al. (2007) demonstrated that low doses of the synthetic CB1R agonist, Win 55212-2, elicit antidepressant behavior in the FST and enhance serotonergic neuronal firing in the DRN through CB1R mechanism. The antidepressant efficacy of Win 55212-2 was abolished by depletion of 5-HT, supporting the hypothesis that CB1R agonists act to increase serotonergic signaling . Enhancement of DRN neuronal firing by systemic administration of Win 55212-2 was abolished by transection of the mPFC, and both the increased firing and decreased FST immobility were mimicked by direct injection mPFC of Win 55212-2. Taken together, these results support the hypothesis that ECS in the PFC increases excitatory drive on DRN serotonergic neurons, resulting in antidepressant-like efficacy. As described above, an inhibitory, multi-synaptic projection from mPFC to the DRN has been described; an untested hypothesis is that ECS decreases activation of this projection through inhibition of glutamate release in the mPFC.
Mice with a genetic deletion of FAAH (therefore, high AEA tone) exhibit reduced responsiveness of mPFC pyramidal cells to a 5-HT2A/2C agonist, which is consistent with increased firing of serotonergic neurons, enhanced 5-HT release in the mPFC and subsequent down-regulation of 5-HT2A/2C receptors (Bambico et al. 2010 ). Interestingly, treatment of rats with a single dose of THC does not alter FST behavior and exerts complex effects on DRN firing while repeated THC exposure (5 days) decreases immobility in the FST and enhances 5-HT1A receptor activity in the hippocampus (Bambico et al. 2012 ). THC is a partial agonist of CB1R (Kearn et al. 1999 ), which could contribute to its lower effectiveness.
A recent biochemical study supports the interaction of ECS and serotonergic circuits in regulating the behavioral response to FST in rats (McLaughlin et al. 2012 ). These investigators found that PFC AEA content is decreased and FAAH activity increased immediately following exposure to FST. When an inhibitor of FAAH was injected into the mPFC, active coping in the FST was increased, as was firing of DRN serotonergic neurons (McLaughlin et al. 2012 ). The effect of FAAH inhibition to increase active coping was blocked by depletion of 5-HT . These data suggest that PFC AEA content is a critical determinant of the firing of serotonergic DRN neurons, thus, is a critical determinant of the behavioral coping mechanism employed.
The role of the CB1R in the regulation of FST behavior is less clear in mice. For example, treatment of mice with moderate doses of THC increases immobility in the FST, an effect that requires 5-HT1A receptors (Egashira et al. 2008 ) and CB1R antagonism can produce antidepressant-like effects (Griebel et al. 2005 ; Shearman et al. 2003 ). A recent study provides some insight into the discrepant data (Haring et al. 2013 ). In this study, low doses of THC (0.1 and 0.5 mg/kg) decreased immobility in the FST which was blocked by inhibition of 5HT1A receptors and depletion of 5-HT. High doses (3 and 10 mg/kg) of the CB1R antagonist, rimonabant, also decreased immobility but this effect was unaffected by suppression of 5-HT signaling . Instead, the antidepressant-like effect of CB1R blockade was reversed by inhibition of catecholamine synthesis and did not occur in mice lacking CB1Rs on forebrain GABAergic neurons. It is possible that the prevailing ECS tone within serotonergic circuits differs among animal species.
9.7.3 Anxiety and Other Emotional Behaviors
Both serotonergic and CB1R signaling are important regulators of anxiety and there is evidence that changes in serotonergic signaling contribute to the effects of ECS on anxiety. Acute treatment of rats with low doses of THC or the AEA clearance inhibitor, AM 404 (Beltramo et al. 1997 ) results in decreased anxiety-like behaviors that is significantly reduced by pretreatment with a 5-HT1A antagonist (Braida et al. 2007). Similarly, FAAH null mice exhibit decreased anxiety compared to wild types (Bambico et al. 2010 ). FAAH null mice also have a greater than 30 % increase in the firing of DRN neurons that is inhibited in a subset of neurons by CB1R antagonism. In addition, these animals have increased basal 5-HT tone in the frontal cortex, but not in the hippocampus, that is reduced by CB1R antagonist treatment (Cassano et al. 2011 ). Taken together, these studies indicate that AEA-mediated CB1R signaling exerts antianxiety effects through interactions with serotonergic signaling in the PFC.
9.7.4 Impact of Changes in Serotonergic Signaling on ECS
The preceding section indicates that ECS modulates serotonergic signaling . There are also data consistent with the reciprocal relationship between these two systems; that is, that serotonergic signaling alters ECS. Although activation of 5-HT2 receptors in the inferior olive induce ECS through PLC-mediated increases in 2-AG (Best and Regehr 2008 ), other data indicate that acute alterations in 5-HT tone do not alter ECS. For example, the behavioral effects of acute exposure to selective serotonin reuptake inhibitors (SSRIs) are preserved in CB1R−/− mice and in the presence of CB1R antagonists (Gobshtis et al. 2007 ; Steiner et al. 2008 ), suggesting that ECS is not required for SSRI efficacy in the FST.
On the other hand, there is evidence that long-lasting elevation of 5-HT tone, as occurs with chronic SSRI treatment, does change ECS. Treatment of rats for 21 days with imipramine (which inhibits reuptake of both NE and 5-HT) decreases CB1R binding site density in hypothalamus , midbrain, and ventral striatum (Hill et al. 2008a ). Similarly, chronic fluoxetine treatment decreases CB1R messenger ribonucleic acid (mRNA) expression in the caudate-putamen (Oliva et al. 2005 ) and decreases CB1R function in cerebellum (Mato et al. 2010 ). Chronic citalopram treatment reduces CB1R coupling in hypothalamus and hippocampus (Hesketh et al. 2008 ). Chronic treatment with imipramine produces a significant increase in 2-AG contents in the hypothalamus and midbrain (Hill et al. 2008a ), which could result in agonist-induced desensitization and/or down-regulation of the CB1R.
In contrast to the effects of SSRIs to decrease CB1R signaling in subcortical regions outlined above, CB1R binding site density is increased in the amygdala by chronic imipramine (Hill et al. 2008a ) and in the PFC by chronic fluoxetine (Hill et al. 2008b). Similarly, chronic fluoxetine increases CB1R function in the frontal cortex (Mato et al. 2010 ). Chronic fluoxetine treatment results in decreased expression of mRNA for the 5-HT2C receptor in the PFC while increasing its expression in the hippocampus (Barbon et al. 2011 ), raising the possibility that region-specific changes in 5-HT2C expression by antidepressants underlie the region-specific changes in ECS.
9.7.5 Models of Depression
Several studies have demonstrated that ECS is dysregulated in animal models of depression and that the dysregulation can be reversed by SSRI treatment. Chronic, unpredictable stress (CUS) results in behavioral changes that mirror those seen in depressed humans. CUS down-regulates ECS in several brain regions, including the hippocampus, hypothalamus , and ventral striatum (Hill et al. 2008a ; Hill et al. 2005). Imipramine reverses the effects of CUS on ECS in all of these brain regions (Hill et al. 2008a). Similarly, CUS exposure decreases CB1R-mediated inhibition of glutamate release in accumbens slices which is reversed by in vivo treatment with fluoxetine (Wang et al. 2010 ). These results suggest that stress-induced decreases in 5-HT tone contribute to the down-regulation of ECS in these brain regions. These studies also suggest that decreased ECS contributes to the symptoms of depression, perhaps through a feed-forward cycle that enhances suppression of 5-HT release.
Further evidence that reduced 5-HT tone affects ECS comes from studies of rats with bilateral olfactory bulbectomy (OBX), which induces behavioral, neurochemical, and structural abnormalities that are similar to human depression . Adaptive changes in the serotonergic system play an important role in the OBX syndrome; for example, serotonergic neurons in the DRN degenerate, resulting in permanent reductions in 5-HT secretion in the hippocampus and amygdala (van der Stelt et al. 2005 ). Male rats with bilateral OBX exhibit significant increases in CB1R density and coupling to GDP/GTP exchange in PFC which is reversed by chronic treatment with fluoxetine (Rodriguez-Gaztelumendi et al. 2009 ). CUS also produces an increase in CB1R density in the PFC; however, this change is not altered by imipramine treatment while CUS-induced decreases in CB1R density were normalized by this antidepressant (Hill et al. 2008a ). The PFC of human suicides also exhibit increased CB1R density and function (Hungund et al. 2004 ; Vinod et al. 2005 ), which provides relevance to these observations.
9.7.6 Summary
There is clear evidence that ECS regulates the release of 5-HT in limbic brain regions through multiple mechanisms. CB1R signaling regulates a bi-directional circuit between the mPFC and DRN; this is likely the substrate for the antidepressant effects of systemic CB1R agonists in rats. Chronic exposure of rats to SSRIs decreases CB1R expression and function in subcortical, limbic regions, but increases CB1R expression in PFC and amygdala; the function of these changes in each brain region are not well understood. Paradoxically, rodents exposed to CUS and OBX, treatments that induce depressive-like behaviors, exhibit the same pattern of changes in CB1R density. Perhaps the ECS changes caused by CUS and OBX are an attempt to recover homeostasis and oppose the depressive symptoms. In any case, it is clear that ECS and serotonergic signaling are both altered significantly by chronic stress through mechanisms that are not currently clear.
9.8 Interactions Between ECS and NE Signaling: Mechanisms
There are two major sources of noradrenergic neurons in the brain: the locus coeruleus (LC) , which gives rise to the dorsal noradrenergic bundle and the lateral tegmentum, which gives rise to the ventral noradrenergic bundle (Dahlstrom and Fuxe 1964 ; Weinshenker and Schroeder 2007 ). Noradrenergic neurons from the LC project widely to all limbic and cortical regions, including the amygdala, hippocampus, neocortex, cingulate, and striatum (Berridge and Waterhouse 2003 ). The LC noradrenergic pathway is involved in the regulation of attention, arousal, cognitive processes, and sleep as a result of NE gating and tuning postsynaptic target neurons (Aston-Jones and Cohen 2005 ). LC neurons receive inputs from other neurotransmitter systems, including GABAergic, glutamatergic, and serotonergic (Berridge and Waterhouse 2003 ) and are also regulated by negative feedback through collateral noradrenergic terminals within the LC (Aghajanian et al. 1977 ). Dysregulation of the LC-NE system is thought to contribute to cognitive, emotional, and attentive dysfunctions that are associated with neuropsychiatric illnesses (Berridge and Waterhouse 2003 ). In light of data that Cannabis in humans alters attention and focus and other aspects of executive function (Pattij et al. 2008 ; Solowij et al. 1995 ; Viveros et al. 2005 ), a “noradrenergic hypothesis” has been put forth which posits that cannabinoids impair attention and cognition via modulation of central noradrenergic transmission (Carvalho and Van Bockstaele 2012 ). In support of this hypothesis, pretreatment of healthy humans with the beta-adrenergic antagonist, propranolol, prevented the acute effects of Cannabis to impair learning (Sulkowski et al. 1977 ).
CB1R protein and mRNA can be detected in the LC and NTS (Derbenev et al. 2004 ; Herkenham et al. 1991b ; Mailleux and Vanderhaeghen 1992 ; Matsuda et al. 1993 ). Analysis of the subcellular distribution of the CB1R protein in the LC reveals the presence of the receptors on both axon terminals and cell bodies, with a majority present in somato-dendritic profiles (Scavone et al. 2010 ). The presynaptic CB1R were more likely to be at symmetrical synapses, suggesting that ECS can inhibit GABA-mediated suppression of noradrenergic neurons. Indeed, Win 55212-2 suppresses the inhibition of LC firing induced by activation of the major GABAergic afferent to the LC (Muntoni et al. 2006 ). In further support of this hypothesis, systemic administration of CB1R agonists (Muntoni et al. 2006 ) and FAAH inhibitors (Gobbi et al. 2005 ) increase the firing rate of unstimulated noradrenergic neurons in the LC in a CB1R-dependent manner. CB1R agonists also increase c fos expression in the LC (Oropeza et al. 2005 ; Patel and Hillard 2003 ); enhance NMDA-induced firing of LC neurons (Mendiguren and Pineda 2004 ); and increase NE synthesis (Moranta et al. 2009 ) and release (Oropeza et al. 2005 ) in terminal regions. Taken together, these results are consistent with a mechanism by which activation of CB1R on GABA terminals in the LC inhibit GABA release and increase firing of noradrenergic neurons. However, neither local administration of CB1R agonists into the LC nor CB1R treatment of LC-containing brain slice preparations alter the spontaneous firing of LC neurons (Mendiguren and Pineda 2006 ). These findings point to an indirect effect of CB1R agonists on LC firing, perhaps through increased peripheral afferent activity into the LC. While further studies are required to elucidate the site of action, studies consistently demonstrate that CB1R agonists increase activity of noradrenergic, LC neurons.
Interestingly, more than 80 % of the CB1R immunoreactivity in the LC is post-synaptic, and this pool of CB1R is localized to the cytosol and not plasma membrane (Scavone et al. 2010 ). The authors of this study speculate that intracellular CB1R could function in an autocrine fashion to regulate LC activity (Carvalho et al. 2010a ). They argue that since the post-synaptic CB1R in the LC are in close proximity to asymmetrical synaptic inputs, it is possible that glutamate release could drive eCB synthesis post-synaptically resulting in activation of CB1R within the noradrenergic neuron in an autocrine manner.
Cannabinoid-mediated increases in LC noradrenergic neuron activity would be predicted to increase NE release in terminal fields; and multiple studies confirm this prediction. Systemic administration of CB1R agonists increase the release of NE in frontal cortex and nucleus accumbens (Jentsch et al. 1997 ; Oropeza et al. 2005 ). Local administration into the frontal cortex of a high concentration of the CB1R antagonist, rimonabant, blocked the effect of systemic CB1R agonist to increase NE release while having no effect alone (Page et al. 2008 ). Systemic administration of CB1R agonists increases the activity of the rate limiting enzyme in NE synthesis, tyrosine hydroxylase (TH), in several brain regions, including LC, hippocampus, and cortex (Moranta et al. 2004 ). Since NE synthesis is tightly coupled to release, these data support the hypothesis that CB1R activation increases NE release. In an excellent recent review of this topic, Kirilly et al. 2012 review other mechanisms that could also contribute to cannabinoid-mediated increases in LC neuronal discharge rate.
On the other hand, there is evidence that ECS suppresses NE release through direct effects on noradrenergic axon terminals. CB1R have been located on noradrenergic axon terminals in the frontal cortex (Oropeza et al. 2007 ). One-third of axon terminals positive for CB1R immunoreactivity were also positive for the NE synthesis enzyme, dopamine-ß-hydroxylase (DßH); other arrangements of CB1R and DßH were also seen that support the hypothesis that NE and eCBs can regulate each other’s function in the frontal cortex. In the nucleus accumbens , CB1R are present on a small fraction (less than 8 %) of axon terminals of noradrenergic neurons (identified by immunoreactivity against DßH and the noradrenergic reuptake transporter, NET) (Carvalho et al. 2010a ). The same study also found that approximately 6 % of noradrenergic axons were apposed to profiles that were immunoreactive for CB1R.
Studies have examined whether CB1R on noradrenergic terminals inhibit NE release. Incubation of synaptosomes with low concentrations of THC results in small but significant reductions in NE release (Poddar and Dewey 1980 ). Systemic administration of rimonabant (a CB1R antagonist) increases microdialysate NE concentrations in anterior hypothalamus and mPFC but not accumbens in freely moving, male Wistar rats (Tzavara et al. 2003 ; Tzavara et al. 2001). Similarly, rimonabant increases electrically-evoked release of NE from human and guinea pig hippocampal slices while agonists have the opposite effect, i.e., inhibit release (Schlicker et al. 1997 ). These data indicate that CB1R on presynaptic terminals can inhibit NE release and that they could be tonically active.
The identity of triggers of eCB mobilization that target CB1R of NE axon terminals is unknown. One possibility is that NE induces 2-AG synthesis in postsynaptic neurons via activation of alpha1 adrenergic receptors which couple to Gq heterotrimeric proteins (Insel and Hammond 1993 ) and, thereby, could trigger monoacylglycerol synthesis (Kano et al. 2009 ). In support of this mechanism, activation of alpha1 adrenergic receptors activates CB1R signaling in a cell-based system (Turu et al. 2009 ). Of course, eCB synthesis and release in these brain regions could also be triggered by the “usual suspects”, i.e., glutamate, membrane depolarization, and glucocorticoids .
Further support for the hypothesis that ECS modulates noradrenergic circuits in the brain comes from studies of chronic cannabinoid agonist treatment. Repeated dosing of male rats with Win 55212-2 increases TH protein expression in the LC accompanied by potentiated NE efflux in response to an acute injection of Win 55212-2 without a change in baseline NE efflux (Page et al. 2007 ). Chronic treatment with CB1R agonists reduces the binding site density of ß1 adreno receptors in neocortex (Hillard and Bloom 1982 ; Reyes et al. 2009 ), and both alpha2 and ß1 adrenoceptors in the accumbens (Carvalho et al. 2010a ). Chronic Win 55212-2 treatment completely abolishes the ability of clonidine to induce an increase in excitability of PFC neurons (Reyes et al. 2012 ). Taken together, these data indicate that sustained CB1R activation results in a sustained increase in NE release which induces down-regulation of adrenergic receptors . Tolerance, at the level of the CB1R, could ultimately develop to the effects of sustained CB1R agonists treatment to stimulate NE synthesis (Moranta et al. 2009 ); so this process is likely highly time and dose dependent.
In summary, convergent data indicate that CB1R activation increases activity in LC neurons, resulting in increased release NE in terminal regions. However, there is also evidence for other paradigms of interaction between ECS and noradrenergic signaling. In particular, CB1Rs in the terminal regions of LC projections can inhibit NE release, likely through presynaptic CB1R on axon terminals of noradrenergic neurons. Since systemic treatment of healthy animals with CB1R antagonists increases NE release, it is possible that ECS has greater ongoing tone at terminal, release-inhibitory CB1R. Data from studies using chronic administration of tricyclic antidepressants (TCAs) suggest that noradrenergic signaling can also regulate ECS, at least when noradrenergic signaling is sustained. There is a paucity of data exploring the effects of acute changes in NE concentrations on ECS in the brain.
9.9 Interactions Between ECS and NE Signaling: Stress Context
9.9.1 ECS Mediates Contributions of Noradrenergic Signaling to Stress Effects
Like the ECS, central noradrenergic signaling is significantly and rapidly increased in response to stress (Cassens et al. 1980 ). Although the ECS dampens stress responses through inhibition of HPA axis activity (described above), ECS also contributes in a positive manner to behavioral changes that result from stress and glucocorticoids . Intriguingly, there is accumulating evidence that the stress-promoting effects of ECS occur through increased noradrenergic signaling . Taken together, the studies described next support the hypothesis that stress and/or glucocorticoids recruit ECS which enhances noradrenergic signaling and contributes to stress responses.
High doses of CB1R agonists increase HPA axis responsivity to stress (Murphy et al. 1998 ; Patel et al. 2004 ) and pretreatment of rats with antagonists of either ß-adrenergic or alpha1-adrenergic receptors significantly attenuates this response (McLaughlin et al. 2009 ). While NE signaling is not the only process involved, these data are consistent with ECS acting up-stream of noradrenergic signaling to potentiate stress-induced HPA axis activation.
Cannabinoid agonists exert biphasic effects on anxiety, with low doses of CB1R agonists generally producing anxiolytic effects and high doses increasing anxiety (Patel and Hillard 2006 ; Patel and Hillard 2009b ). In addition, high doses of CB1R agonists induce conditioned place aversion in rodents (Mallet and Beninger 1998 ; McGregor et al. 1996 ; Pandolfo et al. 2009 ; Sanudo-Pena et al. 1997 ). A recent study demonstrated that a relatively high dose of Win 55212-2 (3 mg/kg) produced aversion in conditioned place preference that was abolished by toxin-induced depletion of NE in nucleus accumbens but not bed nucleus of the stria terminalis (BNST) (Carvalho et al. 2010b ). The role of NE was specific for aversive behaviors since toxin treatment did not alter the responses to the same dose of Win 55212-2 in assays of locomotor activity, spatial memory or elevated zero maze (Carvalho et al. 2010b ). Injection of a ß-adrenergic antagonist into the accumbens abolished Win 55212-2-induced conditioned place aversion (Carvalho and Van Bockstaele 2011 ), further support for noradrenergic signaling acting as a down-stream effector of exogenous CB1R activation to produce aversion. Since the NTS is a source of NE innervation of the accumbens (Delfs et al. 1998 ) and CB1R activate NTS noradrenergic neurons (Carvalho et al. 2010a ; Chen et al. 2010 ), increased NTS outflow could be the mechanism for this effect. Interestingly, Win 55212-2 place aversion does not occur in spontaneously hypertensive rats (SHR) (Pandolfo et al. 2009 ) which have reduced NTS CB1R binding site density and attenuated responsiveness to cannabinoid agonists injected into the NTS (Brozoski et al. 2009). Together, these studies suggest that NTS-accumbens projections are modulated by ECS in the NTS and contribute to the learning of aversion.
Evidence from both rats and mice indicate that increased noradrenergic signaling is required for stress-induced reinstatement to cocaine seeking (Erb et al. 2000 ; Leri et al. 2002 ; Mantsch et al. 2010 ). CB1R antagonism blocked stress-induced but not cocaine-induced reinstatement of cocaine place preference in mice (Vaughn et al. 2012 ). Furthermore, the combination of nonreinstating doses of a CB1R agonist and an alpha2 adrenergic antagonist produced significant reinstatement of cocaine place preference (Vaughn et al. 2012 ). These data suggest that ECS and NE are both required for stress-induced relapse to drug seeking and act synergistically to produce the behavioral effect.
It has been argued that stress-induced increases in ECS potentiate stress-induced memory consolidation through increased NE signaling (Campolongo et al. 2009 ). Although interactions between ECS and noradrenergic signaling in this context have not been demonstrated directly, ECS inhibits GABA signaling in the basolateral amygdala (BLA) during stress (Sumislawski et al. 2011 ) and inhibition of GABAergic activity in the BLA enhances memory consolidation by increasing NE signaling (Hatfield et al. 1999 ). These and other observations (Campolongo et al. 2009 ) are consistent with a model in which stress produces behavioral changes through recruitment of both ECS and NE signaling.
As was discussed above, CB1R agonists increase NE release in the PFC in unstressed rodents. However, Win 55212-2 suppresses stress-induced NE release and increases immobility and decreases climbing in the FST (Reyes et al. 2012 ). The reduction in immobility and reduced non-swimming active behaviors are consistent with decreased NE release in the PFC (Detke et al. 1995 ). Stress also reverses the effect of chronic Win 55212-2 to block clonidine-induced increases in PFC excitability (Reyes et al. 2012 ). These studies indicate that ECS regulation of NE signaling is significantly altered by stress and suggest the intriguing possibility that ECS is a pivot point with regard to the effects of stress on NE signaling.
9.9.2 Sustained CB1R Activation Alters Noradrenergic Signaling
As was described above, serotonergic signaling plays an important role in the regulation of active and passive coping behavioral paradigms employed by rodents in the FST. In a preceding section, data that CB1R agonists exert antidepressant-like effects in the FST through increased 5-HT signaling in rats were presented. Like 5-HT, elevations in NE also produce antidepressant effects in the FST. Acute treatment of rodents with antidepressants that inhibit NE uptake, such as desipramine, reduce immobility and promote the active coping behavior of struggling and climbing rather than swimming in the FST (Detke et al. 1995 ). Chronic treatment of rats with a synthetic CB1R agonist, HU210, reduces immobility and increases struggling without affecting swimming in the FST, consistent with increased NE signaling (Morrish et al. 2009 ). In support of this mechanism, the effect of chronic HU210 was attenuated by a ß-adrenergic receptor antagonist and, to a lesser extent, by an alpha-adrenergic receptor antagonist. In light of the data that chronic treatment with Win 55212-2 increases TH expression in the LC and increases CB1R-agonist induced NE release (Page et al. 2007 ), a possible mechanism is that repeated exposure to CB1R agonists enhances NE synthesis and neurotransmission as a result. An alternative mechanism is that chronic CB1R agonist treatment results in down-regulation of CB1R on NE terminals (Hillard and Bloom 1982 ; Reyes et al. 2009 ), resulting in dysinhibition of NE release. The second possibility is supported by data that acute treatment with a CB1R antagonist produces immobility in the FST through a mechanism that requires catecholamines (Haring et al. 2013 ). If a subset of tonically active CB1R inhibit NE release, acute CB1R antagonism could enhance NE release and thereby increase active coping in FST.
Moranta et al. 2009 investigated the role of enhanced monoaminergic signaling in the aversive effects of withdrawal from chronic cannabinoid agonist treatment. Antagonist-induced withdrawal in rats chronically treated with Win 55212-2 was accompanied by decreased TH activity, likely because of desensitization of alpha2 autoreceptors regulating the synthesis of NE in cortex, hippocampus, and cerebellum. Similar adaptations of alpha2 autoreceptors have been demonstrated in morphine- and ethanol-dependent rats (Esteban et al. 2002 ; Sastre-Coll et al. 2002 ) and could contribute to the somatic symptoms of withdrawal. Lesion data indicate that noradrenergic inputs to the BNST are not involved in these effects (Carvalho et al. 2010b ).
9.9.3 Sustained Changes in NE Signaling Alter ECS
Chronic treatment of rats with the tricyclic antidepressant (TCA) desipramine (Hill et al. 2006 ) but not fluoxetine (Hill et al. 2008b) produces significant increases in the density of CB1R in the hypothalamus and hippocampus. Desipramine is an antidepressant whose primary mechanism is inhibition of NE reuptake (Frazer 1997 ). Chronic desipramine treatment also results in reduced HPA axis activation by stress, evidenced by reductions in both FST-induced c fos expression in the PVN and circulating corticosterone (Hill et al. 2006 ). Acute treatment with rimonabant before FST completely abolished the effect of chronic desipramine treatment to suppress HPA axis activation. Since decreased ECS in the hypothalamus is associated with HPA axis hyperresponsiveness (Cota et al. 2007 ; Patel et al. 2004 ), the authors of this study hypothesized that desipramine-induced, increased CB1R expression in the hypothalamus has the opposite effect, i.e., decreases HPA axis responsivity (Hill et al. 2006 ). It is interesting that TCAs are more efficacious than SSRIs in reducing HPA axis reactivity in humans (Connor et al. 2000 ; Duncan et al. 1996 ). Perhaps this is because of the differential effects of these two drug classes on ECS in the hypothalamus. The mechanism by which desipramine increases CB1R expression is not known, but could be related to its effect to decrease alpha1 adrenoceptor expression (Subhash et al. 2003 ), which could result in decreased 2-AG synthesis and up-regulation of CB1R. The effect of desipramine on CB1R expression could oppose the effect of the TCA to increase NE concentrations in the synapse. Up-regulation of CB1R specifically on noradrenergic terminals could result in reduced NE release, which would normalize NE synaptic concentrations. However, this mechanism is speculative and needs to be tested experimentally.
9.9.4 Summary
The biphasic effects of exogenous CB1R agonists on anxiety, place preference and HPA axis activation by stress are well appreciated. High doses of exogenously administered CB1R agonists produce increased stress-induced activation of the HPA axis and induce conditioned place aversion rather than preference; these effects require intact NE signaling to occur. In addition, the effects of stress to induce cocaine reinstatement require both ECS and NE signaling. Recent data showing that CB1R agonist treatment exerts opposite effects on NE concentrations and signaling in the PFC in the absence and presence of stress indicates that ECS regulation of NE signaling is context dependent. Chronic treatment with CB1R agonists decreases expression and/or function of most adrenoceptors in the brain, likely as a result of sustained increases in NE concentrations. On the other hand, chronic treatment with the TCA desipramine causes increased CB1R density in hypothalamus and hippocampus but not in the PFC, changes that contribute to the decreased HPA axis reactivity following chronic desipramine treatment.
9.10 Interactions Between ECS and DA Signaling: Mechanisms
Dopaminergic cell bodies are present in the substantia nigra (SN) and ventral tegmental area (VTA) of the midbrain. Dopaminergic projections from the SN innervate the dorsal striatum via the nigrostriatal circuit while those from the VTA terminate in ventral striatum (nucleus accumbens) and PFC and form the mesocorticolimbic dopaminergic circuit. The amygdala also receives dopaminergic innervation from the ventral mesencephalon via a mesoamygdaloid projection (Fallon et al. 1978 ; Ungerstedt 1971 ). The tuberoinfundibular dopaminergic pathway projects from the hypothalamus to the pituitary and is relevant for the effects of DA ligands on endocrine function. Considerable experimental evidence demonstrates that ECS and dopaminergic signaling influence each other in a complex and bidirectional manner. I will focus here on the mechanisms of interaction in the mesocorticolimbic dopaminergic circuit; recent excellent reviews summarize the interactions of ECS and DA in the nigrostriatal circuit (El Khoury et al. 2012 ; Fitzgerald et al. 2012 ; Lovinger and Mathur 2012 ).
Dopaminergic afferents from the VTA play a central role in motivation, reward-related behaviors and cognition (Schultz 2002 ), thus the mesocorticolimbic DA circuit is often considered the “reward circuit”. The mesocorticolimbic dopaminergic circuit provides the brain with information about the emotional valence of all sensory stimulation, not just rewarding stimuli (Laviolette and Grace 2006 ). While the circuit underlies the rewarding effects of drugs, deficits are thought to underlie the compromised ability of those with schizophrenia or addictions to accurately assign emotional significance to sensory input. There are considerable data to support the contentions that cannabinoid-induced effects on the mesocorticolimbic circuit could contribute to several of the consequences of Cannabis intoxication in humans, including its rewarding effects (Danovitch and Gorelick 2012 ); impairment of working memory (Volk and Lewis 2010 ) and, at high doses, psychosis (Solowij and Michie 2007 ). In addition, considerable evidence demonstrates that loss of CB1R function reduces the rewarding effects of most abused substances and natural rewards (Serrano and Parsons 2011 ), suggesting that the regulation of the mesocorticolimbic circuit by ECS has broad implications for abuse and addiction . Indeed, recent experience with the use of the CB1R antagonist, rimonabant, in humans suggests that ECS is vital for the maintenance of hedonia, particularly in individuals prone to depression (Nathan et al. 2011 ).
Although the density is low relative to other brain regions, CB1R are present in the VTA on presynaptic terminals of both glutamatergic and GABAergic neurons (Matyas et al. 2008 ). Excitatory, afferent inputs to VTA dopaminergic neurons from the PFC and pontine tegmental nuclei and GABAergic projections from the accumbens have been shown to express CB1R (Marsicano and Lutz 1999 ; Matsuda et al. 1993 ). The axon terminals containing CB1R impinge on VTA neurons expressing TH, evidence that they are dopaminergic (Fitzgerald et al. 2012 ; Matyas et al. 2008 ). The 2-AG synthesizing enzyme, diacylglycerol lipase (DGL) alpha is detected in regions adjacent to the postsynaptic areas targeted by CB1R-containing axons, which provides further support for the hypothesis that ECS regulates synaptic activity at these synapses (Matyas et al. 2008 ). Although CB1R expression in TH positive neurons in the VTA has been detected using light microscopy (Wenger et al. 2003 ), electron microscopy studies indicate that very few TH-positive neurons coexpress CB1R (Fitzgerald et al. 2012 ).
Electrophysiological studies of the synapse between GABAergic interneurons and presumptive dopaminergic neurons in VTA brain slices demonstrate that CB1R agonists inhibit GABA release at a subset of synapses (Riegel and Lupica 2004 ; Szabo et al. 2002 ). CB1R-mediated inhibition of excitatory inputs to VTA dopaminergic neurons has also been described (Melis et al. 2004b ; Riegel and Lupica 2004 ). Increased postsynaptic activity induces ECS at both GABAergic and glutamatergic synapses onto DA neurons in the VTA (Riegel and Lupica 2004 ). ECS in the VTA requires both depolarization and mGluR1 activation and is blocked by inhibitors of DGL and PLC activity, indicating that 2-AG is the likely eCB involved (Melis et al. 2004a ). Morphological data that DGL alpha is in close proximity to CB1R axon terminals (Matyas et al. 2008 ) support this hypothesis. In vivo evidence that stimulation of the PFC recruits ECS in the VTA (Melis et al. 2004a ) is consistent with the hypothesis that ECS in the VTA is triggered by glutamate. Thus, the role of ECS in tuning VTA output likely depends on context: under control conditions, ECS could maintain VTA output through inhibition of GABA release while during times of stress or high emotional tone when the PFC is active, ECS reduces the responsiveness of the VTA through inhibition of glutamate release.
CB1R-dependent DSE is significantly reduced by the D2 dopamine receptor (D2R) antagonist, eticlopride, suggesting that this form of eCB-mediated plasticity is augmented by depolarization-induced release of DA (Melis et al. 2004b ). In addition, chronic cocaine treatment recruits CB1R-mediated long-term depression of inhibition (I-LTD) in the VTA through a mechanism that requires group 1 mGluR and D2R (Pan et al. 2008a, b ). The combination of increased D2R and CB1R activation produces a long-term suppression of GABA release through inhibition of protein kinase A (Pan et al. 2008a ). This effect of chronic cocaine is particularly interesting as some hypothesize that overactive reward circuits could underlie addictive behaviors following chronic drug exposure (Vanderschuren and Kalivas 2000 ).
Exogenous administration of CB1R agonists produces intense burst firing of VTA dopaminergic neurons (Diana et al. 1998 ; French 1997 ; French et al. 1997; Gessa et al. 1998 ; Wu and French 2000 ) and a concomitant increase in DA release in the accumbens (Cheer et al. 2004 ; Chen et al. 1990b; Tanda et al. 1997 ) and the PFC (Chen et al. 1990a ). These pharmacological effects are consistent with cannabinoid-mediated dysinhibiton of GABAergic interneurons within the VTA. Support for this mechanism comes from a recent study of the roles of CB1R on GABA and glutamatergic terminals in the VTA on voluntary wheel running (Dubreucq et al. 2012 ), a rewarding activity in mice that alters neuronal activity of dopaminergic neurons in the VTA (Novak et al. 2012 ). Conditional deletion of CB1R selectively from GABAergic neurons in brain decreases wheel running performance in mice, which is consistent with tonic and permissive effect of CB1R activation to increase DA release in the striatum which promotes reward and/or causes increased locomotor activity (Dubreucq et al. 2012 ).
It is possible that exogenous cannabinoids act in extra-VTA regions of the circuit to increase VTA-ventral striatum outflow. For example, a c fos study from our laboratory suggests that activation of excitatory, noradrenergic afferents from the LC contributes to the effects of exogenous CB1R agonists to activate VTA (Patel and Hillard 2003 ). As is discussed further below, CB1R activation in the PFC also increases the firing rate of PFC neurons projecting to the VTA (Pistis et al. 2001 ), providing an additional, indirect mechanism for increased VTA firing.
In sum, available evidence indicates ECS can significantly alter the discharge pattern of dopaminergic neurons in the VTA. It has been hypothesized that ECS in the VTA, through regulation of glutamate and GABA release, could fine-tune phasic versus tonic DA release in the mesocorticolimbic circuit (Melis et al. 2004a ). As a result, ECS functions as a modulator of the mesocorticolimbic circuit.
A primary projection of VTA dopaminergic neurons is the shell of the nucleus accumbens (NAc) . This region of the accumbens also receives glutamatergic input from the cortex which converges on many of the same medium spiny neurons as the dopaminergic projections from the VTA (Sesack and Pickel 1990 ). Glutamatergic and dopaminergic inputs to the accumbens interact in a bidirectional manner and together regulate activity of the medium spiny neurons.
CB1R are abundantly expressed in the accumbens (Herkenham 1992 ) on both GABAergic terminals that are likely collaterals of medium spiny neurons and on glutamatergic afferents from cortex (Pickel et al. 2004 ; Pickel et al. 2006). CB1R are not present on dopaminergic projections (Fitzgerald et al. 2012 ; Julian et al. 2003 ). As expected from their localization, CB1R activation suppresses evoked release of GABA (Centonze et al. 2007 ; Hoffman and Lupica 2001 ) and glutamate (Pistis et al. 2002b ; Robbe et al. 2001 ) in the accumbens. Stimulation of prelimbic cortical afferents to the accumbens using naturally occurring frequencies induces CB1R-dependent long-term depression (LTD) of excitatory synaptic transmission (Robbe et al. 2002 ) and LTD is absent in rats chronically treated with THC (Hoffman et al. 2003 ). OBX reduces glutamatergic inputs to the ventral striatum and also reduces ECS in this brain region—an effect that could contribute to the reduced responsivity to novel stimuli observed in animals with OBX (Eisenstein et al. 2010). In contrast to the dorsal striatum, D2R activation is not required for CB1R-mediated LTD in the amygdala (Robbe et al. 2002 ). Thus, current evidence indicates that the role of presynaptic ECS in the accumbens is to regulate glutamatergic input, which likely also has an indirect effect on the dopaminergic input to medium spiny neurons.
CB1R are also expressed by striatal neurons (Ferre et al. 2009 ; Kearn et al. 2005 ; Kofalvi et al. 2005; Pickel et al. 2006 ), suggesting that CB1R have postsynaptic effects in this brain region. Interestingly, CB1R can be colocalized with D2R in striatal neurons (Pickel et al. 2006 ) and recent electrophysiological data suggests that postsynaptic CB1R could enhance DR function, particularly when dopamine concentrations are low (Seif et al. 2011 ). In addition, there is evidence that D2R and CB1R can form heterodimers (Kearn et al. 2005 ) and that dual activation of D2R and CB1R results in a stimulatory effect on cAMP, although both CB1R and D2R signal individually through Gi/o family G proteins (Glass and Felder 1997 ; Jarrahian et al. 2004 ; Kearn et al. 2005 ). Thus, ECS and dopaminergic signaling have the ability to influence each other at the level of the receptor.
The mesocortical DA system modulates activity of pyramidal neurons of the PFC. Early studies found a predominant effect of DA to inhibit pyramidal neuron firing through enhanced release of GABA from inhibitory interneurons (Gellman and Aghajanian 1993 ; Pistis et al. 2001 ) likely through D1R activation (Arnsten 2011 ). Since CB1R are present on GABAergic terminals in the PFC and inhibit GABA release (Hill et al. 2011b ), the ECS is in a position to oppose the inhibitory effect of dopamine in this circuit. Indeed, systemic CB1R agonist treatment reverses the inhibitory effects of VTA activation on PFC activity in rodents (Pistis et al. 2001 ), however, whether this occurs through CB1R activation in the PFC or in the VTA is not clear.
Synergistic effects between ECS and DA have also been demonstrated in PFC. In particular, CB1R and D2R are colocalized on GABAergic terminals and simultaneous activation of both triggers CB1R-mediated LTD of GABA release (I-LTD) (Chiu et al. 2010 ). Further evidence in this study argues against D2R-mediated mobilization of eCBs, rather supports synergism between signaling cascades activated by CB1R and D2R. As was discussed above for the synergistic effects of DA and ECS in the VTA (Pan et al. 2008a ), it is possible that CB1R and D2R activation in the PFC suppresses PKA below a threshold that allows for induction of I-LTD.
Biochemical data suggest bidirectional interactions between ECS and dopaminergic signaling at the level of ligand availability; however, none of the available data are specific to the mesocorticolimbic DA system. For example, acute treatment of mice with systemic methylphenidate, GBR 12909 and D1R agonist individually reduce AEA content in the forebrain (Patel et al. 2003 ). Similarly, mice that do not express the dopamine transporter (DAT) have significantly lower striatal AEA concentrations (Tzavara et al. 2006 ). On the other hand, forebrain and striatal AEA concentrations are increased by D2R activation (Centonze et al. 2004 ; Giuffrida et al. 1999 ; Patel et al. 2003 ). CB1R activation increases DA release (Pistis et al. 2002a ; Tanda et al. 1997 ) and genetic deletion of the CB1R results in D2R overexpression, which is consistent with decreased DA tone in the absence of ECS (Houchi et al. 2005 ).
In summary, the interactions between dopaminergic and CB1R signaling are complex and occur in the VTA, accumbens and PFC. There is little evidence in rodent models that ECS regulates the release of DA, although the situation could be different in humans since CB1R are present on DA terminals in human neocortex and inhibit DA release from human synaptosomes (Steffens et al. 2004 ). There is evidence from morphological and electrophysiological studies that ECS regulation of glutamate release indirectly alters responses to DA in the mesocorticolimbic circuit. Intriguingly, there is also evidence that postsynaptic CB1R are important in this circuit and participate in a bidirectional modulation of postreceptor signaling with D2R. These interactions clearly contribute to the role of ECS in regulation of reward and could also underlie the propsychotic effects of Cannabis use in humans.
9.11 Interactions Between ECS and DA Signaling: Stress Context
The VTA is activated by stress and DA concentrations are increased in the mPFC, accumbens and dorsal striatum in response to restraint stress (Deutch et al. 1991 ). Considerable preclinical and clinical data demonstrate that DA, through both D1R and D2R receptors, is one of the most important neuromodulators of fear and anxiety (LeDoux 2000 ). ECS is also activated by stress, and CB1R signaling regulates DA neuronal activation in the mesencephalon; release of DA in projection sites; and can affect dopamine receptor signaling (previous section). Therefore, it seems quite probable that ECS modulates and/or contributes to the dopaminergic role in stress responding. However, there is not much experimental evidence that tests this proposition directly.
9.11.1 Dysregulation of Stress Reactivity by Chronic Exposure to Drugs of Abuse
Dysregulation of stress responses is a major, long-term adverse consequence of drug addiction and it plays a primary role in relapse to drug use following abstinence (Sidhpura and Parsons 2011 ). As is outlined wonderfully in a review by Sidhpura and Parsons (2011 ), the characteristics of ECS, particularly its role in both reward and stress responses, support the hypothesis that chronic drug use causes dysregulated stress responsivity as a result of alterations in ECS-mediated synaptic plasticity. As was described above, ECS is activated by glucocorticoids and long-term drug use results in sensitization of responses to HPA axis activation (Koob and Kreek 2007 ), which suggests the hypothesis that chronic drug exposure could alter glucocorticoid-ECS signaling . To my knowledge, this hypothesis has not been tested.
9.11.2 Anxiety
While systemic administration of CB1R agonists produces a biphasic effect on behavior in the elevated plus maze (Patel and Hillard 2006 ), activation of ECS through administration of indirect agonists primarily exerts anxiolytic effects in this assay particularly when stress is high (Patel and Hillard 2006 ; Sciolino et al. 2011 ). Similarly, blockade of ECS with CB1R antagonists results in anxiogenic like effects (Patel and Hillard 2006 ), implicating ECS as a feedback system that limits the expression of anxiety under stressful circumstances. Acute exposure to psychostimulants produces increased anxiety (Lodge and Grace 2011 ), an effect that is reversed by prior CB1R activation. In particular, exposure of male mice to a single, high dose of psychostimulant results in an anxiogenic phenotype in the elevated plus maze that lasts for at least 5 days (Hayase et al. 2005 ). Treatment of the mice 1 h prior with several CB1R agonists reverses the anxiogenic effects, suggesting that suppression of ECS could contribute to the anxiogenic effects of the psychostimulants. Previous studies have demonstrated that methylphenidate in particular decreases forebrain tissue contents of both AEA and 2-AG (Patel et al. 2003 ), it is possible that a long-lasting suppression of ECS tone contributes to the sustained anxiogenic effects of psychostimulant exposure. On the other hand, the ability of amphetamine to induce long-term depression in the lateral amygdala is dependent upon CB1R and not DA receptors (Huang et al. 2003 ). Thus, prior exposure to CB1R agonists could abrogate the effect of the psychostimulant to induce plasticity in the amygdala.
There is substantial evidence that exposure to most drugs of abuse affects synaptic plasticity in many brain regions and that in many cases, these changes are subsequent to disruption of ECS-mediated plasticity, particularly at glutamatergic synapses (Wolf et al. 2004 ). The bed nucleus of the stria terminalis (BNST) is a region of the extended amygdala that is stress-responsive and a key relay between limbic cognitive centers and reward, stress , and anxiety nuclei (McElligott and Winder 2009 ). ECS-mediated LTD occurs in the BNST and is reduced following multiple exposures of mice to cocaine (Grueter et al. 2006). Since the BNST projects to the VTA (Dong and Swanson 2006), changes in synaptic plasticity in the BNST will alter the mesocorticolimbic circuit and alter motivated behavior (Sidhpura and Parsons 2011 ).
On the other hand, there is considerable evidence that hyperactive ECS could contribute to the anxiogenic effects of withdrawal from addictive drugs. Withdrawal from chronic exposure to drugs and alcohol induces anxiety that is mediated by increases in amygdalar CRF concentrations and can be blocked by antagonism of CRF receptors in the central nucleus of the amygdala (Koob and Le Moal 2008 ). The anxiogenic effects of alcohol withdrawal are reduced in CB1R null mice (Racz et al. 2003 ) and are inhibited by CB1R antagonists (Rubio et al. 2008 ) as are the anxiogenic effects of withdrawal from diazepam and cocaine (Kupferschmidt et al. 2012 ; Onaivi 2008 ). These results suggest that, as a stressor, withdrawal has increased ECS and that this contributes to the anxiogenic effects. There is a high degree of co-localization between mRNA for the CB1R and message for both CRF and CRFR1 in stress-regulated brain regions (Hermann and Lutz 2005 ) and recent data demonstrate that the effects of i.c.v. CRF to induce anxiogenic responses in the elevated plus maze are blocked by CB1R antagonism (Kupferschmidt et al. 2012 ). CRF1 receptors are expressed in both glutamatergic projection neurons and VTA dopaminergic neurons and exert a bi-directional influence on anxiety behaviors (Refojo et al. 2011). ECS modulates these neurons as well, suggesting that they could be the substrate for interactions between CRF and ECS signaling. Withdrawal from alcohol in rats is associated with decreased VTA DA concentrations, which is reversed by rimonabant (Rubio et al. 2008 ).
9.11.3 Emotional Learning
An important role for the mesolimbic dopamine circuit is to associate emotional significance to sensory information, called “emotional learning” (Laviolette and Grace 2006 ). Most animal studies of emotional learning utilize fear conditioning approaches in which a neutral stimulus is paired with a painful stimulus, usually a mild foot shock. Emotional learning is measured as the ability of either the conditioned stimulus (cue) or placing the animal back into the training environment (context) to provoke a fear response (usually freezing). Emotional learning is dependent upon the DA activity in mesocorticolimbic circuit and an intact amygdala. A recent study indicates that ECS in D1R expressing neurons is involved in emotional learning (Terzian et al. 2011 ). In this study, mice with a selective deletion of the CB1R in neurons expressing the D1R were exposed to foot shock with an auditory conditioned stimulus. Both cue and context-induced freezing were significantly increased on the first day after conditioning in the knock out compared to wild type mice, suggesting that ECS opposes the effect of D1R activation to evoke emotional learning. Earlier studies found that CB1R agonists also functionally oppose the effects of D1R agonists on motor behaviors (Martin et al. 2008 ), leading to the hypothesis that CB1R coexpressed in D1R containing neurons opposes the effects of D1R activation. Infusion of AEA into the accumbens before conditioning decreased freezing behavior to the context 24 h later (Pedroza-Llinas et al. 2013 ). Taken together, these data suggest that ECS in the accumbens functions to dampen the magnitude of emotional learning plasticity perhaps through activation of signaling cascades that oppose those of the D1R.
9.11.4 Chronic Stress and Striatal EC
Exposure to inescapable stress alters synaptic plasticity in many brain regions. In particular, social defeat stress suppresses CB1R-mediated inhibition of GABA release in the striatum (Rossi et al. 2008 ). It is likely that the mechanism involves decreased CB1R expression or function since exogenous agonists were ineffective at inhibition of GABA release as well. CB1R regulation of glutamate release was unaffected in the striatum. Importantly, the effect of chronic stress to suppress CB1R signaling was reversed when stressed mice were given access to running wheels, sucrose, or cocaine (Rossi et al. 2008 ). These data suggest that reduced ECS is the result of the anhedonic effects of stress and that reduced ECS contributes to the reward deficit of chronic stress. These data are consistent with biochemical data that dopamine increases striatal CB1R expression (Centonze et al. 2004 ) and that chronic treatment of unstressed rats with the MAO inhibitor, tranylcypromine increases CB1R density in PFC and hippocampus (Hill et al. 2008b ). Taken together, these data indicate a positive relationship between DA signaling and CB1R density. Recent data DA-regulation of brain-derived neurotrophic factor (BDNF) could alter CB1R expression through a mechanism that involves changes in cholesterol metabolism that alter CB1R distribution to lipid rafts (De Chiara et al. 2010 ).
9.11.5 Summary
The interactions between ECS and dopaminergic systems are complex, bidirectional, and affected by stress. With regard to regulation of anxiety, both the anxiogenic and anxiolytic effects of CB1R activation come into play in the dopaminergic circuit. While activation of CB1R opposes the anxiogenic effects of acute psychostimulant exposure, blockade of CB1R ameliorates the anxiogenic effects of withdrawal. Since DA signaling is also highly dysregulated during withdrawal, it is possible that the state of DA signaling is an important determinant of ECS. Around 10 years ago, Sachin Patel and I suggested, based on the dose-response relationship between CB1R agonist and c fos expression in the VTA, that effects of cannabinoids to activate specific subregions of the VTA could contribute to both the reward-promoting and stress-promoting effects of CB1R agonists (Patel and Hillard 2003 ). Activation of dopaminergic neurons within the parabrachial pigmented and paranigral regions of the VTA exhibits a steep and bell-shaped dose response relationship that we hypothesized matched well the rewarding effects of cannabinoids while activation in the caudal linear region exhibited a more classical relationship with agonist dose and could subserve the aversive and anxiogenic effects of CB1R activation. Until better tools are developed or applied to study subregional roles of ECS, it will be impossible to completely understand the interactions of these two very important systems.
9.12 Concluding Remarks
A recent genetic association study provides strong evidence that CB1R function plays an important role in stress adaptation in humans (Agrawal et al. 2012 ). This study examined a synonymous polymorphism in exon 4 of the gene encoding the CB1R in two different cohorts of individuals exposed to childhood physical abuse. In both samples, individuals reporting physical abuse in childhood were significantly more likely to report anhedonia. However, in both cohorts, those with one or more copies of the minor allele at rs1049353 were nearly 50 % less likely to have anhedonia as an adult. Thus, this study suggests that CB1R genotype can buffer the effects of childhood physical abuse on major depression . This study, along with other accumulating data that differences in genes associated with ECS influence the likelihood for substance use and mood disorders , suggest that ECS function contributes to human psychiatric illness (Hillard et al. 2012). Thus, enhanced understanding of the many roles of ECS in the regulation of mood, emotion, and reward is an important goal.
The studies described in this review indicate that we are just beginning to understand the intersection of ECS with monoaminergic systems that are also vital for mood, reward, and processing of emotions. However, there is much work to be done. Although we are fortunate to have excellent morphological maps of the subregional and sometimes even subcellular distribution of the CB1R, our understanding of the roles of individual pools of CB1R in the responses to systemic agonist and antagonist treatment is primitive in most cases, which, along with the complexity of the monoamine systems, results in a clouded picture at this stage of our knowledge. However, the development of selective knock out mouse models, and increased use of local injections along with the promise of other more precise techniques, such as optogenetics and live brain imaging, will ultimately improve our focus and allow for the development and use of ECS-targeted therapeutic approaches in a rationale manner.
References
Aghajanian GK, Cedarbaum JM, Wang RY (1977) Evidence for norepinephrine-mediated collateral inhibition of locus coeruleus neurons. Brain Res 136:570–577
Agrawal A, Nelson EC, Littlefield AK, Bucholz KK, Degenhardt L, Henders AK, Madden PA, Martin NG, Montgomery GW, Pergadia ML, Sher KJ, Heath AC, Lynskey MT (2012) Cannabinoid receptor genotype moderation of the effects of childhood physical abuse on anhedonia and depression. Arch Gen Psychiatry 69:732–740
Arnsten AF (2011) Catecholamine influences on dorsolateral prefrontal cortical networks. Biol Psychiatry 69:e89–99
Aso E, Renoir T, Mengod G, Ledent C, Hamon M, Maldonado R, Lanfumey L, Valverde O (2009) Lack of CB(1) receptor activity impairs serotonergic negative feedback. J Neurochem 109:935–944
Aston-Jones G, Cohen JD (2005) An integrative theory of locus coeruleus-norepinephrine function: adaptive gain and optimal performance. Annu Rev Neurosci 28:403–450
Bambico FR, Katz N, Debonnel G, Gobbi G (2007) Cannabinoids elicit antidepressant-like behavior and activate serotonergic neurons through the medial prefrontal cortex. J Neurosci 27:11700–11711
Bambico FR, Hattan PR, Garant JP, Gobbi G (2012) Effect of delta-9-tetrahydrocannabinol on behavioral despair and on pre- and postsynaptic serotonergic transmission. Prog Neuropsychopharmacol Biol Psychiatry 38:88–96
Bambico FR, Cassano T, Dominguez-Lopez S, Katz N, Walker CD, Piomelli D, Gobbi G (2010) Genetic deletion of fatty acid amide hydrolase alters emotional behavior and serotonergic transmission in the dorsal raphe, prefrontal cortex, and hippocampus. Neuropsychopharmacology 35:2083–2100
Barbon A, Orlandi C, La Via L, Caracciolo L, Tardito D, Musazzi L, Mallei A, Gennarelli M, Racagni G, Popoli M, Barlati S (2011) Antidepressant treatments change 5-HT2C receptor mRNA expression in rat prefrontal/frontal cortex and hippocampus. NeuropsychoBiology 63:160–168
Barnes NM, Sharp T (1999) A review of central 5-HT receptors and their function. Neuropharmacology 38:1083–1152
Beltramo M, Stella N, Calignano A, Lin SY, Makriyannis A, Piomelli D (1997) Functional role of high-affinity anandamide transport, as revealed by selective inhibition. Science 277:1094–1097
Bergamaschi MM, Queiroz RH, Chagas MH, Oliveira DC de, De Martinis BS, Kapczinski F, Quevedo J, Roesler R, Schroder N, Nardi AE, Martin-Santos R, Hallak JE, Zuardi AW, Crippa JA (2011) Cannabidiol reduces the anxiety induced by simulated public speaking in treatment-naive social phobia patients. Neuropsychopharmacology 36:1219–1226
Berridge CW, Waterhouse BD (2003) The locus coeruleus-noradrenergic system: modulation of behavioral state and state-dependent cognitive processes. Brain Res Brain Res Rev 42:33–84
Best AR, Regehr WG (2008) Serotonin evokes endocannabinoid release and retrogradely suppresses excitatory synapses. J Neurosci 28:6508–6515
Bland ST, Hargrave D, Pepin JL, Amat J, Watkins LR, Maier SF (2003) Stressor controllability modulates stress-induced dopamine and serotonin efflux and morphine-induced serotonin efflux in the medial prefrontal cortex. Neuropsychopharmacology 28:1589–1596
Braida D, Limonta V, Malabarba L, Zani A, Sala M (2007) 5-HT1A receptors are involved in the anxiolytic effect of Delta9-tetrahydrocannabinol and AM 404, the anandamide transport inhibitor, in Sprague-Dawley rats. Eur J Pharmacol 555:156–163
Brozoski DT, Dean C, Hopp FA, Hillard CJ, Seagard JL (2009) Differential endocannabinoid regulation of baroreflex-evoked sympathoinhibition in normotensive versus hypertensive rats. Auton Neurosci 150(1–2):82–93
Campolongo P, Roozendaal B, Trezza V, Hauer D, Schelling G, McGaugh JL, Cuomo V (2009) Endocannabinoids in the rat basolateral amygdala enhance memory consolidation and enable glucocorticoid modulation of memory. Proc Natl Acad Sci U S A 106:4888–4893
Carvalho AF, Van Bockstaele EJ (2011) Direct intra-accumbal infusion of a beta-adrenergic receptor antagonist abolishes WIN 55,212-2-induced aversion. Neurosci Lett 500:82–85
Carvalho AF, Van Bockstaele EJ (2012) Cannabinoid modulation of noradrenergic circuits: implications for psychiatric disorders. Prog Neuropsychopharmacol Biol Psychiatry 38:59–67
Carvalho AF, Mackie K, Van Bockstaele EJ (2010a) Cannabinoid modulation of limbic forebrain noradrenergic circuitry. Eur J Neurosci 31:286–301
Carvalho AF, Reyes AR, Sterling RC, Unterwald E, Van Bockstaele EJ (2010b) Contribution of limbic norepinephrine to cannabinoid-induced aversion. Psychopharmacology (Berl) 211:479–491
Cassano T, Gaetani S, Macheda T, Laconca L, Romano A, Morgese MG, Cimmino CS, Chiarotti F, Bambico FR, Gobbi G, Cuomo V, Piomelli D (2011) Evaluation of the emotional phenotype and serotonergic neurotransmission of fatty acid amide hydrolase-deficient mice. Psychopharmacology (Berl) 214:465–476
Cassens G, Roffman M, Kuruc A, Orsulak PJ, Schildkraut JJ (1980) Alterations in brain norepinephrine metabolism induced by environmental stimuli previously paired with inescapable shock. Science 209:1138–1140
Celada P, Puig MV, Casanovas JM, Guillazo G, Artigas F (2001) Control of dorsal raphe serotonergic neurons by the medial prefrontal cortex: involvement of serotonin-1A, GABA(A), and glutamate receptors. J Neurosci 21:9917–9929
Centonze D, Battista N, Rossi S, Mercuri NB, Finazzi-Agro A, Bernardi G, Calabresi P, Maccarrone M (2004) A critical interaction between dopamine D2 receptors and endocannabinoids mediates the effects of cocaine on striatal gabaergic transmission. Neuropsychopharmacology 29:1488–1497
Centonze D, Rossi S, De Chiara V, Prosperetti C, Battista N, Bernardi G, Mercuri NB, Usiello A, Maccarrone M (2007) Chronic cocaine sensitizes striatal GABAergic synapses to the stimulation of cannabinoid CB1 receptors. Eur J Neurosci 25:1631–1640
Cheer JF, Wassum KM, Heien ML, Phillips PE, Wightman RM (2004) Cannabinoids enhance subsecond dopamine release in the nucleus accumbens of awake rats. J Neurosci 24:4393–4400
Chen J, Paredes W, Lowinson JH, Gardner EL (1990a) Delta 9-tetrahydrocannabinol enhances presynaptic dopamine efflux in medial prefrontal cortex. Eur J Pharmacol 190:259–262
Chen JP, Paredes W, Li J, Smith D, Lowinson J, Gardner EL (1990b) Delta 9-tetrahydrocannabinol produces naloxone-blockable enhancement of presynaptic basal dopamine efflux in nucleus accumbens of conscious, freely-moving rats as measured by intracerebral microdialysis. Psychopharmacology (Berl) 102:156–162
Chen CY, Bonham AC, Dean C, Hopp FA, Hillard CJ, Seagard JL (2010) Retrograde release of endocannabinoids inhibits presynaptic GABA release to second-order baroreceptive neurons in NTS. Auton Neurosci 158:44–50
Chiu CQ, Puente N, Grandes P, Castillo PE (2010) Dopaminergic modulation of endocannabinoid-mediated plasticity at GABAergic synapses in the prefrontal cortex. J Neurosci 30:7236–7248
Connor TJ, Kelliher P, Shen Y, Harkin A, Kelly JP, Leonard BE (2000) Effect of subchronic antidepressant treatments on behavioral, neurochemical, and endocrine changes in the forced-swim test. Pharmacol Biochem Behav 65:591–597
Cota D, Steiner MA, Marsicano G, Cervino C, Herman JP, Grubler Y, Stalla J, Pasquali R, Lutz B, Stalla GK, Pagotto U (2007) Requirement of cannabinoid receptor type 1 for the basal modulation of hypothalamic-pituitary-adrenal axis function. Endocrinology 148:1574–1581
Cravatt BF, Giang DK, Mayfield SP, Boger DL, Lerner RA, Gilula NB (1996) Molecular characterization of an enzyme that degrades neuromodulatory fatty-acid amides. Nature 384:83–87
Cryan JF, Valentino RJ, Lucki I (2005) Assessing substrates underlying the behavioral effects of antidepressants using the modified rat forced swimming test. Neurosci Biobehav Rev 29:547–569
Dahlstrom A, Fuxe K (1964) Localization of monoamines in the lower brain stem. Experientia 20:398–399
Danovitch I, Gorelick DA (2012) State of the art treatments for cannabis dependence. Psychiatr Clin North Am 35:309–326
Darmani NA, Janoyan JJ, Kumar N, Crim JL (2003) Behaviorally active doses of the CB1 receptor antagonist SR 141716A increase brain serotonin and dopamine levels and turnover. Pharmacol Biochem Behav 75:777–787
De Chiara V, Angelucci F, Rossi S, Musella A, Cavasinni F, Cantarella C, Mataluni G, Sacchetti L, Napolitano F, Castelli M, Caltagirone C, Bernardi G, Maccarrone M, Usiello A, Centonze D (2010) Brain-derived neurotrophic factor controls cannabinoid CB1 receptor function in the striatum. J Neurosci 30:8127–8137
Dean C (2011) Endocannabinoid modulation of sympathetic and cardiovascular responses to acute stress in the periaqueductal gray of the rat. Am J Physiol Regul Integr Comp Physiol 300:R771–779
Delfs JM, Zhu Y, Druhan JP, Aston-Jones GS (1998) Origin of noradrenergic afferents to the shell subregion of the nucleus accumbens: anterograde and retrograde tract-tracing studies in the rat. Brain Res 806:127–140
Derbenev AV, Stuart TC, Smith BN (2004) Cannabinoids suppress synaptic input to neurones of the rat dorsal motor nucleus of the vagus nerve. J Physiol 559:923–938
Detke MJ, Rickels M, Lucki I (1995) Active behaviors in the rat forced swimming test differentially produced by serotonergic and noradrenergic antidepressants. Psychopharmacology 121:66–72
Deutch AY, Lee MC, Gillham MH, Cameron DA, Goldstein M, Iadarola MJ (1991) Stress selectively increases fos protein in dopamine neurons innervating the prefrontal cortex. Cereb Cortex 1:273–292
Devane WA, Hanus L, Breuer A, Pertwee RG, Stevenson LA, Griffin G, Gibson D, Mandelbaum A, Etinger A, Mechoulam R (1992) Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science 258:1946–1949
Devlin MG, Christopoulos A (2002) Modulation of cannabinoid agonist binding by 5-HT in the rat cerebellum. J Neurochem 80:1095–1102
Di S, Malcher-Lopes R, Halmos KC, Tasker JG (2003) Nongenomic glucocorticoid inhibition via endocannabinoid release in the hypothalamus: a fast feedback mechanism. J Neurosci 23:4850–4857
Di S, Malcher-Lopes R, Marcheselli VL, Bazan NG, Tasker JG (2005) Rapid glucocorticoid-mediated endocannabinoid release and opposing regulation of glutamate and GABA inputs to hypothalamic magnocellular neurons. Endocrinology 146:4292–4301
Diana M, Melis M, Gessa GL (1998) Increase in meso-prefrontal dopaminergic activity after stimulation of CB1 receptors by cannabinoids. Eur J Neurosci 10:2825–2830
Diorio D, Viau V, Meaney MJ (1993) The role of the medial prefrontal cortex (cingulate gyrus) in the regulation of hypothalamic-pituitary-adrenal responses to stress. J Neurosci 13:3839–3847
Dong HW, Swanson LW (2006) Projections from bed nuclei of the stria terminalis, dorsomedial nucleus: implications for cerebral hemisphere integration of neuroendocrine, autonomic, and drinking responses. J Comp Neurol 494:75–107
Dubreucq S, Durand A, Matias I, Benard G, Richard E, Soria-Gomez E, Glangetas C, Groc L, Wadleigh A, Massa F, Bartsch D, Marsicano G, Georges F, Chaouloff F (2012) Ventral tegmental area cannabinoid type-1 receptors control voluntary exercise performance. Biol Psychiatry 73(9):895–903
Duncan GE, Knapp DJ, Johnson KB, Breese GR (1996) Functional classification of antidepressants based on antagonism of swim stress-induced fos-like immunoreactivity. J Pharmacol Exp Ther 277:1076–1089
Egashira N, Mishima K, Katsurabayashi S, Yoshitake T, Matsumoto Y, Ishida J, Yamaguchi M, Iwasaki K, Fujiwara M (2002) Involvement of 5-hydroxytryptamine neuronal system in Delta(9)-tetrahydrocannabinol-induced impairment of spatial memory. Eur J Pharmacol 445:221–229
Egashira N, Matsuda T, Koushi E, Higashihara F, Mishima K, Chidori S, Hasebe N, Iwasaki K, Nishimura R, Oishi R, Fujiwara M (2008) Delta(9)-tetrahydrocannabinol prolongs the immobility time in the mouse forced swim test: involvement of cannabinoid CB(1) receptor and serotonergic system. Eur J Pharmacol 589:117–121
Eisenstein SA, Clapper JR, Holmes PV, Piomelli D, Hohmann AG (2010) A role for 2-arachidonoylglycerol and endocannabinoid signaling in the locomotor response to novelty induced by olfactory bulbectomy. Pharmacol Res 61:419–429
El Khoury MA, Gorgievski V, Moutsimilli L, Giros B, Tzavara ET (2012) Interactions between the cannabinoid and dopaminergic systems: evidence from animal studies. Prog Neuropsychopharmacol Biol Psychiatry 38:36–50
Erb S, Hitchcott PK, Rajabi H, Mueller D, Shaham Y, Stewart J (2000) Alpha-2 adrenergic receptor agonists block stress-induced reinstatement of cocaine seeking. Neuropsychopharmacology 23:138–150
Esteban S, Moranta D, Sastre-Coll A, Miralles A, Garcia-Sevilla JA (2002) Withdrawal from chronic ethanol increases the sensitivity of presynaptic 5-HT(1A) receptors modulating serotonin and dopamine synthesis in rat brain in vivo. Neurosci Lett 326:121–124
Evanson NK, Tasker JG, Hill MN, Hillard CJ, Herman JP (2010) Fast feedback inhibition of the HPA axis by glucocorticoids is mediated by endocannabinoid signaling. Endocrinology 151:4811–4819
Fallon JH, Koziell DA, Moore RY (1978) Catecholamine innervation of the basal forebrain. II. Amygdala, suprarhinal cortex and entorhinal cortex. J Comp Neurol 180:509–532
Ferre S, Goldberg SR, Lluis C, Franco R (2009) Looking for the role of cannabinoid receptor heteromers in striatal function. Neuropharmacology 56(Suppl 1):226–234
Fitzgerald ML, Shobin E, Pickel VM (2012) Cannabinoid modulation of the dopaminergic circuitry: implications for limbic and striatal output. Prog Neuropsychopharmacol Biol Psychiatry 38:21–29
Frazer A (1997) Pharmacology of antidepressants. J Clin Psychopharmacol 17(Suppl 1):2S–18S
French ED (1997) Delta9-Tetrahydrocannabinol excites rat VTA dopamine neurons through activation of cannabinoid CB1 but not opioid receptors. Neurosci Lett 226:159–162
French ED, Dillon K, Wu X (1997) Cannabinoids excite dopamine neurons in the ventral tegmentum and substantia nigra. Neuroreport 8:649–652
Freund TF, Katona I, Piomelli D (2003) Role of endogenous cannabinoids in synaptic signaling. Physiol Rev 83:1017–1066
Gardiner SM, March JE, Kemp PA, Bennett T (2005) Involvement of CB1-receptors and beta-adrenoceptors in the regional hemodynamic responses to lipopolysaccharide infusion in conscious rats. Am J Physiol Heart Circ Physiol 288:H2280–2288
Gellman RL, Aghajanian GK (1993) Pyramidal cells in piriform cortex receive a convergence of inputs from monoamine activated GABAergic interneurons. Brain Res 600:63–73
Gessa GL, Melis M, Muntoni AL, Diana M (1998) Cannabinoids activate mesolimbic dopamine neurons by an action on cannabinoid CB1 receptors. Eur J Pharmacol 341:39–44
Giuffrida A, Parsons LH, Kerr TM, Rodriguez de Fonseca F, Navarro M, Piomelli D (1999) Dopamine activation of endogenous cannabinoid signaling in dorsal striatum. Nat Neurosci 2:358–363
Glass M, Felder CC (1997) Concurrent stimulation of cannabinoid CB1 and dopamine D2 receptors augments cAMP accumulation in striatal neurons: evidence for a Gs linkage to the CB1 receptor. J Neurosci 17:5327–5333
Gobbi G, Bambico FR, Mangieri R, Bortolato M, Campolongo P, Solinas M, Cassano T, Morgese MG, Debonnel G, Duranti A, Tontini A, Tarzia G, Mor M, Trezza V, Goldberg SR, Cuomo V, Piomelli D (2005) Antidepressant-like activity and modulation of brain monoaminergic transmission by blockade of anandamide hydrolysis. Proc Natl Acad Sci U S A 102:18620–18625
Gobshtis N, Ben-Shabat S, Fride E (2007) Antidepressant-induced undesirable weight gain: prevention with rimonabant without interference with behavioral effectiveness. Eur J Pharmacol 554:155–163
Greenwood BN, Foley TE, Day HE, Campisi J, Hammack SH, Campeau S, Maier SF, Fleshner M (2003) Freewheel running prevents learned helplessness/behavioral depression: role of dorsal raphe serotonergic neurons. J Neurosci 23:2889–2898
Griebel G, Rodgers RJ, Perrault G, Sanger DJ (1997) Risk assessment behaviour: evaluation of utility in the study of 5-HT-related drugs in the rat elevated plus-maze test. Pharmacol Biochem Behav 57:817–827
Griebel G, Stemmelin J, Scatton B (2005) Effects of the cannabinoid CB1 receptor antagonist rimonabant in models of emotional reactivity in rodents. Biol Psychiatry 57:261–267
Grueter BA, Gosnell HB, Olsen CM, Schramm-Sapyta NL, Nekrasova T, Landreth GE, Winder DG (2006) Extracellular-signal regulated kinase 1-dependent metabotropic glutamate receptor 5-induced long-term depression in the bed nucleus of the stria terminalis is disrupted by cocaine administration. J Neurosci 26:3210–3219
Haj-Dahmane S, Shen RY (2005) The wake-promoting peptide orexin-B inhibits glutamatergic transmission to dorsal raphe nucleus serotonin neurons through retrograde endocannabinoid signaling. J Neurosci 25:896–905
Haj-Dahmane S, Shen RY (2009) Endocannabinoids suppress excitatory synaptic transmission to dorsal raphe serotonin neurons through the activation of presynaptic CB1 receptors. J Pharmacol Exp Ther 331:186–196
Haj-Dahmane S, Shen RY (2011) Modulation of the serotonin system by endocannabinoid signaling. Neuropharmacology 61:414–420
Haring M, Marsicano G, Lutz B, Monory K (2007) Identification of the cannabinoid receptor type 1 in serotonergic cells of raphe nuclei in mice. Neuroscience 146:1212–1219
Haring M, Grieb M, Monory K, Lutz B, Moreira FA (2013) Cannabinoid CB(1) receptor in the modulation of stress coping behavior in mice: the role of serotonin and different forebrain neuronal subpopulations. Neuropharmacology 65:83–89
Haroon E, Raison CL, Miller AH (2012) Psychoneuroimmunology meets neuropsychopharmacology: translational implications of the impact of inflammation on behavior. Neuropsychopharmacology 37:137–162
Hatfield T, Spanis C, McGaugh JL (1999) Response of amygdalar norepinephrine to footshock and GABAergic drugs using in vivo microdialysis and HPLC. Brain Res 835:340–345
Hayase T, Yamamoto Y, Yamamoto K (2005) Persistent anxiogenic effects of a single or repeated doses of cocaine and methamphetamine: interactions with endogenous cannabinoid receptor ligands. Behav Pharmacol 16:395–404
Herkenham M (1992) Cannabinoid receptor localization in brain: relationship to motor and reward systems. Ann N Y Acad Sci 654:19–32
Herkenham M, Groen BG, Lynn AB, De Costa BR, Richfield EK (1991a) Neuronal localization of cannabinoid receptors and second messengers in mutant mouse cerebellum. Brain Res 552:301–310
Herkenham M, Lynn AB, Johnson MR, Melvin LS, Costa BR de, Rice KC (1991b) Characterization and localization of cannabinoid receptors in rat brain: a quantitative in vitro autoradiographic study. J Neurosci 11:563–583
Hermann H, Lutz B (2005) Coexpression of the cannabinoid receptor type 1 with the corticotropin-releasing hormone receptor type 1 in distinct regions of the adult mouse forebrain. Neurosci Lett 375:13–18
Hermann H, Marsicano G, Lutz B (2002) Coexpression of the cannabinoid receptor type 1 with dopamine and serotonin receptors in distinct neuronal subpopulations of the adult mouse forebrain. Neuroscience 109:451–460
Hesketh SA, Brennan AK, Jessop DS, Finn DP (2008) Effects of chronic treatment with citalopram on cannabinoid and opioid receptor-mediated G-protein coupling in discrete rat brain regions. Psychopharmacology (Berl) 198:29–36
Hill MN, Gorzalka BB (2005) Pharmacological enhancement of cannabinoid CB(1) receptor activity elicits an antidepressant-like response in the rat forced swim test. Eur Neuropsychopharmacol 15:593–599
Hill MN, Gorzalka BB (2009) The endocannabinoid system and the treatment of mood and anxiety disorders. CNS Neurol Disord Drug Targets 8:451–458
Hill MN, McEwen BS (2009) Endocannabinoids: the silent partner of glucocorticoids in the synapse. Proc Natl Acad Sci U S A 106:4579–4580
Hill MN, Patel S, Carrier EJ, Rademacher DJ, Ormerod BK, Hillard CJ, Gorzalka BB (2005) Downregulation of endocannabinoid signaling in the hippocampus following chronic unpredictable stress. Neuropsychopharmacology 30:508–515
Hill MN, Ho WS, Sinopoli KJ, Viau V, Hillard CJ, Gorzalka BB (2006) Involvement of the endocannabinoid system in the ability of long-term tricyclic antidepressant treatment to suppress stress-induced activation of the hypothalamic-pituitary-adrenal axis. Neuropsychopharmacology 31:2591–2599
Hill MN, Carrier EJ, McLaughlin RJ, Morrish AC, Meier SE, Hillard CJ, Gorzalka BB (2008a) Regional alterations in the endocannabinoid system in an animal model of depression: effects of concurrent antidepressant treatment. J Neurochem 106:2322–2336
Hill MN, Ho WS, Hillard CJ, Gorzalka BB (2008b) Differential effects of the antidepressants tranylcypromine and fluoxetine on limbic cannabinoid receptor binding and endocannabinoid contents. J Neural Transm 115:1673–1679
Hill MN, Hillard CJ, Bambico FR, Patel S, Gorzalka BB, Gobbi G (2009a) The therapeutic potential of the endocannabinoid system for the development of a novel class of antidepressants. Trends Pharmacol Sci 30:484–493
Hill MN, McLaughlin RJ, Morrish AC, Viau V, Floresco SB, Hillard CJ, Gorzalka BB (2009b) Suppression of amygdalar endocannabinoid signaling by stress contributes to activation of the hypothalamic-pituitary-adrenal axis. Neuropsychopharmacology 34:2733–2745
Hill MN, Karatsoreos IN, Hillard CJ, McEwen BS (2010a) Rapid elevations in limbic endocannabinoid content by glucocorticoid hormones in vivo. Psychoneuroendocrinology 35:1333–1338
Hill MN, McLaughlin RJ, Bingham B, Shrestha L, Lee TT, Gray JM, Hillard CJ, Gorzalka BB, Viau V (2010b) Endogenous cannabinoid signaling is essential for stress adaptation. Proc Natl Acad Sci U S A 107:9406–9411
Hill MN, McLaughlin RJ, Pan B, Fitzgerald ML, Roberts CJ, Lee TT, Karatsoreos IN, Mackie K, Viau V, Pickel VM, McEwen BS, Liu QS, Gorzalka BB, Hillard CJ (2011a) Recruitment of prefrontal cortical endocannabinoid signaling by glucocorticoids contributes to termination of the stress response. J Neurosci 31:10506–10515
Hill MN, McLaughlin RJ, Pan B, Fitzgerald ML, Roberts CJ, Lee TT, Karatsoreos IN, Mackie K, Viau V, Pickel VM, McEwen BS, Liu QS, Gorzalka BB, Hillard CJ (2011b) Recruitment of prefrontal cortical endocannabinoid signaling by glucocorticoids contributes to termination of the stress response. J Neurosci 31:10506–10515
Hillard CJ, Bloom AS (1982) Delta 9-Tetrahydrocannabinol-induced changes in beta-adrenergic receptor binding in mouse cerebral cortex. Brain Res 235:370–377
Hillard CJ, Weinlander KM, Stuhr KL (2012) Contributions of endocannabinoid signaling to psychiatric disorders in humans: genetic and biochemical evidence. Neuroscience 204:207–229
Ho WV, Hill MN, Miller GE, Gorzalka BB, Hillard CJ (2012) Serum contents of endocannabinoids are correlated with blood pressure in depressed women. Lipids Health Dis 11:32
Hoffman AF, Lupica CR (2001) Direct actions of cannabinoids on synaptic transmission in the nucleus accumbens: a comparison with opioids. J Neurophysiol 85:72–83
Hoffman AF, Oz M, Caulder T, Lupica CR (2003) Functional tolerance and blockade of long-term depression at synapses in the nucleus accumbens after chronic cannabinoid exposure. J Neurosci 23:4815–4820
Hohmann AG, Suplita RL, Bolton NM, Neely MH, Fegley D, Mangieri R, Krey JF, Walker JM, Holmes PV, Crystal JD, Duranti A, Tontini A, Mor M, Tarzia G, Piomelli D (2005) An endocannabinoid mechanism for stress-induced analgesia. Nature 435:1108–1112
Houchi H, Babovic D, Pierrefiche O, Ledent C, Daoust M, Naassila M (2005) CB1 receptor knockout mice display reduced ethanol-induced conditioned place preference and increased striatal dopamine D2 receptors. Neuropsychopharmacology 30:339–349
Huang YC, Wang SJ, Chiou LC, Gean PW (2003) Mediation of amphetamine-induced long-term depression of synaptic transmission by CB1 cannabinoid receptors in the rat amygdala. J Neurosci 23:10311–10320
Hungund BL, Vinod KY, Kassir SA, Basavarajappa BS, Yalamanchili R, Cooper TB, Mann JJ, Arango V (2004) Upregulation of CB1 receptors and agonist-stimulated [35S]GTPgammaS binding in the prefrontal cortex of depressed suicide victims. Mol Psychiatry 9:184–190
Insel PA, Hammond HK (1993) Beta-adrenergic receptors in heart failure. J Clin Invest 92:2564
Ishac EJ, Jiang L, Lake KD, Varga K, Abood ME, Kunos G (1996) Inhibition of exocytotic noradrenaline release by presynaptic cannabinoid CB1 receptors on peripheral sympathetic nerves. Br J Pharmacol 118:2023–2028
Jarrahian A, Watts VJ, Barker EL (2004) D2 dopamine receptors modulate Galpha-subunit coupling of the CB1 cannabinoid receptor. J Pharmacol Exp Ther 308:880–886
Jentsch JD, Andrusiak E, Tran A, Bowers MB Jr, Roth RH (1997) Delta 9-tetrahydrocannabinol increases prefrontal cortical catecholaminergic utilization and impairs spatial working memory in the rat: blockade of dopaminergic effects with HA966. Neuropsychopharmacology 16:426–432
Julian MD, Martin AB, Cuellar B, Rodriguez De Fonseca F, Navarro M, Moratalla R, Garcia-Segura LM (2003) Neuroanatomical relationship between type 1 cannabinoid receptors and dopaminergic systems in the rat basal ganglia. Neuroscience 119:309–318
Kamprath K, Romo-Parra H, Haring M, Gaburro S, Doengi M, Lutz B, Pape HC (2011) Short-term adaptation of conditioned fear responses through endocannabinoid signaling in the central amygdala. Neuropsychopharmacology 36:652–663
Kano M, Ohno-Shosaku T, Hashimotodani Y, Uchigashima M, Watanabe M (2009) Endocannabinoid-mediated control of synaptic transmission. Physiol Rev 89:309–380
Kearn CS, Greenberg MJ, DiCamelli R, Kurzawa K, Hillard CJ (1999) Relationships between ligand affinities for the cerebellar cannabinoid receptor CB1 and the induction of GDP/GTP exchange. J Neurochem 72:2379–2387
Kearn CS, Blake-Palmer K, Daniel E, Mackie K, Glass M (2005) Concurrent stimulation of cannabinoid CB1 and dopamine D2 receptors enhances heterodimer formation: a mechanism for receptor cross-talk? Mol Pharmacol 67:1697–1704
Kim J, Isokawa M, Ledent C, Alger BE (2002) Activation of muscarinic acetylcholine receptors enhances the release of endogenous cannabinoids in the hippocampus. J Neurosci 22:10182–10191
Kirilly E, Hunyady L, Bagdy G (2012) Opposing local effects of endocannabinoids on the activity of noradrenergic neurons and release of noradrenaline: relevance for their role in depression and in the actions of CB(1) receptor antagonists. J Neural Transm 120(1):177–186
Klaassen T, Riedel WJ, Praag HM van, Menheere PP, Griez E (2002) Neuroendocrine response to meta-chlorophenylpiperazine and ipsapirone in relation to anxiety and aggression. Psychiatry Res 113:29–40
Kofalvi A, Rodrigues RJ, Ledent C, Mackie K, Vizi ES, Cunha RA, Sperlagh B (2005) Involvement of cannabinoid receptors in the regulation of neurotransmitter release in the rodent striatum: a combined immunochemical and pharmacological analysis. J Neurosci 25:2874–2884
Koob G, Kreek MJ (2007) Stress, dysregulation of drug reward pathways, and the transition to drug dependence. Am J Psychiatry 164:1149–1159
Koob GF, Le Moal M (2008) Review. Neurobiological mechanisms for opponent motivational processes in addiction. Philos Trans R Soc Lond B Biol Sci 363:3113–3123
Kupferschmidt DA, Newman AE, Boonstra R, Erb S (2012) Antagonism of cannabinoid 1 receptors reverses the anxiety-like behavior induced by central injections of corticotropin-releasing factor and cocaine withdrawal. Neuroscience 204:125–133
Lanfumey L, Mongeau R, Cohen-Salmon C, Hamon M (2008) Corticosteroid-serotonin interactions in the neurobiological mechanisms of stress-related disorders. Neurosci Biobehav Rev 32:1174–1184
Lau T, Schloss P (2008) The cannabinoid CB(1) receptor is expressed on serotonergic and dopaminergic neurons. Eur J Pharmacol 578:137–141
Laviolette SR, Grace AA (2006) The roles of cannabinoid and dopamine receptor systems in neural emotional learning circuits: implications for schizophrenia and addiction. Cell Mol Life Sci 63:1597–1613
LeDoux JE (2000) Emotion circuits in the brain. Annu Rev Neurosci 23:155–184
Leri F, Flores J, Rodaros D, Stewart J (2002) Blockade of stress-induced but not cocaine-induced reinstatement by infusion of noradrenergic antagonists into the bed nucleus of the stria terminalis or the central nucleus of the amygdala. J Neurosci 22:5713–5718
Lodge DJ, Grace AA (2011) Developmental pathology, dopamine, stress and schizophrenia. Int J Dev Neurosci 29:207–213
Lovinger DM, Mathur BN (2012) Endocannabinoids in striatal plasticity. Parkinsonism Relat Disord 18(Suppl 1):S132–S134
Lowry CA (2002) Functional subsets of serotonergic neurones: implications for control of the hypothalamic-pituitary-adrenal axis. J Neuroendocrinol 14:911–923
Mackie K, Hille B (1992) Cannabinoids inhibit N-type calcium channels in neuroblastoma-glioma cells. Proc Natl Acad Sci 89:3825–3829
Mailleux P, Vanderhaeghen J-J (1992) Distribution of neuronal cannabinoid receptor in the adult rat brain: a comparative receptor binding autoradiography and in situ hybridization histochemistry. Neuroscience 48:655–668
Mallet PE, Beninger RJ (1998) Delta9-tetrahydrocannabinol, but not the endogenous cannabinoid receptor ligand anandamide, produces conditioned place avoidance. Life Sci 62:2431–2439
Malone DT, Taylor DA (1998) Modulation of delta9-tetrahydrocannabinol-induced hypothermia by fluoxetine in the rat. Br J Pharmacol 124:1419–1424
Malone DT, Taylor DA (2001) Involvement of somatodendritic 5-HT(1A) receptors in Delta(9)-tetrahydrocannabinol-induced hypothermia in the rat. Pharmacol Biochem Behav 69:595–601
Mantsch JR, Weyer A, Vranjkovic O, Beyer CE, Baker DA, Caretta H (2010) Involvement of noradrenergic neurotransmission in the stress- but not cocaine-induced reinstatement of extinguished cocaine-induced conditioned place preference in mice: role for beta-2 adrenergic receptors. Neuropsychopharmacology 35:2165–2178
Marco EM, Perez-Alvarez L, Borcel E, Rubio M, Guaza C, Ambrosio E, File SE, Viveros MP (2004) Involvement of 5-HT1A receptors in behavioural effects of the cannabinoid receptor agonist CP 55,940 in male rats. Behav Pharmacol 15:21–27
Marsicano G, Lutz B (1999) Expression of the cannabinoid receptor CB1 in distinct neuronal subpopulations in the adult mouse forebrain. Eur J Neurosci 11:4213–4225
Martin AB, Fernandez-Espejo E, Ferrer B, Gorriti MA, Bilbao A, Navarro M, Rodriguez de Fonseca F, Moratalla R (2008) Expression and function of CB(1) receptor in the rat striatum: localization and effects on D(1) and D(2) dopamine receptor-mediated motor behaviors. Neuropsychopharmacology 33:1667–1679
Mato S, Aso E, Castro E, Martin M, Valverde O, Maldonado R, Pazos A (2007) CB1 knockout mice display impaired functionality of 5-HT1A and 5-HT2A/C receptors. J Neurochem 103:2111–2120
Mato S, Vidal R, Castro E, Diaz A, Pazos A, Valdizan EM (2010) Long-term fluoxetine treatment modulates cannabinoid type 1 receptor-mediated inhibition of adenylyl cyclase in the rat prefrontal cortex through 5-hydroxytryptamine 1A receptor-dependent mechanisms. Mol Pharmacol 77:424–434
Matsuda LA, Lolait SJ, Brownstein MJ, Young AC, Bonner TI (1990) Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature 346:561–564
Matsuda LA, Bonner TI, Lolait SJ (1993) Localization of cannabinoid receptor mRNA in rat brain. J Comp Neurol 327:535–550
Matyas F, Urban GM, Watanabe M, Mackie K, Zimmer A, Freund TF, Katona I (2008) Identification of the sites of 2-arachidonoylglycerol synthesis and action imply retrograde endocannabinoid signaling at both GABAergic and glutamatergic synapses in the ventral tegmental area. Neuropharmacology 54:95–107
McElligott ZA, Winder DG (2009) Modulation of glutamatergic synaptic transmission in the bed nucleus of the stria terminalis. Prog Neuropsychopharmacol Biol Psychiatry 33:1329–1335
McEwen BS (2008) Central effects of stress hormones in health and disease: understanding the protective and damaging effects of stress and stress mediators. Eur J Pharmacol 583:174–185
McGregor IS, Issakidis CN, Prior G (1996) Aversive effects of the synthetic cannabinoid CP 55,940 in rats. Pharmacol Biochem Behav 53:657–664
McLaughlin RJ, Hill MN, Morrish AC, Gorzalka BB (2007) Local enhancement of cannabinoid CB1 receptor signalling in the dorsal hippocampus elicits an antidepressant-like effect. Behav Pharmacol 18:431–438
McLaughlin RJ, Hill MN, Gorzalka BB (2009) Monoaminergic neurotransmission contributes to cannabinoid-induced activation of the hypothalamic-pituitary-adrenal axis. Eur J Pharmacol 624(1–3):71–76
McLaughlin RJ, Hill MN, Bambico FR, Stuhr KL, Gobbi G, Hillard CJ, Gorzalka BB (2012) Prefrontal cortical anandamide signaling coordinates coping responses to stress through a serotonergic pathway. Eur Neuropsychopharmacol 22:664–671
Mechoulam R, Ben-Shabat S, Hanus L, Ligumsky M, Kaminski NE, Schatz AR, Gopher A, Almog S, Martin BR, Compton DR et al (1995) Identification of an endogenous 2-monoglyceride, present in canine gut, that binds to cannabinoid receptors. Biochem Pharmacol 50:83–90
Melis M, Perra S, Muntoni AL, Pillolla G, Lutz B, Marsicano G, Di Marzo V, Gessa GL, Pistis M (2004a) Prefrontal cortex stimulation induces 2-arachidonoyl-glycerol-mediated suppression of excitation in dopamine neurons. J Neurosci 24:10707–10715
Melis M, Pistis M, Perra S, Muntoni AL, Pillolla G, Gessa GL (2004b) Endocannabinoids mediate presynaptic inhibition of glutamatergic transmission in rat ventral tegmental area dopamine neurons through activation of CB1 receptors. J Neurosci 24:53–62
Mendiguren A, Pineda J (2004) Cannabinoids enhance N-methyl-d-aspartate-induced excitation of locus coeruleus neurons by CB(1) receptors in rat brain slices. Neurosci Lett 363:1–5
Mendiguren A, Pineda J (2006) Systemic effect of cannabinoids on the spontaneous firing rate of locus coeruleus neurons in rats. Eur J Pharmacol 534:83–88
Mendiguren A, Pineda J (2009) Effect of the CB(1) receptor antagonists rimonabant and AM251 on the firing rate of dorsal raphe nucleus neurons in rat brain slices. Br J Pharmacol 158:1579–1587
Merroun I, Errami M, Hoddah H, Urbano G, Porres JM, Aranda P, Llopis J, Lopez-Jurado M (2009) Influence of intracerebroventricular or intraperitoneal administration of cannabinoid receptor agonist (WIN 55,212-2) and inverse agonist (AM 251) on the regulation of food intake and hypothalamic serotonin levels. Br J Nutr 101:1569–1578
Moranta D, Esteban S, Garcia-Sevilla JA (2004) Differential effects of acute cannabinoid drug treatment, mediated by CB(1) receptors, on the in vivo activity of tyrosine and tryptophan hydroxylase in the rat brain. Naunyn Schmiedebergs Arch Pharmacol 369:516–524
Moranta D, Esteban S, Garcia-Sevilla JA (2009) Chronic treatment and withdrawal of the cannabinoid agonist WIN 55,212-2 modulate the sensitivity of presynaptic receptors involved in the regulation of monoamine syntheses in rat brain. Naunyn Schmiedebergs Arch Pharmacol 379:61–72
Morrish AC, Hill MN, Riebe CJ, Gorzalka BB (2009) Protracted cannabinoid administration elicits antidepressant behavioral responses in rats: role of gender and noradrenergic transmission. Physiol Behav 98:118–124
Munro S, Thomas KL, Abu-Shaar M (1993) Molecular characterization of a peripheral receptor for cannabinoids. Nature 365:61–65
Muntoni AL, Pillolla G, Melis M, Perra S, Gessa GL, Pistis M (2006) Cannabinoids modulate spontaneous neuronal activity and evoked inhibition of locus coeruleus noradrenergic neurons. Eur J Neurosci 23:2385–2394
Murphy LL, Munoz RM, Adrian BA, Villanua MA (1998) Function of cannabinoid receptors in the neuroendocrine regulation of hormone secretion. Neurobiol Dis 5:432–446
Nakazi M, Bauer U, Nickel T, Kathmann M, Schlicker E (2000) Inhibition of serotonin release in the mouse brain via presynaptic cannabinoid CB1 receptors. Naunyn Schmiedebergs Arch Pharmacol 361:19–24
Nathan PJ, O’Neill BV, Napolitano A, Bullmore ET (2011) Neuropsychiatric adverse effects of centrally acting antiobesity drugs. CNS Neurosci Ther 17:490–505
Novak CM, Burghardt PR, Levine JA (2012) The use of a running wheel to measure activity in rodents: relationship to energy balance, general activity, and reward. Neurosci Biobehav Rev 36:1001–1014
Oliva JM, Uriguen L, Perez-Rial S, Manzanares J (2005) Time course of opioid and cannabinoid gene transcription alterations induced by repeated administration with fluoxetine in the rat brain. Neuropharmacology 49:618–626
Onaivi ES (2008) An endocannabinoid hypothesis of drug reward and drug addiction. Ann N Y Acad Sci 1139:412–421
Oropeza VC, Page ME, Van Bockstaele EJ (2005) Systemic administration of WIN 55,212-2 increases norepinephrine release in the rat frontal cortex. Brain Res 1046:45–54
Oropeza VC, Mackie K, Van Bockstaele EJ (2007) Cannabinoid receptors are localized to noradrenergic axon terminals in the rat frontal cortex. Brain Res 1127:36–44
Osei-Owusu P, James A, Crane J, Scrogin KE (2005) 5-Hydroxytryptamine 1A receptors in the paraventricular nucleus of the hypothalamus mediate oxytocin and adrenocorticotropin hormone release and some behavioral components of the serotonin syndrome. J Pharmacol Exp Ther 313:1324–1330
Page ME, Oropeza VC, Sparks SE, Qian Y, Menko AS, Van Bockstaele EJ (2007) Repeated cannabinoid administration increases indices of noradrenergic activity in rats. Pharmacol Biochem Behav 86:162–168
Page ME, Oropeza VC, Van Bockstaele EJ (2008) Local administration of a cannabinoid agonist alters norepinephrine efflux in the rat frontal cortex. Neurosci Lett 431:1–5
Pan B, Hillard CJ, Liu QS (2008a) D2 dopamine receptor activation facilitates endocannabinoid-mediated long-term synaptic depression of GABAergic synaptic transmission in midbrain dopamine neurons via cAMP-protein kinase A signaling. J Neurosci 28:14018–14030
Pan B, Hillard CJ, Liu QS (2008b) Endocannabinoid signaling mediates cocaine-induced inhibitory synaptic plasticity in midbrain dopamine neurons. J Neurosci 28:1385–1397
Pandolfo P, Vendruscolo LF, Sordi R, Takahashi RN (2009) Cannabinoid-induced conditioned place preference in the spontaneously hypertensive rat-an animal model of attention deficit hyperactivity disorder. Psychopharmacology (Berl) 205:319–326
Parolaro D, Realini N, Vigano D, Guidali C, Rubino T (2010) The endocannabinoid system and psychiatric disorders. Exp Neurol 224:3–14
Patel S, Hillard CJ (2003) Cannabinoid-induced Fos expression within A10 dopaminergic neurons. Brain Res 963:15–25
Patel S, Hillard CJ (2006) Pharmacological evaluation of cannabinoid receptor ligands in a mouse model of anxiety: further evidence for an anxiolytic role for endogenous cannabinoid signaling. J Pharmacol Exp Ther 318:304–311
Patel S, Hillard CJ (2008) Adaptations in endocannabinoid signaling in response to repeated homotypic stress: a novel mechanism for stress habituation. Eur J Neurosci 27:2821–2829
Patel S, Hillard CJ (2009a) Endocannabinoids as modulators of synaptic signaling. In: Reggio PH (ed) The Cannabinoid Receptors. Humana Press, New York, pp 281–308
Patel S, Hillard CJ (2009b) Role of endocannabinoid signaling in anxiety and depression. In: Alexander DKaS (ed) Behavioral Neurobiology of the Endocannabinoid System. Springer Verlag, Berlin, pp 347–372
Patel S, Rademacher DJ, Hillard CJ (2003) Differential regulation of the endocannabinoids anandamide and 2-arachidonylglycerol within the limbic forebrain by dopamine receptor activity. J Pharmacol Exp Ther 306:880–888
Patel S, Roelke CT, Rademacher DJ, Cullinan WE, Hillard CJ (2004) Endocannabinoid signaling negatively modulates stress-induced activation of the hypothalamic-pituitary-adrenal axis. Endocrinology 145:5431–5438
Patel S, Roelke CT, Rademacher DJ, Hillard CJ (2005) Inhibition of restraint stress-induced neural and behavioural activation by endogenous cannabinoid signalling. Eur J Neurosci 21:1057–1069
Patel S, Kingsley PJ, Mackie K, Marnett LJ, Winder DG (2009) Repeated homotypic stress elevates 2-arachidonoylglycerol levels and enhances short-term endocannabinoid signaling at inhibitory synapses in basolateral amygdala. Neuropsychopharmacology 34:2699–2709
Pattij T, Wiskerke J, Schoffelmeer AN (2008) Cannabinoid modulation of executive functions. Eur J Pharmacol 585:458–463
Pedroza-Llinas R, Mendez-Diaz M, Ruiz-Contreras AE, Prospero-Garcia O (2013) CB1 receptor activation in the nucleus accumbens core impairs contextual fear learning. Behav Brain Res 237:141–147
Pertwee RG, Howlett AC, Abood ME, Alexander SP, Di Marzo V, Elphick MR, Greasley PJ, Hansen HS, Kunos G, Mackie K, Mechoulam R, Ross RA (2010) International Union of Basic and Clinical Pharmacology. LXXIX. Cannabinoid receptors and their ligands: beyond CB1 and CB2. Pharmacol Rev 62:588–631
Pfitzer T, Niederhoffer N, Szabo B (2005) Search for an endogenous cannabinoid-mediated effect in the sympathetic nervous system. Naunyn Schmiedebergs Arch Pharmacol 371:9–17
Pickel VM, Chan J, Kash TL, Rodriguez JJ, MacKie K (2004) Compartment-specific localization of cannabinoid 1 (CB1) and mu-opioid receptors in rat nucleus accumbens. Neuroscience 127:101–112
Pickel VM, Chan J, Kearn CS, Mackie K (2006) Targeting dopamine D2 and cannabinoid-1 (CB1) receptors in rat nucleus accumbens. J Comp Neurol 495:299–313
Pistis M, Porcu G, Melis M, Diana M, Gessa GL (2001) Effects of cannabinoids on prefrontal neuronal responses to ventral tegmental area stimulation. Eur J Neurosci 14:96–102
Pistis M, Ferraro L, Pira L, Flore G, Tanganelli S, Gessa GL, Devoto P (2002a) Delta(9)-tetrahydrocannabinol decreases extracellular GABA and increases extracellular glutamate and dopamine levels in the rat prefrontal cortex: an in vivo microdialysis study. Brain Res 948:155–158
Pistis M, Muntoni AL, Pillolla G, Gessa GL (2002b) Cannabinoids inhibit excitatory inputs to neurons in the shell of the nucleus accumbens: an in vivo electrophysiological study. Eur J Neurosci 15:1795–1802
Poddar MK, Dewey WL (1980) Effects of cannabinoids on catecholamine uptake and release in hypothalamic and striatal synaptosomes. J Pharmacol Exp Ther 214:63–67
Racz I, Bilkei-Gorzo A, Toth ZE, Michel K, Palkovits M, Zimmer A (2003) A critical role for the cannabinoid CB1 receptors in alcohol dependence and stress-stimulated ethanol drinking. J Neurosci 23:2453–2458
Rademacher DJ, Meier SE, Shi L, Ho WS, Jarrahian A, Hillard CJ (2008) Effects of acute and repeated restraint stress on endocannabinoid content in the amygdala, ventral striatum, and medial prefrontal cortex in mice. Neuropharmacology 54:108–116
Radley JJ, Sawchenko PE (2011) A common substrate for prefrontal and hippocampal inhibition of the neuroendocrine stress response. J Neurosci 31:9683–9695
Refojo D, Schweizer M, Kuehne C, Ehrenberg S, Thoeringer C, Vogl AM, Dedic N, Schumacher M, Wolff G von, Avrabos C, Touma C, Engblom D, Schutz G, Nave KA, Eder M, Wotjak CT, Sillaber I, Holsboer F, Wurst W, Deussing JM (2011) Glutamatergic and dopaminergic neurons mediate anxiogenic and anxiolytic effects of CRHR1. Science 333:1903–1907
Reyes BA, Rosario JC, Piana PM, Van Bockstaele EJ (2009) Cannabinoid modulation of cortical adrenergic receptors and transporters. J Neurosci Res 87:3671–3678
Reyes BA, Szot P, Sikkema C, Cathel AM, Kirby LG, Van Bockstaele EJ (2012) Stress-induced sensitization of cortical adrenergic receptors following a history of cannabinoid exposure. Exp Neurol 236:327–335
Riegel AC, Lupica CR (2004) Independent presynaptic and postsynaptic mechanisms regulate endocannabinoid signaling at multiple synapses in the ventral tegmental area. J Neurosci 24:11070–11078
Robbe D, Alonso G, Duchamp F, Bockaert J, Manzoni OJ (2001) Localization and mechanisms of action of cannabinoid receptors at the glutamatergic synapses of the mouse nucleus accumbens. J Neurosci 21:109–116
Robbe D, Kopf M, Remaury A, Bockaert J, Manzoni OJ (2002) Endogenous cannabinoids mediate long-term synaptic depression in the nucleus accumbens. Proc Natl Acad Sci U S A 99:8384–8388
Rodriguez-Gaztelumendi A, Rojo ML, Pazos A, Diaz A (2009) Altered CB receptor-signaling in prefrontal cortex from an animal model of depression is reversed by chronic fluoxetine. J Neurochem 108:1423–1433
Rossi S, De Chiara V, Musella A, Kusayanagi H, Mataluni G, Bernardi G, Usiello A, Centonze D (2008) Chronic psychoemotional stress impairs cannabinoid-receptor-mediated control of GABA transmission in the striatum. J Neurosci 28:7284–7292
Rubio M, Fernandez-Ruiz J, Miguel R de, Maestro B, Walker JM, Ramos JA (2008) CB1 receptor blockade reduces the anxiogenic-like response and ameliorates the neurochemical imbalances associated with alcohol withdrawal in rats. Neuropharmacology 54:976–988
Sakamoto Y, Mashiko K, Obata T, Matsumoto H, Hara Y, Kutsukata N, Yamamoto Y (2008) Effectiveness of continuous hemodiafiltration using a polymethylmethacrylate membrane hemofilter after polymyxin B-immobilized fiber column therapy of septic shock. ASAIO J 54:129–132
Sanudo-Pena MC, Tsou K, Delay ER, Hohman AG, Force M, Walker JM (1997) Endogenous cannabinoids as an aversive or counter-rewarding system in the rat. Neurosci Lett 223:125–128
Sapolsky RM, Meaney MJ, McEwen BS (1985) The development of the glucocorticoid receptor system in the rat limbic brain. III. Negative-feedback regulation. Brain Res 350:169–173
Sastre-Coll A, Esteban S, Garcia-Sevilla JA (2002) Supersensitivity of 5-HT1A autoreceptors and alpha2-adrenoceptors regulating monoamine synthesis in the brain of morphine-dependent rats. Naunyn Schmiedebergs Arch Pharmacol 365:210–219
Scavone JL, Mackie K, Van Bockstaele EJ (2010) Characterization of cannabinoid-1 receptors in the locus coeruleus: relationship with mu-opioid receptors. Brain Res 1312:18–31
Schlicker E, Timm J, Zentner J, Gothert M (1997) Cannabinoid CB1 receptor-mediated inhibition of noradrenaline release in the human and guinea-pig hippocampus. Naunyn Schmiedebergs Arch Pharmacol 356:583–589
Schlosburg JE, Blankman JL, Long JZ, Nomura DK, Pan B, Kinsey SG, Nguyen PT, Ramesh D, Booker L, Burston JJ, Thomas EA, Selley DE, Sim-Selley LJ, Liu QS, Lichtman AH, Cravatt BF (2010) Chronic monoacylglycerol lipase blockade causes functional antagonism of the endocannabinoid system. Nat Neurosci 13:1113–1119
Schultz W (2002) Getting formal with dopamine and reward. Neuron 36:241–263
Sciolino NR, Zhou W, Hohmann AG (2011) Enhancement of endocannabinoid signaling with JZL184, an inhibitor of the 2-arachidonoylglycerol hydrolyzing enzyme monoacylglycerol lipase, produces anxiolytic effects under conditions of high environmental aversiveness in rats. Pharmacol Res 64:226–234
Seagard JL, Dean C, Patel S, Rademacher DJ, Hopp FA, Schmeling WT, Hillard CJ (2004) Anandamide content and interaction of endocannabinoid/GABA modulatory effects in the NTS on baroreflex-evoked sympathoinhibition. Am J Physiol Heart Circ Physiol 286:H992–H1000
Seif T, Makriyannis A, Kunos G, Bonci A, Hopf FW (2011) The endocannabinoid 2-arachidonoylglycerol mediates D1 and D2 receptor cooperative enhancement of rat nucleus accumbens core neuron firing. Neuroscience 193:21–33
Serrano A, Parsons LH (2011) Endocannabinoid influence in drug reinforcement, dependence and addiction-related behaviors. Pharmacol Ther 132:215–241
Sesack SR, Pickel VM (1990) In the rat medial nucleus accumbens, hippocampal and catecholaminergic terminals converge on spiny neurons and are in apposition to each other. Brain Res 527:266–279
Shearman LP, Rosko KM, Fleischer R, Wang J, Xu S, Tong XS, Rocha BA (2003) Antidepressant-like and anorectic effects of the cannabinoid CB1 receptor inverse agonist AM251 in mice. Behav Pharmacol 14:573–582
Shimizu T, Lu L, Yokotani K (2010) Possible inhibitory roles of endogenous 2-arachidonoylglycerol during corticotropin-releasing factor-induced activation of central sympatho-adrenomedullary outflow in anesthetized rats. Eur J Pharmacol 641:54–60
Sidhpura N, Parsons LH (2011) Endocannabinoid-mediated synaptic plasticity and addiction-related behavior. Neuropharmacology 61:1070–1087
Sim L, Hampson R, Deadwyler S, Childers S (1996) Effects of chronic treatment with delta-9-tetrahydrocannabinol in cannabinoid-stimulated [35S]GTPgS autoradiography in rat brain. J Neurosci 16:8057–8066
Solowij N, Michie PT (2007) Cannabis and cognitive dysfunction: parallels with endophenotypes of schizophrenia? J Psychiatry Neurosci 32:30–52
Solowij N, Michie PT, Fox AM (1995) Differential impairments of selective attention due to frequency and duration of cannabis use. Biol Psychiatry 37:731–739
Steffens M, Engler C, Zentner J, Feuerstein TJ (2004) Cannabinoid CB1 receptor-mediated modulation of evoked dopamine release and of adenylyl cyclase activity in the human neocortex. Br J Pharmacol 141:1193–1203
Steiner MA, Wanisch K, Monory K, Marsicano G, Borroni E, Bachli H, Holsboer F, Lutz B, Wotjak CT (2008) Impaired cannabinoid receptor type 1 signaling interferes with stress-coping behavior in mice. Pharmacogenomics J 8:196–208
Stelt HM van der, Breuer ME, Olivier B, Westenberg HG (2005) Permanent deficits in serotonergic functioning of olfactory bulbectomized rats: an in vivo microdialysis study. Biol Psychiatry 57:1061–1067
Subhash MN, Nagaraja MR, Sharada S, Vinod KY (2003) Cortical alpha-adrenoceptor downregulation by tricyclic antidepressants in the rat brain. Neurochem Int 43:603–609
Sugiura T, Kondo S, Sukagawa A, Nakane S, Shinoda A, Itoh K, Yamashita A, Waku K (1995) 2-Arachidonoylglycerol: a possible endogenous cannabinoid receptor ligand in brain. Biochem Biophys Res Commun 215:89–97
Sulkowski A, Vachon L, Rich ES Jr (1977) Propranolol effects on acute marihuana intoxication in man. Psychopharmacology 52:47–53
Sumislawski JJ, Ramikie TS, Patel S (2011) Reversible gating of endocannabinoid plasticity in the amygdala by chronic stress: a potential role for monoacylglycerol lipase inhibition in the prevention of stress-induced behavioral adaptation. Neuropsychopharmacology 36:2750–2761
Szabo B, Siemes S, Wallmichrath I (2002) Inhibition of GABAergic neurotransmission in the ventral tegmental area by cannabinoids. Eur J Neurosci 15:2057–2061
Tanda G, Pontieri FE, Di Chiara G (1997) Cannabinoid and heroin activation of mesolimbic dopamine transmission by a common mu1 opioid receptor mechanism. Science 276:2048–2050
Terzian AL, Drago F, Wotjak CT, Micale V (2011) The dopamine and cannabinoid interaction in the modulation of emotions and cognition: assessing the role of cannabinoid CB1 receptor in neurons expressing dopamine D1 receptors. Front Behav Neurosci 5:49
Tournier M, Sorbara F, Gindre C, Swendsen JD, Verdoux H (2003) Cannabis use and anxiety in daily life: a naturalistic investigation in a non-clinical population. Psychiatry Res 118:1–8
Turu G, Varnai P, Gyombolai P, Szidonya L, Offertaler L, Bagdy G, Kunos G, Hunyady L (2009) Paracrine transactivation of the CB1 cannabinoid receptor by AT1 angiotensin and other Gq/11 protein-coupled receptors. J Biol Chem 284:16914–16921
Tzavara ET, Perry KW, Rodriguez DE, Bymaster FP, Nomikos GG (2001) The cannabinoid CB(1) receptor antagonist SR141716A increases norepinephrine outflow in the rat anterior hypothalamus. Eur J Pharmacol 426:R3–4
Tzavara ET, Davis RJ, Perry KW, Li X, Salhoff C, Bymaster FP, Witkin JM, Nomikos GG (2003) The CB1 receptor antagonist SR141716A selectively increases monoaminergic neurotransmission in the medial prefrontal cortex: implications for therapeutic actions. Br J Pharmacol 138:544–553
Tzavara ET, Li DL, Moutsimilli L, Bisogno T, Di Marzo V, Phebus LA, Nomikos GG, Giros B (2006) Endocannabinoids activate transient receptor potential vanilloid 1 receptors to reduce hyperdopaminergia-related hyperactivity: therapeutic implications. Biol Psychiatry 59:508–515
Ungerstedt U (1971) Stereotaxic mapping of the monoamine pathways in the rat brain. Acta Physiol Scand Suppl 367:1–48
Vanderschuren LJ, Kalivas PW (2000) Alterations in dopaminergic and glutamatergic transmission in the induction and expression of behavioral sensitization: a critical review of preclinical studies. Psychopharmacology 151:99–120
Varga K, Wagner JA, Bridgen DT, Kunos G (1998) Platelet- and macrophage-derived endogenous cannabinoids are involved in endotoxin-induced hypotension. Faseb J 12:1035–1044
Varma N, Carlson GC, Ledent C, Alger BE (2001) Metabotropic glutamate receptors drive the endocannabinoid system in hippocampus. J Neurosci 21:RC188
Vaughn LK, Mantsch JR, Vranjkovic O, Stroh G, Lacourt M, Kreutter M, Hillard CJ (2012) Cannabinoid receptor involvement in stress-induced cocaine reinstatement: potential interaction with noradrenergic pathways. Neuroscience 204:117–124
Vinod KY, Arango V, Xie S, Kassir SA, Mann JJ, Cooper TB, Hungund BL (2005) Elevated levels of endocannabinoids and CB1 receptor-mediated G-protein signaling in the prefrontal cortex of alcoholic suicide victims. Biol Psychiatry 57:480–486
Viveros MP, Marco EM, File SE (2005) Endocannabinoid system and stress and anxiety responses. Pharmacol Biochem Behav 81:331–342
Volk DW, Lewis DA (2010) Prefrontal cortical circuits in schizophrenia. Curr Top Behav Neurosci 4:485–508
Wang Y, Liu Y, Ito Y, Hashiguchi T, Kitajima I, Yamakuchi M, Shimizu H, Matsuo S, Imaizumi H, Maruyama I (2001) Simultaneous measurement of anandamide and 2-arachidonoylglycerol by polymyxin b-selective adsorption and subsequent high-performance liquid chromatography analysis: increase in endogenous cannabinoids in the sera of patients with endotoxic shock. Anal Biochem 294:73–82
Wang W, Sun D, Pan B, Roberts CJ, Sun X, Hillard CJ, Liu QS (2010) Deficiency in endocannabinoid signaling in the nucleus accumbens induced by chronic unpredictable stress. Neuropsychopharmacology 35:2249–2261
Wang J, Shen RY, Haj-Dahmane S (2012a) Endocannabinoids mediate the glucocorticoid-induced inhibition of excitatory synaptic transmission to dorsal raphe serotonin neurons. J Physiol 590:5795–5808
Wang M, Hill MN, Zhang L, Gorzalka BB, Hillard CJ, Alger BE (2012b) Acute restraint stress enhances hippocampal endocannabinoid function via glucocorticoid receptor activation. J Psychopharmacol 26:56–70
Weinshenker D, Schroeder JP (2007) There and back again: a tale of norepinephrine and drug addiction. Neuropsychopharmacology 32:1433–1451
Wenger T, Moldrich G, Furst S (2003) Neuromorphological background of cannabis addiction. Brain Res Bull 61:125–128
Wilson RI, Nicoll RA (2001) Endogenous cannabinoids mediate retrograde signalling at hippocampal synapses. Nature 410:588–592
Wolf ME, Sun X, Mangiavacchi S, Chao SZ (2004) Psychomotor stimulants and neuronal plasticity. Neuropharmacology 47(Suppl 1):61–79
Wu X, French ED (2000) Effects of chronic delta9-tetrahydrocannabinol on rat midbrain dopamine neurons: an electrophysiological assessment. Biochem J 346:835–840
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer Science+Business Media New York
About this chapter
Cite this chapter
Hillard, C.J. (2013). Endocannabinoids, Monoamines and Stress. In: Van Bockstaele, E. (eds) Endocannabinoid Regulation of Monoamines in Psychiatric and Neurological Disorders. Springer, New York, NY. https://doi.org/10.1007/978-1-4614-7940-6_9
Download citation
DOI: https://doi.org/10.1007/978-1-4614-7940-6_9
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4614-7939-0
Online ISBN: 978-1-4614-7940-6
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)