
Abstract
The liver is known to have an expansive role in the synthesis, degradation, and regulation of pathways involved in the metabolism of carbohydrates, proteins, lipids, trace elements, and vitamins. Subsequently, abnormalities that affect these pathways as well as specific enzyme deficiencies often affect the liver either primarily or secondarily. The spectrum of liver injury results when there is absence or blockage of an enzyme in the metabolic pathway often resulting in accumulation of unmetabolized toxic substrates and/or deficiencies in essential substances normally produced distal to the dysfunctional enzyme. Clinical presentations of children with metabolic liver disease vary from acute, life-threatening illness in the neonatal period to a more indolent, chronic disease process that becomes evident in adolescence or adulthood. Disease progression often results from the failure of metabolic functions resulting in hepatocyte injury or cellular derangement and hepatomegaly with advancement to cirrhosis and/or tumor development. A detailed history can often elicit the possibility of a metabolic dysfunction and assist in the guidance of further clinical investigations while treatment is dependent of the particular metabolic defect that is identified.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Bile Acid
- Glycogen Storage Disease
- Zellweger Syndrome
- Progressive Familial Intrahepatic Cholestasis
- Hereditary Fructose Intolerance
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
Introduction
The liver is known to have an expansive role in the synthesis, degradation, and regulation of pathways involved in the metabolism of carbohydrates, proteins, lipids, trace elements, and vitamins. Subsequently, abnormalities that affect these pathways as well as specific enzyme deficiencies often affect the liver either primarily or secondarily. The spectrum of liver injury results when there is absence or blockage of an enzyme in the metabolic pathway often resulting in accumulation of unmetabolized toxic substrates and/or deficiencies in essential substances normally produced distal to the dysfunctional enzyme. Clinical presentations of children with metabolic liver disease vary from acute, life-threatening illness in the neonatal period to a more indolent, chronic disease process that becomes evident in adolescence or adulthood. Disease progression often results from the failure of metabolic functions resulting in hepatocyte injury or cellular derangement and hepatomegaly with advancement to cirrhosis and/or tumor development. A detailed history can often elicit the possibility of a metabolic dysfunction and assist in the guidance of further clinical investigations while treatment is dependent of the particular metabolic defect that is identified.
Disorders of Bilirubin Metabolism
Bilirubin is formed by the breakdown of heme-containing products – most notably red blood cells (RBCs). This two-step process begins with the degradation of heme into carbon monoxide and biliverdin. Biliverdin is then metabolized to bilirubin by the enzyme biliverdin reductase. The majority of bilirubin is bound to albumin and transported to the liver where it is taken up by hepatic cells for secretion into the biliary ductal system after undergoing conjugation via the enzyme uridine diphosphogluconurate glucuronosyltransferase (UGT). The majority of neonatal jaundice is non-pathologic and is due to normal changes in bilirubin metabolism that result in increased production, decreased clearance, and increased intrahepatic circulation.
Breast Milk Jaundice
Introduction
Breast milk jaundice is a term used to describe the jaundice that results from the normal physiologic mechanisms contributing to elevated circulating bilirubin levels.
Pathogenesis
Breast milk jaundice is thought to result from several physiologic mechanisms unique to the newborn infant: Increased bilirubin production results from the increased breakdown and turnover of RBCs [1]. This results from elevated hematocrit levels with accompanying decreased RBC half-life [2]. UGT, the enzyme responsible for bilirubin conjugation and subsequent secretion into the biliary system, is deficient in newborns, often operating at 0.1–1 % of the adult liver capacity and not reaching full efficiency until 14 weeks of life [3, 4]. Increased intrahepatic circulation occurs in the newborn secondary to relative sterility of the neonatal gut. Under normal conditions, gut flora is responsible for converting the conjugated bilirubin secreted in bile into stercobilin – the material that gives stool its brown color. In the absence of sufficient intestinal flora, bilirubin is de-conjugated by beta-glucuronidase and reabsorbed resulting in increased intrahepatic circulation. Breast milk composition provides additional elements that contribute to hyperbilirubinemia. Both progesterone metabolites and lipoprotein lipase have been isolated in breast milk and shown to inhibit the action of UGT thus leading to decreased bilirubin clearance [5, 6].
Clinical Manifestations and Diagnosis
Secondary to placental clearance, infants do not exhibit jaundice over the first day or two of life. However, as the production begins to exceed the infants’ ability to process and clear the bilirubin, the stereotypical yellowing of the skin and sclera becomes clinically evident. Jaundice progresses in a cephalocaudal manner with symptoms appearing first over the head and progressing to the palms of the hands and soles of the feet. Bilirubin elevation can be correlated with degree of infant jaundice with whole body jaundice occurring with levels >15 mg/dL. Moderate jaundice (>12 mg/dL) occurs in at least 12 % of breast-fed infants with severe jaundice (>15 mg/dL) occurring in 2 % [7]. Kernicterus, a bilirubin-derived brain dysfunction resulting from deposition of the neurotoxic bilirubin in the grey matter of the newborn brain, is not thought to be associated with breast milk jaundice. However, kernicterus has been reported in otherwise healthy newborn infants with no other identified etiology for their jaundice [8]. Measurement of the total serum bilirubin concentration allows quantification of jaundice with a diagnosis of hyperbilirubinemia given to infants whose total bilirubin levels are elevated on the hour-specific Bhutani nomogram (see Fig. 8.1) [9].

Bhutani nomogram. Hour-specific nomogram developed to predict infants most at risk for developing severe hyperbilirubinemia (Reprinted with permission from publisher of Peds (American Academy of Pediatrics))
Treatment, Management, and Outcomes
The hyperbilirubinemia associated with breast milk jaundice will improve with cessation of breast-feeding for 24–48 h. However, given the lack of specificity and the breadth of potential etiologies for elevated serum bilirubin, jaundice should always be considered a sign of potentially more serious disease. Immediate investigations should be considered as to the etiology when [1] jaundice occurs before 36 h of age [2], jaundice persists beyond 10 days of age [4], serum bilirubin >12 mg/dL, and [10] elevation of the direct reacting fraction of bilirubin [1].
Gilbert Syndrome
Introduction
Gilbert syndrome (GS) is the most common inherited disorder of bilirubin glucuronidation and is characterized by chronic and recurrent elevations in unconjugated bilirubin in the setting of normal liver function and normal aminotransferases. The typical inheritance pattern is autosomal recessive as only individuals who are homozygous for promoter gene mutations express symptoms (see Pathogenesis). However, individuals who have heterozygous mutations do average higher bilirubin levels when compared to the general population [11].
Pathogenesis
Uridinediphosphoglucuronate glucuronosyltransferase 1 (UGT1) is a gene whose enzyme is responsible for the glucuronidation of several compounds in the body, including bilirubin. Bilirubin-specific UGT (UGT1A1 or BUGT) activity is decreased in patients with GS often secondary to mutations appreciated on the gene’s promoter region (see Fig. 8.2), leading to elevations in serum unconjugated bilirubin. The expression of the GS phenotype is likely multifactorial as not all individuals who carry homozygous mutations develop unconjugated hyperbilirubinemia [11].

Bilirubin metabolism. Mutations in bilirubin-uridine diphosphate glucuronosyltransferase (UGT1A1) are responsible for genetic errors in bilirubin conjugation that cause Gilbert syndrome as well as Crigler-Najjar syndromes 1 and 2. Rotor syndrome is caused by defective binding anions with deficient intracellular storage capacity of bilirubin
Clinical Manifestations and Diagnosis
Patients diagnosed with GS present in adolescence or young adulthood with mild, intermittent elevations in unconjugated bilirubin levels without evidence additional liver disease or dysfunction. Conjugated bilirubin levels seldom rise above 4 mg/dL. Often, patients are first identified on screening blood chemistries, or mild jaundice is appreciated during periods of fasting or in accompaniment of a viral illness. The majority of patients are asymptomatic. Physical exam findings may exhibit only mild scleral icterus, and yellowing of the skin is rare in patients with GS. Critical to the diagnosis of GS is the clinician’s suspicion and the avoidance of unnecessary invasive and investigatory testing. A presumptive diagnosis can be made when patients exhibit (1) unconjugated hyperbilirubinemia on repeat testing; (2) a normal complete blood count, blood smear, and reticulocyte count; and (3) no additional evidence of liver dysfunction or liver disease. While several tests can be used when GS is suspected, such as fasting or nicotinic acid administration with subsequent measurement of unconjugated bilirubin levels, clinical awareness and recognition remains the most important factor when entertaining a diagnosis of GS.
Treatment, Management, and Outcomes
GS is considered a benign condition. No specific treatment is required for patients with GS, and the most important aspect of the diagnosis is a recognition of the disorder and the understanding of its favorable outcomes.
Crigler-Najjar Syndrome
Introduction
Crigler-Najjar syndrome (CN) is a rare autosomal recessive disease characterized by elevated unconjugated bilirubin levels. Bilirubin elevations in CN result from defects in the gene that is responsible for bilirubin conjugation – bilirubin-uridine diphosphate glucuronosyltransferase (UGT1A1). Two distinct forms of the disease have been identified depending of the severity of UGT1A1 enzyme dysfunction (See Fig. 8.2).
Type 1 disease is more severe and associated with impressive, early jaundice with neurologic impairment and the development of kernicterus. Type 2 disease is often milder, with lower bilirubin levels and with improved patient survival.
Pathogenesis
UGT1A1 is the gene responsible for conjugation of bilirubin in the hepatocyte. The degree of genetic disruption of UGT1A1 results in the clinical severity that defines CNI and CNII. In the more severe CNI, there is complete absence of functional UGT1A1 activity, whereas in CNII UGT1A1 activity is markedly reduced. While mutations in the promoter region of the UGT1A1 gene result in the Gilbert’s phenotype, the mutations associated with CN are located primarily in the exon-coding regions of the gene. Patients with CN thus have a profound decrease in their ability to conjugate and subsequently excrete bilirubin in the bile.
Clinical Manifestations and Diagnosis
Patients with CN often present in the first few days of life with jaundice and serum bilirubin elevations, often greater than 20 mg/dL. Bilirubin elevations are primarily attributable to the unconjugated fraction. Pigmented stool is present as CN is not associated with biliary obstruction; however, fecal urobilinogen excretion is decreased secondary to reduced bilirubin conjugation and stool presence [12]. With exception of elevated bilirubin levels, liver function tests in children are usually normal and liver histology may only reveal deposition of bile pigments. Bilirubin-induced brain dysfunction, or kernicterus, is always a concern in children with CN. Bilirubin deposition in the grey matter of various central nervous system structures can result in a myriad of acute symptoms including lethargy, decreased feeding, hypo- or hypertonia, a high-pitched cry, spasmodic torticollis, opisthotonus, fever, seizures, and death.
Differentiating between CNI and CNII can be difficult. One major differing characteristic is the response to drugs that stimulate hyperplasia of the endoplasmic reticulum – such as phenobarbital. Serum unconjugated bilirubin can be reduced by the administration of phenobarbital in most patients with CNII but not CNI.
CN should be suspected in any infant with persistently elevated unconjugated bilirubin levels after evaluation for more common etiologies such as hypothyroidism, infection, and hemolysis. Genetic testing can be performed to assess UGT1A1 mutations, and liver biopsy can be used to obtain tissue for UGT1A1 function assays.
Treatment, Management, and Outcomes
The mainstay of treatment in patients with CN is phototherapy and plasmapheresis in order to lower serum bilirubin levels and prevent neurological sequelae. Additional therapeutic options are the oral administration of binding agents such as cholestyramine, agar, or calcium phosphate that trap unconjugated bilirubin in the gut and prevent enterohepatic recirculation [1]. Patients with CNII may respond to daily administration of phenobarbital; however, the response is often variable.
While bilirubin conjugation is abnormal, additional liver synthetic function is preserved in children with CN making them ideal candidates for orthotopic liver transplantation. Future therapies may include isolated hepatocyte cell transplant with donor hepatocytes containing functional UGT1A1; however, this therapy remains investigational.
The effects of elevated bilirubin concentrations on the central nervous system and the development of kernicterus remains the most important indicator of patient outcome. Patients with CNII whose UGT1A1 function may be preserved often are spared the neurological devastation seen in CNI. However, long-term effects of chronic bilirubin elevations have been shown to cause variable degrees of brain damage in older patients with CN [13].
Rotor Syndrome
Introduction
Rotor syndrome is a benign, autosomal recessive disorder resulting in chronic elevations of both conjugated and unconjugated bilirubin levels without evidence of hemolysis. The primary defect in Rotor syndrome is in the liver’s ability to store conjugated bilirubin resulting in elevated plasma concentrations.
Pathogenesis
While the exact abnormality resulting in Rotor syndrome remains unknown, the primary deficiency is thought to be related to a defective intracellular storage capacity of the liver for binding anions involved in the excretion of conjugated bilirubin (See Fig. 8.2).
The resulting deficiency of hepatocellular biliary storage allows for seepage of bilirubin back into the plasma resulting in the elevations of conjugated bilirubin that define Rotor syndrome.
Clinical Manifestations and Diagnosis
Rotor syndrome should be suspected in children presenting with jaundice and scleral icterus in the setting of normal liver function tests and no evidence of biliary obstruction or hemolysis. On laboratory evaluation, the conjugated bilirubin fraction is over half of the total serum bilirubin concentration. Total bilirubin levels generally reside in the 2–7 mg/dL range but occasionally may reach 20 mg/dL. A confirmatory diagnosis can be made when urinary coproporphyrin levels are measured to be 2.5–5 times higher than normal [14].
Treatment, Management, and Outcomes
Patients with Rotor syndrome require no specific therapy. Elevated serum bilirubin with jaundice may be chronic findings; however, no treatment is needed, and only reassurance and education should be offered.
Disorders of Carbohydrate Metabolism
A primary function of the liver is the management of glucose homeostasis. Glucose production can occur through a variety of metabolic pathways including gluconeogenesis as well as degradation of glycogen – the major storage form of glucose. Glucose along with galactose and fructose constitute the majority of dietary carbohydrates that are absorbed and processed to produce the energy required to sustain cellular function. Deficiencies in the metabolism of carbohydrates result in significant morbidity and mortality often affecting infants and neonates.
Galactosemia
Introduction
Galactosemia is an autosomal recessive disorder, with and incidence of 1:60,000 live births, characterized by an inability to affectively metabolize galactose. Galactosemia results from an enzymatic deficiency in one of three proteins in the metabolic pathway through which galactose is converted to glucose: galactokinase (GALK), galactose-1-phosphate uridyltransferase (GALT), and uridine diphosphate galactose-4-epimerase (GALE).
Pathogenesis
Galactose is metabolized into glucose-1-phosphate by a series of reactions involving the enzymes GALK, GALT, and GALE (See Fig. 8.3).

Galactose metabolism. Galactokinase (GALK), galactose-1-phosphate uridyltransferase (GALT), and uridine diphosphate galactose 4-epimerase (GALE) function to metabolize glucose from galactose. “Classical” galactosemia results from GALT deficiency
Although galactosemia can result from dysfunction of any enzyme involved in the pathway, “classical” galactosemia constituting the majority of cases involves a complete deficiency of GALT activity often due to the missense mutation on chromosome 9. Dysfunctional metabolism results in accumulation of the toxic by-products galactose-1-phosphate and galactitol that produce the pathological changes found in the liver, brain, kidney, and lens of the eye [1].
Clinical Manifestations and Diagnosis
Classic galactosemia presents in the first weeks of life after ingestion of breast milk or milk-based formulas containing lactose. Failure to thrive and jaundice are the most common presenting symptoms often accompanied by difficulty feeding, vomiting, and diarrhea. The range of presentation can be from an acute, life-threatening decompensation often associated with E. coli sepsis to a more subtle appearance with nonspecific manifestations. Physical exam reveals jaundice with hepatomegaly, ascites, edema, excessive bruising, hypotonia, and a full fontanelle. Laboratory investigations show multisystem abnormalities with direct hyperbilirubinemia, aminotransferase elevation, hypoalbuminemia, and coagulopathy.
While newborn screening (NBS) has allowed early detection of children with suspected galactosemia, the genetic complexity and nonstandardization of screening techniques has complicated matters for general pediatricians who are often left to interpret the results. Although classic galactosemia is indeed a medical emergency, most positive NBS results occur due to false positives and Duarte variants – a genotypic variant that results in varying degrees of GALT phenotypic activity [2]. The demonstration of complete absence of GALT activity via a quantitative assay remains the gold standard for diagnosis [4].
Treatment, Management, and Outcomes
The immediate management of a child with galactosemia is urgent removal of galactose from the diet. Further acute management should be aimed at treating comorbidities present at diagnosis – such as jaundice, infection, coagulopathy, and acidosis. Antibiotics, intravenous fluids, blood products, and vitamin K are often needed. Acute symptoms often correct soon after galactose restriction. Infants should be placed on an appropriate nutritional alternative such as a soy-based formula with referral to a dietary specialist. Once solids are introduced, continued avoidance of lactose-containing foods is recommended.
Patients with galactosemia should be followed to ensure dietary compliance and assess end-organ damage. Most affected children will have some degree of intellectual deficit with speech and language delays. Abnormalities in coordination, gait, and balance are also seen [10].
Hereditary Fructose Intolerance
Introduction
Hereditary fructose intolerance (HFI) is an autosomal recessive disorder, with an incidence of 1:20,000, characterized by an inability to effectively metabolize fructose-1-phosphate. HFI results from a deficiency of the catalytic enzyme fructose-1-phosphate aldolase, otherwise known as aldolase B, located in the liver, kidney, and small intestine.
Pathogenesis
Fructose is a widely available monosaccharide often found naturally but also from the hydrolysis of the disaccharide sucrose into fructose and glucose. Once absorbed, fructose is taken up mostly by the liver (75 %) but also by the kidney and small intestines (remaining 25 %) where enzymes are able to further metabolize the sugar (See Fig. 8.4).

Fructose metabolism. Metabolic pathway of fructose to pyruvate. Deficiency of aldose B results in toxic accumulation of fructose-1-phosphate and hereditary fructose intolerance
The biochemical toxicity results from a deficiency of aldose B and subsequent accumulation of fructose-1-phosphate [1]. Increased fructose-1-phosphate inhibits both gluconeogenesis and glycogenolysis evident in the manifestation of hypoglycemia, renal, and hepatic dysfunction in affected patients.
Clinical Manifestations and Diagnosis
In general, children with HFI are healthy until they ingest fructose and/or sucrose. Patients commonly present with vomiting, hepatomegaly, decreased oral intake, and failure to thrive. A detailed nutritional history is critical as symptom onset frequently correlates with intake of fructose-containing foods. Laboratory investigations may reveal evidence of acute liver failure, proximal renal tubular dysfunction, and hypoglycemia. The presence of reducing substances in the urine may result from fructosuria. More than 95 % of patients with HFI can be diagnosed with noninvasive DNA amplification with a limited number of allele-specific oligonucleotides [15].
Treatment, Management, and Outcomes
Treatment consists of fructose and sucrose removal from the diet. Particular attention should be paid to food additives, pill coatings, and medication suspensions. Complete elimination is rarely a therapeutic goal given the wide distribution and high concentration of fructose in foods. Optimal levels of restriction have not been established, and while some patients are able to demonstrate normalization of hepatic and renal function with low fructose intake, others may suffer with chronic, nonspecific symptoms despite treatment [16]. Multivitamin supplementation, particularly forms with ascorbic acid and folates, should be prescribed due to the need for fruit and vegetable restriction [17]. Once appropriate dietary restriction is implemented, clinical progression is rarely encountered and the majority of children exhibit normal growth and intellectual development.
Fructose-1,6-Bisphosphatase Deficiency
Introduction
Fructose-1,6-bisphosphatase (FDPase) deficiency (also known as fructose 1,6-diphosphatase deficiency) is a rare autosomal recessive disorder of gluconeogenesis with an estimated incidence of 1:350,000.
Pathogenesis
FDPase is important in the processes of gluconeogenesis, and its deficiency results in impaired formation of glucose from precursors – including fructose. Importantly, glycogenolysis is not affected in these individuals (See Fig. 8.5).

Fructose-1,6-bisphosphatase. Fructose metabolism resulting in gluconeogenesis and glycogenolysis. Fructose-1,6-bisphosphatase is involved in gluconeogenesis. Deficiency results in fructose-1,6-biphosphatase (FDPase) deficiency
Clinical Manifestations and Diagnosis
Infants with FDPase deficiency often present within the first days of life with hypoglycemia, lactic acidosis, and compensatory hyperventilation, while older children can present with acute ataxia and lethargy [18]. Hepatomegaly may be present, but transaminases are normal. Fasting may trigger attacks of hypoglycemia. Lactate, pyruvate, and ketones may be elevated. Aminoaciduria with 2-oxoglutaric acid is seen on urine biochemical analysis. Diagnosis can be made with DNA analysis [17]. If results are inconclusive, a liver biopsy confirming decreased enzyme activity or mutation analysis is diagnostic.
Treatment, Management, and Outcomes
In acute crisis, patients with FDPase deficiency respond rapidly to intravenous or oral glucose with sodium bicarbonate administration to correct acidosis. Long-term therapy consists of fasting avoidance. Frequent feedings and the use of slowly absorbed carbohydrates such as cornstarch are helpful [17]. With appropriate treatment, complications are often avoided while growth and development is normal.
Glycogen Storage Diseases
Introduction
Glycogen is a polysaccharide that constitutes the primary carbohydrate storage compound and is the principal source of energy providing substrates for the generation of ATP. Although particularly prevalent in liver and muscle tissues, glycogen is present in nearly all animal cells and is processed during fasting resulting in release of glucose needed to sustain cellular processes and glucose homeostasis. The complexity of this process is underscored by the multitude of enzymes involved, deficiencies in which result in the 12 recognized forms of glycogen storage diseases (GSD). The overall incidence of GSD is estimated to be 1:20,000–40,000 cases per live birth. The discussion here is limited to types I, III, and IV because their clinical expression primarily involves the liver (See Fig. 8.6).

Glycogen metabolism. Metabolic pathway of glycogen metabolism. Deficiencies in glucose-6-phosphatase, debranching enzyme, and branching enzyme result in the clinical features of glycogen storage diseases I, III, and IV whose clinical features primarily involve the liver
GSD I (Von Gierke Disease)
Pathogenesis
GSD I is an autosomal recessive disease of glycogen metabolism with an incidence of approximately 1:100,000 live births. Several enzymatic defects have been identified resulting in the subclassification of GSD I. Types Ia and Ib constitute the majority of cases, while Ic and Id are extremely rare and will not be discussed in this chapter. Patients with GSD Ia are noted to have complete absence of glucose-6-phosphatase – an enzyme involved in the final process of both gluconeogenesis and glycogenolysis. Patients with GSD Ib have a defect in the enzyme glucose-6-phosphatase translocase which transports glucose-6-phosphatase from the cytoplasm into microsomes [19].
Clinical Manifestations and Diagnosis
Children with GSD1 typically present in early infancy with profound hepatomegaly and hypoglycemia associated with fasting. Oftentimes, symptoms may be first appreciated once a child begins to sleep through the night – a consequence of prolonged fasting. Other presenting signs and symptoms include protuberant abdomen due to hepatomegaly, metabolic derangement, seizures, growth failure, recurrent infections, muscular hypotonia, epistaxis, and delayed psychomotor development [20]. In addition to profound hypoglycemia, other laboratory findings include lactic acidosis, hyperuricemia, hypophosphatemia, hyperlipidemia, and platelet dysfunction. Neutropenia is a unique manifestation of GSD Ib. Confirming the diagnosis with DNA testing for common mutations is recommended. In the absence of identifiable genetic abnormality, liver enzyme activity can be measured.
Treatment, Management, and Outcomes
The main goal of therapy in the management of GSD Ia is to maintain normal blood glucose concentrations and correct metabolic abnormalities. A carbohydrate-balanced diet with frequent feedings over the day paired with continuous nocturnal feeds and/or the addition of uncooked cornstarch is the mainstay of treatment. Cornstarch is known to provide a “time-release” source of glucose given its slow degradation to glucose by α-amylase. However, decreased α-amylase levels in young infants may lead to suboptimal management with resulting growth failure; therefore, high-starch meals while awake coupled with continuous overnight enteral feeds with a high-glucose, low-fat formula should be used. Published guidelines discussing appropriate biochemical targets are available to assist in the management of patients with GSD I [21]. Further treatment strategies may be required for the management of secondary complications such as hyperuricemia, lactic acidosis, hyperlipidemia, and hypertension. Management of GSD Ib is identical to Ia with the addition of granulocyte colony-stimulating factor (G-CSF) to correct neutropenia and defects in phagocytic cell function.
Current treatment strategies have allowed for patients with GSD to survive well into the third decade with normal growth and pubertal development. Hepatocellular adenomas may develop in patients with GSD I, and the malignant degeneration of the adenomas has been reported. Annual serum alpha-fetoprotein levels and liver ultrasounds are recommended. While adequate metabolic control can result in normal neurological development, repeated episodes of hypoglycemia and lactic acidosis can also result in mental handicaps. Additionally, GSD 1 patients have increased rates of cardiovascular disease, pancreatitis, and renal insufficiency.
GSD III (Forbes Disease, Cori Disease, Glycogen Debrancher Deficiency)
Pathogenesis
GSD III is an autosomal recessive disease resulting from mutations in the gene that codes for amylo-1, 6-glucosidase – a glycogen debranching enzyme. The two main subclassifications are GSD IIIa that involves both the liver and muscle and constitutes the majority of cases and GSD IIIb that affects the liver only.
Clinical Manifestations and Diagnosis
Children with GSD III often have similar clinical presentations to those with GSD I with hepatomegaly, hypoglycemia, hyperlipidemia, and acidosis [22]. Muscular involvement manifesting as weakness, muscle wasting, and cardiac dysfunction can be used to clinically distinguish GSD I and IIIa; however, the onset of muscular symptoms usually does not occur until adulthood. Furthermore, children with GSD IIIb do not develop muscular manifestations as the affected RNA isoform is only expressed in the liver.
Biochemically, children with GSD III will have similar, but less severe, abnormalities compared to children with GSD I with the exception that GSD III can develop hyperuricemia and elevated lactate levels. Elevated creatinine kinase levels indicating muscular involvement are present in GSD IIIa.
Although DNA testing of the aminoglucosidase gene is available, the diagnosis should be confirmed with direct measurement of amylo-1, 6-glucosidase activity in liver (IIIa/b) and muscle (IIIa) tissue samples. Liver biopsy may reveal the presence of fibrous septa and the paucity of fat that is unique to GSD III [1].
Treatment, Management, and Outcomes
Treatment of GSD III is directed toward avoidance of hypoglycemia. Just as the manifestations of GSD III are less severe than GSD I, so too is the demands to maintain strict dietary measures. As gluconeogenesis is not affected in GSD III, high-protein diets can be used in order to manage the disease [23].
Overall, hepatic involvement in GSD III is considered mild and self-limiting and improves with age usually with resolution by the second decade of life [17]. Hepatocellular carcinoma and cirrhosis have been reported, and practitioners should monitor with at least annual serum alpha-fetoprotein and liver ultrasound.
GSD IV (Andersen Disease, Glycogen Branching Enzyme Deficiency)
Pathogenesis
GSD IV is an autosomal recessive disorder resulting from mutations in the gene encoding the glycogen branching enzyme (GBE). GBE catalyzes the attachment of glucosyl chains to glycogen. GSD IV results from a functional deficiency of GBE and subsequent abnormal glycogen structure. Various mutations result in a spectrum of disease ranging from intrauterine hydrops to perinatal hypotonia and cardiomyopathy. Four main phenotypic variants have been described based on age of presentation [24] (see below).
Clinical Manifestations and Diagnosis
The earliest manifestation may be in the perinatal period with fetal akinesia deformation sequence (FADS) that is characterized by contractures, hydrops fetalis, cardiac dysfunction, and death. A congenital form is defined by hypotonia, muscle atrophy, cardiomyopathy, and rapid progression to death. A juvenile variant has been described with muscular cardiomyopathy. An adult form of GSD IV is described primarily with neuromuscular manifestations such as myopathy, upper and lower neuron dysfunction, and dementia.
GSD IV is a unique disorder of glycogenolysis in that hypoglycemia is uncommon. Elevated transaminases are accompanied by elevations in alkaline phosphatase. A diagnosis of GSD IV is confirmed by the demonstration of absent branching enzyme activity in skin fibroblasts. Mutations analysis of the GBE gene can also reveal the diagnosis.
Treatment, Management, and Outcomes
Liver transplantation is the sole medical treatment for patients with diagnosed GSD IV. Without prompt diagnosis and transplant, most patients rapidly progress to end-stage liver disease, cirrhosis, and death by the third year of life.
Disorders of Amino Acid and Organic Acid Metabolism
Introduction
Although rare, these disorders may be encountered in practice, and an approach to the diagnosis, management, and outcomes is essential. The most common and important disorders that have hepatic manifestations include disorders of tyrosine, with its fulminant presentation in early infancy, and methylmalonic acid, isovaleric acid, and maple syrup urine disease which may manifest as recurrent episodes of liver dysfunction and alterations in neurocognitive function in infancy or early childhood.
Type I Tyrosinemia
Introduction
Type I tyrosinemia is a key element in the differential diagnosis of neonatal fulminant hepatic failure. Inherited as an autosomal recessive trait, it has an incidence of 1:100,000 with varying incidence by geography and an incidence of 1:1,846 in the Saguenay-Lac-Saint-Jean region of Quebec. Although historically type 1 tyrosinemia was uniformly fatal, the development of a treatment strategy with NTBC [2-(2-nitro-4-trifluromethylbenzoyl)-1,3-cyclohexanedione] has dramatically changed the long-term prognosis for these patients if identified and treated early in the course.
Pathogenesis
The clinic-pathologic manifestations of the disease relate to the metabolic defect which is deficiency of the enzyme fumarylacetoacetate hydrolase, the last enzyme in the pathway of tyrosine degradation. The consequent accumulation of fumarylacetoacetate and maleylacetoacetate and their by-products, succinylacetone and succinylacetoacetate, is responsible for the hepatocellular injury, propensity to hepatocellular carcinoma (HCC), and the development of neurotoxicity (see Fig. 8.7).

Tyrosine degradation pathway including site of action of NTBC [2-(2-nitro-4-trifluromethylbenzoyl)-1,3-cyclohexanedione]
Clinical Manifestations
Most patients present with the acute form of disease characterized by findings of acute liver failure with poor growth, vomiting, ascites, coagulopathy, hypoglycemia, hypoalbminemia, and hyperbilirubinemia. This condition must be differentiated from other forms of acute liver failure since life-saving therapy is available for tyrosinemia. Patients with the chronic form have growth failure; renal tubular dysfunction including Fanconi syndrome; and neurologic crises with pain and paresthesias associated with evidence of liver dysfunction including hepatomegaly, transaminasemia, hyperbilirubinemia, and coagulopathy with long-term risk of HCC. The diagnosis is made by identifying elevated urinary succinylacetone. Serum α-fetoprotein may be markedly elevated at diagnosis and hemolytic anemia may be present. Renal tubular dysfunction is common with increased excretion of phosphate, glucose, protein and amino acids. Liver histologic findings in acute disease include fatty infiltration and changes of pseudoacinar formation of lobules found in metabolic disorders, hepatocellular necrosis. Chronic forms lead to micronodular cirrhosis with regenerative nodules which may include dysplasia or HCC.
Treatment, Management, and Outcomes
Neonates presenting with acute liver failure should be treated aggressively to prevent and treat complications such as hypoglycemia, sepsis, and coagulopathy. Active bleeding should be treated with fresh frozen plasma, platelets and cryoprecipitate based upon the laboratory findings. Infusions of intravenous glucose should be given to prevent hypoglycemia. Dietary restriction of tyrosine with formulas such as Tyrex® or Tyros® should be used. NTBC ((2-(2-nitro-4-trifluromethylbenzoyl)-1,3-cyclohexanedione, nitisinone) which inhibits 4-hydroxyphenyl dioxygenase, the enzyme proximal to the deficient enzyme in type I tyrosinemia, is effective in the majority of patients with reversal of hepatic and renal dysfunction. Patients should be monitored with annual eye exams to assess eye changes associated with treatment and periodic liver chemistries and semiannual abdominal ultrasound and α-fetoprotein to assess for evidence of neoplasia. Survival with NTBC treatment is excellent with remarkable improvement over a <40 % survival at 1 year with diet alone in those presenting at less than age 2 months [25–27]. Liver transplantation should be considered for patients unresponsive to NTBC with excellent overall 1-year survival rates exceeding 85 % [28].
Methylmalonic Acid, Isovaleric Acid, HMG-CoA Lyase, and Maple Syrup Urine Disease
Introduction
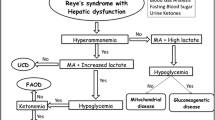
These disorders are collectively quite rare but important to the practicing gastroenterologists and pediatricians because affected infants/children may present with recurrent episodes of vomiting, acidosis, modest transaminasemia, hyperammonemia, and variable neurocognitive dysfunction. Collectively, these conditions, the urea cycle defects, and defects of fatty acid oxidation were conditions that previously were part of the differential diagnosis of Reye syndrome and might present to the gastroenterologist or practicing pediatrician.
Pathophysiology
Each of the conditions has its unique metabolic profile found in serum and urine. As examples, accumulation of methylmalonic acid (MMA) occurs in methylmalonic academia; isovaleric acid, 3-hydroxyvaleric acid, N-isovalerylglycine, and isovalerylcarnitine in isovaleric academia; branched chain amino acids (valine, leucine, and isoleucine) in maple syrup urine disease; and 3-hydroxy-3-methylglutaric, 3- methylglutaconic, 3-methylglutaric, and 3-hydroxyisovalaeric acids in HMG-CoA lyase deficiency.
Clinical Manifestations
The diagnosis of each of these conditions is usually made in the newborn period (although some present later in life) by identifying typical urinary organic acid or serum amino acid patterns in the presence of acidosis, hypoglycemia, and alterations in consciousness. Older infants and children may present with recurrent episodes of alterations in consciousness. Liver chemistries may be abnormal with elevated serum aminotransferases and normal serum bilirubin. Acidosis, hypoglycemia, and hyperammonemia are commonly present. In patients with isovaleric acidemia, the patient has a “sick sweet” smell of sweaty feet or cheese during episodes. These conditions are commonly part of the newborn screen performed in all US states. All are inherited as autosomal recessive traits and family history may be revealing.
Treatment, Management, and Outcomes
Treatment for each of these conditions is dictated by the specific defect. Typically, for each condition intravenous glucose is administered with the intent of minimizing ketosis. Protein restriction with avoidance of catabolism is essential in the management of these conditions. If marked elevations of serum ammonia are found, use of intravenous sodium phenylacetate, sodium benzoate, and arginine should be considered or even hemodialysis for refractory episodes. In selected circumstances of MMA, patients are responsive to vitamin B12, and this should be considered when a patient is acutely ill with MMA. Chronic management includes diets tailored for specific defects. The prognosis of these conditions is guarded with morbidity and mortality from the acute episodes and associated morbidities accompanying the diseases. Because of recurrent metabolic crises and associated neurodevelopmental delay associated with repeated central nervous system injury, liver transplantation has been considered an option for some patients.
Fatty Acid Oxidation Defects
Mitochondrial fatty acid oxidation (FAO) defects are a group of inherited metabolic disorders that contribute greatly to pediatric morbidity and mortality. FAO provides most of the energy supplied to the heart and skeletal muscle and is crucial in the maintenance of glucose homeostasis during periods of fasting. The end product of FAO is ketones that constitute an important secondary energy source for tissues when glucose supplies are depleted.
MCAD (Medium-Chain Acyl-CoA Dehydrogenase) Deficiency
Introduction
MCAD is a nucleus-encoded mitochondrial matrix enzyme that catalyzes the initial dehydrogenation step in the β-oxidation of medium-chain fatty acids. MCAD deficiency is inherited in an autosomal recessive pattern with a prevalence of 1 in 10,000–25,000. It is considered to be an important disorder of FAO and is the most common β-oxidation defect.
Pathogenesis
MCAD is responsible for the initial metabolism of acyl-CoAs with chain lengths of 4–12 carbon atoms. Most patients with MCAD deficiency have a missense mutation with ensuing enzyme inactivity. MCAD deficiency leads to fasting-induced hypoglycemia and accumulation of toxic acyl-CoA compounds.
Clinical Manifestations and Diagnosis
The major role of β-oxidation is to supplement energy during fasting. Historically, the inherited deficiency of MCAD with resulting dysfunction of the β-oxidation process often presented in the first months to years of life with recurrent emesis, lethargy, coma, and even death provoked by fasting or illness. MCAD deficiency is now part of newborn screening. Hepatomegaly may be present during acute decompensation, which is also characterized by hypoketosis and hypoglycemia, anion gap acidosis, hyperuricemia, elevated liver enzymes, and hyperammonemia. Increased metabolites (acylcarnitines) may be present. Characteristic urine gas chromatographic findings include elevated levels of sebacic, sebaric, and adipic acid. Liver biopsy reveals microvesicular fatty changes. Fibroblast enzymatic assays and/or genetic testing of the MCAD gene can make a confirmatory diagnosis [1].
Treatment, Management, and Outcomes
The mainstay of treatment for patients with MCAD deficiency is avoidance of fasting. Supplementation of the diet with carbohydrates or glucose can also assist in the avoidance of catabolism and symptom development during acute illness [29]. Unidentified patients with this disorder have a significant risk of sudden death in early childhood, and survivors have a significant risk of developmental disability and chronic somatic illnesses [30].
LCHAD (Long-Chain 3-Hydroxyacyl-CoA Dehydrogenase) Deficiency
Introduction
LCHAD deficiency is a rare, autosomal recessive inborn error of fatty acid metabolism. Long-chain 3-hydroxyacyl-CoA dehydrogenase (LCHAD) is an enzyme that constitutes part of the mitochondrial trifunctional (TFP) complex. This complex is composed of 3 separate enzymes whose main function is to metabolize long-chain fatty acids often found in milk and oils. Complete TFP deficiency is rare and more commonly patients are found to have isolated LCHAD deficiency with the remaining two enzymes of the TFP complex functioning at or above 60 % or normal [31].
Pathogenesis
The multienzyme mitochondrial TFP complex consists of 4 α- and 4 β-subunits that together work to catalyze the last three steps in β-oxidation of long-chain fatty acids. The α-subunit contains the LCHAD enzyme as well as the long-chain enoyl-CoA hydratase enzyme, while the β-subunit contains the long-chain 3-ketothiolase (LKAT) enzyme [32]. The most common genetic mutation associated with LCHAD deficiency is the mutation in the HADHA gene that results in isolated LCHAD dysfunction. The resulting pathology in patients with LCHAD deficiency results from inadequate energy supply and toxic accumulation of metabolites.
Clinical Manifestations and Diagnosis
The classic presentation of a child with LCHAD deficiency is a newborn with acute onset feeding difficulties, lethargy, hypotonia, and hypoketotic hypoglycemia. Neuropathy and retinopathy are two unique sequelae of LCHAD deficiency (these are also found in TFP complex deficiency) that often occur in older individuals, and LCHAD deficiency in a fetus is now well recognized to predispose mothers to gestational complications such as HELLP (hemolysis, liver dysfunction, and low platelets) syndrome and AFLP (acute fatty liver of pregnancy).
Newborn screening programs now test for LCHAD deficiency. Confirmation of the diagnosis is possible by measuring LCHAD activity in lymphocytes, fibroblasts, and muscle or liver biopsies and by mutational analysis [33].
Treatment, Management, and Outcomes
The primary treatment of LCHAD deficiency is dietary restriction with a low-fat, carbohydrate-rich diet. Supplementation with a medium-chain triglyceride-enriched diet can also be beneficial. L-carnitine has been given to patients with LCHAD deficiency; however, there have been several reports of adverse effects of this treatment strategy due to accumulation of long-chain acyl-CoA esters. Long-term complications include progressive peripheral neuropathy and retinopathy. Despite early recognition and dietary treatments, the morbidity in LCHAD deficiency remains high.
HMG (3-Hydroxy-3-Methylglutaryl) CoA Synthase Deficiency
Introduction
Mitochondrial HMG-CoA synthase is an enzyme that regulates the formation of ketone bodies. It is the only disorder exclusively effecting hepatic ketogenesis. Deficiency of HMG-CoA synthase is inherited in an autosomal recessive pattern.
Pathogenesis
HMG-CoA synthase catalyzes the rate-limiting step in the formation of ketones. Under normal conditions, when the body undergoes stressors such as acute illness or fasting, ketone production is increased, and the resulting product is utilized as an alternative energy source by the brain. In patients with HMG-CoA synthase deficiency, the ensuing enzyme inactivity results in failure to produce ketones during stress with resultant hypoketotic hypoglycemia often progressing to encephalopathy and coma.
Clinical Manifestations and Diagnosis
Patients often present initially with hepatomegaly, encephalopathy, and hypoketotic hypoglycemia often associated with acute illness or prolonged fasting. In the majority of cases, patients are asymptomatic prior to their acute decompensation. Clinically, these patients can present similarly to those with MCAD deficiency; however, in HMG-CoA synthase deficiency, lactate is often normal as are liver transaminases. Currently, few diagnostic tests indicate the diagnosis of HMG-CoA synthase deficiency, and high clinical suspicion is required. While the combination of normal acylcarnitine and absence of urinary ketones suggests the diagnosis, definitive testing can only be accomplished with molecular testing [34].
Treatment, Management, and Outcomes
Prompt clinical recognition along with patient and family education on the importance of fasting avoidance is critical in the management of patients with HMG-CoA synthase deficiency. No long-term organ dysfunction has been appreciated in patients with HMG-CoA synthase deficiency.
CACT (Carnitine-Acylcarnitine Translocase) Deficiency
Introduction
Carnitine-acylcarnitine translocase (CACT) deficiency is a rare β-oxidation defect often associated with cardiomyopathy and early childhood death.
Pathogenesis
CACT is one of the enzymes that constitute the carnitine shuttle needed to transport long-chain fatty acids from the cytosol into the mitochondria where β-oxidation occurs. CACT is the enzyme responsible for the transfer of fatty acids into the mitochondria in exchange for free carnitine. Subsequent oxidation of long-chain fatty acids produces ATP used for energy or is converted to ketone bodies. Enzyme dysfunction impairs mitochondrial fatty acid oxidation.
Clinical Manifestations and Diagnosis
Patients often present during the neonatal period with hypoketotic hypoglycemia and significant cardiomyopathy often resulting in death. Additional manifestations include hepatic dysfunction, ventricular arrhythmias, seizures, and apnea [1]. There have been few reports of a milder phenotype often associated with the absence of significant cardiac involvement. Laboratory investigations often reveal hyperammonemia, elevated creatinine kinase, elevated transaminases, dicarboxylic aciduria, decreased carnitine, and elevations of long-chain acylcarnitines. Many states now include CACT analysis on their newborn screens. A definitive diagnosis is made with mutational analysis of the CACT gene.
Treatment, Management, and Outcomes
Treatment strategies include low-fat diet, medium-chain triglyceride supplementation, intravenous glucose to assist in the prevention of lipolysis, and carnitine supplementation [1]. Medium-chain triglycerides are utilized because they do not require transport via CACT across the mitochondrial membrane. Morbidity in patients with CACT deficiency when accompanied cardiac involvement remains high, and most children will die within the first year of life. In contrast, children with CACT deficiency without cardiac involvement have been reported to survive the neonatal period and into childhood [35].
Urea Cycle Defects
Introduction
Ureagenesis is an essential hepatic function that allows degradation of protein to its end product of ammonia to be rendered nontoxic (See Fig. 8.8).

Urea cycle with depiction of pathway within mitochondrion and cytoplasm with highlighted enzymatic steps commonly identified in inborn errors of ureagenesis
Ammonia and other precursor metabolites are known to be neurotoxic with the developing central nervous system particularly susceptible to injury. These conditions have an incidence of 1:8,200–1:30,000. All are inherited in autosomal recessive manner except ornithine transcarbamylase (OTC) deficiency which is inherited as a sex-linked trait.
Pathophysiology
Disorders of ureagenesis lead to accumulation of ammonia and glutamine and other products proximal to the point of block of the cycle associated with each disorder. The accumulation of ammonia and its by-products leads to hepatocellular dysfunction with associated liver enzyme elevation and, in some cases, synthetic dysfunction with profound effects on the central nervous system with direct toxic effects on the brain with attendant brain swelling.
Clinical Manifestations
The most common disorders of ureagenesis including ornithine transcarbamylase deficiency (OTC), carbamoyl phosphate synthetase (CPS), and argininosuccinate synthetase (ASS) or citrullinemia commonly present in the neonatal period (see Fig. 8.8). Infants with urea cycle defects are normal at birth and rapidly develop hyperammonemia and cerebral edema and progressive coma in the first days of life. These conditions are commonly mistaken to be septic because of lethargy, hypothermia, hyper- or hypoventilation, and seizures. Repeated episodes of hyperammonemia can lead to variable long-term neurologic impairment ranging from mild cognitive dysfunction to severe cerebral palsy. There may be a family history of neonatal deaths particularly in males with OTC deficiency. In older children, particularly in females who have hemizygous ornithine transcarbamylase deficiency, recurrent episodes of vomiting with variable alterations in consciousness or aversion to protein may be presenting complaints. During episodes of hyperammonemia, there may be mild elevation in the aminotransferases with minimal evidence of other hepatic dysfunction [36, 37]. Some older children may present with marked elevation of aminotransferases without hyperbilirubinemia and coagulopathy suggestive of acute liver failure. The diagnosis is made by a high index of suspicion in the presence of elevated plasma ammonia levels and reduced blood urea nitrogen and serum amino acid patterns typical of specific defects and the presence of orotic acid with OTC deficiency (Fig. 8.9). Definitive diagnosis of a urea cycle defect can be made by molecular genetic testing of measurement of enzyme activity in the liver. Molecular genetic testing is available for all of the urea cycle enzyme defects and is the diagnostic method of choice. Many states have newborn screening for selected urea cycle defects; however, CPS1 and OTC deficiencies are not reliably detected and manifestations of disease may precede the return of the results to the physician. For pregnancies at risk for an affected infant with a family history, prenatal testing via testing of DNA from amniocentesis or chorionic villus sampling.

Algorithm depicting the approach to the assessment of the infant/child with hyperammonemia
Treatment, Management, and Outcomes
Treatment should be tailored to the specific urea cycle disorder and should be directed by a metabolic specialist at a referral center. Initial therapy should be directed at returning elevated plasma ammonia levels to the normal range. Reversing catabolism is essential, and nutritional support with hypertonic glucose should be initiated immediately. Searches for causes of catabolic state such as sepsis should be made and presumptive treatment given. Initial pharmacologic interventions with alternative pathway excretion of excessive nitrogen should be considered using intravenously administered sodium phenylacetate and sodium benzoate with arginine. Neomycin and lactulose tend not to be very effective and should not be used. If rapid improvement in plasma ammonia is not observed or if plasma ammonia is markedly increased, immediate treatment with hemodialysis should be considered. If dialysis is initiated, it should be continued until plasma ammonia falls below 150 μmol/L. Once the acute hyperammonemic crisis is resolved, feeding should be commenced with a protein restricted formula with at least ½ of protein as essential amino acids. Sodium phenylbutyrate (Buphenyl®) should be initiated, and a newer product glycerol butyrate, which is more palatable than Buphenyl, may be available soon. Citrulline or arginine should be added based upon the location of the block in the urea cycle. Long-term support should include surveillance of illnesses that may precipitate catabolism and hyperammonemia protein restriction. In patients with severe, recurrent episodes of hyperammonemia, liver transplantation is an effective means to prevent future episodes. Currently, no effective protocols for gene therapy exist. After transplantation, there should be no further deterioration in neurologic dysfunction. With effective alternate pathway therapy, the 1-year survival for urea cycle defects is 92 %. Of these survivors, 79 % have mental retardation with a mean intelligence quotient of 43, 46 % have cerebral palsy, and 4 % are blind [38]. The IQ is correlated with the duration of elevated plasma ammonia not the peak concentration [39]. Hemizygote OTC females have a good prognosis but may have recurrent episodes of hyperammonemia which will compromise neurocognitive function.
Bile Acid and Biliary Transport Defects
Biliary Transport Defects
Introduction
Normal bile flow is dependent in part upon specific membrane transporters found in the liver and intestine. Inherited defects in the genes for some of these transporters lead to cholestasis and as a group comprise conditions termed progressive familial intrahepatic cholestasis (PFIC). PFIC diseases are inherited as autosomal recessive traits and generally present as cholestasis in the first year of life leading to progressive liver injury associated with a family history of similarly affected infants/children/adults.
Pathophysiology
Three conditions comprise the currently known group of diseases referred to as PFIC 1, 2, and 3. Patients with PFIC 1 have mutations in the FIC1 gene (ATP8B1) which causes progressive disease. This disease was initially described as two distinct clinical entities, Byler disease and benign recurrent intrahepatic cholestasis (BRIC). Both diseases are the result of abnormalities in FIC1 but differ in severity of the underlying defect in FIC1, with milder defects being present in BRIC. Other gene modifiers may also be responsible for variations in the PFIC phenotype. FIC1 mediates the flipping of aminophospholipids from the outer to inner hemi-leaflet of the canalicular membrane. The exact nature of how FIC1 deficiency causes disease is not known. FIC1 is located on other tissues including the pancreas and intestine leading to other extrahepatic symptoms and signs.
Patients with PFIC 2 deficiency have defects in the canalicular bile salt export pump caused by mutation in ABCB11. BRIC-like disease (BRIC2) and intrahepatic cholestasis of pregnancy (ICP) have been observed in children and adults with genetic defects in bile salt excretory pump (BSEP). BSEP is responsible for transporting bile acids from inside the hepatocyte into the bile canaliculus.
PFIC 3 is caused by mutations in ABC B4 which encodes multidrug resistance-associated protein 3 (MDR3) and mediates the flopping of aminophospholipids from the inner to outer hemi-leaflet of the canalicular lipid bilayer. ABCB4 mutations have also been associated with low phospholipid-associated cholelithiasis syndrome and ICP.
Pathophysiology
Liver disease in PFIC results from the effects of hepatocellular accumulation of bile acids. The liver disease may be mild or severe, depending on the specific gene defect present. In FIC1 deficiency, biliary excretion of bile acids is diminished. The rate of progression to end-stage liver disease in FIC1 deficiency may be slower than in BSEP deficiency. In severe forms of BSEP deficiency, BSEP expression and function are completely absent. Hepatocellular bile acids accumulation is pronounced causing rapidly progressive liver disease. End-stage liver disease in severe BSEP deficiency can occur in the first 1–2 years of life. In MDR3 deficiency, phospholipids in canalicular bile are either deficient or absent leading to the formation of a toxic bile that likely causes the pathogenesis of a progressive intrahepatic cholangiopathy. The resulting liver disease is a consequence of the cholestasis and inflammatory response generated by this cholangiopathy. Hepatocellular injury in MDR3 deficiency also results from intracellular bile salt accumulation.
Clinical Manifestations
Patients with severe forms of these diseases present with jaundice and/or pruritus. Life-threatening hemorrhage, secondary to cholestasis-related vitamin K deficiency, can also be a dramatic early presentation of PFIC. Profound, medical therapy-resistant pruritus is one of the most common early manifestations of all three of these forms of PFIC. Typically scratching begins between ages 6 and 12 months. Patients with PFIC I and 2 have markedly elevated serum bile acid levels with mildly elevated serum bilirubin, normal serum gamma glutamyl transpeptidase (gGTP), normal serum cholesterol, and only mild elevation in serum aminotransferase values. FIC1 deficiency is a systemic condition, and affected children may have growth failure, hearing problems, recurrent respiratory problems, elevated sweat chloride, recurrent pancreatitis, and diarrhea that is independent of the cholestasis. PFIC 2 has a strong association with hepatocellular carcinoma even in the first years of life. Affected patients with BRIC related to defects in BSEP may be associated with cholelithiasis [40–42]. MDR3 deficiency should be suspected in children with progressive cholestasis who have an elevated serum gGTP and no evidence of extrahepatic biliary tract disease. In patients with severe disease, biliary phospholipid concentrations are markedly reduced. PFIC 3 may present with intrahepatic cholestasis of pregnancy, drug-induced cholestasis, and a form of benign recurrent intrahepatic cholestasis. Low phospholipid-associated cholestasis syndrome is caused by a mutation in MDR3 and presents as cholesterol gallstones and intrahepatic cholelithiasis in adults younger than 40 years [43]. As liver disease in children with PFIC I, 2, and 3 progresses, the biochemical parameters become more typical for chronic liver disease and can include elevated bilirubin and aminotransferase values. Children with PFIC develop end-stage liver disease like other forms of progressive cholestatic liver disease. Portal hypertension develops secondary to the development of biliary cirrhosis. Definitive diagnosis of a specific form of PFIC is dependent upon identification of characteristic genetic defects. CLIA-certified molecular testing laboratories for genes of interest may be found at www.genetests.org. Histologic and ultrastructural analysis of the liver may be useful in distinguishing FIC1 from BSEP deficiency. Hepatocytes in FIC1 deficiency tend to be tidy and compact as opposed to BSEP deficiency in which there is more pronounced hepatocellular disarray, edema, giant-cell change, and hepatocellular necrosis (“neonatal hepatitis”). Portal and lobular fibrosis is more often seen at presentation in BSEP deficiency, but both FIC1 and BSEP deficiencies can progressively develop increasing amounts of fibrosis and a subset result in cirrhosis. Transmission electron microscopy in FIC1 deficiency may identify coarsely granular “Byler bile.” MDR3 deficiency can display expanded portal tracts and ductular proliferation with mixed inflammatory infiltrate mimicking a biliary obstruction pattern of injury or extensive portal fibrosis and biliary cirrhosis.
Treatment, Management, and Outcomes
The treatment of PFIC includes standard nutritional approaches for fat and fat-soluble vitamin malabsorption due to cholestasis and therapies for end-stage liver disease. Certain aspects of the management are unique. Initially, the pruritus associated with PFIC may be quite debilitating. Standard treatment including ursodeoxycholic acid, cholestyramine, rifampin, and opioid antagonists may not be successful. Surgical interruption of the enterohepatic circulation of bile acids with either ileal bypass or diversion may be effective with improvement in pruritus, serum liver chemistries, and prevention of progression of liver disease. Ursodeoxycholic acid is effective therapy in milder cases of MDR3 deficiency with many patients normalizing liver function with therapy. Ursodeoxycholic acid is successful in resolution of gallstones in adult patients with low phospholipid-associated cholelithiasis syndrome.
The prognosis for children with PFIC can be quite variable and influenced by the genetic abnormality (both the specific gene mutated and the impact of the mutation on the transporter protein) and the therapeutic approaches used. Severe defects in BSEP are associated with a high risk of hepatocellular carcinoma. Severe defects in BSEP and MDR3 deficiency are typically associated with an unremitting form of cholestasis that are unresponsive to medical and surgical therapies and eventuate in liver transplantation. End-stage liver disease typically evolves in the first 5–10 years of life. Recurrent BSEP deficiency has been described after liver transplant. FIC1 is a systemic disease, and the posttransplant course can be problematic including intractable diarrhea, liver steatosis leading to cirrhosis, and recurrent pancreatitis [44]. Liver transplantation, while effective for pruritus, may not be an optimal therapy for children with FIC1 deficiency. Surgical interruption of the enterohepatic circulation appears to be preferable to liver transplantation in FIC1 deficiency. Overall, with optimal surgical intervention, the long-term prognosis for children with PFIC is excellent.
Inborn Errors of Bile Acid Metabolism
Introduction
Bile acids are the natural detergents that the liver makes that help with bile flow and efficient fat and fat-soluble vitamin absorption. The primary bile acids, cholic acid and chenodeoxycholic acid, are synthesized by the liver from cholesterol through a series of 17 enzymatic steps, which results in the production taurine or glycine conjugates. An alteration in each of these steps can lead to the blockage of the bile acid production pathway with failure to produce “normal bile acids” and the accumulation of intermediary metabolites above the point at which the block in the pathway occurs. Because each of the enzymes in the pathway is regulated by a gene, it is believed that abnormalities in any of the steps of the pathway are inherited. Inborn errors of bile acid metabolism cause liver disease or may be a portion of more generalized diseases which include neurologic disease (See Fig. 8.10).

Classical and acidic pathways of bile acid synthesis including enzymatic defects associated with inborn errors of bile acid metabolism (Reprinted with permission from publisher of Liver Disease in Children, 3e (Cambridge University Press))
Pathophysiology
For all of the defects for which the genes have been identified, the inheritance of the bile acid defect is believed to be autosomal recessive. Blockage in the bile acid production pathway leads to accumulation of materials in the pathway prior to the block and few normal bile acids produced. This may result in mild to severe liver disease depending upon which of the enzymes is affected. Some conditions are associated with jaundice and significantly impaired function, while with others there may be mild changes in liver enzymes with poor bile flow and associated malabsorption of fat and fat-soluble vitamins with attendant complications (Δ4-3-oxosteroid 5-β-reductase deficiency and 3β-OH steroid dehydrogenase/isomerase deficiency [3-HSD]). Some are associated predominantly with fat-soluble vitamin malabsorption and growth failure (bile acid-acyl transferase deficiency). In some conditions, there is liver disease associated with neurologic dysfunction (racemase deficiency, cerebrotendinous xanthomatosis {CTX}). Patients with conditions caused by peroxisomal diseases including Zellweger and neonatal adrenoleukodystrophy have abnormalities in bile acid production because part of the production of bile acids requires steps within the peroxisome. These disorders affect many body organs since peroxisomes are present throughout the body. Liver disease may be variable in severity and is accompanied by neurologic disease which may include mental retardation, seizures, deafness, blindness, and muscular weakness.
There are two principal causes of liver injury associated with inborn errors of bile acid metabolism. The first relates to the failure to make “normal” bile acids which are a major force propelling bile out of the liver. This leads to impaired bile flow with attendant reduction of biliary excretion of individual components including cholesterol and other fats, proteins, drugs, and environmental toxins out of the liver. Secondly, the intermediary metabolites produced because of the block in the bile acid production pathway may themselves lead to liver injury. The combination of poor bile flow coupled with the production of potential toxic bile acid intermediates most likely is responsible for the injury to the liver seen in these conditions.
Clinical Manifestations
The most common defects including 3-HSD and Δ4 -3-oxosteroid 5-β-reductase deficiencies commonly present with neonatal jaundice, poor growth, liver or spleen enlargement, bleeding, rickets (vitamin D deficiency), or liver disease of unknown cause. Liver chemistries measured in the blood are abnormal and serum vitamin levels may be reduced. There is elevation in the serum ALT and AST with normal gGTP concentrations. Pruritus is usually absent. There may be variable alterations in the prothrombin time and serum 25-OH vitamin D and retinol and tocopherol levels. Serum bile acids measured by standard clinical methods are either normal or low. Patients with CTX may present in the neonatal period with neonatal cholestasis or diarrhea in infancy prior to any of the neurologic deterioration or xanthomata accumulation seen in adulthood. Patients with conjugation defects (bile acid acyl transferase deficiency) may present with transient neonatal cholestasis or infancy and childhood with growth failure and fat-soluble vitamin deficiencies. Diagnosis of these rare conditions requires a high index of suspicion by the treating physician. In the right clinical setting in which an infant or child has jaundice, liver disease of unknown cause, or abnormalities of fat or fat-soluble vitamin absorption, the treating physician needs to consider the diagnosis. In the presence of jaundice, elevated serum bile acid levels usually exclude the diagnosis. If serum bile acids are low or normal, urine should be sent for analysis to specialized laboratories for measurement of urinary bile acids by methods such as fast atom bombardment mass spectrometry (FAB-MS). These techniques allow identification of the “profile” of the bile acids in urine that would determine the potential presence of an inborn error of bile acid metabolism. If the urine FAB-MS study is suggestive of an abnormality, additional evaluation (including measurement of bile acids in blood and bile and evaluation of the microscopic appearance of the liver) would be necessary in order to establish the diagnosis. Liver biopsy findings for the most common conditions including 3-HSD, Δ4 -3-oxosteroid 5-β-reductase deficiencies do not show disease-specific findings but show evidence of cholestasis with canalicular bile plugs, giant-cell transformation, extramedullary hematopoiesis, and mild inflammation without bile duct proliferation [45–49].
Treatment, Management, and Outcomes
The treatment for inborn errors of bile acid metabolism not due to conjugation defects focuses on restoring normal bile flow with reduction in the injury to the liver by the toxic intermediates of the bile acid production pathway. Bile acid therapy with a specific bile acid, cholic acid, is currently available as an investigational drug approved by the US Food and Drug Administration. Another bile acid, ursodeoxycholic acid, is a drug approved by the FDA but not for this indication. UDCA is not an ideal treatment because it does not suppress synthesis of abnormal, potentially toxic metabolites, as cholic acid does. It may be prescribed by physicians as an “off label” indication which means that a drug approved by the FDA for a specific disease may be used for other diseases at the discretion of the treating physician. When first diagnosed, patients with inborn errors of metabolism malabsorb fat and fat-soluble vitamins (vitamins A, D, E, and K) and infant formulas containing medium-chain triglycerides (Alimentum®, Pregestimil®) and supplemental vitamins may be prescribed to overcome initial deficits of these nutrients. Although most affected patients have a complete resolution of the jaundice and liver biochemistry abnormalities with bile acid treatment, the liver disease may progress to end-stage liver disease with cirrhosis and a liver transplant may be required. Patients treated with orally administered bile acids (cholic acid and ursodeoxycholic acid) usually correct all abnormalities of liver function including jaundice, growth failure, fat and vitamin malabsorption, and blood liver chemistry abnormalities. This occurs over a period of several weeks after starting therapy. Cholic acid is the preferred treatment since it suppresses the production of toxic bile acid intermediates whereas ursodeoxycholic acid does not. Occasionally, patients have worsening of liver disease despite treatment with the development of cirrhosis and liver failure. Patients who are not treated with bile acids have progressive worsening until cirrhosis develops. If cirrhosis develops, the only effective therapy is liver transplantation. Patients with conjugation defects respond to treatment with glycocholic acid, which is available under an IND from the FDA. The liver disease in these patients tends to be mild, but they commonly have fat-soluble vitamin deficiency and growth failure that is corrected with bile acid therapy. With bile acid therapy, the prognosis for children with conjugation defects is excellent [50].
Lysosomal Storage Disorders
Introduction
Lysosomal storage disorders are encountered by the pediatric gastroenterologist as hepatosplenomegaly with other systemic manifestations. The most common and important of these include Gaucher disease, Niemann-Pick type C disease, and lysosomal acid lipase deficiency (cholesterol ester storage disease and Wolman disease).
Gaucher Disease
Introduction
Gaucher disease (GD) is the most common lysosomal storage disease and is inherited as an autosomal recessive trait that affects the recycling of cellular glycolipids. It occurs in approximately 1/75,000 births worldwide but is more prevalent in individuals of Ashkenazi Jewish descent. Approximately 90 % of patients have type 1 Gaucher disease (GD1), the nonneuronopathic form, and discussion will be limited to it.
Pathophysiology
Gaucher disease results from deficiency of a lysosomal enzyme glucocerebrosidase (also known as glucosylceramide or acid beta-glucosidase, GBA) leading to accumulation glucocerebroside and other glycolipids within the lysosomes of macrophages in the spleen, liver, bone marrow, bone, and other tissues/organs.
Clinical Manifestations
GD1 is characterized by variability in signs, symptoms, severity, and progression even among siblings with the same genotype and monozygotic twins. Symptomatic patients have visceral involvement, bone disease, and bleeding and may present to the gastroenterologist with hepatosplenomegaly. Splenomegaly is the most common presenting sign. The spleen can be markedly enlarged. Hepatomegaly is universal, but the liver size increases relatively less than the spleen. Hepatic fibrosis commonly occurs, but hepatic failure, cirrhosis, or portal hypertension is uncommon. Liver biopsy findings include macrophages filled with lipid material known as Gaucher cells which have a characteristic appearance of wrinkled tissue paper. Thrombocytopenia and anemia typically are commonly present. Liver chemistries may be mildly elevated. The diagnosis of GD is confirmed by the finding of reduced glucocerebrosidase activity, usually in peripheral leukocytes, in a patient with clinical features consistent with GD. Mutation analysis provides additional confirmation of the diagnosis and can help predict clinical manifestations and identify undiagnosed affected family members and heterozygote carriers. Prenatal diagnosis can be performed by enzyme analysis by chorionic villus sampling or amniocentesis.
Treatment, Management, and Outcome
The goals of treatment of GD disease are elimination of symptoms, prevention of irreversible damage, and improvement in the overall health and quality of life. Treatment of GD is tailored to the individual patient because of the variability in the manifestations, severity, and progression of the disease. The decision to offer enzyme replacement therapy (ERT) for GD1 is based upon disease severity and disease progression. ERT with recombinant glucocerebrosidases (imiglucerase or velaglucerase alfa) is recommended for all symptomatic children. ERT is individualized. The availability and efficacy of ERT has limited the indications for splenectomy and hematopoietic cell transplantation. Splenectomy is indicated if other measures fail to control life-threatening thrombocytopenia [51, 52].
Niemann-Pick Disease
Introduction
Niemann-Pick disease type C (NPD-C) is an autosomal recessive disease caused by mutations of the NPC1 and NPC2 genes. NPD-C can present in infants, children, or adults with an estimated prevalence of 1:150,000 in Europe.
Pathophysiology
Mutations in the NPC1 and NPC2 genes result in impaired cellular processing and transport of low-density lipoprotein (LDL) cholesterol. Abnormal lipid trafficking and storage of lipids in lysosomes of affected cells results in progressive neurocognitive and visceral disease.
Clinical Manifestations
More than 90 % of affected individuals have splenomegaly and or hepatomegaly particularly those presenting in infancy and early childhood. Over half present with infantile cholestatic liver disease similar to neonatal hepatitis of PFIC 1, 2, or 3. A small fraction present with liver failure. Older patients may have cerebellar involvement characterized by clumsiness and gait problems progressing to frank ataxia, and slow cognitive deterioration. Progressive dystonia, dysarthria, and dysphagia occur, eventually impairing oral feeding, and approximately one-third of patients develop seizures. Death typically occurs from aspiration pneumonia in the second or third decade of life. The laboratory findings may include lipid abnormalities such as decreased high-density lipoprotein (HDL) cholesterol, hypertriglyceridemia, and increased LDL cholesterol. Large lipid-laden foam cells are seen in the reticuloendothelial system of the spleen, liver, bone marrow, lymph nodes, blood vessels, Schwann cells in peripheral nerves, CNS, and retinal cells [53–55]. The diagnosis of NPD-C is suspected on the basis of the clinical features and confirmed by fibroblast cell culture. Genetic testing is commercially available. A mutation in the NPCI gene is found in approximately 90 % with an NPC2 gene mutation found in less than 5 % of cases of NPD-C.
Management, Outcome, and Prognosis
There is no specific treatment for NPD-C and treatments are symptomatic and directed to neurologic and psychiatric symptoms. Lipid lowering treatment and bone marrow transplantation have not been shown to affect outcome. Similarly, liver transplantation improves hepatic function but does not alter the natural history of the neurological disease.
Lysosomal Acid Lipase Deficiency (Wolman Disease and Cholesteryl Ester Storage Disease) (CESD)
Introduction
Lysosomal acid lipase deficiency results in two diseases with considerably different phenotypes. Both are associated with accumulation of cholesteryl esters, triglycerides, and other lipids in lysosomes, and disease manifestation is heavily dependent upon how much acid lipase enzyme activity is present in the affected patient. The gene for lysosomal acid lipase (LIPA) codes for acid lipase activity for which mutations lead to minimal or absent enzyme activity in Wolman disease with mutations in cholesterol ester storage disease resulting in variable amounts of residual enzyme activity.
Pathophysiology
Absence of lysosomal acid lipase activity leads to accumulation of cholesterol esters, triglycerides, and other lipids in lysosomes in liver and other affected organs including the intestine in CESD and lymph nodes, bone marrow, intestine, liver, spleen, and adrenal glands in Wolman disease leading to remarkable variations in organ dysfunction.
Clinical Manifestations
Wolman disease is a severe disease with rapidly progressive course. Infants typically present in the first weeks of life with steatorrhea, failure to thrive, hepatosplenomegaly, and jaundice with death in the first year of life. Calcifications of the adrenal gland are evident on plain film of the abdomen. There is variable liver dysfunction with cholestasis, serum aminotransferase elevation, and worsening evidence of synthetic dysfunction as the disease progresses. Plasma lipids are generally normal, and liver biopsy has a characteristic orange-yellow in appearance and found to contain triglycerides and cholesterol esters on microscopic examination. Acid lipase activity in liver, leukocytes, or fibroblasts is markedly reduced. CESD is characterized by hepatomegaly without splenomegaly and hyperbetalipoproteinemia identified in older children, adolescent, or adult. There may be mild increases in aminotransferases without jaundice or evidence of synthetic dysfunction. Characteristically, the liver is orange macroscopically and there is accumulation of cholesterol ester and triglycerides on light microscopy.
Management, Outcome, and Prognosis
Limited success has been observed in patients with Wolman disease treated with hematopoietic cell transplantation [56]. Early success has been demonstrated using a plant-derived human lysosomal acid lipase for treatment in mice [57]. Currently, there is no FDA-approved therapy for CESD or Wolman disease. Phase 3 trials of recombinant lysosomal acid lipase began in 2012. Without specific treatment, Wolman disease is uniformly fatal in the first year of life, and only symptomatic therapy may be of any benefit. Patients with CESD have a normal life expectancy with some developing fibrosis but not end-stage liver disease in adulthood. Some adults may benefit from diets low in cholesterol and triglycerides and the use of HMG-CoA reductase inhibitors [58].
Peroxisomal Disorders
Introduction
Disorders of peroxisomal biogenesis affect multiple organ systems. Zellweger syndrome, neonatal adrenoleukodystrophy, and infantile Refsum disease comprise the majority of patients with disorders of biogenesis. With each of these conditions, the numbers of peroxisomes in liver are markedly reduced or absent.
Pathophysiology
Absence or marked reduction in the numbers of peroxisomes in multiple organs leads to characteristic manifestations. Peroxisomes play a role in multiple metabolic pathways including β-oxidation of fatty acids, α-oxidation of plant products, bile acid synthesis, ether lipid biosynthesis, cholesterol and isoprenoid biosynthesis, amino acid metabolism, and catalases for decomposing H2O2. Accumulation of by-products above the block in the pathway related to peroxisomal metabolism or inability to complete the steps in the pathway may be responsible for the clinical manifestations of disease.
Clinical Manifestations
Zellweger syndrome (ZS), neonatal adrenoleukodystrophy, and infant Refsum disease form a spectrum of features from ZS being most severe to infantile Refsum disease as the mildest. ZS includes typical craniofacial features such as wide anterior fontanelle, prominent forehead, anteverted nostrils, epicanthal folds, and narrow upper lip. Severe neurocognitive function is present with hypotonia, mental retardation, and seizures. Visual and hearing impairment are common. Typically, no peroxisomes can be identified in the liver. Infants with ZS commonly have cholestatic liver disease with varying degrees of synthetic dysfunction with progression to end-stage liver disease at variable rates. Infants with neonatal adrenoleukodystrophy have dysmorphic features, deafness, psychomotor retardation, hypotonia, and seizures. A few residual peroxisomes may be found in the liver. The liver disease tends to be milder than ZS with better long-term prognosis. Patients with infantile Refsum disease have dysmorphic features but milder than ZS. Similarly, hypotonia is not as severe as in ZS. There is psychomotor delay and deafness. Hepatomegaly is common and liver disease may manifest as neonatal cholestasis. Liver disease may progress over time. Hepatic peroxisomes are absent or very few in number. The diagnosis of a peroxisomal disorder may be made in specialized laboratories by identifying metabolites in urine and serum that are present because of absent or diminished peroxisomal function such as very long-chain fatty acids, pipecolic acid, phytanic acid, and pristanic acid [59]. Skin fibroblast analysis may be performed to better characterize the metabolic pathways affected through examination of de novo plasmalogen synthesis, fatty acid beta-oxidation, phytanic acid alpha oxidation, and catalase immunofluorescence. Qualitative analysis of urine by fast atom bombardment mass spectrometry can identify characteristic bile acids with long acyl side chains of the sterol such as trihydroxycholestanoic acid (THCA) and dihydrocholestanoic acid (DHCA).
Treatment, Management, and Outcomes
There remain no specific treatment options for any of the defects of peroxisomal biogenesis. Limited experience with the use of cholic acid has suggested that there may be some benefit in treating cholestatic liver disease in patients with biogenesis defects. Clinical and biochemical benefits for non-hepatic manifestations of these conditions have been suggested with the use of docosahexaenoic acid (DHA) or induction of peroxisomal proliferation with agents such as 4-phenylbutyrate [60, 61]. Prognosis for these conditions is variable with death in ZS patients in the first years of life and longer survival with the other disorders.
Mitochondrial Hepatopathies
Mitochondria are intracellular organelles that contain the respiratory chain complex, the site of oxidative phosphorylation. A main function of the mitochondria is to produce adenosine triphosphate (ATP) by the respiratory chain located on the inner membrane. ATP subsequently functions to transport chemical energy within cells for metabolism and is critical for all intracellular processes. As a metabolically active organ, hepatocytes require continuous synthesis of ATP and contain elevated amounts of mitochondria to achieve this demand [62]. Alterations within the mitochondrial structure and function can result in clinical hepatopathies often presenting in children. The overall incidence of all respiratory chain defects is approximately 1 in 20,000 children with liver involvement occurring in about 10–20 %.
Neonatal Liver Failure
Introduction
Mitochondrial hepatopathies presenting as acute liver failure in the neonatal period can often be attributed to defects in the respiratory chain complex.
Pathogenesis
The respiratory chain protein complex of the mitochondria reduces equivalents derived from glycolysis, fatty acid oxidation, and the tricarboxylic acid (TCA) cycle and produces ATP via a system of electron carriers (protein complexes I through IV, coenzyme Q, and cytochrome c) in the inner mitochondrial membrane. This results in the generation of a transmembrane proton gradient that drives the synthesis of ATP by complex V [61]. Most commonly, a deficiency in complex IV of the respiratory chain complex is causative; however, decreased activity of complexes I and III has also been associated with the development of neonatal liver failure.
Clinical Manifestations and Diagnosis
Neonatal liver failure often presents within the first weeks of life with transient hypoglycemia, neurological involvement with hypotonia, seizure activity, and psychomotor retardation in the setting of early hepatic dysfunction and failure [63]. Biochemical analysis reveals marked serum lactate elevations with a lactate-pyruvate ratio often >30. Although respiratory chain complex analysis will generally show decreased activity of complex I, III, or IV, no definitive diagnostic test is available.
Treatment, Management, and Outcomes
Mortality remains high in children presenting with neonatal liver failure, and most children progress to death within weeks to months after presentation.
Navajo Neurohepatopathy
Introduction
Navajo neurohepatopathy (NNH) is an autosomal recessive sensorimotor neuropathy associated with progressive liver disease that affects full-blooded Navajo children [64].
Pathogenesis
NNH results from a depletion of mitochondrial DNA (mtDNA) whose product, MPV 17, is involved in the regulation of oxidative phosphorylation and subsequent ATP production.
Clinical Manifestations and Diagnosis
There are three clinical presentations of NNH. The classical NNH presentation develops in Navajo children and presents with dominant, progressive neurologic disease accompanied by liver dysfunction. The neurological symptoms most associated with NNH are weakness, hypotonia, areflexia, and loss of extremity sensations. The infantile presentation is demonstrated in children who develop failure to thrive, jaundice, and progressive liver failure over the first 2 years of life. These children may or may not have associated neurological findings. A childhood presentation clinically appears between 1 and 5 years of age with hepatic dysfunction and rapid progression to liver failure [62].
Treatment, Management, and Outcomes
The liver disease in children with NNH is progressive, often advancing to liver failure and death within several months to years after diagnosis.
Alpers-Huttenlocher Syndrome
Introduction
Alpers-Huttenlocher syndrome is disorder of oxidative metabolism related to mitochondrial dysfunction.
Pathogenesis
Alpers-Huttenlocher syndrome is caused by a mutation in the POLG gene whose product, DNA polymerase-γ, is essential for mtDNA replication and repair
Clinical Manifestations and Diagnosis
Children with Alpers-Huttenlocher syndrome typically present with jaundice, hepatomegaly, hypoglycemia, and coagulopathy between 2 and 8 months of life [65]. Liver disease is often found in association with failure to thrive, emesis, ataxia, hypotonia, and seizure activity. [62] A diagnosis of Alpers-Huttenlocher syndrome can be made clinically with (1) the presence of refractory seizures that have a focal component, (2) psychomotor regression often triggered by infection, and (3) hepatopathy with or without acute liver failure. Molecular testing for POLG mutations is also available.
Treatment, Management, and Outcomes
No specific treatments have shown to be effective in the management of Alpers-Huttenlocher syndrome. However, it should be emphasized that while multiple antiepileptic medications may be indicated in order to control the refractory seizures, valproic acid should be strictly avoided in children with Alpers-Huttenlocher syndrome secondary to exacerbations of the hepatic dysfunction and the risk of precipitating fulminant liver failure.
Pearson Syndrome
Introduction
Pearson syndrome is a generalized mitochondriopathy that presents in infancy and is attributed to defects in mtDNA.
Pathogenesis
Pearson syndrome is now known to result from mtDNA rearrangements that result in dysfunctional protein products. The most common deletion results in dysfunction of the respiratory chain complexes I, IV, and V.
Clinical Manifestations and Diagnosis
The clinical presentation of children with Pearson syndrome involves multiple organs including the hematopoietic system, exocrine pancreas, liver, and kidneys. Hepatic disease is manifested by liver enlargement, steatosis, hemosiderosis, and cirrhosis [62].
Treatment, Management, and Outcomes
No effective therapy exists for the treatment of Pearson syndrome, and death generally occurs in infancy secondary to metabolic disorders or infection. Symptomatic management with RBC transfusions and carbohydrate restriction has been utilized in some patients.
References
Suchy FJ, Sokol RJ, Balistreri WF. Liver disease in children. 3rd ed. Cambridge/New York: Cambridge University Press; 2007.
Beutler E, Baluda MC, Sturgeon P, Day R. A new genetic abnormality resulting in galactose-1-phosphate uridyltransferase deficiency. Lancet. 1965;1:353–4.
Kawade N, Onishi S. The prenatal and postnatal development of UDP-glucuronyltransferase activity towards bilirubin and the effect of premature birth on this activity in the human liver. Biochem J. 1981;196:257–60.
Ridel KR, Leslie ND, Gilbert DL. An updated review of the long-term neurological effects of galactosemia. Pediatr Neurol. 2005;33:153–61.
Arias IM, Gartner LM, Seifter S, Furman M. Prolonged neonatal unconjugated hyperbilirubinemia associated with breast feeding and a steroid, pregnane-3(alpha), 20(beta)-diol, in maternal milk that inhibits glucuronide formation in vitro. J Clin Invest. 1964;43:2037–47.
Poland RL, Schultz GE, Garg G. High milk lipase activity associated with breast milk jaundice. Pediatr Res. 1980;14:1328–31.
Schneider 2nd AP. Breast milk jaundice in the newborn. A real entity. JAMA. 1986;255:3270–4.
Maisels MJ, Newman TB. Kernicterus in otherwise healthy, breast-fed term newborns. Pediatrics. 1995;96:730–3.
Bhutani VK, Johnson L, Sivieri EM. Predictive ability of a predischarge hour-specific serum bilirubin for subsequent significant hyperbilirubinemia in healthy term and near-term newborns. Pediatrics. 1999;103:6–14.
Waggoner DD, Buist NR, Donnell GN. Long-term prognosis in galactosaemia: results of a survey of 350 cases. J Inherit Metab Dis. 1990;13:802–18.
Bosma PJ, Chowdhury JR, Bakker C, Gantla S, De Boer A, Oostra BA, Lindhout D, Tytgat GN, Jansen PL, Oude Elferink RP, et al. The genetic basis of the reduced expression of bilirubin UDP-glucuronosyltransferase 1 in Gilbert’s syndrome. N Engl J Med. 1995;333:1171–5.
Crigler Jr JF, Najjar VA. Congenital familial nonhemolytic jaundice with kernicterus. Pediatrics. 1952;10:169–80.
Van Der Veere CN, Sinaasappel M, Mcdonagh AF, Rosenthal P, Labrune P, Odievre M, Fevery J, Otte JB, Mcclean P, Burk G, Masakowski V, Sperl W, Mowat AP, Vergani GM, Heller K, Wilson JP, Shepherd R, Jansen PL. Current therapy for Crigler-Najjar syndrome type 1: report of a world registry. Hepatology. 1996;24:311–5.
Wolkoff AW, Wolpert E, Pascasio FN, Arias IM. Rotor’s syndrome. A distinct inheritable pathophysiologic entity. Am J Med. 1976;60:173–9.
Cross NC, De Franchis R, Sebastio G, Dazzo C, Tolan DR, Gregori C, Odievre M, Vidailhet M, Romano V, Mascali G, et al. Molecular analysis of aldolase B genes in hereditary fructose intolerance. Lancet. 1990;335:306–9.
Stanbury JB. The metabolic basis of inherited disease. 5th ed. New York: McGraw-Hill; 1983.
Mayatepek E, Hoffmann B, Meissner T. Inborn errors of carbohydrate metabolism. Best Pract Res Clin Gastroenterol. 2010;24:607–18.
Saudubray JM, Sedel F, Walter JH. Clinical approach to treatable inborn metabolic diseases: an introduction. J Inherit Metab Dis. 2006;29:261–74.
Hiraiwa H, Pan CJ, Lin B, Moses SW, Chou JY. Inactivation of the glucose 6-phosphate transporter causes glycogen storage disease type 1b. J Biol Chem. 1999;274:5532–6.
Rake JP, Visser G, Labrune P, Leonard JV, Ullrich K, Smit GP. Glycogen storage disease type I: diagnosis, management, clinical course and outcome. Results of the European Study on Glycogen Storage Disease Type I (ESGSD I). Eur J Pediatr. 2002;161 Suppl 1:S20–34.
Rake JP, Visser G, Labrune P, Leonard JV, Ullrich K, Smit GP. Guidelines for management of glycogen storage disease type I – European Study on Glycogen Storage Disease Type I (ESGSD I). Eur J Pediatr. 2002;161 Suppl 1:S112–9.
Geberhiwot T, Alger S, Mckiernan P, Packard C, Caslake M, Elias E, Cramb R. Serum lipid and lipoprotein profile of patients with glycogen storage disease types I, III and IX. J Inherit Metab Dis. 2007;30:406.
Goldberg T, Slonim AE. Nutrition therapy for hepatic glycogen storage diseases. J Am Diet Assoc. 1993;93:1423–30.
Bruno C, Van Diggelen OP, Cassandrini D, Gimpelev M, Giuffre B, Donati MA, Introvini P, Alegria A, Assereto S, Morandi L, Mora M, Tonoli E, Mascelli S, Traverso M, Pasquini E, Bado M, Vilarinho L, Van Noort G, Mosca F, Dimauro S, Zara F, Minetti C. Clinical and genetic heterogeneity of branching enzyme deficiency (glycogenosis type IV). Neurology. 2004;63:1053–8.
Mckiernan PJ. Nitisinone in the treatment of hereditary tyrosinaemia type 1. Drugs. 2006;66:743–50.
Holme E, Lindstedt S. Tyrosinaemia type I and NTBC (2-(2-nitro-4-trifluoromethylbenzoyl)-1,3-cyclohexanedione). J Inherit Metab Dis. 1998;21:|507–17.
Masurel-Paulet A, Poggi-Bach J, Rolland MO, Bernard O, Guffon N, Dobbelaere D, Sarles J, De Baulny HO, Touati G. NTBC treatment in tyrosinaemia type I: long-term outcome in French patients. J Inherit Metab Dis. 2008;31:81–7.
Mohan N, Mckiernan P, Preece MA, Green A, Buckels J, Mayer AD, Kelly DA. Indications and outcome of liver transplantation in tyrosinaemia type 1. Eur J Pediatr. 1999;158 Suppl 2:S49–54.
Matern D, Rinaldo P. Medium-chain acyl-coenzyme A dehydrogenase deficiency. In: Pagon RA, editor. Gene reviews. Seattle: University of Washington; 1993.
Iafolla AK, Thompson Jr RJ, Roe CR. Medium-chain acyl-coenzyme A dehydrogenase deficiency: clinical course in 120 affected children. J Pediatr. 1994;124:409–15.
Hayes B, Lynch B, O’keefe M, Monavari AA, Treacy EP. Long chain fatty acid oxidation defects in children: importance of detection and treatment options. Ir J Med Sci. 2007;176:189–92.
Olpin SE, Clark S, Andresen BS, Bischoff C, Olsen RK, Gregersen N, Chakrapani A, Downing M, Manning NJ, Sharrard M, Bonham JR, Muntoni F, Turnbull DN, Pourfarzam M. Biochemical, clinical and molecular findings in LCHAD and general mitochondrial trifunctional protein deficiency. J Inherit Metab Dis. 2005;28:533–44.
Wanders RJ, IJlst L, Van Gennip AH, Jakobs C, De Jager JP, Dorland L, Van Sprang FJ, Duran M. Long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency: identification of a new inborn error of mitochondrial fatty acid beta-oxidation. J Inherit Metab Dis. 1990;13:311–4.
Zschocke J, Penzien JM, Bielen R, Casals N, Aledo R, Pie J, Hoffmann GF, Hegardt FG, Mayatepek E. The diagnosis of mitochondrial HMG-CoA synthase deficiency. J Pediatr. 2002;140:778–80.
Morris AA, Olpin SE, Brivet M, Turnbull DM, Jones RA, Leonard JV. A patient with carnitine-acylcarnitine translocase deficiency with a mild phenotype. J Pediatr. 1998;132:514–6.
Summar ML, Dobbelaere D, Brusilow S, Lee B. Diagnosis, symptoms, frequency and mortality of 260 patients with urea cycle disorders from a 21-year, multicentre study of acute hyperammonaemic episodes. Acta Paediatr. 2008;97:1420–5.
Tuchman M, Lee B, Lichter-Konecki U, Summar ML, Yudkoff M, Cederbaum SD, Kerr DS, Diaz GA, Seashore MR, Lee HS, Mccarter RJ, Krischer JP, Batshaw ML. Cross-sectional multicenter study of patients with urea cycle disorders in the United States. Mol Genet Metab. 2008;94:397–402.
Enns GM, Berry SA, Berry GT, Rhead WJ, Brusilow SW, Hamosh A. Survival after treatment with phenylacetate and benzoate for urea-cycle disorders. N Engl J Med. 2007;356:2282–92.
Krivitzky L, Babikian T, Lee HS, Thomas NH, Burk-Paull KL, Batshaw ML. Intellectual, adaptive, and behavioral functioning in children with urea cycle disorders. Pediatr Res. 2009;66:96–101.
Davit-Spraul A, Fabre M, Branchereau S, Baussan C, Gonzales E, Stieger B, Bernard O, Jacquemin E. ATP8B1 and ABCB11 analysis in 62 children with normal gamma-glutamyl transferase progressive familial intrahepatic cholestasis (PFIC): phenotypic differences between PFIC1 and PFIC2 and natural history. Hepatology. 2010;51:1645–55.
Folmer DE, Van Der Mark VA, Ho-Mok KS, Oude Elferink RP, Paulusma CC. Differential effects of progressive familial intrahepatic cholestasis type 1 and benign recurrent intrahepatic cholestasis type 1 mutations on canalicular localization of ATP8B1. Hepatology. 2009;50:1597–605.
Strautnieks SS, Byrne JA, Pawlikowska L, Cebecauerova D, Rayner A, Dutton L, Meier Y, Antoniou A, Stieger B, Arnell H, Ozcay F, Al-Hussaini HF, Bassas AF, Verkade HJ, Fischler B, Nemeth A, Kotalova R, Shneider BL, Cielecka-Kuszyk J, Mcclean P, Whitington PF, Sokal E, Jirsa M, Wali SH, Jankowska I, Pawlowska J, Mieli-Vergani G, Knisely AS, Bull LN, Thompson RJ. Severe bile salt export pump deficiency: 82 different ABCB11 mutations in 109 families. Gastroenterology. 2008;134:1203–14.
Davit-Spraul A, Gonzales E, Baussan C, Jacquemin E. The spectrum of liver diseases related to ABCB4 gene mutations: pathophysiology and clinical aspects. Semin Liver Dis. 2010;30:134–46.
Englert C, Grabhorn E, Richter A, Rogiers X, Burdelski M, Ganschow R. Liver transplantation in children with progressive familial intrahepatic cholestasis. Transplantation. 2007;84:1361–3.
Setchell KD, Heubi JE. Defects in bile acid biosynthesis – diagnosis and treatment. J Pediatr Gastroenterol Nutr. 2006;43 Suppl 1:S17–22.
Heubi JE, Setchell KD, Bove KE. Inborn errors of bile acid metabolism. Semin Liver Dis. 2007;27:282–94.
Sundaram SS, Bove KE, Lovell MA, Sokol RJ. Mechanisms of disease: inborn errors of bile acid synthesis. Nat Clin Pract Gastroenterol Hepatol. 2008;5:456–68.
Gonzales E, Gerhardt MF, Fabre M, Setchell KD, Davit-Spraul A, Vincent I, Heubi JE, Bernard O, Jacquemin E. Oral cholic acid for hereditary defects of primary bile acid synthesis: a safe and effective long-term therapy. Gastroenterology. 2009;137(1310–1320):e1311–3.
Clayton PT. Disorders of bile acid synthesis. J Inherit Metab Dis. 2011;34:593–604.
Setchell KDR, Heubi JE, Shah S, Levine JE, Suskind D, Al-Edreesi M, Potter C, Russell DW, O’Connell NC, Wolfe B, Jha P, Zhang W, Bove KE, Knisely A, Hofmann AF, Rosenthal P, Bull LN. Genetic defects in bile acid conjugation cause fat-soluble vitamin deficiency. Gastroenterology. 2013;144(5):945–55.e6; quiz e14–5.
Grabowski GA. Recent clinical progress in Gaucher disease. Curr Opin Pediatr. 2005;17:519–24.
Grabowski GA, Andria G, Baldellou A, Campbell PE, Charrow J, Cohen IJ, Harris CM, Kaplan P, Mengel E, Pocovi M, Vellodi A. Pediatric non-neuronopathic Gaucher disease: presentation, diagnosis and assessment. Consensus statements. Eur J Pediatr. 2004;163:58–66.
Vanier MT, Wenger DA, Comly ME, Rousson R, Brady RO, Pentchev PG. Niemann-Pick disease group C: clinical variability and diagnosis based on defective cholesterol esterification. A collaborative study on 70 patients. Clin Genet. 1988;33:331–48.
Fink JK, Filling-Katz MR, Sokol J, Cogan DG, Pikus A, Sonies B, Soong B, Pentchev PG, Comly ME, Brady RO, et al. Clinical spectrum of Niemann-Pick disease type C. Neurology. 1989;39:1040–9.
Yerushalmi B, Sokol RJ, Narkewicz MR, Smith D, Ashmead JW, Wenger DA. Niemann-pick disease type C in neonatal cholestasis at a North American Center. J Pediatr Gastroenterol Nutr. 2002;35:44–50.
Tolar J, Petryk A, Khan K, Bjoraker KJ, Jessurun J, Dolan M, Kivisto T, Charnas L, Shapiro EG, Orchard PJ. Long-term metabolic, endocrine, and neuropsychological outcome of hematopoietic cell transplantation for Wolman disease. Bone Marrow Transplant. 2009;43:21–7.
Du H, Cameron TL, Garger SJ, Pogue GP, Hamm LA, White E, Hanley KM, Grabowski GA. Wolman disease/cholesteryl ester storage disease: efficacy of plant-produced human lysosomal acid lipase in mice. J Lipid Res. 2008;49:1646–57.
Leone L, Ippoliti PF, Antonicelli R. Use of simvastatin plus cholestyramine in the treatment of lysosomal acid lipase deficiency. J Pediatr. 1991;119:1008–9.
Moser HW, Moser AB. Peroxisomal disorders: overview. Ann N Y Acad Sci. 1996;804:427–41.
Paker AM, Sunness JS, Brereton NH, Speedie LJ, Albanna L, Dharmaraj S, Moser AB, Jones RO, Raymond GV. Docosahexaenoic acid therapy in peroxisomal diseases: results of a double-blind, randomized trial. Neurology. 2010;75:826–30.
Wei H, Kemp S, Mcguinness MC, Moser AB, Smith KD. Pharmacological induction of peroxisomes in peroxisome biogenesis disorders. Ann Neurol. 2000;47:286–96.
Lee WS, Sokol RJ. Mitochondrial hepatopathies: advances in genetics and pathogenesis. Hepatology. 2007;45:1555–65.
Cormier-Daire V, Chretien D, Rustin P, Rotig A, Dubuisson C, Jacquemin E, Hadchouel M, Bernard O, Munnich A. Neonatal and delayed-onset liver involvement in disorders of oxidative phosphorylation. J Pediatr. 1997;130:817–22.
Holve S, Hu D, Shub M, Tyson RW, Sokol RJ. Liver disease in Navajo neuropathy. J Pediatr. 1999;135:482–93.
Narkewicz MR, Sokol RJ, Beckwith B, Sondheimer J, Silverman A. Liver involvement in Alpers disease. J Pediatr. 1991;119:260–7.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer Science+Business Media New York
About this chapter
Cite this chapter
Squires, J.E., Heubi, J.E. (2014). Metabolic Liver Disease: Part 1. In: Murray, K., Horslen, S. (eds) Diseases of the Liver in Children. Springer, New York, NY. https://doi.org/10.1007/978-1-4614-9005-0_8
Download citation
DOI: https://doi.org/10.1007/978-1-4614-9005-0_8
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4614-9004-3
Online ISBN: 978-1-4614-9005-0
eBook Packages: MedicineMedicine (R0)