
Abstract
Background
Mitochondrial β oxidation plays a major role in energy production. Long chain fatty acid oxidation defects include deficiency of the trifunctional protein (rare) or more commonly defects of the long chain 3-hydroxy acyl-CoA dehydrogenase enzyme (LCHAD). These long chain defects have variable presentations, they may present in the neonate or infant with sudden death, hepatopathy (Reyes disease), hypoketotic hypoglycaemia, rhabdomyolysis, myopathy, cardiomyopathy and with late complications such as peripheral neuropathy, pigmentary retinopathy, retinal degeneration and progressive visual loss. The correct diagnosis at presentation is not only life saving but also allows for the appropriate dietary and other intervention, which may have major effects on outcome.
Aim
Three case reports of patients with long chain fatty acid oxidation defects who have shown significant benefits from treatment are reported.
Conclusions
These paediatric presentations illustrate the clinical heterogeneity of long chain fatty acid oxidation defects and opportunities for effective management if correctly diagnosed.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
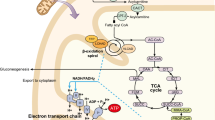
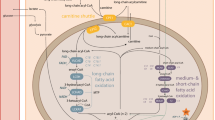
More than 20 enzyme and transport defects of mitochondrial oxidation of saturated fatty acids involved in mitochondrial beta oxidation are known [1]. Long chain fatty acid oxidation defects include deficiency of the trifunctional protein, which is rare or more commonly defects of the long chain 3-hydroxy acyl-CoA dehydrogenase enzyme (LCHAD) [1–3]. The trifunctional protein is an enzyme complex that catalyses the final three steps of β-oxidation (Fig. 1). Trifunctional protein deficiency (TFP) presents with a deficiency of all three enzymes, long chain 2-enoyl CoA dehydratase, 3-ketoacylCoA thiolase (LKAT), and long chain 3-hydroxyacyl CoA dehydrogenase (LCHAD). General trifunctional protein deficiency results in a wide spectrum of disease ranging from severe infantile cardiomyopathy to a milder late onset presentation with episodic rhabdomyolysis and chronic progressive polyneuropathy [1, 3]. In isolated LCHAD deficiency there is a deficiency of the LCHAD enzyme with activity of the other two enzymes being greater than 60% [1]. LCHAD deficiency was first described in 1983. There is considerable clinical heterogeneity described with these disorders. Classically they present in the neonate or infant with sudden death, hepatopathy (Reyes disease) and hypoketotic hypoglycaemia [1–7]. Prompt recognition is critical as treatment prevents mortality and major morbidity. Recently there is an increased awareness that presenting symptoms may be subtle and chronic in nature. This is particularly true of trifunctional protein deficiency, which often has a delayed presentation with neuromyopathic symptoms. Late complications not commonly observed in other fatty acid oxidation disorders may include peripheral neuropathy, pigmentary retinopathy, retinal degeneration and progressive visual loss [2, 5–9]. The diagnosis of LCHAD or TFP deficiency is suggested by the finding of characteristic organic acid urine profiles (increased excretion of 3-hydroxy-dicarboxylic acids) or by an abnormal acylcarnitine profile [1]. A confirmatory diagnosis is made by elucidation of enzymatic deficiency in lymphocytes, fibroblasts, and/or muscle or liver biopsies followed by mutation analysis.

Trifunctional protein deficiency pathway
The management of long chain fatty acid oxidation defects is largely dietary control with the avoidance of fasting and strenuous exercise and minimisation of energy production from long-chain fatty acids [1, 10], achieved by providing a high carbohydrate diet with restriction of long chain fats and supplementation with medium chain triglycerides to ensure all essential fatty acids are provided. While this prevents mortality there is still considerable associated morbidity. Recently it has been shown that patients with LCHAD have a profound brain deficiency of Docosahexaenoic acid ethyl ester, the long-chain polyunsaturated fatty acid (DHA; 22:6n-3). Low DHA concentrations have been documented in all tissues and in blood [11]. Progressive visual loss is present in over 70% of cases with LCHAD deficiency [11]. Cataracts are also reported [12]. An association with retinopathy and DHA deficiency has been demonstrated with preliminary reports demonstrating improvement of visual function with oral supplementation of DHA [5, 11]. In addition Tein et al. [13]have reported a response of the peripheral neuropathy with cod liver oil supplementation.
The early chorioretinopathy leads to progressive atrophy of the posterior choroid, retinal pigment, epithelium and retina as the retinopathy progresses, with loss of night vision, followed by total loss of vision and legal blindness [11]. The etiology of the chorioretinopathy proposed to be associated with the toxic properties of the accumulating metabolites, such as long chain 3-hydroxyacylcarnitines or deficiency of DHA [2, 11]. It may also result from reduced energy production secondary to impaired mitochondrial fatty acid β oxidation [14]. These cases of long chain fatty acid oxidation defects who have shown significant benefits from treatment are reported.
Case 1
Female, birth weight 3.19 kg, currently age 15 years. There was an uneventful prenatal and neonatal period, with normal developmental milestones. She presented at 5 months of age with hypotonia, hepatomegaly and irritability following an episode of gastroenteritis. Hypoketotic hypoglycaemia was noted with an abnormal urine organic acid profile (elevated long chain hydroxyl-dicarboxylic acids) and elevated hydroxyacylcarnitines in blood. An ultrasound of the liver demonstrated increased echogenicity, a cardiac echocardiogram demonstrated hypertrophic cardiomyopathy, pigmentary retinopathy was also noted. The highest CPK recorded was 20,000 IU/l. Enzyme analysis on cells obtained from a fibroblast culture demonstrated LCHAD deficiency (3-hydroxyacyl-CoA dehydrogenase activity of 9.5 nmol/mgmin−1 (control 50)). Mutation analysis for the LCHAD gene, demonstrated homozygosity for the common LCHAD mutation G1528C. An emergency dietary regime of decreased long chain fats and increased carbohydrate with avoidance of fasting was commenced. The patient responded within 2 weeks with no further decompensations, the cardiomyopathy had fully resolved at 5 years of age. Long-term dietary intervention prevented further decompensation with clearance of organic acid metabolites. However, progressive pigmentary retinopathy developed, which was refractory to dietary treatment. At 13 years of age there was reduced visual acuity noted to 6/9 in each eye. An ERG performed showed reduced amplitude and increased latency in rod and maximal responses. The cone response was shown to be of normal latency with an abnormal β wave amplitude. The pattern ERG showed poorly defined waveforms with an abnormal pattern visual evolved response. Overall, there was noted to be evidence of diffuse rod and cone dysfunction with macular involvement.
DHA (100 mg/day) was commenced at age 14 following documentation of low DHA levels in erythrocytes. There was also noted stabilisation of visual function. DHA levels in erythrocytes measured 2 months after replacement were within normal limits. The patient now is attending mainstream secondary school with no academic difficulties.
Case 2
Female, currently age 14 years. There was a normal birth history and delivery, birth weight 3.5 kg. A sibling died suddenly at 8 weeks of age following a viral illness. High risk metabolic screening for a possible fatty acid oxidation defect was performed within the first 2 weeks of life, glucose monitoring and organic acid analyses were found to be within normal limits. Following discharge, she developed a respiratory arrest with RSV bronchiolitis. Hepatomegaly and non-ketotic hypoglycaemia was noted with a CPK of 679 IU/l. She was placed on an emergency intravenous high dextrose infusion with stabilisation over the ensuing next few days. A marked LCHAD deficiency was noted on enzymatic analysis of skin fibroblasts, LCHAD activity of 10 nmol/mgmin−1 (26% of controls). Analysis of DNA derived from lymphocytes subsequently demonstrated heterozygosity for the LCHAD mutation, G1528C. Pigmentary retinopathy was noted at 5 months of age. She had vesicoureteric reflux and numerous associated urinary tract infections, which required frequent hospital admissions between the age of 1 and 3 years. All episodes of decompensation with rhabdomyolysis resolved with institution of unwell dietary regimes and aggressive treatment of co-existing infection. At 3 years of age she presented with a severe metabolic decompensation following a minor viral illness, with markedly decreased orientation and generalized tonic clonic seizures. Over the next 24 h, she developed acute hepatic and cardiac failure. 2 days post admission her condition had stabilized with emergency dietary treatment with no long-term consequences. The maximum CPK level noted was 10,524 IU/l. A psychological assessment at age 7 years showed significant learning difficulties with both verbal and non-verbal functioning requiring a special schooling placement. Recently she has been noted to have a peripheral neuropathy with absent deep tendon reflexes. Nerve conduction studies demonstrated a polyneuropathy with selective impact on sensory amplitude and latency. She currently has bilateral pigmentary retinopathy with low myopia and 6/9 vision in each eye. Dark adaptation on ERG recently performed shows normal rod and maximal response. The cone response is normal in latency with subnormal amplitude. A PERG performed indicates macular dysfunction. Testing showed rod bilateral para-central scotomata on visual field testing. She has recently been commenced on DHA at 100 mg/day.
Case 3
Male currently aged 12 years. There was a normal birth history and delivery, birth weight 3.01 kg. He was the first infant born to non-consanguineous parents. He first presented at aged 15 months with gait abnormalities, muscle weakness and delayed motor milestones. The weakness was predominantly noted in his lower limbs, both proximally and distally. He sat at 10 months without support and started to walk at 15 months. However, he was noted to have persisting gait abnormalities that tended to fluctuate. At this time also he was noted to have Achilles tendon tightness, which continued to progress and required surgery at 4 years of age. An echocardiogram and initial ophthalmologic review were normal. Nerve conduction studies confirmed a peripheral motor and sensory neuropathy with moderately reduced compound motor action potential amplitude with mild loss of conduction velocity and complete absence of sensory nerve action potentials in the upper and lower extremity. Clinically, he had symptoms of a combination of both proximal and distal weakness. Cranial and spinal MRI imaging was found to be normal. Molecular genetics studies for common mutations, for spinal muscular atrophy, Hereditary motor-sensory neuropathy and Duchenne muscular dystrophy were negative. A muscle biopsy taken showed an increase of number of subsarcolemmal mitochondria but was otherwise unremarkable. Respiratory chain enzymology was performed from the sample and was found to be within normal limits.
At 10 years of age significant progression of clinical symptoms was noted. He demonstrated difficulty climbing stairs and getting up from sitting position and needed a wheelchair for ambulation. He presented with three episodes of acute rhabdomyolysis, with a rise in creatine kinase (CPK) of up to 26,000 IU/l on one occasion and an episode of visionary hallucinations. Poor energy levels and extreme fatigability was also noted. Mild pigmentary retinopathy was seen on eye examination. Blood acylcarnitine analysis showed a mild increase in 3-hydroxyacyl species. Enzymatic analysis of the long chain fatty acid oxidation enzymes in muscle subsequently demonstrated a moderate trifunctional protein deficiency with a mutation of the HADHB gene (341 A→G) encoding the β subunit of the protein.
He was commenced on dietary therapy of a high carbohydrate, low fat diet with cornstarch supplementation at night, avoidance of fasting and limited exercise regime. This led to stabilisation of his condition with no further episodes of rhabdomyolysis. However, extreme fatigability persisted resulting in him being practically bed bound. One year ago he was commenced on a DHA dietary supplement (130 mg/day) after documentation of DHA deficiency in erythrocytes (DHA level: 12.60 pm/106 cells (n = 15.2–37.6). He has subsequently shown marked improvement in mobility and general strength. He is now capable of climbing stairs and playing limited soccer. Repeat nerve conduction studies performed 1 year after commencement of therapy showed moderate improvement in function.
Discussion
These three cases illustrate the phenotypic heterogeneity associated with long chain fatty acid oxidation defects with presentations ranging from acute Reye-like illness during intercurrent minor illness to a more chronic presentation with neuromyopathic symptoms. In all cases once diagnosis was confirmed prompt commencement of dietary intervention led to stabilization of the condition and prevention of death. When presented with the typical history of acute decompensation a metabolic disorder was considered. However, presentation of fatty acid oxidation defects with more chronic symptomatology may be less easily recognisable, such as progressive peripheral neuropathy, a significant feature of myopathic TFP deficiency. In most cases the neuropathic features precede rhabdomyolysis. Unexplained cardiomyopathy may also be a common late presenting feature.
These case reports illustrate the need for clinicians to have a high index of suspicion for fatty acid oxidation disorders in patients with chronic non-specific symptoms of unknown aetiology with the potential for treatment intervention. While survival with these conditions is vastly improved by current therapeutic regimes, morbidity still remains high.
The clinical and biochemical data supporting supplementation with DHA appear very promising. Results to date on two of our children currently on DHA supplements agree with this observation- one child demonstrating stabilization of visual function and the other noting markedly increased energy levels.
In summary, these paediatric cases illustrate the clinical heterogeneity of long chain fatty acid oxidation defects and opportunities for effective management if diagnosed.
References
Roe CR, Ding J (2001) Mitochondrial fatty acid oxidation disorders. In: Scriver CR, Beaudet A, Valle D, Vogelstezn B, Childs B (eds) The metabolic and molecular bases of inherited disease, 8th edn. McGraw Hill, New York, pp 2297–2326
den Boer ME, Wanders RJ, Morris AA, IJlst L, Heymans HS, Wijburg FA (2002) Long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency: clinical presentation and follow-up of 50 patients. Pediatrics 109(1):99–104
Olpin SE, Clark S, Andresen BS, et al (2005) Biochemical, clinical and molecular findings in LCHAD and general mitochondrial trifunctional protein deficiency. J Inherit Metab Dis 28(4):533–44
Bertini E, Dionisi-Vici C, Garavaglia B, et al (1992) Peripheral sensory-motor polyneuropathy, pigmentary retinopathy, and fatal cardiomyopathy in long-chain 3-hydroxy-acyl-CoA dehydrogenase deficiency. Eur J Pediatr 151(2):121–6
Boles RG, Buck EA, Blitzer MG, et al (1998) Retrospective biochemical screening of fatty acid oxidation disorders in postmortem livers of 418 cases of sudden death in the first year of life. J Pediatr 132(6):924–33
Spiekerkoetter U, Bennett MJ, Ben-Zeev B, Strauss AW, Tein I (2004) Peripheral neuropathy, episodic myoglobinuria, and respiratory failure in deficiency of the mitochondrial trifunctional protein. Muscle Nerve 29(1):66–72
Tyni T, Palotie A, Viinikka L, et al (1997) Long-chain 3-hydroxyacyl-coenzyme A dehydrogenase deficiency with the G1528C mutation: clinical presentation of thirteen patients. J Pediatr 130(1):67–76
Russell-Eggitt IM, Leonard JV, Lund AM, Manoj B, Thompson DA, Morris AA (2003) Cataract in long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency (LCHADD). Ophthalmic Genet 24(1):49–57
Tyni T, Kivela T, Lappi M, Summanen P, Nikoskelainen E, Pihko H (1998) Ophthalmologic findings in long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency caused by the G1528C mutation: a new type of hereditary metabolic chorioretinopathy. Ophthalmology 105(5):810–24
Gillingham MB, Connor WE, Matern D, et al (2003) Optimal dietary therapy of long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency. Mol Genet Metab 79(2):114–23
Harding CO, Gillingham MB, van Calcar SC, Wolff JA, Verhoeve JN, Mills MD (1999) Docosahexaenoic acid and retinal function in children with long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency. J Inherit Metab Dis 22(3):276–80
Russell-Eggitt IM, Leonard JV, Lund AM, Manoj B, Thompson DA, Morris AA (2003) Cataract in long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency (LCHADD). Ophthalmic Genet 21(1):49–57
Tein I, Vajsar J, MacMillan L, Sherwood WG (1999) Long-chain L-3-hydroxyacyl-coenzyme A dehydrogenase deficiency neuropathy: response to cod liver oil. Neurology 52(3):640–3
Tyni T, Paetau A, Strauss AW, Middleton B, Kivela T (2004) Mitochondrial fatty acid beta-oxidation in the human eye and brain: implications for the retinopathy of long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency. Pediatr Res 56(5):744–50
Acknowledgments
Dr. Eileen Naughten and Dr. G. Fox are thanked for their expert care of these patients.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Hayes, B., Lynch, B., O’Keefe, M. et al. Long chain fatty acid oxidation defects in children: importance of detection and treatment options. Ir J Med Sci 176, 189–192 (2007). https://doi.org/10.1007/s11845-007-0025-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11845-007-0025-y