
Abstract
Host–bacteria interactions are highly diverse in humans and animals in general. In the gastrointestinal tract they can range from mutualistic to pathogenic interactions. Host and intestinal symbionts form a superorganism where co-evolution has set up a dynamic but fragile homeostasis. Symbiotic bacteria are contained in the intestinal lumen by tightly controlled innate immune mechanisms referred as “physiological inflammation”, which is tightly regulated by sustained mechanisms of innate immune tolerance responding to host–bacteria cross talks that remain to be fully deciphered. Conversely, pathogenic bacteria need to be quickly perceived and discriminated from symbiotic bacteria in order for the host to develop rapid and efficient bactericidal responses referred as “pathologic inflammation”. Recognition of pathogen/microbe-associated molecular patterns (P/MAMP) by pathogen recognition receptors (PRR) can hardly account for discriminating bacterial symbionts from bacterial pathogens, which largely share similar PAMPs. Unlike bacterial symbionts, the pathogens engage the host epithelial surface by tightly adhering to cells, possibly invading them, multiplying intracellularly, introducing massive amounts of PAMPs in their cytosol and altering their membranes by the secretion of pore-forming toxins and various secretory translocators. A large part of these “aggressive” events is recognized by dedicated systems, for instance, the toll-like receptors (TLR) or the cytosolic NOD-like receptors (NLR) that activate major proinflammatory pathways such as the NF-κB cascade and the inflammasome, leading to the release of the potent inflammatory cytokine IL-1β. These danger signals are amplified by endogenous signals, the damage-associated molecular patterns (DAMPs), derived from the damage induced to the host and mediated by PRRs and other receptors such as purinergic receptors for ATP. On the top of the PRR-associated first wave of signalling, this second wave of signalling that is strongly related to pathogen-associated dangers will complete the discrimination and innate inflammatory response to pathogens. However, bacterial classification in symbionts versus pathogens is far too simple and may not reflect the reality of host–bacteria interactions. The existence of pathobionts as well as the emergence of inflammatory bowel diseases associated with a loss of bacterial mutualism indicates that there is a spectrum of situations between the mutualistic and pathogenic poles.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Pathogenic Bacterium
- Innate Immune System
- Host Immune System
- Symbiotic Bacterium
- Inflammasome Activation
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
Introduction
The human gastrointestinal tract is continuously exposed to microorganisms, ranging from symbionts to pathogens. These bacteria establish complex and dynamic interactions with the intestinal mucosa. Symbiotic bacteria have adapted to selective pressure exerted by the host through evolution and activate innate immune responses, leading to “physiological inflammation” and contributing actively to intestinal immune homeostasis. Pathogens can also interact with the intestinal mucosa to promote invasion, thus compromising intestinal homeostasis. They penetrate intestinal host tissues, proliferate and disseminate to other hosts, inducing “pathological inflammation” and resulting in damage or death. Thus the mammalian innate immune system has to deal with symbiotic and pathogenic bacteria, in sickness and in health, in order to maintain or re-establish intestinal homeostasis. As proposed by Janeway [1], microbial structures called pathogen/microbe-associated molecular patterns (P/MAMP) are recognized by host pathogen recognition receptors (PRRs) of the innate immune system. This recognition can lead to activation or regulation of the innate and adaptive immune systems. However both symbionts and pathogens produce these molecular structures such as lipopolysaccharide (LPS) or peptidoglycan (PGN). This recognition raises the question of the discrimination between symbionts and pathogens by the host immune system. How does the intestinal mucosa face the challenge to be simultaneously tolerant to symbiotic bacteria that populate the gut lumen and release PAMPs and antigenic molecules but also highly responsive to the occurrence of pathogenic bacteria that have evolved strategies for attaching to and invading mucosal surfaces?
In this chapter we will mainly focus on the discrimination between symbionts and pathogens by the host immune system. We first discuss how host and bacteria interact to enable the establishment of a stable intestinal microbiota that participates in host fitness. This microbiota is beneficial to the host as it constitutes a physical barrier against pathogenic infection but also stimulates the development of the innate immune system. However this physiological inflammation that contributes to the containment of the microbiota in the intestinal lumen is not sufficient to counteract the attack by true pathogens. These bacteria indeed developed virulence strategies to cross the intestinal epithelial barrier, breach innate immune host defences, manipulate host signalling and invade deeper host tissues. Thus the innate immune system has to mount rapid and efficient responses to limit the infection. We will then focus on the recognition of this pathogenic threat by the host innate immune system and we will try to decipher the mechanisms that permit the discrimination between symbionts and pathogens necessary to set up a specific and adapted response. We will consider that, in addition to recognition of PAMPs, the immune system responds to other signals associated with infection or inflammation, allowing the discrimination between harmless and virulent bacteria. The host immune system would then adapt its response to the level of encountered threat. In particular, damaged cells can release in the extracellular milieu endogenous molecules that signal the danger such as molecules associated to cell death [2, 3]. These molecules, called DAMPs (damage-associated molecular patterns), are thus not strictly specific to pathogenic infection but translate the emergence of a danger coming from either a pathogen or a “harmful symbiont”, namely, pathobionts, but also from a sterile inflammation. Indeed these DAMPs are not necessarily due to the pathogens themselves but can be released after the induction of damage by the host innate immune response and the activation of pathologic inflammation.
Host–Symbionts Interactions: Adapted to Live with Our Best Enemies
Until recently, microbes associated with humans were largely described as aggressors engaging host surfaces, controlling host immune defences, expanding their population and propagating to other hosts. Microorganisms were essentially considered as pathogens and interactions between host and bacteria were mostly associated with infection [4]. Studies of the host immune system were thus based on the dichotomy between recognition and elimination of microorganisms and tolerance of self-molecules to maintain host homeostasis. However it is increasingly recognized that interactions between microorganisms and their hosts are not exclusively detrimental. Indeed these interactions can range from a symbiotic association to a deadly infection. “Illness is the exception rather than the rule”, state Scott Merrell and Stanley Falkow [5]. As a matter of fact, the dominant forms of human–bacteria interactions are those in which microorganisms—now collectively called microbiota—do not cause harm (commensal interactions) or even benefit to the host (mutualistic interactions). Obligate and facultative symbionts associate with eukaryotes and both partners can take advantage from these interactions, forming superorganisms in which homeostasis is preserved. We thus share a complex and subtle relationship with our microbiota.
Humans acquire a resident microbiota at birth. A complex and dense microbial flora colonizes the adult intestinal tract, with its highest density in the terminal ileum and in the colon. The human gastrointestinal tract harbours from 10 to 100 trillion microorganisms, most of which are anaerobic bacteria. Many studies have focused on determining the core elements of the microbiota that are far from being fully defined. It was estimated that more than 500 bacterial species are present in the intestine [6]. The number and composition change along the gastrointestinal tract. Predominant communities belong to two major groups, the Firmicutes (Gram-positive anaerobes) and the Bacteroidetes (Gram-negative anaerobes), suggesting that selective pressure may participate to this diversity. Despite the high variability of bacterial abundance and variety, metagenomic sequencing points out the existence of a common core of microbial genes, shared among at least 50 % of individuals [7, 8]. Moreover other vertebrates are colonized by related but distinct microbiota from those described in humans. These symbionts contribute to many functions that are beneficial to the host, especially by their metabolic capacities [9]. They achieve fermentation of non-digestible substrates, produce essential vitamins, generate short-chain fatty acids from glycans and contribute to ionic absorption of calcium or magnesium. Furthermore they shape the gut immune system, affect components of the enteric nervous system, contribute to oral tolerance to food antigens and play a role in wound repair of the intestinal mucosa after epithelial damage. Finally, these symbionts participate in immune homeostasis and protect the host against colonization and invasion of pathogenic bacteria by secreting bactericidal substances and competing for nutrients and niche colonization [10].
Since the most ancient time, the bacterial communities have been selected, meaning that competing organisms underwent biological co-evolution and co-adaptation to persist in specific host niches. Each host has co-evolved with its own microbiota. Mechanisms have been selected to promote and maintain mutualistic interactions between bacteria and eukaryotes, particularly in the gastrointestinal tract [11]. In the intestinal lumen, symbiotic bacteria benefit from a situation of active host tolerance. Indeed co-evolution has selected host immune mechanisms at mucosal surfaces that control immune overresponse to the microbiota and help maintaining a low level of responsiveness that is nevertheless sufficient to contain the symbionts intraluminally (i.e. physiological inflammation), while keeping the ability to recognize and fight pathogens. Compelling evidences show that the microbiota can influence the host immune response and elicit immune mechanisms that modify the balance between proinflammatory and regulatory responses [12]. In case of dysbiosis, the microbiota composition is altered, leading to inappropriate host immune response and possibly to inflammatory disorders [13, 14]. The “hygiene hypothesis” [15] or the “disappearing microbiota” hypothesis [16] state that alterations in the human microbiota, mainly due to hygiene and antibiotics, could be an important factor increasing the incidence of some diseases such as allergy, asthma, inflammatory bowel diseases, obesity and diabetes. A major issue is now to decipher the role of indigenous microbial communities in human health and disease. A cellular microbiology of symbiosis is quickly emerging which necessitates the development of a novel array of cell and animal models.
The Law of the Strongest: The Pathogenic Attack
Pathogenic bacteria constitute only a small proportion of bacterial species. Historically however, these bacteria have been the cause of deadly and widespread epidemics like the medieval “Great Plague”, or “Black Death”, and pox that marked the spirit of mankind. Work by Louis Pasteur, Robert Koch and their schools, at the end of the nineteenth century, enlightened the beginning of host–pathogen interaction studies. In spite of the implementation of hygiene, vaccination and antibiotic treatment in the last century that reduced the global morbidity and mortality of infectious diseases, pathogens remain an important public health threat in low-income countries, and more than 300 new infectious diseases emerged since 1940 [17] with 87 “novel” pathogens identified since 1980 [5, 18]. When a pathogen breaches a host anatomic barrier, the innate immune system provides immediate defence against infection to limit bacterial entry, proliferation and propagation. Activation of innate immunity by invading microorganisms needs to be very rapid and bacterial recognition is central to innate immunity [19, 20]. Invading pathogens are rapidly sensed in a non-specific manner by the host innate immune system using a limited set of receptors. Pathogen recognition leads to the immediate activation of humoral and cellular components of the innate immune system. This participates in bacterial clearance and favours immune cell recruitment to sites of infection but also crucial activation of the adaptive immune system, which confers final eradication capacity and long-lasting immune memory [21].
The host immune system and pathogens have co-evolved, leading to an evolutionary arm race [22] that can be deleterious for both players. A tempting hypothesis has been proposed by May and Anderson [23], according to which virulence was the first step on the way to a symbiotic interaction. It was also assumed that immune processes evolved to avoid overreaction to pathogens to eventually achieve mutualism [24]. The best example of this ultimate symbiosis could be the mitochondria. According to the endosymbiotic theory, this organelle may derive from primitive bacteria, contributing to oxidative metabolism when oxygen appeared on earth [25].
Pathogenic and symbiotic bacteria may belong to the same genus or species, but are different by the relationships they maintain with the host. Symbiotic bacteria provide a first line of defence against pathogenic bacteria as they compete with pathogens for nutrients and constitute a colonization barrier [22]. However, most enteric pathogens are not as well metabolically equipped as symbionts [26]. Indeed their genomes contain a limited number of genes involved in saccharide uptake and hydrolysis compared to symbiotic bacteria. Unnecessary or detrimental metabolic pathways may have been lost to confer advantages in a selective niche. Therefore they may be less adapted to compete with symbionts for nutrients and the virulence strategies they developed allow them to gain access to host tissues, thus possibly resolving these nutritional issues and replicating [27, 28]. In parallel, symbionts are devoid of genes that promote invasion and subversion of host tissues [29].
Pathogenic bacteria can cross the intestinal epithelial barrier, breach innate immune host defences and invade deeper host tissues. The main differences between symbionts and pathogenic bacteria thus reside in the latter producing effectors mediating adherence or penetration of the intestinal epithelium, but also innate immune response evasion (Box 14.1).
Pathogens inherited specific pathogenicity genes usually organized in pathogenicity islands [30]. The gene clusters present on pathogenicity islands encode for adhesins, invasins, secretory apparatus such as the type-three secretion system (TTSS) and their dedicated effectors, enzymes, toxins and hemolysins. These bacterial factors are necessary for the pathogens to adhere to and colonize the epithelial surface or to breach the epithelial barrier. Further they participate in host-cell manipulation and subversion of immune responses to promote bacterial survival, colonization, proliferation and dissemination to other sites. Pathogens are able to dampen innate immune defences at any stage of their progression, such as inhibiting expression of antimicrobial peptides and mucins, evading or suppressing phagocytic killing. This ability to overcome and/or manipulate host innate immune responses also defines the identity of being a pathogen. Intracellular colonization by pathogens contributes to mucosal inflammation as invaded epithelial cells produce proinflammatory molecules that recruit immune cells, and ultimately to epithelial destruction.
Microbial Sensing by Host Receptors: The Crossroad Between Inflammation and Tolerance
Innate immunity is the initial step of defence against bacterial infection. The front-line target cells need to recognize microorganisms with prokaryote-specific receptors to induce antimicrobial innate immune responses. Invariant microbial structures called pathogen-associated molecular patterns (PAMP) are recognized by a variety of germ line-encoded pattern recognition receptors (PRR) [31]. Receptor activation induces a response characterized by a burst of inflammation in the infection site, tissue destruction and recruitment of immune cells such as phagocytes and antigen-presenting cells. PRRs play an important role in costimulation of the adaptive immune system. They recognize foreign organisms, i.e. viruses, bacteria, parasites and fungi. Different types of ligands activate these receptors such as LPS, PGN, non-methylated DNA, RNA, flagellins, lipopeptides, toxins and fimbriae. Four families of PRRs cooperate to recognize microorganisms: the toll-like receptors (TLR), the nucleotide oligomerization domain-like (NOD) receptors (NLR), the RIG-I-like receptors (RLR) and the C-type lectin receptors (CLR). They may be secreted in the extracellular fluid (such as mannan-binding lectin, C-reactive protein or serum amyloid protein), intracellular (such as members of the NLR family, MDA-5 or RIG-1) or membrane-anchored such as the TLRs that are expressed either at the cell surface or associated with endosomes.
TLRs represent a family of highly conserved transmembrane molecules with an intracellular domain similar to the cytoplasmic domain of the Interleukin-1 receptor (IL-1R) [32]. The extracellular domain is the recognition site. It is characterized by leucine-rich repeats (LRR) and determines the ligand specificity. Some TLRs are present at the surface membrane (TLR1, 2, 6, 4 and 5) whereas others are defined to intracellular endosomal compartments (TLR3, 7, 8 and 9). The ligand specificity also depends on TLRs association, as they may homo- or heterodimerize. TLR specificities have been widely studied. One of the most described is TLR4 that recognizes LPS of the outer cell membrane of Gram-negative bacteria. TLR expression was first detected on blood monocytes, but intestinal epithelial cells (IECs) also express at least a subset of these TLRs.
Bacterial ligands are moreover recognized intracellularly by NLRs [33]. These multidomain proteins usually contain an LRR domain that determines the recognition specificity, a nucleotide-binding and self-oligomerization domain called NACHT domain and effector-binding domains that can be either caspase recruitment domains (CARD) for the NLRC family that comprises NOD1 and NOD2 or pyrin effector domains (PYR) for the NLRP subclass. This binding domain interacts with adaptor molecules to initiate signalling cascades following receptor activation [34]. NOD1 and NOD2 are key cytosolic sensors. They recognize muropeptides that are fragments of the PGN, a component of the bacterial cell wall. PGN consists of carbohydrate chains of β (1–4) linked, alternating N-acetylglucosamine and N-acetylmuramic acid sugars, cross-linked by short peptide chains. NOD1 is found in all cell types and binds to a diaminopimelate containing the GlcNAc-MurNAc tripeptide (GM-TriDAP or DAP) found in mainly Gram-negative bacterial PGN, whereas NOD2 detects muramyl dipeptide (MDP), the minimal bioactive PGN motif shared by all bacteria, in myelomonocytic and IECs [35–37].
Activation of most of these PRRs initiates signal transduction pathways that converge on the key transcription factor Nuclear Factor-κB (NF-κB) and leads to antimicrobial and proinflammatory gene expression and also recruitment of immune cells to the site of infection [34]. NOD1 and NOD2 are the only NLR members able to activate the NF-κB pathway upon stimulation. Other members of the NLR family, the NLRPs, participate in the activation of caspase-1 by the inflammasome (described in chapter “Genetic Overlap Between Inflammatory Bowel Disease and Other Diseases”).
Symbiotic and pathogenic bacteria share many striking similarities. Notably, the microbial structures that are recognized by the host innate immune system are common. The molecular motifs recognized by NLRs or TLRs are ubiquitously present in intestinal bacteria and are therefore important not only for mucosal host defence but also for intestinal homeostasis. As these immune activators are found in symbiotic and pathogenic bacteria, it was proposed to rename these components MAMPs for microbial-associated molecular patterns. These similarities make them, in general, unlikely candidates for discrimination between symbiotic and pathogenic microorganisms. How does the intestinal mucosa avoid the continuous activation of inflammatory responses by symbionts that are a source of proinflammatory PAMPs? The association found between mutations affecting NOD2 and Crohn’s disease illustrates this question [38, 39]. PRRs do not generally efficiently discriminate between pathogenic and symbiotic microorganisms because none of the PAMPs is specific to one category, and they do not recognize pathogenicity-specific components. TLRs expressed by IECs can sense mucosal surfaces to monitor bacterial densities reflected by MAMPs concentration [40, 41]; thus, they play important functions to maintain intestinal epithelial homeostasis [42]. Innate immune recognition of symbiotic bacteria by TLRs at the mucosal surface is also necessary under steady-state conditions to limit inflammation within the intestine [43]. Similarly, NOD1 and NOD2 participate to host defence but also to intestinal homeostasis [44, 45].
Symbionts and Pathogens: Persona Non Grata Beyond the Mucosal Barrier
Controlling bacterial interactions at the intestinal mucosal surfaces is an important host strategy to limit bacterial contacts and prevent bacterial invasion. Symbiotic bacteria are contained in the intestinal lumen and tolerated. A general view is that most of the microbiota is maintained, as a complex population, away from the epithelial surface, by a process earlier qualified as “physiological inflammation” (see Fig. 14.1a). It is a complex mixture of a physical barrier, largely achieved by the mucus, a great majority of the bacteria residing in its softer outer layer and of chemical compounds, such as NOS, ROS and antimicrobial peptides, embedded in the denser lattice of mucins that forms the inner mucus layer in close apposition to the epithelial surface [12, 34]. The mucus layer, composed of mucin glycoproteins produced by goblet cells, covers the intestinal epithelium and protects the mucosal surface from invasion, defining a relatively “germ-free” zone [46]. Bacteria are thus prevented to adhere directly to the intestinal epithelium [47]. Mucin 2-deficient mice do not exhibit this bacteria-free area and develop spontaneous colonic mucosal inflammation [48, 49]. Similarly, mucin upregulation is observed after infection with enteropathogenic bacteria such as Salmonella, Yersinia or Shigella [50, 51].

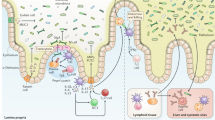
Host–bacteria interactions in health and disease. The human gastrointestinal tract is continuously exposed to microorganisms, ranging from symbionts to pathogens, including pathobionts. These bacteria establish complex and dynamic interactions with the intestinal mucosa. (a) Symbiotic bacteria participate in immune homeostasis and protect the host against colonization and invasion of pathogenic bacteria by secreting bactericidal substances and competing for nutrients and niche colonization. Symbionts communicate among themselves by releasing small molecules such as autoinducers. They are contained in the intestinal lumen by tightly controlled innate immune mechanisms referred as “physiological inflammation”, contributing to intestinal immune homeostasis. Intestinal epithelial cells (IECs) are attached together by tight junctions that form a sealed barrier to the luminal environment, preventing bacterial penetration. The production of mucus by goblet cells, NOS, ROS, antimicrobial peptides and microvillus-derived vesicles (MDV) containing catalytically active intestinal alkaline phosphatase by IECs and IgA by subepithelial B cells prevents the overt stimulation of intestinal innate immunity by symbionts and limit mucosal inflammation. Microbial structures called PAMP are recognized by host PRRs of the innate immune system such as toll-like receptors (TLR) and NOD-like receptors (NLR), inducing regulatory signalling cascades that participate in intestinal homeostasis by inducing the production of regulatory cytokines and chemokines. Several subtypes of differentiated T cells are associated with the epithelial layer and participate to the mucosal immune response, the same as dendritic cells [117]. They produce effector cytokines necessary to contain bacteria to the intestine and stimulate macrophages located in the subepithelial area of the lamina propria to quickly phagocytose and kill the symbionts that would cross the epithelial barrier. To avoid overt immune responses, PRRs expression is restricted and limited to certain cell populations and the localization of the receptors on the cells is restricted. Host cells may also modify PAMPs to limit their agonist function on PRRs. Symbiotic bacteria may directly participate to the tolerogenic process by producing weakly stimulatory PAMPs. (b) Bacteria venturing towards the epithelial surface are quickly killed unless they can resist surface defence molecules. This is the case of a subcategory of symbionts called the pathobionts, illustrated by the segmented filamentous bacteria (SFB) Clostridium. Pathobionts are contained by innate immune mechanisms. They may be considered “good bacteria” as they achieve maturation of the mucosal immune system, inducing the maturation of naïve T cells into inflammatory (i.e. Th17) lymphocytes that are essential to the maintenance of mucosal innate immune protection. However, if the density of pathobionts is not balanced by sufficient density and diversity of symbionts, they may express their pathogenic potential and account for chronic inflammation of the gut. (c) Pathogenic bacteria engage the host epithelial surface by tightly adhering to cells, possibly invading them, multiplying intracellularly, introducing massive amounts of PAMPs in their cytosol and altering their membranes. Tight-junction opening, secretion of pore-forming toxins (PFT), insertion of secretory translocators such as the type-three secretion system (TTSS) and secretion of toxins and effectors (mucinases, adhesins, invasins) are necessary for host tissue invasion. Activation of PRRs initiates signal transduction pathways that lead to proinflammatory gene expression but also recruitment and activation of immune cells to the site of infection such as DCs, macrophages, T cells and polymorphonuclear neutrophils (PMN). Endogenous signals amplify this response. Indeed, damage-associated molecular patterns (DAMPs) derived from the damage induced to the host such as cell lysis or cytoskeleton modifications and mediated by PRRs and other receptors such as P2X7, the receptor for ATP, participate in the formation of molecular scaffolds named inflammasomes that induce a caspase-1-dependent cellular death called pyroptosis, but also cytokine activation such as IL-1β (beta) and IL-18
IECs play an important role in maintaining intestinal homeostasis. These cells produce and secrete microbicidal molecules such as antimicrobial proteins, mainly defensins or cathelicidins [50–53]. Some of these molecules are constitutively produced, but others are controlled by bacterial activation. IECs lining the gut lumen also produce microvillus-derived vesicles that are released and accumulate in the lumen. These vesicles contain catalytically active intestinal alkaline phosphatase, an enzyme responsible for LPS dephosphorylation, preventing intestinal inflammation [54–57]. These vesicles cluster on the luminal bacteria, inhibiting bacterial attachment to the host cells and limiting bacterial growth in the intestinal lumen [58]. In addition to their production of bactericidal molecules that keep the cellular apical environment relatively clear of symbiotic bacteria, IECs are attached together by tight junctions that form a sealed barrier to the luminal environment, preventing bacterial penetration. Another epithelial cell, the Paneth cell, secretes antimicrobial proteins at the base of the small intestinal crypts. Paneth cells contain microbicidal granules that they discharge when they sense bacteria, controlling the invasion by symbiotic and pathogenic bacteria.
Adaptive immunological responses are also induced to protect the intestinal surface and limit bacterial interactions with the mucosal surface. It is largely based upon IgA production by subepithelial B cells located in the lamina propria. Mice lacking IgA exhibit an increase in mucosa-associated bacteria [59]. Dendritic cells located in intestinal lymphoid structures called Peyer’s patches or solitary nodules in the colon sample bacteria translocated through epithelial M cells. They interact with B cells that differentiate into plasma cells to produce IgA that transcytose across the epithelium [60]. In parallel, several subtypes of differentiated T cells are associated with the epithelial layer and participate to the mucosal immune response. They produce effector cytokines necessary to contain bacteria to the intestine [61] and stimulate macrophages located in the subepithelial area of the lamina propria to quickly phagocytose and kill the symbionts that would cross the epithelial barrier.
Bacteria venturing towards the epithelial surface are therefore quickly killed unless they can resist surface defence molecules. This is the case of a subcategory of symbionts called the pathobionts, illustrated by the segmented filamentous bacteria (SFB) Clostridium in the murine intestine [62], and probably Enterobacteriaceae and Enterococci in humans (see Fig. 14.1b). Pathobionts are contained by these innate immune mechanisms but may quickly trespass the mucosal barrier in case of immune failure such as immunosuppressive chemotherapies for cancer, leukemias and organ transplantation [63]. From their “distant” site of residence, symbionts communicate among themselves as any complex microbial population in various environments, releasing small molecules such as autoinducers [64] and PAMPs. This collection of molecules is sensed by the front line of IECs, which are likely, on this basis, to gauge the bacterial density and accordingly adjust its response that encompasses the dual necessity to fine tune its bactericidal response to the exact level of threat, and to elicit the signals maintaining the tolerogenic process. Microbiota detection therefore activates host mechanisms involved in mucosal homeostasis [65].
All the strategies that control the symbiotic luminal content are also involved in the fight against pathogenic infection, particularly the sensing of PAMPs, thus reemphasizing the need to better understand how the host can discriminate between symbionts, including their more adventurous companions, the pathobionts and the true pathogens. A characteristics of the pathogens is their capacity to engage the epithelial barrier and possibly to achieve its subversion and invasion [66]. One could therefore operationally discriminate symbionts and pathogens on the capacity of the latter to trespass the epithelial barrier, thus generating a systemic immune response, instead of the controlled mucosally localized response that is characteristic of the microbiota [67].
Mesenteric Lymph Nodes as Immune Firewalls Between Tolerance and Ignorance
Symbionts do not induce a strong systemic inflammatory reaction, indicating that the host immune system is highly adapted to the microbiota. On the contrary host–pathogen interaction leads to a strong systemic response. A major problem is again to understand how the immune system can distinguish pathogenic bacteria from symbiotic bacteria. The development of the mucosal immune system depends on colonization by the microbiota. Comparisons between germ-free and specific pathogen-free (SPF) animals indicate that the mucosal immune system is underdeveloped in germ-free animals. They exhibit a strong decrease in IgA-producing plasma cells in the lamina propria, a reduction in some subclasses of intraepithelial lymphocytes and hypoplastic lymphoid follicles. Interestingly, expansion of the structures of the spleen and lymph nodes is also dependent upon the presence of the microbiota, even if these organs are not in direct contact with the microbiota [68]. While the microbiota is restricted to the intestinal lumen, some symbionts can be sampled by mucosal lymphoid organs such as Peyer’s patches where they interact with dendritic cells. A local immune response is induced but these primed dendritic cells are not found farther than the draining mesenteric lymph nodes (MLN). This indicates that these primed dendritic cells are limited to the mucosal immune system, thus restricting symbiont dissemination. Symbiotic bacteria normally do not penetrate beyond the draining MLNs that form a “firewall” between the mucosal and the systemic immune system. As symbionts do not go further, they are poorly recognized by the systemic immune system. The intravenous injection of symbiotic bacteria in mice induces a systemic immune response [69]. In parallel their ignorance by the adaptive systemic immune response disappears when MLNs are surgically removed [59]. This indicates that symbiotic bacteria normally do not prime the systemic immune response, mucosal immunity being sufficient to their containment. This highly compartmentalized immune response preserves the host capacity to mount an efficient systemic response against bacteria breaching the epithelial barriers. Conversely, pathogenic bacteria are able to trespass the draining MLN “barrier”. Infection is not confined to mucosal tissues; therefore, pathogenic bacteria prime the adaptive systemic immune system.
The TLR-MYD88 (myeloid differentiation primary-response protein 88) signalling pathway can sense symbiotic bacteria and is important for the establishment and the maintenance of host–symbionts homeostasis. Classically the activation of this pathway leads to the induction of an inflammatory response that participates in bacterial clearance. This cascade is involved in many diverse processes such as the production of antimicrobial proteins like RegIIIγ, enlightening the importance of symbiotic bacteria in the activation of the host innate immune response [70]. Interestingly, innate immune defects such as a deficiency in TLR signalling adaptor molecules can result in the priming of adaptive immune system by symbiotic bacteria. Mice lacking MYD88 or TRIF (TIR-domain-containing adaptor protein inducing IFN-β) adaptors exhibit a complete loss of host–symbionts compartmentalization and produce IgG responses against symbionts, probably because many symbiotic bacteria cross the epithelial barrier and are not efficiently eliminated by phagocytes [71]. The systemic adaptive immune system can thus compensate a loss of the mucosal innate immunity. Therefore the activation of the systemic immune response is essentially a matter of balance. This balance is disrupted in the presence of pathogens or in the absence of an efficient mucosal immunity necessary to contain the symbionts upstream the MLNs.
Active Immune Tolerance Towards Symbiotic Bacteria
To avoid the overt stimulation of intestinal innate immune receptors by symbiotic PAMPs and to limit mucosal inflammation, several sophisticated strategies have been developed by symbiotic bacteria and host tissues.
PRRs expression is restricted and limited to certain cell populations. For example, intestinal macrophages show reduced PRRs expression contrary to macrophages present in other tissues [72–74]. Also TLR4 expression is restricted to crypt epithelial cells, limiting its activation by bacterial ligands. Paneth cells located at the base of these intestinal crypts express high levels of TLR4 and NOD2, protecting the crypts and probably the stem cells from bacteria [75, 76].
Second, the localization of the receptors on the cells can be restricted. Indeed intracellular localization of some receptors such as NOD2 requires the transport of the ligands into IECs or microbial invasion of the cytosol, thus likely avoiding major receptor activation by extracellular muropeptides and non-invasive bacteria [44, 77]. Similarly, TLRs distribution is restricted at the apical surface of IECs. TLR5, responsible for bacterial flagellin recognition, is only exposed at the basolateral side of differentiated IECs [78, 79].
Third, signalling regulations may prevent or modify immune activation. Indeed many negative regulators of the TLR signalling pathways have been described. For example, the negative regulator Tollip is expressed in IECs and directly interacts with IRAK-1, preventing its autophosphorylation and inhibiting any further signalling [80, 81]. Host cells may also modify PAMPs to limit their agonist function on PRRs as exemplified by IECs producing hydrolases, cleaving acyl chains from lipid A, the endotoxin moiety of LPS in Gram-negative bacteria [82, 83] or producing an alkaline phosphatase that dephosphorylates the lipid A [54], thus in both cases attenuating endotoxin activity on TLR4.
In parallel, symbiotic bacteria may directly participate to the tolerogenic process by producing weakly stimulatory PAMPs such as hypoacylated lipid A in Bacteroides fragilis [84, 85]. Symbiotic bacteria may also actively suppress epithelial proinflammatory signalling by interfering with NF-κB activation. For example, Bacteroides thetaiotaomicron promotes nuclear export of the NF-κB p65 subunit in a peroxisome proliferator-activated receptor γ-dependent manner [86]. Also the probiotic species Lactobacillus casei inhibits the degradation of the inhibitor I-κB in order to avoid proinflammatory gene induction [87].
Invasion, Damage and Inflammation Are Hallmarks of Pathogenic Bacteria
Intestinal homeostasis that prevails between the intestinal mucosa and symbiotic bacteria is fragile and can be subverted by pathogens (see Fig. 14.1c). Indeed enteropathogens have the capacity to disturb the intestinal epithelium, invade host cells and induce the inflammatory destruction of the intestinal mucosa with the help of virulence factors that are specific to pathogenic bacteria [66]. Pathogen recognition by host cells mainly occurs through PAMPs and PRRs. However this recognition may not be straightforward due to the recognition of harmless bacteria by PRRs or to PAMPs modifications by pathogens to subvert immune recognition, making difficult the discrimination between symbionts and pathogens. As a matter of fact, other signals are clearly necessary to activate a full immune response in case of true pathogens. In 1994, Polly Matzinger pioneered the concept of “danger signal” [3], suggesting that the host perceives the presence of pathogens or pathological conditions as much as the cellular damage they cause. The molecules that signal tissue and host damage (i.e. “danger molecules”) may have two different origins: (1) host molecules released during the infectious process and called DAMPs or alarmins and (2) cellular structures affected by pathogenic factors [88]. Consequently tissues that undergo destruction and loss of integrity would trigger the immune system. These danger signals recruit and activate the innate immune system and participate in the restoration of the destroyed tissue. Notably some alarmins can signal through TLRs and NLRs to induce inflammatory and immune responses, suggesting that these receptors can also sense “self-ligands” [89].
Cells dying by necrosis release DAMPs like ATP, uric acid, DNA and DNA-binding proteins such as high-mobility group box-1 protein (HMGB1) into the extracellular milieu [88]. These secondary stimulatory molecules have a high proinflammatory impact. Necrotic cell lysates are a source of endogenous factors such as HSPs or uric acid that induce dendritic cell activation. Extracellular HSPs interact with several receptors including TLRs, leading to secretion of proinflammatory cytokines. HMGB-1 is a nuclear protein that binds to the nucleosome. When cells die by necrosis, this protein is released extracellularly. HMGB-1 has chemotactic activities on immune cells such as monocytes, macrophages, neutrophils and dendritic cells, but also proangiogenic and immunostimulatory activities. Similarly an increase of extracellular ATP concentration reflects the presence of dying cells and leads to its binding to the ligand-gated ion channel P2X purinergic receptor 7 (P2X7). This receptor is important for ion transport and allows potassium efflux after activation [90, 91]. Tissue architecture disorder during bacterial infection may also send signals to the immune system [92]. Blood vessel rupture and inflammation induce the extravascular relocation of fibrinogen that can activate macrophages through TLR4. Disruption of the extracellular matrix and basement membranes by bacterial proteases are also sensed like danger signals. Soluble fragments of heparin-sulphate proteoglycans released from cell surface and basement membranes can also activate TLR4 of dendritic cells. Host membrane recruitment is also considered as a danger signal, the same as membrane integrity disruption by the insertion of secretory systems such as the TTSS of enteropathogens. Indeed it may lead to expression of proinflammatory cytokines [93].
The innate immune system recognizes invading microbes, tissue damage or stress through conserved receptors such as the TLRs and NLRs that are activated by PAMPs or DAMPs. These PRRs activate signalling cascades that converge in the transcription of cytokines, chemokines and proteins involved in bacterial clearance [94]. Among the NLRs, NOD1 and NOD2 play major roles in the intestinal epithelium as they detect intracellular ligands and activate NF-κB signalling leading to the transcription of proinflammatory genes. However other NLRs are also important for the innate immune response to activate inflammation and limit microbial invasion. Especially some NLRs are involved in the post-translational activation of inflammatory caspases and participate in the formation of molecular scaffolds named inflammasomes [93]. These large multiprotein complexes are activated by pathogen-associated signatures or endogenous molecules of similar structure produced after tissue damage. Inflammasome activation leads to autocatalytic cleavage and activation of caspase-1, and processing and secretion of proinflammatory cytokines such as IL-1β and IL-18 [95]. Whereas IL-1β induces IL-17 release from Th17 cells to amplify early effector responses, IL-18 stimulates CD8+ T cells and Th1 cells to secrete IFN-γ [96, 97]. Inflammasome activation induces a caspase-1-dependent cellular death called pyroptosis, but also autophagy and bacterial degradation [98]. Several types of inflammasome may be distinguished according to the NLR that is involved and the ligand that activates this platform. For example, the NLRP1 inflammasome is activated after MDP or anthrax lethal toxin recognition and the NLRC4 inflammasome after flagellin sensing. The most studied inflammasome is the one containing NLRP3. This complex is activated by different stimuli such as crystals, pore-forming toxins, bacteria and viruses. Some cytosolic proteins like AIM2 and RIG-I, which do not belong to the NLR family, also form inflammasomes in response to cytosolic DNA and virus respectively. Bacterial infection can activate several inflammasomes [99]. For example, Listeria monocytogenes induces via listeriolysin O, flagellin and bacterial DNA the NLRP3, NLRC4 and AIM2 inflammasomes, whereas Shigella flexneri and Salmonella typhimurium activate the NLRP3 and NLRC4 platforms [95]. However, the contribution or redundancy of these different complexes is not clear. Interestingly, deregulated activation of inflammasomes is associated with autoinflammatory syndromes and other pathologies [99].
Host Responses Manipulation by Pathogens
Enteric bacteria have developed sophisticated strategies to overcome the innate immune system and successfully hijack host signalling. Manipulation is the hallmark of pathogens. Pathogenic bacteria secrete factors that first favour infection and bacterial survival inside the cells. These virulence factors target cytoskeletal components, host cell receptors and signalling molecules. For instance, Shigella flexneri secretes IpaB that prevents rapid epithelial turnover to maintain its replicative niche but also inhibits IEC detachment by delivering the OspE effector through the TTSS, reinforcing epithelial adhesion to the basal lamina [100, 101]. Some bacterial factors manipulate host membrane and regulate actin cytoskeleton remodelling to permit bacterial adhesion, invasion and propagation. Rho GTPases regulation enables tight junction opening, barrier function reduction and bacterial entry, for example. The bacterial effectors SopB, SopE, SopE2 and SipA participate in tight junction disruption during Salmonella typhimurium infection, for example, but also induce an inflammatory response [102–104]. Bacterial effectors can affect post-translational modifications on host cells by mimicking the corresponding activities such as ubiquitination or sumoylation to hijack signalling pathways. Some intracellular bacteria have also the capacities to escape from the early vacuole after invasion in order to avoid the unfriendly environment present in this compartment. Then they hijack host cytoskeleton to move intracellularly and disseminate.
In parallel, pathogenic bacteria have expanded mechanisms to avoid recognition by the host innate immune response. First they developed strategies to modify their PGN and thus avoid recognition by the innate immune receptors. Many modifications of the PGN are possible to ensure resistance to antibiotics or host degradative enzymes that target the cell wall but also impairment of detection by PRRs to avoid innate immune signalling. During the course of a bacterial infection, the structure and composition of PGN is likely to be modified through the action of bacterial enzymes. For instance, SltY, a bacterial lytic transglycosylase involved in PGN processing, is highly up-regulated during the infection of IECs by Shigella flexneri [105]. Many modifications are observed in pathogenic species, mainly GlcNAc N-deacetylation and MurNAc O-acetylation. Listeria monocytogenes is a gram-positive bacteria that plays on PGN structure. This pathogen produces and secretes autolysins important for the virulence such as the endopeptidase p60 and the N-acetylmuramidase NamA. These two enzymes cleave the PGN to generate the NOD2 agonist, thus modulating the inflammatory response and the microbial recognition [106].
Several mechanisms to circumvent and overcome innate immune responses also exist. Among these strategies, the secretion of bacterial effectors through the TTSS enables bacteria to manipulate a broad array of host pathways by regulating or mimicking host proteins in order to survive and colonize host tissues [107]. Some pathogens have evolved strategies to avoid phagocytosis by delivering effectors that impair the signalling downstream of phagocytic receptors such as YopH, YopE and YopT effectors secreted by Yersinia to inhibit phagocytosis [108]. Many bacterial effectors can directly modulate host proinflammatory pathways to suppress detrimental inflammation during invasion steps. Given the role of the NF-κB pathway in the immune responses, enteric bacteria have developed mechanisms to interfere with this cascade [109]. First pathogenic factors contribute to the inhibition of the cascade at the level of the TLRs by host protein mimicking. Indeed pathogenic effectors can share sequence or structural homology with the TLR domain necessary for the signalling, which is the TIR domain responsible for adaptation recruitment, interfering with TLR signalling and preventing thus further innate host defence. An example of bacterial mimicry is Salmonella effector TIR-like protein A (TlpA) that impairs TLR4-mediated NF-κB activation [110]. Secondly bacterial effectors act at different levels of the pathway to prevent its activation by regulating phosphorylation, acetylation, ubiquitination and neddylation of host proteins required for NF-κB activation [111]. This modulation can be focused on a specific protein or affect a more general mechanism such as the ubiquitin pathway. A major strategy to regulate inflammation and maintain this control for long periods may be epigenetic regulation by bacterial effectors. OspF, an effector produced by Shigella, directly inhibits in the nucleus the activation of NF-κB target genes. Indeed OspF is a phosphothreonine lyase that dephosphorylates irreversibly and inactivates the enzymes responsible for histone H3 phosphorylation in the nucleus, the MAPKs p38 and ERK2 [112]. Finally, enteric pathogens have evolved stratagems to block deleterious inflammation and to evade inflammasome activation. Enteropathogenic Yersinia enterocolitica bacteria exploit several processes to prevent caspase-1 activation and secretion of IL-1β and IL-18 after inflammasome activation [113]. Symbiotic bacteria have also developed means to control their recognition by the host and the activation of the host immune system. However these mechanisms are much less developed and do not enable such bacteria to invade tissues and proliferate in the case of immune homeostatic balance.
Symbionts, Pathobionts and Pathogens: Evolutionary Distant Relatives?
Host–bacteria interactions have evolved thanks to their high capacities for genomic modifications. Compared to multicellular eukaryotes, bacteria have higher generation times and highly efficient properties to increase their genetic variability, mainly by horizontal gene transfer. These characteristics allow bacteria to evolve and continuously adapt to the selective pressure imposed by environmental changes, particularly in the host. A Manichean view of the microbial world often classifies bacteria into two different types: the “good bacteria” versus the “bad bacteria” and symbionts versus pathogens. However this classification is far too simplistic. There is increasing evidence for a continuous and linear evolution between the “bona fide” symbionts and the “bona fide” pathogens. Between these two ends of the spectrum, a vast “grey zone” comprises species like Escherichia coli that affect a large number of intermediate isolates between strictly symbiotic or pathogenic strains, depending upon the number, variety and complementarity of pathogenic or metabolic traits present in their respective genomes, in addition to their common core genome [114].
In this “grey zone”, emerge a category of microorganisms called the pathobionts [115]. Pathobionts may still be considered “good bacteria” as they achieve maturation of the mucosal immune system. For instance, in mice, the segmented filamentous bacterium clostridial strain SFB adheres to Peyer’s patches in the healthy terminal ileum where it induces IgA production and activates B cells [116]. It also induces the maturation of naïve T cells into inflammatory (i.e. Th17) lymphocytes that are essential to the maintenance of mucosal innate immune protection [14, 117]. However, if the density of pathobionts is not balanced by sufficient density and diversity of symbionts, they may express their pathogenic potential and account for chronic inflammation of the gut [118]. In this context, the immune status of the host is essential to determine whether bacteria will be harmful or harmless to the host. Immunodeficient mice (SCID mice) reconstituted with CD4 + CD45Rbhigh T cells and colonized with SFB develop severe colitis and intestinal inflammation [119]. SFB does not only affect the gastrointestinal tract, it may affect the whole fitness of the host as SFB reconstitution in germ-free mice also increases rheumatoid arthritis and multiple sclerosis susceptibility [62, 120]. Similarly, Helicobacter hepaticus colonizes mucosal surfaces of the gastrointestinal tract and the liver. These bacteria trigger in wild-type animals inflammatory and tolerogenic responses that participate in physiological inflammation. However immunocompromised mice such as SCID and Il10 −/− mice reconstituted with CD4 + CD45Rbhigh T cells or Rag2 −/− mice exhibit pathogenic inflammation after H. hepaticus infection and can develop rapid colitis or even colon cancer [121–123]. In immunocompromised animals, H. hepaticus induces a deregulated inflammatory response that leads to pathological inflammation. The type VI secretion system of H. hepaticus mediates these effects as deletion of this apparatus leads to higher colonization and elevated inflammatory response in immunocompromised hosts, suggesting that it is involved in the tolerogenic responses [63]. However the secreted effectors involved in this process have not been identified. Host interactions with Bacteroides fragilis have been better deciphered. This pathobiont closely associates to mucosa and produces an anti-inflammatory capsular polysaccharide A (PSA) that induces the differentiation of IL-10 secreting Foxp3 (+) Treg cells [124, 125]. These bacteria thus protect mice from H. hepaticus-induced colitis [124]. Enterotoxigenic B. fragilis (ETBF) has been recently characterized [126]. This strain secretes a proinflammatory toxin called BFT that stimulates colonic inflammation in predisposed multiple intestinal neoplasia (MIN) mice [127, 128].
An example of pathobiont in humans is Helicobacter pylori. This species interacts with the gastric mucosa and is responsible for gastritis, peptic ulcer and gastric adenocarcinoma. Although found in 50 % of the human population, however, only a few percent of this population will develop these pathologies, and the advantageous aspects of colonization by H. pylori remain to be demonstrated, even if colonization with this strain seems to decrease the risk of oesophageal carcinomas and asthma [129–131]. Virulent strains of H. pylori translocate into gastric epithelial cells the virulence factor CagA that hijacks host signalling pathways involved in inflammation and oncogenesis [132, 133]. However this bacterial protein is not sufficient to induce pathogenesis as asymptomatic carriers exist, suggesting that other factors are required such as host genetic polymorphisms for the IL-1β proinflammatory gene [134]. Other bacteria such as γ-proteobacteria, particularly Enterobacteriaceae, also present proinflammatory characteristics that may turn out to be deleterious for the host in case of host immunological failure or dysbiosis [135].
Conclusion and Perspectives
Compelling evidence shows that regulation of bacterial interactions with the intestinal mucosal surface is a critical stage for the establishment and maintenance of intestinal homeostasis. Discrimination between harmful and harmless bacteria is a matter of survival for the host. PRRs play an essential role in PAMPs and DAMPs recognition, leading to activation of immune defence mechanisms. In a healthy host, this recognition leads to physiological or pathological inflammation, in the case of symbiotic bacteria or pathogenic bacteria respectively. However, the existence of pathobionts indicates the existence of a continuum in between. The recognition of host damage is a more efficient mechanism to detect harmful bacteria rather than the discrimination between symbionts and pathogens.
Many questions remain unanswered concerning host–bacteria interactions. Immunity qualitatively and quantitatively shapes the microbiota. However, despite many metagenomic studies, we still know little about microbiota composition and how mucosa-associated bacterial species are distinct from those that are in the lumen. Virulence factors that are used by pathogens are well described. On the contrary, colonization or symbiosis factors are poorly known. Moreover bacteria–bacteria interactions are even less known and are highly difficult to grasp and to decipher in the context of the intestinal mucosa. These bacteria have co-evolved into a “community behaviour”, that could be important to understand, for instance, in the context of mechanisms triggering inflammatory bowel diseases. Interpreting the behaviour and the fate of host–bacteria interactions will provide clues and biomarkers to anticipate and predict the evolution of host–bacteria interactions, and as a consequence the evolution of infectious and inflammatory processes. This ecosystem may also be considered a “gold mine” to identify novel bioactive molecules, from host and microbial origin that will contribute to the development of original therapeutic and preventive approaches. The field of prebiotics, probiotics and postbiotics is likely to undergo a revolution, thanks to the global application of cellular microbiology principles to the study of the symbiosis-to-pathogenesis transition.
References
Janeway CA Jr (1989) Approaching the asymptote? Evolution and revolution in immunology. Cold Spring Harb Symp Quant Biol 54(pt 1):1–13, Epub 1989/01/01
Kono H, Rock KL (2008) How dying cells alert the immune system to danger. Nat Rev Immunol 8(4):279–289, Epub 2008/03/15
Matzinger P (1994) Tolerance, danger, and the extended family. Annu Rev Immunol 12:991–1045, Epub 1994/01/01
Casadevall A, Pirofski LA (2000) Host-pathogen interactions: basic concepts of microbial commensalism, colonization, infection, and disease. Infect Immun 68(12):6511–6518, Epub 2000/11/18
Merrell DS, Falkow S (2004) Frontal and stealth attack strategies in microbial pathogenesis. Nature 430(6996):250–256, Epub 2004/07/09
Eckburg PB, Bik EM, Bernstein CN, Purdom E, Dethlefsen L, Sargent M et al (2005) Diversity of the human intestinal microbial flora. Science 308(5728):1635–1638, Epub 2005/04/16
Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C et al (2010) A human gut microbial gene catalogue established by metagenomic sequencing. Nature 464(7285):59–65, Epub 2010/03/06
Tap J, Mondot S, Levenez F, Pelletier E, Caron C, Furet JP et al (2009) Towards the human intestinal microbiota phylogenetic core. Environ Microbiol 11(10):2574–2584, Epub 2009/07/16
Backhed F, Ding H, Wang T, Hooper LV, Koh GY, Nagy A et al (2004) The gut microbiota as an environmental factor that regulates fat storage. Proc Natl Acad Sci U S A 101(44):15718–15723, Epub 2004/10/27
Lotz M, Menard S, Hornef M (2007) Innate immune recognition on the intestinal mucosa. Int J Med Microbiol 297(5):379–392, Epub 2007/04/27
Nishiguchi MK, Hirsch AM, Devinney R, Vedantam G, Riley MA, Mansky LM (2008) Deciphering evolutionary mechanisms between mutualistic and pathogenic symbioses. Vie Milieu Paris 58(2):87–106, Epub 2008/01/01
Hooper LV (2009) Do symbiotic bacteria subvert host immunity? Nat Rev Microbiol 7(5):367–374, Epub 2009/04/17
Cerf-Bensussan N, Gaboriau-Routhiau V (2010) The immune system and the gut microbiota: friends or foes? Nat Rev Immunol 10(10):735–744, Epub 2010/09/25
Gaboriau-Routhiau V, Rakotobe S, Lecuyer E, Mulder I, Lan A, Bridonneau C et al (2009) The key role of segmented filamentous bacteria in the coordinated maturation of gut helper T cell responses. Immunity 31(4):677–689, Epub 2009/10/17
Bach JF (2002) The effect of infections on susceptibility to autoimmune and allergic diseases. N Engl J Med 347(12):911–920, Epub 2002/09/20
Blaser MJ, Falkow S (2009) What are the consequences of the disappearing human microbiota? Nat Rev Microbiol 7(12):887–894, Epub 2009/11/10
Jones KE, Patel NG, Levy MA, Storeygard A, Balk D, Gittleman JL et al (2008) Global trends in emerging infectious diseases. Nature 451(7181):990–993, Epub 2008/02/22
Woolhouse M, Gaunt E (2007) Ecological origins of novel human pathogens. Crit Rev Microbiol 33(4):231–242, Epub 2007/11/23
Mogensen TH (2009) Pathogen recognition and inflammatory signaling in innate immune defenses. Clin Microbiol Rev 22(2):240–273, Table of contents. Epub 2009/04/16
Vance RE, Isberg RR, Portnoy DA (2009) Patterns of pathogenesis: discrimination of pathogenic and nonpathogenic microbes by the innate immune system. Cell Host Microbe 6(1):10–21, Epub 2009/07/21
Macpherson AJ, Geuking MB, McCoy KD (2005) Immune responses that adapt the intestinal mucosa to commensal intestinal bacteria. Immunology 115(2):153–162, Epub 2005/05/12
Van den Abbeele P, Van de Wiele T, Verstraete W, Possemiers S (2011) The host selects mucosal and luminal associations of coevolved gut microorganisms: a novel concept. FEMS Microbiol Rev 35(4):681–704, Epub 2011/03/03
May RM, Anderson RM (1983) Epidemiology and genetics in the coevolution of parasites and hosts. Proc R Soc Lond B Biol Sci 219(1216):281–313, Epub 1983/10/22
Levin BR, Bergstrom CT (2000) Bacteria are different: observations, interpretations, speculations, and opinions about the mechanisms of adaptive evolution in prokaryotes. Proc Natl Acad Sci U S A 97(13):6981–6985, Epub 2000/06/22
Dyall SD, Brown MT, Johnson PJ (2004) Ancient invasions: from endosymbionts to organelles. Science 304(5668):253–257, Epub 2004/04/10
Rohmer L, Hocquet D, Miller SI (2011) Are pathogenic bacteria just looking for food? Metabolism and microbial pathogenesis. Trends Microbiol 19(7):341–348, Epub 2011/05/24
Stecher B, Macpherson AJ, Hapfelmeier S, Kremer M, Stallmach T, Hardt WD (2005) Comparison of Salmonella enterica serovar Typhimurium colitis in germfree mice and mice pretreated with streptomycin. Infect Immun 73(6):3228–3241, Epub 2005/05/24
Stecher B, Robbiani R, Walker AW, Westendorf AM, Barthel M, Kremer M et al (2007) Salmonella enterica serovar typhimurium exploits inflammation to compete with the intestinal microbiota. PLoS Biol 5(10):2177–2189, Epub 2007/09/01
Xu J, Bjursell MK, Himrod J, Deng S, Carmichael LK, Chiang HC et al (2003) A genomic view of the human-Bacteroides thetaiotaomicron symbiosis. Science 299(5615):2074–2076, Epub 2003/03/29
Hacker J, Blum-Oehler G, Muhldorfer I, Tschape H (1997) Pathogenicity islands of virulent bacteria: structure, function and impact on microbial evolution. Mol Microbiol 23(6):1089–1097, Epub 1997/03/01
Janeway CA Jr, Medzhitov R (2002) Innate immune recognition. Annu Rev Immunol 20:197–216, Epub 2002/02/28
Kawai T, Akira S (2010) The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol 11(5):373–384, Epub 2010/04/21
Elinav E, Strowig T, Henao-Mejia J, Flavell RA (2011) Regulation of the antimicrobial response by NLR proteins. Immunity 34(5):665–679, Epub 2011/05/28
Kufer TA, Sansonetti PJ (2011) NLR functions beyond pathogen recognition. Nat Immunol 12(2):121–128, Epub 2011/01/20
Chamaillard M, Hashimoto M, Horie Y, Masumoto J, Qiu S, Saab L et al (2003) An essential role for NOD1 in host recognition of bacterial peptidoglycan containing diaminopimelic acid. Nat Immunol 4(7):702–707, Epub 2003/06/11
Girardin SE, Boneca IG, Carneiro LA, Antignac A, Jehanno M, Viala J et al (2003) Nod1 detects a unique muropeptide from gram-negative bacterial peptidoglycan. Science 300(5625):1584–1587, Epub 2003/06/07
Girardin SE, Boneca IG, Viala J, Chamaillard M, Labigne A, Thomas G et al (2003) Nod2 is a general sensor of peptidoglycan through muramyl dipeptide (MDP) detection. J Biol Chem 278(11):8869–8872, Epub 2003/01/16
Hugot JP, Chamaillard M, Zouali H, Lesage S, Cezard JP, Belaiche J et al (2001) Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature 411(6837):599–603, Epub 2001/06/01
Ogura Y, Bonen DK, Inohara N, Nicolae DL, Chen FF, Ramos R et al (2001) A frameshift mutation in NOD2 associated with susceptibility to Crohn’s disease. Nature 411(6837):603–606, Epub 2001/06/01
Brandl K, Plitas G, Schnabl B, DeMatteo RP, Pamer EG (2007) MyD88-mediated signals induce the bactericidal lectin RegIII gamma and protect mice against intestinal Listeria monocytogenes infection. J Exp Med 204(8):1891–1900, Epub 2007/07/20
Vaishnava S, Behrendt CL, Ismail AS, Eckmann L, Hooper LV (2008) Paneth cells directly sense gut commensals and maintain homeostasis at the intestinal host-microbial interface. Proc Natl Acad Sci U S A 105(52):20858–20863, Epub 2008/12/17
Rakoff-Nahoum S, Paglino J, Eslami-Varzaneh F, Edberg S, Medzhitov R (2004) Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell 118(2):229–241, Epub 2004/07/21
Round JL, Lee SM, Li J, Tran G, Jabri B, Chatila TA et al (2011) The Toll-like receptor 2 pathway establishes colonization by a commensal of the human microbiota. Science 332(6032):974–977, Epub 2011/04/23
Kobayashi KS, Chamaillard M, Ogura Y, Henegariu O, Inohara N, Nunez G et al (2005) Nod2-dependent regulation of innate and adaptive immunity in the intestinal tract. Science 307(5710):731–734, Epub 2005/02/05
Maeda S, Hsu LC, Liu H, Bankston LA, Iimura M, Kagnoff MF et al (2005) Nod2 mutation in Crohn’s disease potentiates NF-kappaB activity and IL-1beta processing. Science 307(5710):734–738, Epub 2005/02/05
McGuckin MA, Linden SK, Sutton P, Florin TH (2011) Mucin dynamics and enteric pathogens. Nat Rev Microbiol 9(4):265–278, Epub 2011/03/17
Swidsinski A, Weber J, Loening-Baucke V, Hale LP, Lochs H (2005) Spatial organization and composition of the mucosal flora in patients with inflammatory bowel disease. J Clin Microbiol 43(7):3380–3389, Epub 2005/07/08
Van der Sluis M, De Koning BA, De Bruijn AC, Velcich A, Meijerink JP, Van Goudoever JB et al (2006) Muc2-deficient mice spontaneously develop colitis, indicating that MUC2 is critical for colonic protection. Gastroenterology 131(1):117–129, Epub 2006/07/13
Velcich A, Yang W, Heyer J, Fragale A, Nicholas C, Viani S et al (2002) Colorectal cancer in mice genetically deficient in the mucin Muc2. Science 295(5560):1726–1729, Epub 2002/03/02
Mantle M, Thakore E, Hardin J, Gall DG (1989) Effect of Yersinia enterocolitica on intestinal mucin secretion. Am J Physiol 256(2 pt 1):G319–G327, Epub 1989/02/01
Nutten S, Sansonetti P, Huet G, Bourdon-Bisiaux C, Meresse B, Colombel JF et al (2002) Epithelial inflammation response induced by Shigella flexneri depends on mucin gene expression. Microbes Infect 4(11):1121–1124, Epub 2002/10/04
Bevins CL (2006) Paneth cell defensins: key effector molecules of innate immunity. Biochem Soc Trans 34(pt 2):263–266, Epub 2006/03/21
Hornef MW, Putsep K, Karlsson J, Refai E, Andersson M (2004) Increased diversity of intestinal antimicrobial peptides by covalent dimer formation. Nat Immunol 5(8):836–843, Epub 2004/07/06
Bates JM, Akerlund J, Mittge E, Guillemin K (2007) Intestinal alkaline phosphatase detoxifies lipopolysaccharide and prevents inflammation in zebrafish in response to the gut microbiota. Cell Host Microbe 2(6):371–382, Epub 2007/12/15
Goldberg RF, Austen WG Jr, Zhang X, Munene G, Mostafa G, Biswas S et al (2008) Intestinal alkaline phosphatase is a gut mucosal defense factor maintained by enteral nutrition. Proc Natl Acad Sci U S A 105(9):3551–3556, Epub 2008/02/23
Koyama I, Matsunaga T, Harada T, Hokari S, Komoda T (2002) Alkaline phosphatases reduce toxicity of lipopolysaccharides in vivo and in vitro through dephosphorylation. Clin Biochem 35(6):455–461, Epub 2002/11/05
Ramasamy S, Nguyen DD, Eston MA, Alam SN, Moss AK, Ebrahimi F et al (2011) Intestinal alkaline phosphatase has beneficial effects in mouse models of chronic colitis. Inflamm Bowel Dis 17(2):532–542, Epub 2010/07/21
Shifrin DA Jr, McConnell RE, Nambiar R, Higginbotham JN, Coffey RJ, Tyska MJ (2012) Enterocyte microvillus-derived vesicles detoxify bacterial products and regulate epithelial-microbial interactions. Curr Biol 22(7):627–631, Epub 2012/03/06
Macpherson AJ, Uhr T (2004) Induction of protective IgA by intestinal dendritic cells carrying commensal bacteria. Science 303(5664):1662–1665, Epub 2004/03/16
Macpherson AJ, Hunziker L, McCoy K, Lamarre A (2001) IgA responses in the intestinal mucosa against pathogenic and non-pathogenic microorganisms. Microbes Infect 3(12):1021–1035, Epub 2001/10/03
Spits H, Di Santo JP (2011) The expanding family of innate lymphoid cells: regulators and effectors of immunity and tissue remodeling. Nat Immunol 12(1):21–27, Epub 2010/11/30
Lee YK, Menezes JS, Umesaki Y, Mazmanian SK (2011) Proinflammatory T-cell responses to gut microbiota promote experimental autoimmune encephalomyelitis. Proc Natl Acad Sci U S A 108(Suppl 1):4615–4622, Epub 2010/07/28
Chow J, Mazmanian SK (2010) A pathobiont of the microbiota balances host colonization and intestinal inflammation. Cell Host Microbe 7(4):265–276, Epub 2010/04/24
Miller MB, Bassler BL (2001) Quorum sensing in bacteria. Annu Rev Microbiol 55:165–199, Epub 2001/09/07
Fujiya M, Musch MW, Nakagawa Y, Hu S, Alverdy J, Kohgo Y et al (2007) The Bacillus subtilis quorum-sensing molecule CSF contributes to intestinal homeostasis via OCTN2, a host cell membrane transporter. Cell Host Microbe 1(4):299–308, Epub 2007/11/17
Cossart P (2004) Bacterial invasion: a new strategy to dominate cytoskeleton plasticity. Dev Cell 6(3):314–315, Epub 2004/03/20
Hooper LV, Littman DR, Macpherson AJ (2012) Interactions between the microbiota and the immune system. Science 336(6086):1268–1273, Epub 2012/06/08
Mueller C, Macpherson AJ (2006) Layers of mutualism with commensal bacteria protect us from intestinal inflammation. Gut 55(2):276–284, Epub 2006/01/13
Macpherson AJ, Gatto D, Sainsbury E, Harriman GR, Hengartner H, Zinkernagel RM (2000) A primitive T cell-independent mechanism of intestinal mucosal IgA responses to commensal bacteria. Science 288(5474):2222–2226, Epub 2000/06/24
Vaishnava S, Yamamoto M, Severson KM, Ruhn KA, Yu X, Koren O et al (2011) The antibacterial lectin RegIIIgamma promotes the spatial segregation of microbiota and host in the intestine. Science 334(6053):255–258, Epub 2011/10/15
Slack E, Hapfelmeier S, Stecher B, Velykoredko Y, Stoel M, Lawson MA et al (2009) Innate and adaptive immunity cooperate flexibly to maintain host-microbiota mutualism. Science 325(5940):617–620, Epub 2009/08/01
Lotz M, Gutle D, Walther S, Menard S, Bogdan C, Hornef MW (2006) Postnatal acquisition of endotoxin tolerance in intestinal epithelial cells. J Exp Med 203(4):973–984, Epub 2006/04/12
Rogler G, Brand K, Vogl D, Page S, Hofmeister R, Andus T et al (1998) Nuclear factor kappaB is activated in macrophages and epithelial cells of inflamed intestinal mucosa. Gastroenterology 115(2):357–369, Epub 1998/07/25
Smith PD, Ochsenbauer-Jambor C, Smythies LE (2005) Intestinal macrophages: unique effector cells of the innate immune system. Immunol Rev 206:149–159, Epub 2005/07/29
Ogura Y, Lala S, Xin W, Smith E, Dowds TA, Chen FF et al (2003) Expression of NOD2 in Paneth cells: a possible link to Crohn’s ileitis. Gut 52(11):1591–1597, Epub 2003/10/23
Ortega-Cava CF, Ishihara S, Rumi MA, Kawashima K, Ishimura N, Kazumori H et al (2003) Strategic compartmentalization of Toll-like receptor 4 in the mouse gut. J Immunol 170(8):3977–3985, Epub 2003/04/12
Viala J, Chaput C, Boneca IG, Cardona A, Girardin SE, Moran AP et al (2004) Nod1 responds to peptidoglycan delivered by the Helicobacter pylori cag pathogenicity island. Nat Immunol 5(11):1166–1174, Epub 2004/10/19
Gewirtz AT, Navas TA, Lyons S, Godowski PJ, Madara JL (2001) Cutting edge: bacterial flagellin activates basolaterally expressed TLR5 to induce epithelial proinflammatory gene expression. J Immunol 167(4):1882–1885, Epub 2001/08/08
Reed KA, Hobert ME, Kolenda CE, Sands KA, Rathman M, O’Connor M et al (2002) The Salmonella typhimurium flagellar basal body protein FliE is required for flagellin production and to induce a proinflammatory response in epithelial cells. J Biol Chem 277(15):13346–13353, Epub 2002/02/01
Burns K, Clatworthy J, Martin L, Martinon F, Plumpton C, Maschera B et al (2000) Tollip, a new component of the IL-1RI pathway, links IRAK to the IL-1 receptor. Nat Cell Biol 2(6):346–351, Epub 2000/06/15
Zhang G, Ghosh S (2002) Negative regulation of toll-like receptor-mediated signaling by Tollip. J Biol Chem 277(9):7059–7065, Epub 2001/12/26
Lu M, Zhang M, Takashima A, Weiss J, Apicella MA, Li XH et al (2005) Lipopolysaccharide deacylation by an endogenous lipase controls innate antibody responses to Gram-negative bacteria. Nat Immunol 6(10):989–994, Epub 2005/09/13
Munford RS (2005) Detoxifying endotoxin: time, place and person. J Endotoxin Res 11(2):69–84, Epub 2005/06/14
Alhawi M, Stewart J, Erridge C, Patrick S, Poxton IR (2009) Bacteroides fragilis signals through Toll-like receptor (TLR) 2 and not through TLR4. J Med Microbiol 58(pt 8):1015–1022, Epub 2009/06/17
Weintraub A, Zahringer U, Wollenweber HW, Seydel U, Rietschel ET (1989) Structural characterization of the lipid A component of Bacteroides fragilis strain NCTC 9343 lipopolysaccharide. Eur J Biochem 183(2):425–431, Epub 1989/08/01
Kelly D, Campbell JI, King TP, Grant G, Jansson EA, Coutts AG et al (2004) Commensal anaerobic gut bacteria attenuate inflammation by regulating nuclear-cytoplasmic shuttling of PPAR-gamma and RelA. Nat Immunol 5(1):104–112, Epub 2003/12/24
Tien MT, Girardin SE, Regnault B, Le Bourhis L, Dillies MA, Coppee JY et al (2006) Anti-inflammatory effect of Lactobacillus casei on Shigella-infected human intestinal epithelial cells. J Immunol 176(2):1228–1237, Epub 2006/01/06
Piccinini AM, Midwood KS (2010) DAMPening inflammation by modulating TLR signalling. Mediators Inflamm 2010:1–21. doi: 10.1155/2010/672395, Epub 2010/08/14
Pedra JH, Cassel SL, Sutterwala FS (2009) Sensing pathogens and danger signals by the inflammasome. Curr Opin Immunol 21(1):10–16, Epub 2009/02/19
Mariathasan S, Weiss DS, Newton K, McBride J, O’Rourke K, Roose-Girma M et al (2006) Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 440(7081):228–232, Epub 2006/01/13
Pelegrin P, Surprenant A (2006) Pannexin-1 mediates large pore formation and interleukin-1beta release by the ATP-gated P2X7 receptor. EMBO J 25(21):5071–5082, Epub 2006/10/13
Skoberne M, Beignon AS, Bhardwaj N (2004) Danger signals: a time and space continuum. Trends Mol Med 10(6):251–257, Epub 2004/06/05
Lamkanfi M, Dixit VM (2011) Modulation of inflammasome pathways by bacterial and viral pathogens. J Immunol 187(2):597–602, Epub 2011/07/08
Santaolalla R, Abreu MT (2012) Innate immunity in the small intestine. Curr Opin Gastroenterol 28(2):124–129, Epub 2012/01/14
Franchi L, Munoz-Planillo R, Nunez G (2012) Sensing and reacting to microbes through the inflammasomes. Nat Immunol 13(4):325–332, Epub 2012/03/21
Nakanishi K, Yoshimoto T, Tsutsui H, Okamura H (2001) Interleukin-18 is a unique cytokine that stimulates both Th1 and Th2 responses depending on its cytokine milieu. Cytokine Growth Factor Rev 12(1):53–72, Epub 2001/04/20
Sutton CE, Lalor SJ, Sweeney CM, Brereton CF, Lavelle EC, Mills KH (2009) Interleukin-1 and IL-23 induce innate IL-17 production from gammadelta T cells, amplifying Th17 responses and autoimmunity. Immunity 31(2):331–341, Epub 2009/08/18
Franchi L, Eigenbrod T, Munoz-Planillo R, Nunez G (2009) The inflammasome: a caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat Immunol 10(3):241–247, Epub 2009/02/18
Lamkanfi M, Walle LV, Kanneganti TD (2011) Deregulated inflammasome signaling in disease. Immunol Rev 243(1):163–173, Epub 2011/09/03
Iwai H, Kim M, Yoshikawa Y, Ashida H, Ogawa M, Fujita Y et al (2007) A bacterial effector targets Mad2L2, an APC inhibitor, to modulate host cell cycling. Cell 130(4):611–623, Epub 2007/08/28
Kim M, Ogawa M, Fujita Y, Yoshikawa Y, Nagai T, Koyama T et al (2009) Bacteria hijack integrin-linked kinase to stabilize focal adhesions and block cell detachment. Nature 459(7246):578–582, Epub 2009/06/03
Boyle EC, Brown NF, Finlay BB (2006) Salmonella enterica serovar Typhimurium effectors SopB, SopE, SopE2 and SipA disrupt tight junction structure and function. Cell Microbiol 8(12):1946–1957, Epub 2006/07/28
Bruno VM, Hannemann S, Lara-Tejero M, Flavell RA, Kleinstein SH, Galan JE (2009) Salmonella typhimurium type III secretion effectors stimulate innate immune responses in cultured epithelial cells. PLoS Pathog 5(8):e1000538, Epub 2009/08/08
Muller AJ, Hoffmann C, Galle M, Van Den Broeke A, Heikenwalder M, Falter L et al (2009) The S. typhimurium effector SopE induces caspase-1 activation in stromal cells to initiate gut inflammation. Cell Host Microbe 6(2):125–136
Bartoleschi C, Pardini MC, Scaringi C, Martino MC, Pazzani C, Bernardini ML (2002) Selection of Shigella flexneri candidate virulence genes specifically induced in bacteria resident in host cell cytoplasm. Cell Microbiol 4(9):613–626, Epub 2002/10/23
Lenz LL, Mohammadi S, Geissler A, Portnoy DA (2003) SecA2-dependent secretion of autolytic enzymes promotes Listeria monocytogenes pathogenesis. Proc Natl Acad Sci U S A 100(21):12432–12437, Epub 2003/10/07
Ham H, Sreelatha A, Orth K (2011) Manipulation of host membranes by bacterial effectors. Nat Rev Microbiol 9(9):635–646, Epub 2011/07/19
Viboud GI, Bliska JB (2005) Yersinia outer proteins: role in modulation of host cell signaling responses and pathogenesis. Annu Rev Microbiol 59:69–89, Epub 2005/04/26
Neish AS, Naumann M (2011) Microbial-induced immunomodulation by targeting the NF-kappaB system. Trends Microbiol 19(12):596–605, Epub 2011/10/01
Newman RM, Salunkhe P, Godzik A, Reed JC (2006) Identification and characterization of a novel bacterial virulence factor that shares homology with mammalian Toll/interleukin-1 receptor family proteins. Infect Immun 74(1):594–601, Epub 2005/12/22
Krachler AM, Woolery AR, Orth K (2011) Manipulation of kinase signaling by bacterial pathogens. J Cell Biol 195(7):1083–1092, Epub 2011/11/30
Arbibe L, Kim DW, Batsche E, Pedron T, Mateescu B, Muchardt C et al (2007) An injected bacterial effector targets chromatin access for transcription factor NF-kappaB to alter transcription of host genes involved in immune responses. Nat Immunol 8(1):47–56, Epub 2006/12/13
Schotte P, Denecker G, Van Den Broeke A, Vandenabeele P, Cornelis GR, Beyaert R (2004) Targeting Rac1 by the Yersinia effector protein YopE inhibits caspase-1-mediated maturation and release of interleukin-1beta. J Biol Chem 279(24):25134–25142, Epub 2004/04/03
Brzuszkiewicz E, Bruggemann H, Liesegang H, Emmerth M, Olschlager T, Nagy G et al (2006) How to become a uropathogen: comparative genomic analysis of extraintestinal pathogenic Escherichia coli strains. Proc Natl Acad Sci U S A 103(34):12879–12884, Epub 2006/08/17
Round JL, Mazmanian SK (2009) The gut microbiota shapes intestinal immune responses during health and disease. Nat Rev Immunol 9(5):313–323, Epub 2009/04/04
Talham GL, Jiang HQ, Bos NA, Cebra JJ (1999) Segmented filamentous bacteria are potent stimuli of a physiologically normal state of the murine gut mucosal immune system. Infect Immun 67(4):1992–2000, Epub 1999/03/20
Ivanov II, Atarashi K, Manel N, Brodie EL, Shima T, Karaoz U et al (2009) Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell 139(3):485–498, Epub 2009/10/20
Littman DR, Pamer EG (2011) Role of the commensal microbiota in normal and pathogenic host immune responses. Cell Host Microbe 10(4):311–323, Epub 2011/10/25
Stepankova R, Powrie F, Kofronova O, Kozakova H, Hudcovic T, Hrncir T et al (2007) Segmented filamentous bacteria in a defined bacterial cocktail induce intestinal inflammation in SCID mice reconstituted with CD45RBhigh CD4+ T cells. Inflamm Bowel Dis 13(10):1202–1211, Epub 2007/07/04
Wu HJ, Ivanov II, Darce J, Hattori K, Shima T, Umesaki Y et al (2010) Gut-residing segmented filamentous bacteria drive autoimmune arthritis via T helper 17 cells. Immunity 32(6):815–827, Epub 2010/07/14
Cahill RJ, Foltz CJ, Fox JG, Dangler CA, Powrie F, Schauer DB (1997) Inflammatory bowel disease: an immunity-mediated condition triggered by bacterial infection with Helicobacter hepaticus. Infect Immun 65(8):3126–3131, Epub 1997/08/01
Erdman SE, Poutahidis T, Tomczak M, Rogers AB, Cormier K, Plank B et al (2003) CD4+ CD25+ regulatory T lymphocytes inhibit microbially induced colon cancer in Rag2-deficient mice. Am J Pathol 162(2):691–702, Epub 2003/01/28
Kullberg MC, Ward JM, Gorelick PL, Caspar P, Hieny S, Cheever A et al (1998) Helicobacter hepaticus triggers colitis in specific-pathogen-free interleukin-10 (IL-10)-deficient mice through an IL-12- and gamma interferon-dependent mechanism. Infect Immun 66(11):5157–5166, Epub 1998/10/24
Mazmanian SK, Round JL, Kasper DL (2008) A microbial symbiosis factor prevents intestinal inflammatory disease. Nature 453(7195):620–625, Epub 2008/05/30
Round JL, Mazmanian SK (2010) Inducible Foxp3+ regulatory T-cell development by a commensal bacterium of the intestinal microbiota. Proc Natl Acad Sci U S A 107(27):12204–12209, Epub 2010/06/23
Sears CL (2009) Enterotoxigenic Bacteroides fragilis: a rogue among symbiotes. Clin Microbiol Rev 22(2):349–369, Table of contents. Epub 2009/04/16
Housseau F, Sears CL (2010) Enterotoxigenic Bacteroides fragilis (ETBF)-mediated colitis in Min (Apc+/-) mice: a human commensal-based murine model of colon carcinogenesis. Cell Cycle 9(1):3–5, Epub 2009/12/17
Wu S, Rhee KJ, Albesiano E, Rabizadeh S, Wu X, Yen HR et al (2009) A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses. Nat Med 15(9):1016–1022, Epub 2009/08/25
Chow WH, Blaser MJ, Blot WJ, Gammon MD, Vaughan TL, Risch HA et al (1998) An inverse relation between cagA + strains of Helicobacter pylori infection and risk of esophageal and gastric cardia adenocarcinoma. Cancer Res 58(4):588–590, Epub 1998/03/04
Dorer MS, Talarico S, Salama NR (2009) Helicobacter pylori’s unconventional role in health and disease. PLoS Pathog 5(10):e1000544, Epub 2009/10/27
Sonnenberg A, Lash RH, Genta RM (2010) A national study of Helicobacter pylori infection in gastric biopsy specimens. Gastroenterology 139(6):1894–1901.e2, quiz e12. Epub 2010/08/24
Odenbreit S, Puls J, Sedlmaier B, Gerland E, Fischer W, Haas R (2000) Translocation of Helicobacter pylori CagA into gastric epithelial cells by type IV secretion. Science 287(5457):1497–1500, Epub 2000/02/26
Polk DB, Peek RM Jr (2010) Helicobacter pylori: gastric cancer and beyond. Nat Rev Cancer 10(6):403–414, Epub 2010/05/25
El-Omar EM, Carrington M, Chow WH, McColl KE, Bream JH, Young HA et al (2000) Interleukin-1 polymorphisms associated with increased risk of gastric cancer. Nature 404(6776):398–402, Epub 2000/04/04
Mukhopadhya I, Hansen R, El-Omar EM, Hold GL (2012) IBD-what role do Proteobacteria play? Nat Rev Gastroenterol Hepatol 9(4):219–230, Epub 2012/02/22
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer Science+Business Media New York
About this chapter
Cite this chapter
Lhocine, N., Sansonetti, P.J. (2013). Host Interactions with Bacteria: From “Entente Cordiale” to “Casus Belli”. In: D'Amato, M., Rioux, J. (eds) Molecular Genetics of Inflammatory Bowel Disease. Springer, New York, NY. https://doi.org/10.1007/978-1-4614-8256-7_14
Download citation
DOI: https://doi.org/10.1007/978-1-4614-8256-7_14
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4614-8255-0
Online ISBN: 978-1-4614-8256-7
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)