
Abstract
Accumulating evidence has suggested that epigenetic alternations are as important as genetic mutations in cancer development. It is proposed that tumors are arisen by “malignant reprogramming” driven by a combination of both genetic and epigenetic changes. It therefore comes as no surprise that histone demethylases, the newest members of the histone modifying enzymes, are found to be targets of mutations and dysregulation in cancer cells. In this review article, we provide an overview of the types of histone demethylases whose genetic structure or expression is altered in cancers, the action of histone demethylases in cancer development and their potential inhibitors. Special emphasis is placed on the roles of histone demethylases in prostate cancer progression.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Histone Lysine Demethylase and Cancer
Chromatin is a highly ordered structure of eukaryotic DNA, which is packed into nucleosomes by core histone protein octamers: H2A, H2B, H3 and H4. Post-translational modifications of histone N-terminal basic tails cause conformational change of the nucleosome, allowing access of regulatory machineries to the DNA for transcription, replication, and repair [1, 2]. Several lysine residues on the histone tails can be mono-, di- or trimethylated. Depending on the position and degree of lysine methylation, the biological outcome is different. For example, histone marks such as H3K9me2, -me3 and H3K27me3, are involved in heterochromatin formation and gene silencing; while H3K4me3 is associated with actively transcribed genes. Because of the importance in gene expression, histone methylation and demethylation play critical roles in several biological processes, and altered histone methylation patterns are linked to human diseases such as neurological disorders and cancer.
Previously, histone methylation was believed to be irreversible. It was not until recently that the enzymes capable of removing methyl groups from histone were identified. As summarized in Table 15.1, histone lysine-specific demethylases (KDMs) have exquisite substrate specificities toward particular lysines on histones. KDMs can be broadly classified into two families: LSD1 (KDM1) and Jumonji C domain-containing (JmjC) family (KDM2 to KDM8). LSD1 family is amine oxidase which catalyzes demethylation through a flavin adenine dinucleotide (FAD)-dependent reaction. The JmjC family demethylase contains a conserved JmjC catalytic domain, which coordinates an electron shuffle between the methylated lysine with the co-factors Fe(II), α-keto-glutarate, and molecular oxygen. This reaction ultimately results in the removal of the methyl group. Dysregulation of these enzymes alters the chromatin conformation and reprograms gene expression, which sometimes leads to malignant transformation of the cells. Table 15.2 presents a summary of recent literature on the topics of KDMs dysregulation in cancer. There are several other excellent reviews on this subject [3–5] which the readers may wish to refer to.
1.1 Dysregulation of Histone Demethylases in Cancer
Global alteration of histone methylation such as the loss of H3K4me2 and H4K20me3 are hallmarks of many cancers and are associated with poor prognosis [6–9]. Aside from the global changes, alterations of histone methylation patterns within specific cancer-causing loci also have consequences in cancer cell proliferation and survival. It is thus not surprising that mutations and aberrant expression of histone demethylases (Table 15.2) have been associated with carcinogenesis.
1.1.1 Aberrant Expression
While some histone demethylases behave like oncogenes, others play tumor suppressive roles. KDM4 family members that are widely overexpressed in several tumor types are believed to be putative oncogenes because of their demethylation activity toward H3K9me3/me2, a histone mark crucial to maintaining the heterochromatin structure. The maintenance of heterochromatic structure not only is essential for gene expression regulation but also plays an important role for protection of chromosome integrity. Narita et al. reported that during senescence, the levels of H3K9me3/me2 are increased at senescence-associated heterochromatic foci (SAHFs), concomitant with the increased binding of Heterochromatin protein 1 (HP1) [10]. The authors showed that some of the E2F target promoters in fact, acquire heterochromatic features during senescence, resulting in a permanent shutdown of these genes. Therefore, dissociation of heterochromatin could result in re-expression of E2F target genes and an escape from cellular senescence. Peters’ study on the other hand, revealed that decrease of H3K9me3 at pericentric chromatin results in loss of the heterochromatin structure, leading to severe chromosome mis-segregation and genomic instability [11]. Together, as is found in prostate cancer [12], dysregulation of KDM4 family therefore may function as oncogenes and contributes to tumorigenesis. Other oncogenic roles such as alteration of cellular signaling pathways, promotion of cell cycle, expression of oncogenes/repression of tumor suppressors, are often linked with overexpression of various demethylases in tumor (Table 15.2). In addition to KDM4, KDM1A, KDM3 and KDM5 families are found to be overexpressed in several types of tumor.
While fewer cases, down-regulation of demethylases with tumor suppressive roles in cancer has also been reported. KDM2A plays a crucial role in sustaining heterochromatin structure and genome stability by repressing transcription of the centromeric satellite repeats [13]. KDM2B on the other hand, transcriptionally represses ribosomal RNA genes whose expressions are in high demand for proliferating cancer cells [14, 15]. Underexpression of KDM2B results in increased cell sizes and proliferation in tumor, suggesting it being a tumor suppressor [14].
Making things more complicated were the observations that the roles of histone demethylases as oncogenes or tumor suppressors are cell context dependent. As described above, KDM1A is overexpressed in many cancer types (Table 15.2), and appears to play oncogenic roles. However, KDM1A is also found to be down-regulated in liver and breast cancer, where it inhibits tumor invasion and metastasis [16, 17]. Similarly, while KDM2B is down-regulated in brain and glioblastoma, and was proposed as a putative tumor suppressor [14], it is found to be highly expressed in various leukemias with an oncogenic function [18]. These findings suggest that the functions of histone demethylases in cancer are dictated by the loci they act upon and the cell-type specific cofactors they associated with. As such, the results have strong therapeutic implications and underscore the importance in understanding the target gene profiles and the associated mechanisms of histone demethylases in particular cancer types.
The mechanisms of dysregulation of histone demethylase expression are multitude including alterations at the level of transcription and post-transcriptional modifications or genomic alterations. Below we will discuss the genetic alterations of KDMs in cancers.
1.1.2 Gene Amplification
Comparative genomic hybridization (CGH) analysis revealed that the 9p23-24 region is frequently amplified in several tumors including esophageal cancer, breast cancer and lymphoma; while KDM4C (GASC1/JMJD2C) gene located at the 9p23-24 amplicon is overexpressed in these tumors [19–24]. Overexpression of KDM4C results in tumorigenic phenotypes, and is found to be associated with aggressive breast tumors [23]. Similarly, KDM5A gene located at 12p11 is found to be amplified in several tumors including breast cancer, and overexpression of which, contributes to cancer proliferation and drug resistance [25].
1.1.3 Gene Translocation
van Zutven et al. first reported chromosome rearrangements involving KDM5A (JARID1A) and NUP98 in acute leukemias [26]. This translocation results in a fusion product consisting of the N-terminus of NUP98 and the C-terminal PHD finger domain of KDM5A. In doing so, the PHD domain targets NUP98 to active chromatin region with H3K4me3 histone mark and prevents the recruitment of the repressive polycomb complex (PRC2) to the promoter, thus, enabling active transcription of the crucial developmental loci and eventually leading to the development of leukemia [27]. In this case, it is not the catalytic of demethylase, but rather the chromatin binding domain which participates in the oncogenic transformation.
1.1.4 Gene Mutation
Inactivating mutations in histone demethylases have been identified. H3K4me3 demethylase KDM5C (JARID1C), suggested to be a tumor suppressor [28], has several nonsense and missense mutations in clear cell renal cell carcinoma patients [29]. Similarly, systematic mutational screen of KDM6A (UTX) reveals that nonsense, frameshift, and deletion mutations are often identified in cancers including leukemia, lymphoma, myeloma, glioblastoma, breast, colorectal, endometrial, lung, esophageal, pancreatic, bladder and clear cell renal cell carcinoma [29–31]. These findings suggest that KDM6A is a tumor suppressor, consistent with its ability to demethylate H3K27me3, a histone mark whose elevation is often associated with malignancy, and to positively regulate Rb tumor suppressor and antagonize Notch signaling pathway [32–35].
1.2 The Functional Roles of Histone Lysine Demethylases in Cancer
Histone lysine demethylases affect a wide spectrum of cellular pathways. Based on the literature cited in Table 15.2, the following oncogenic pathways appear to be the most frequently dysregulated by histone demethylases.
1.2.1 Cell Cycle Regulation
One of the major oncogenic properties of histone lysine demethylases is their role in cell cycle regulation. Overexpression of several demethylases is linked to promoting G1-S progression and inducing tumor cell proliferation through positive regulation of S-phase cyclins and cyclin-dependent kinases (CDKs), and/or negatively regulating CDK inhibitors. Demethylases that are reported to induce expression of the S-phase cyclins, Cyclin D1, include KDM3A, KDM4A, KDM4B and KDM5B [36–40]; and those for Cyclin A1 expression includes KDM4B and KDM8 [41, 42]. Those which negatively regulate CDK inhibitors such as p21 Cip1/Waf1, are KDM1A, KDM5A, KDM5B and KDM8 [25, 43–46]; and p27Kip1 by KDM5A and KDM5B [25, 47]. E2F transcription factors that play crucial roles in G1-S transition are also common targets of histone demethylases [43, 48, 49]. KDM1A positively regulates E2F1 gene expression, and also regulates its transcriptional activity by destabilizing the Rb regulator MYPT1 [43, 50]. PHF8 functions as a co-activator of E2F1 during G1-S transition by forming a complex with it, and upon recruitment to E2F1 target promoters, PHF8 removes the repressive H4K20me1 mark and consequently activates expression of the target genes [51].
Histone demethylase-mediated cell cycle control and proliferation is also found to channel through the p53-Rb axis [46, 48, 52–55] and senescence regulation (see below). In addition to transcriptional regulation, histone demethylase such as KDM4A directly regulates DNA replication by removing heterochromatin marks and increasing chromatin accessibility for replication machinery [56].
1.2.2 Senescence
Senescence is a process of irreversible cell growth arrest important for preventing excessive proliferation and functions as a suppression mechanism of tumorigenesis [57, 58]. INK4b-ARF-INK4a locus encodes p15INK4B, p16INK4A and p14ARF proteins that sense stress signals and function as key regulators of senescence [59]. The INK4b-ARF-INK4a locus is normally silenced with H3K27 methylation by the polycomb complexes PRC1 and PRC2. When cells undergo senescence, the PRC complexes and H3K27me3 are lost from the locus, leading to expression of INK4A, INK4B and ARF [60]. Studies on KDM2B revealed a double regulatory mechanism for senescence and cell proliferation. KDM2B regulates senescence in part, by directly binding to the INK4b-ARF-INK4a locus and demethylating the locus-associated H3K36me2 and H3K4me3 which results in the suppression of INK4a and INK4b [61, 62]. KDM2B also modulates the expression of H3K27 tri-methyltransferase EZH2 by negatively regulating tumor suppressor miRNAs let-7b and miR-101. KDM2B-mediated up-regulation of EZH2 increases the suppressive histone mark of H3K27me3 on INK4b-ARF-INK4a locus and further contributes to silencing. When primary mouse embryonic fibroblasts (MEFs) undergo senescence, the KDM2B-let7-EZH2 pathway presents a feed-forward mechanism to ensure senescence: KDM2B is down-regulated, leading to expression of let-7b and miR-101 which in turn, represses EZH2 expression [63]. The ability of KDM2B overexpression in promoting immortalization and sustained proliferation of both wild type and INK4a-ARF null MEFs suggests its important roles in tumor initiation and development.
Rb and p53 tumor suppressors also play essential roles in senescence. They induce senescence by regulating the expression of CDK inhibitor p21, and formation of heterochromatin on E2F-responsive promoters [10, 64]. During senescence, global changes in histone modifications include increase of transcriptional silencing marks H3K9me3, H3K27me3 and H4K20me3; and loss of active histone mark H3K4me3/me2 [64]. As suggested above, overexpression of demethylases that are involved in the Rb-p53-p21 pathway, or in removing the transcriptional silencing marks globally or loci-specific, may contribute to the loss of the tumor-suppressing senescence mechanism.
1.2.3 Hypoxia
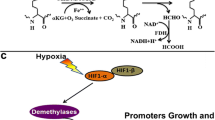
Hypoxia is a stage when a cellular demand of oxygen for metabolism exceeds the local blood supply. It occurs in tumor tissues where cell proliferation outgrows angiogenesis, resulting in local low concentration of oxygen in the high-cell dense regions of tumors. Tumor hypoxia has been reported to associate with poor prognosis and with resistance to radiotherapy and chemotherapy. Therefore understanding the survival mechanisms of tumor cells under hypoxic conditions is of significant importance in the design of therapeutic strategy. Hypoxia-induced transcription factor (HIF) complexes, consisting of α- (HIF-1α, HIF-2α or HIF-3α) and β- subunits, are the predominant hypoxia-responsive regulators that modulate adaptive gene expression aiming at restoring cellular oxygen homeostasis. Recent studies suggest cooperative actions between epigenetic regulators and HIF in response to hypoxia. These regulators include enzymes involved in DNA methylation, chromatin remodeling and histone modifications at the HIF promoter regions. Under hypoxic conditions, global levels of H3K4me3 and H3K9me2 have been demonstrated to be increased, whereas H3K27me3 level decreases [65, 66]. It is speculated that these chromatin modification signatures indicate flexible access of other chromatin modifiers and transcriptional regulators to promoters, in turn facilitating reprogramming of gene expression under transient hypoxia-reoxygenation conditions that often occur in tumor [65]. Several JmjC histone demethylases such as KDM3A, KDM4B, KDM4C, KDM5A and KDM5B are found to be activated in the HIF signaling pathway during hypoxic conditions [67–70]. The importance of hypoxia-induced histone demethylases is best illustrated by KDM3A (JMJD1A/JHDM2A). Upon induction under hypoxia, KDM3A is recruited to HIF target genes, and by removing the repressive H3K9me2 marks, KDM3A facilitates hypoxic gene expression such as adrenomedullin (ADM), differentiation factor 15 (GDF15), and GLUT3 (SLC2A3) [67, 71]. Mimura et al. further demonstrated that the recruitment of KDM3A to the GLUT3 promoter depends on physical interaction between KDM3A and HIF1 [71]. In addition to HIF, other transcription factors such as NFκB, CREB and EGR-1 are involved in hypoxia responses [72–74]. Given that many non-histone proteins including NFκB are found to be methylated, the roles of lysine demethylases in hypoxia may be broader than previously thought. Indeed, KDM2A is found to demethylate NFκB [75] and affects its transcriptional potential.
2 Inhibitors of Histone Demethylases
Given the strong implications of KDMs in cancer, small molecule inhibitors targeting their enzyme activities are being actively investigated in recent years (Table 15.3). Among them, inhibitors for KDM1A are the most extensively pursued; 12 such inhibitors (Compound 1 to 12) are listed in Table 15.3. KDM1A belongs to a superfamily of FAD-containing amine oxidases and the developed inhibitors include substrate analogues, MAO (monoamine oxidase) inhibitor analogues and polyamine analogues. Culhane et al. first reported compound 1 as a suicide inhibitor of KDM1A through substitution of lysine4 in H3 [76]. Through coupling of compound 1 with FAD, the mechanism of KDM1A demethylation was revealed ([77, 78], compound 2). The prototype of amino oxidases is the antidepressant MAO, based on which several inhibitors have been developed. Analogues of MAO inhibitors such as trans-2-phenylcyclopropylamine (compound 3-8) were shown to be effective in inhibiting KDM1A [79–85]. Another class of KDM1A inhibitors is polyamine analogues (compound 9-11) [86–88], inhibition of KDM1A in colon cancer cell by this class of compounds resulted in reexpression of aberrantly silenced genes [86, 87]. Compound 12, a new KDM1A inhibitor, was found to selectively target cancer cells with pluripotent stem cell properties [89]. Although these inhibitors are very useful in exploring the structure and biological function of KDM1A, their IC50s for cell killing are in the range of micromolar to millimolar, too high to be appropriate for clinical trials and hence further optimization is needed.
Other KDMs are JmjC containing demethylases, which are α-ketoglutarate dependent oxygenases. The design of most inhibitors is based on the scaffold of this cofactor. Compound 13, a NOG (N-oxalylglycine) analogue, was identified as a KDM4A inhibitor with IC50 of 3 mM. This compound is not cell permeable and methylation of the two hydroxyl groups is required for cell penetration [90]. Other KDM inhibitors include hydroxamate analogs, pyridine dicarboxylates and bipyridil compounds. Hamada et al. identified a series of hydroxamate analogues as KDM4A and KDM4C inhibitors in low micromole range ([91], compound 14), whereas 3-substituted pyridine 2,4-dicarboxylate was found to be a potent inhibitor of KDM4E ([92], compound 15). Through high-throughput screening, 8-hydroxyquinolines were identified as cell-active KDM4A and KDM4E inhibitors ([93], compound 16). Compound 17, a diazepin-quinazoline-amine derivative, selectively inhibits KDM7A ([94]). Interestingly, 2,4-pyridine-dicarboxylic acid (compound 18) was found to be an active inhibitor of KDM4A, KDM4E and KDM5A, indicating substrate similarities of these KDMs [93, 95]. Recently, catechols were reported to be active KDM4C and KDM6A inhibitors ([96], compound 19). The IC50s of most of these inhibitors are in millimolar or micromolar range, Kruidenier et al. identified compound 20 as a selective KDM6B inhibitor in nanomolar range. Interestingly, this inhibitor modulates the proinflammatory macrophage response ([97], compound 20). Although these inhibitors showed variable activities against purified KDMs, none of these inhibitors inhibit cancer cell growth below micromole range and none has reached clinical trials. Given the important function of KDMs in cancer, development of more potent KDM inhibitors is highly desirable.
3 Histone Lysine Demethylases in Prostate Cancer
Using the Oncomine database, we performed a comparison of several prostate cancer studies to identify histone demethylases that are differentially expressed in normal and tumor samples (Fig. 15.1). Our analysis agrees well with those reported in the literature (Table 15.2), and can serve as a future guide for developing demethylase-targeted therapies. The mechanisms regarding how the demethylases serve as progression factors in prostate cancer are being actively investigated, and below is a summary of the most relevant signaling pathways targeted by histone lysine demethylases.

Expression of histone demethylases that are differentially expressed in prostate cancer versus normal tissues are compared among 16 independent studies from Oncomine databases. In each dataset, the expression data was either based upon mRNA or DNA copy number analysis. Left panel shows the top 11 histone demethylase (half of total demethylases with substrates identified) that are gained/overexpressed in prostate tumors compared to that in normal tissue; right panel illustrates those that are loss/underexpressed in tumor. As described in Oncomine Differential Expression Analyses, each gene in each individual dataset is ranked to indicate its statistical significance of over- or under-expression. The ranking percentile of each demethylase relative to the ranks of all other genes in individual dataset is indicated with different color gradient: top 1 % of the ranks are shown with the darkest color (red, overexpression; blue, underexpression), the lower the ranking percentile, the lighter the color to be indicated. Briefly, the demethylases that are shown to be present with higher ranks (darker color) in more datasets suggests a higher probability of them being overexpressed or underexpressed in prostate tumors
3.1 Targeting Androgen Signaling
Androgen/androgen receptor (AR) signaling is essential for early stage of prostate cancer cell growth and survival, which can be managed by anti-androgen hormone therapy. At a later stage, the majority of the tumors are transitioned into hormone-independent or castration-resistant state (CRPC), for which there is thus far no effective cure. Although most of the CRPCs no longer depend on the external androgen for growth and survival, the AR activity which is often aberrantly activated is still required. Several histone demethylases overexpressed in prostate cancer (Table 15.2) contribute to the aberrant activation of AR and AR associated signaling. This is understandable, as nuclear hormone receptors such as AR are known to form complex with “co-activators” to exert their transcriptional function; these coactivators are often histone-modifying enzymes and chromatin remodeling proteins, to which histone demethylases belong. Their general functions are to generate an open chromatin conformation allowing RNA polymerase and transcriptional complex to engage and to transcribe the target gene. Aberrantly expressed histone methylases thus can cause aberrant activation of AR.
At least seven histone lysine demethylases are found to promote AR transcriptional activity: KDM1A, KDM3A, KDM4A, KDM4C, KDM4D, KDM5B and KDM8. KDM1A was originally described as a specific “eraser” of the active histone mark, H3K4me2/me1, and thus, functions as a transcriptional repressor. Interestingly, when it complexes with AR, and after H3T6 is phosphorylated by PKCβ1, KDM1A switches its demethylation specificity from H3K4me2/me1 to the repressive histone mark H3K9me2/me1 [98, 99], thereby enhancing AR activity on the target genes. Inhibition or silencing of KDM1A results in reduced androgen-dependent proliferation and PSA (an AR target gene) transcription [98, 100]. These findings suggest that KDM1A functions as a coactivator for AR, and plays an important role in prostate cancer. Indeed, KDM1A is up-regulated in prostate tumor tissues, and overexpression of which is associated with higher relapse risk [100]. Wissmann et al. later identified a single complex consisting of KDM1A, KDM4C and AR, and reported KDM4C also as an AR coactivator. KDM4C and KDM1A bind to androgen responsive elements (ARE) located at promoter and enhancer of AR target genes, and upon hormone stimulation, they cooperatively remove the repressive tri-, di- and mono-methylated H3K9 marks [101]. This cooperation action synergistically enhances AR transcriptional activity on PSA enhancer. It is worth noting that different to the scenario of KDM1A-mediated estrogen receptor (ER) coactivation, where recruitment of KDM1A to ER target genes is ligand-dependent [102], chromatin binding of KDM1A and KDM4C to AR targets occurs in the absence of androgen treatment, while their demethylation activity on H3K9 depends on androgen signaling. KDM3A by contrast, displays hormone-dependent interaction with AR as well as chromatin recruitment to AREs [103]. Binding of KDM3A in turn, catalyzes loci specific demethylation of mono- and di-methylated H3K9. Similar to KDM1A, KDM3A and KDM4C are essential for hormone-induction of AR targets and hormone-dependent proliferation [101, 103]. Given their overexpession in prostate tumors and contributing to AR activation [12, 104], KDM3A and KDM4C are potential therapeutic targets for prostate cancer.
As exemplified by the analysis of AR and ER, recent studies have suggested that removal of the repressive H3K9 methylation marks at the promoter of target genes is crucial for nuclear receptor-mediated gene expression [105]. Aside from the KDM1A-KDM4C complex, H3K9me3/me2 demethylases KDM4A and KDM4D are also shown to interact with AR, and their overexpression enhance AR activity on the PSA enhancer [106]. Surprisingly, KDM5B (JARID1B) that demethylates the active histone mark H3K4me3/me2, is found to be overexpressed in prostate cancer and also serves as a coactivator for AR [104, 107]. While the detailed mechanism associated with KDM5B being an AR coactivator is not clear, at least two possibilities can be considered. First, as has been reported in different contexts [108, 109], H3K4me3/me2 may function as a repressive mark in a loci-specific way, and demethylation by KDM5B would activate AR target gene transcription. Second, similar to the case of KDM1A discussed above, association with AR could alter KDM5B’s substrate specificity. The fact that KDM5B is significantly overexpressed in metastatic prostate cancer cells [107] indicates that the KDM5B-mediated AR activation is likely to bypass the androgen requirement, and KDM5B could serve as a potential target for late stage therapeutics. Our unpublished data showed that several other histone demethylases also interact with AR. We found, for instance, KDM8 (JMJD5), a H3K36me2 demethylase [42, 46], is overexpressed in high-grade prostate cancer and forms a complex with KDM4A and AR on chromatin. Ectopic expression of KDM8 and KDM4A synergistically enhanced AR activity with concomitant decrease of H3K36me2 at the target promoter. Because KDM4A is also capable of catalyzing demethylation on H3K36me3/me2 [110, 111], the combination of KDM8 and KDM4A is expected to potently demethylate both H3K36me3/me2 and H3K9me3, allowing effective H3 acetylation. In addition to KDM8, we also identified interactions between KDM1B, KDM2A, KDM4B, KDM5A, KDM5D and AR. The data taken together suggest that histone demethylases either singly or in combination can serve as coactivator of AR to change the chromatin landscape of the AR target genes, thereby augmenting the transcription.
Finally, in addition to changing the local chromatin structure for AR target genes, histone demethylases are found to directly regulate AR. For instance, KDM1A is recruited by AR to an intronic enhancer of AR locus and represses AR expression via the removal of H3K4me3 mark [112]. Unlike the situation in PSA promoter, KDM1A in this case is not complexed with KDM4C and serves as a transcriptional repressor. This autoregulation takes place only when there is abundant androgen and acts as a feedback mechanism to shut off androgen signal. Under androgen-depleted conditions as in the case of CRPC, AR expression is usually increased. The discussion above indicates that histone demethylases, like histone acetylases and deacetylases, are partners of AR, and may play significant role in the dysregulation of AR activity during transition to hormone refractory prostate cancer. What discussed above is almost certainly only the tip of iceberg. More histone demethylases which directly or indirectly affect androgen receptor signaling are likely to be uncovered in the coming years.
3.2 Targeting Other Oncogenic Signals
KDM5C (SMCX/JARID1C) is a H3K4me3/me2 demethylase also found to be overexpressed in prostate cancer. KDM5C physically interacts with TGFβ-downstream transcription factor Smad3, and overexpression of which inhibits Smad3 activity independently of its demethylase activity [113]. Since TGFβ signaling acts as a tumor suppressive pathway in early prostate cancer [114], antagonizing the TGFβ-Smad3 pathway by KDM5C may therefore promote prostate tumor initiation. Another strongly overexpressed demethylase observed in clinical prostate cancer samples is PHF8, whose expression is correlated with high Gleason grade and poor prognosis [104]. PHF8 can demethylate multiple substrates including H3K9me2/me1, H3K27me2 and H4K20me1. Although the mechanism remains unclear, knockdown of PHF8 inhibits proliferation, migration and invasion ability of prostate cancer cells, indicating PHF8 as a potent oncogene for prostate cancer [104].
While a number of demethylases seem to exhibit oncogenic potential, KDM2A was found to be underexpressed in prostate cancer and functions as a tumor suppressor. Frescas et al. showed that KDM2A is required to maintain the centromeric heterochromatin state and also sustain genomic integrity. Underexpression of KDM2A in prostate cancer may thus cause genomic instability, contributing to cellular transformation [13].
3.3 Histone Methylation as Biomarkers for CRPC
Seligson et al. first reported that global levels of histone modification can be used to predict clinical outcome of prostate cancer patients with low Gleason grade [115]. Elevated H3K4me3 and H3K27me3, and reduced level of H3K9me2 in prostate tumor tissue are found to associate with poor prognosis [7, 116, 117]. While H3K4me1, H3K4me2 and H3K4me3 levels are significantly increased in CRPC, higher level of H3K4me1 is more likely to develop recurrence [118]. One of the mechanisms underlying the altered histone methylation-associated malignancy and prostate tumor recurrence is AR -mediated activation of proto-oncogenes and repression of tumor suppressors. Genome-wide analysis revealed that in CRPC cells, H3K4me1 and H3K4me2 are selectively enriched at enhancers of oncogenes such as UBE2C and CDK1, facilitating recruitment of AR for their transcription. Up-regulation of these cell cycle genes in turn, promotes growth of CRPC cells [119]. Similarly, increased H3K4me3 in prostate cancer cell correlates with the expression of oncogenes including FGFR1, BCL2 and HOXC5[120]. By contrast, H3K27me3 mark is enriched at the promoters of tumor suppressor genes, leading to their silencing in metastatic prostate cancer cell [121]. Together, emerging studies have suggested that histone modifications can serve as prognostic markers to predict outcome of prostate cancer. The intervention potential of the possible demethylases and methyltransferases that are responsible for the altered histone methylation is worthy of further consideration.
4 Concluding Remarks
In the past 8 years since the discovery of the first histone demethylase, KDM1A, extraordinary progress has been made in understanding their modes of action on histones and their connections to epigenetic regulation of carcinogenesis. Epigenetic regulation of cancer is important not only during transformation and metastasis processes, but also during therapeutic resistance. As master programmers of epigenetic regulation, histone demethylases are potential targets for intervention. Attentions to this group of genes, especially on understanding of their up- and down-stream signal pathways will only increase in the coming years. A few comments on the future direction of these research activities are provided here. First, the early literatures on histone demethylases have mostly focused on their actions on histone. Yet, we now know that KDMs may have other cellular substrates whose demethylation fuel the carcinogenesis processes. Identification of non-histone substrates of KDMs will be important to fully appreciate KDMs’ modes of action. Second, as exemplified by KDM5C, KDMs may exert their function in a demethylation-independent manner. Hence, small molecules targeting the enzymatic activity may not work in this case. Third, paradoxically, in some cancers, KDMs and their counteracting histone methylases can both be overexpressed and serve as progression factors (e.g., KDM8 and NSD2). This suggests that it is not the global level of the particular histone marks, but rather the loci-specific epigenetic landscape which determines the final outcome. This makes the measurement of therapeutic responses more challenging. The development of histone demethylase inhibitors is still at very early stage; however several promising leads have already surfaced (e.g., KDM1A for prostate cancer). Given the wide range of activities and biological outcomes of histone demethylases, one can envision a tremendous surge of research activities in the related areas with an intensity which may rival those for tyrosine kinases.
Abbreviations
- AR:
-
Androgen receptor
- ARE:
-
Androgen responsive elements
- CDK:
-
Cyclin-dependent kinase
- CGH:
-
Comparative genomic hybridization
- CRPC:
-
Castration-resistant prostate cancer
- ER:
-
Estrogen receptor
- FAD:
-
Flavin adenine dinucleotide
- HIF:
-
Hypoxia-induced transcription factor
- HP1:
-
Heterochromatin protein 1
- KDM:
-
Histone lysine demethylase
- MEF:
-
Mouse embryonic fibroblast
- MAO:
-
Monoamine oxidase
- NOG:
-
N-oxalylglycine
- PRC:
-
Polycomb repressive complex
- SAHF:
-
Senescence-associated heterochromatic foci
References
Berger SL (2007) The complex language of chromatin regulation during transcription. Nature 447(7143):407–412
Kouzarides T (2007) Chromatin modifications and their function. Cell 128(4):693–705
Varier RA, Timmers HT (2011) Histone lysine methylation and demethylation pathways in cancer. Biochim Biophys Acta 1815(1):75–89
Pedersen MT, Helin K (2010) Histone demethylases in development and disease. Trends Cell Biol 20(11):662–671
Geutjes EJ, Bajpe PK, Bernards R (2012) Targeting the epigenome for treatment of cancer. Oncogene 31(34):3827–3844
Fraga MF, Ballestar E, Villar-Garea A, Boix-Chornet M, Espada J, Schotta G et al (2005) Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat Genet 37(4):391–400
Seligson DB, Horvath S, McBrian MA, Mah V, Yu H, Tze S et al (2009) Global levels of histone modifications predict prognosis in different cancers. Am J Pathol 174(5):1619–1628
Elsheikh SE, Green AR, Rakha EA, Powe DG, Ahmed RA, Collins HM et al (2009) Global histone modifications in breast cancer correlate with tumor phenotypes, prognostic factors, and patient outcome. Cancer Res 69(9):3802–3809
Barlesi F, Giaccone G, Gallegos-Ruiz MI, Loundou A, Span SW, Lefesvre P et al (2007) Global histone modifications predict prognosis of resected non small-cell lung cancer. J Clin Oncol 25(28):4358–4364
Narita M, Nunez S, Heard E, Lin AW, Hearn SA, Spector DL et al (2003) Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell 113(6):703–716
Peters AH, O’Carroll D, Scherthan H, Mechtler K, Sauer S, Schofer C et al (2001) Loss of the Suv39h histone methyltransferases impairs mammalian heterochromatin and genome stability. Cell 107(3):323–337
Cloos PA, Christensen J, Agger K, Maiolica A, Rappsilber J, Antal T et al (2006) The putative oncogene GASC1 demethylates tri- and dimethylated lysine 9 on histone H3. Nature 442(7100):307–311
Frescas D, Guardavaccaro D, Kuchay SM, Kato H, Poleshko A, Basrur V et al (2008) KDM2A represses transcription of centromeric satellite repeats and maintains the heterochromatic state. Cell Cycle 7(22):3539–3547
Frescas D, Guardavaccaro D, Bassermann F, Koyama-Nasu R, Pagano M (2007) JHDM1B/FBXL10 is a nucleolar protein that represses transcription of ribosomal RNA genes. Nature 450(7167):309–313
Ruggero D, Pandolfi PP (2003) Does the ribosome translate cancer? Nat Rev Cancer 3(3):179–192
Magerl C, Ellinger J, Braunschweig T, Kremmer E, Koch LK, Holler T et al (2010) H3K4 dimethylation in hepatocellular carcinoma is rare compared with other hepatobiliary and gastrointestinal carcinomas and correlates with expression of the methylase Ash2 and the demethylase LSD1. Hum Pathol 41(2):181–189
Wang Y, Zhang H, Chen Y, Sun Y, Yang F, Yu W et al (2009) LSD1 is a subunit of the NuRD complex and targets the metastasis programs in breast cancer. Cell 138(4):660–672
He J, Nguyen AT, Zhang Y (2011) KDM2b/JHDM1b, an H3K36me2-specific demethylase, is required for initiation and maintenance of acute myeloid leukemia. Blood 117(14): 3869–3880
Han W, Jung EM, Cho J, Lee JW, Hwang KT, Yang SJ et al (2008) DNA copy number alterations and expression of relevant genes in triple-negative breast cancer. Genes Chromosom Cancer 47(6):490–499
Italiano A, Attias R, Aurias A, Perot G, Burel-Vandenbos F, Otto J et al (2006) Molecular cytogenetic characterization of a metastatic lung sarcomatoid carcinoma: 9p23 neocentromere and 9p23-p24 amplification including JAK2 and JMJD2C. Cancer Genet Cytogenet 167(2): 122–130
Savelyeva L, Claas A, An H, Weber RG, Lichter P, Schwab M (1999) Retention of polysomy at 9p23-24 during karyotypic evolution in human breast cancer cell line COLO 824. Genes Chromosomes Cancer 24(1):87–93
Yang ZQ, Imoto I, Fukuda Y, Pimkhaokham A, Shimada Y, Imamura M et al (2000) Identification of a novel gene, GASC1, within an amplicon at 9p23-24 frequently detected in esophageal cancer cell lines. Cancer Res 60(17):4735–4739
Liu G, Bollig-Fischer A, Kreike B, van de Vijver MJ, Abrams J, Ethier SP et al (2009) Genomic amplification and oncogenic properties of the GASC1 histone demethylase gene in breast cancer. Oncogene 28(50):4491–4500
Rui L, Emre NC, Kruhlak MJ, Chung HJ, Steidl C, Slack G et al (2010) Cooperative epigenetic modulation by cancer amplicon genes. Cancer Cell 18(6):590–605
Hou J, Wu J, Dombkowski A, Zhang K, Holowatyj A, Boerner JL et al (2012) Genomic amplification and a role in drug-resistance for the KDM5A histone demethylase in breast cancer. Am J Transl Res 4(3):247–256
van Zutven LJ, Onen E, Velthuizen SC, van Drunen E, von Bergh AR, van den Heuvel-Eibrink MM et al (2006) Identification of NUP98 abnormalities in acute leukemia: JARID1A (12p13) as a new partner gene. Genes Chromosomes Cancer 45(5):437–446
Wang GG, Song J, Wang Z, Dormann HL, Casadio F, Li H et al (2009) Haematopoietic malignancies caused by dysregulation of a chromatin-binding PHD finger. Nature 459(7248):847–851
Niu X, Zhang T, Liao L, Zhou L, Lindner DJ, Zhou M et al (2012) The von Hippel-Lindau tumor suppressor protein regulates gene expression and tumor growth through histone demethylase JARID1C. Oncogene 31(6):776–786
Dalgliesh GL, Furge K, Greenman C, Chen L, Bignell G, Butler A et al (2010) Systematic sequencing of renal carcinoma reveals inactivation of histone modifying genes. Nature 463(7279):360–363
van Haaften G, Dalgliesh GL, Davies H, Chen L, Bignell G, Greenman C et al (2009) Somatic mutations of the histone H3K27 demethylase gene UTX in human cancer. Nat Genet 41(5):521–523
Gui Y, Guo G, Huang Y, Hu X, Tang A, Gao S et al (2011) Frequent mutations of chromatin remodeling genes in transitional cell carcinoma of the bladder. Nat Genet 43(9):875–878
Wang JK, Tsai MC, Poulin G, Adler AS, Chen S, Liu H et al (2010) The histone demethylase UTX enables RB-dependent cell fate control. Genes Dev 24(4):327–332
Herz HM, Madden LD, Chen Z, Bolduc C, Buff E, Gupta R et al (2010) The H3K27me3 demethylase dUTX is a suppressor of Notch- and Rb-dependent tumors in Drosophila. Mol Cell Biol 30(10):2485–2497
Tsai MC, Wang JK, Chang HY (2010) Tumor suppression by the histone demethylase UTX. Cell Cycle 9(11):2043–2044
Terashima M, Ishimura A, Yoshida M, Suzuki Y, Sugano S, Suzuki T (2010) The tumor suppressor Rb and its related Rbl2 genes are regulated by Utx histone demethylase. Biochem Biophys Res Commun 399(2):238–244
Berry WL, Shin S, Lightfoot SA, Janknecht R (2012) Oncogenic features of the JMJD2A histone demethylase in breast cancer. Int J Oncol 41(5):1701–1706
Toyokawa G, Cho HS, Iwai Y, Yoshimatsu M, Takawa M, Hayami S et al (2011) The histone demethylase JMJD2B plays an essential role in human carcinogenesis through positive regulation of cyclin-dependent kinase 6. Cancer Prev Res (Phila) 4(12):2051–2061
Kawazu M, Saso K, Tong KI, McQuire T, Goto K, Son DO et al (2011) Histone demethylase JMJD2B functions as a co-factor of estrogen receptor in breast cancer proliferation and mammary gland development. PLoS One 6(3):e17830
Mitra D, Das PM, Huynh FC, Jones FE (2011) Jumonji/ARID1 B (JARID1B) protein promotes breast tumor cell cycle progression through epigenetic repression of microRNA let-7e. J Biol Chem 286(47):40531–40535
Cho HS, Toyokawa G, Daigo Y, Hayami S, Masuda K, Ikawa N et al (2012) The JmjC domain-containing histone demethylase KDM3A is a positive regulator of the G1/S transition in cancer cells via transcriptional regulation of the HOXA1 gene. Int J Cancer 131(3): E179–E189
Yang J, Jubb AM, Pike L, Buffa FM, Turley H, Baban D et al (2010) The histone demethylase JMJD2B is regulated by estrogen receptor alpha and hypoxia, and is a key mediator of estrogen induced growth. Cancer Res 70(16):6456–6466
Hsia DA, Tepper CG, Pochampalli MR, Hsia EY, Izumiya C, Huerta SB et al (2010) KDM8, a H3K36me2 histone demethylase that acts in the cyclin A1 coding region to regulate cancer cell proliferation. Proc Natl Acad Sci U S A 107(21):9671–9676
Lim S, Janzer A, Becker A, Zimmer A, Schule R, Buettner R et al (2010) Lysine-specific demethylase 1 (LSD1) is highly expressed in ER-negative breast cancers and a biomarker predicting aggressive biology. Carcinogenesis 31(3):512–520
Zeng J, Ge Z, Wang L, Li Q, Wang N, Bjorkholm M et al (2010) The histone demethylase RBP2 Is overexpressed in gastric cancer and its inhibition triggers senescence of cancer cells. Gastroenterology 138(3):981–992
Ishimura A, Minehata K, Terashima M, Kondoh G, Hara T, Suzuki T (2012) Jmjd5, an H3K36me2 histone demethylase, modulates embryonic cell proliferation through the regulation of Cdkn1a expression. Development 139(4):749–759
Oh S, Janknecht R (2012) Histone demethylase JMJD5 is essential for embryonic development. Biochem Biophys Res Commun 420(1):61–65
Dey BK, Stalker L, Schnerch A, Bhatia M, Taylor-Papidimitriou J, Wynder C (2008) The histone demethylase KDM5b/JARID1b plays a role in cell fate decisions by blocking terminal differentiation. Mol Cell Biol 28(17):5312–5327
Nijwening JH, Geutjes EJ, Bernards R, Beijersbergen RL (2011) The histone demethylase Jarid1b (Kdm5b) is a novel component of the Rb pathway and associates with E2f-target genes in MEFs during senescence. PLoS One 6(9):e25235
Hayami S, Yoshimatsu M, Veerakumarasivam A, Unoki M, Iwai Y, Tsunoda T et al (2010) Overexpression of the JmjC histone demethylase KDM5B in human carcinogenesis: involvement in the proliferation of cancer cells through the E2F/RB pathway. Mol Cancer 9:59
Cho HS, Suzuki T, Dohmae N, Hayami S, Unoki M, Yoshimatsu M et al (2011) Demethylation of RB regulator MYPT1 by histone demethylase LSD1 promotes cell cycle progression in cancer cells. Cancer Res 71(3):655–660
Liu W, Tanasa B, Tyurina OV, Zhou TY, Gassmann R, Liu WT et al (2010) PHF8 mediates histone H4 lysine 20 demethylation events involved in cell cycle progression. Nature 466(7305):508–512
Li W, Zhao L, Zang W, Liu Z, Chen L, Liu T et al (2011) Histone demethylase JMJD2B is required for tumor cell proliferation and survival and is overexpressed in gastric cancer. Biochem Biophys Res Commun 416(3–4):372–378
Tsai WW, Nguyen TT, Shi Y, Barton MC (2008) p53-targeted LSD1 functions in repression of chromatin structure and transcription in vivo. Mol Cell Biol 28(17):5139–5146
Kim TD, Oh S, Shin S, Janknecht R (2012) Regulation of tumor suppressor p53 and HCT116 cell physiology by histone demethylase JMJD2D/KDM4D. PLoS One 7(4):e34618
Ishimura A, Terashima M, Kimura H, Akagi K, Suzuki Y, Sugano S et al (2009) Jmjd2c histone demethylase enhances the expression of Mdm2 oncogene. Biochem Biophys Res Commun 389(2):366–371
Black JC, Allen A, Van Rechem C, Forbes E, Longworth M, Tschop K et al (2010) Conserved antagonism between JMJD2A/KDM4A and HP1gamma during cell cycle progression. Mol Cell 40(5):736–748
Braig M, Lee S, Loddenkemper C, Rudolph C, Peters AH, Schlegelberger B et al (2005) Oncogene-induced senescence as an initial barrier in lymphoma development. Nature 436(7051):660–665
Collado M, Gil J, Efeyan A, Guerra C, Schuhmacher AJ, Barradas M et al (2005) Tumour biology: senescence in premalignant tumours. Nature 436(7051):642
Lowe SW, Sherr CJ (2003) Tumor suppression by Ink4a-Arf: progress and puzzles. Curr Opin Genet Dev 13(1):77–83
Bracken AP, Kleine-Kohlbrecher D, Dietrich N, Pasini D, Gargiulo G, Beekman C et al (2007) The Polycomb group proteins bind throughout the INK4A-ARF locus and are disassociated in senescent cells. Genes Dev 21(5):525–530
He J, Kallin EM, Tsukada Y, Zhang Y (2008) The H3K36 demethylase Jhdm1b/Kdm2b regulates cell proliferation and senescence through p15(Ink4b). Nat Struct Mol Biol 15(11):1169–1175
Pfau R, Tzatsos A, Kampranis SC, Serebrennikova OB, Bear SE, Tsichlis PN (2008) Members of a family of JmjC domain-containing oncoproteins immortalize embryonic fibroblasts via a JmjC domain-dependent process. Proc Natl Acad Sci U S A 105(6): 1907–1912
Tzatsos A, Paskaleva P, Lymperi S, Contino G, Stoykova S, Chen Z et al (2011) Lysine-specific demethylase 2B (KDM2B)-let-7-enhancer of zester homolog 2 (EZH2) pathway regulates cell cycle progression and senescence in primary cells. J Biol Chem 286(38): 33061–33069
Chicas A, Kapoor A, Wang X, Aksoy O, Evertts AG, Zhang MQ et al (2012) H3K4 demethylation by Jarid1a and Jarid1b contributes to retinoblastoma-mediated gene silencing during cellular senescence. Proc Natl Acad Sci U S A 109(23):8971–8976
Johnson AB, Denko N, Barton MC (2008) Hypoxia induces a novel signature of chromatin modifications and global repression of transcription. Mutat Res 640(1–2):174–179
Chen H, Yan Y, Davidson TL, Shinkai Y, Costa M (2006) Hypoxic stress induces dimethylated histone H3 lysine 9 through histone methyltransferase G9a in mammalian cells. Cancer Res 66(18):9009–9016
Krieg AJ, Rankin EB, Chan D, Razorenova O, Fernandez S, Giaccia AJ (2010) Regulation of the histone demethylase JMJD1A by hypoxia-inducible factor 1 alpha enhances hypoxic gene expression and tumor growth. Mol Cell Biol 30(1):344–353
Beyer S, Kristensen MM, Jensen KS, Johansen JV, Staller P (2008) The histone demethylases JMJD1A and JMJD2B are transcriptional targets of hypoxia-inducible factor HIF. J Biol Chem 283(52):36542–36552
Sar A, Ponjevic D, Nguyen M, Box AH, Demetrick DJ (2009) Identification and characterization of demethylase JMJD1A as a gene upregulated in the human cellular response to hypoxia. Cell Tissue Res 337(2):223–234
Xia X, Lemieux ME, Li W, Carroll JS, Brown M, Liu XS et al (2009) Integrative analysis of HIF binding and transactivation reveals its role in maintaining histone methylation homeostasis. Proc Natl Acad Sci U S A 106(11):4260–4265
Mimura I, Nangaku M, Kanki Y, Tsutsumi S, Inoue T, Kohro T et al (2012) Dynamic change of chromatin conformation in response to hypoxia enhances the expression of GLUT3 (SLC2A3) by cooperative interaction of hypoxia-inducible factor 1 and KDM3A. Mol Cell Biol 32(15):3018–3032
Koong AC, Chen EY, Giaccia AJ (1994) Hypoxia causes the activation of nuclear factor kappa B through the phosphorylation of I kappa B alpha on tyrosine residues. Cancer Res 54(6):1425–1430
Leonard MO, Howell K, Madden SF, Costello CM, Higgins DG, Taylor CT et al (2008) Hypoxia selectively activates the CREB family of transcription factors in the in vivo lung. Am J Respir Crit Care Med 178(9):977–983
Rong Y, Hu F, Huang R, Mackman N, Horowitz JM, Jensen RL et al (2006) Early growth response gene-1 regulates hypoxia-induced expression of tissue factor in glioblastoma multiforme through hypoxia-inducible factor-1-independent mechanisms. Cancer Res 66(14): 7067–7074
Lu T, Jackson MW, Wang B, Yang M, Chance MR, Miyagi M et al (2010) Regulation of NF-kappaB by NSD1/FBXL11-dependent reversible lysine methylation of p65. Proc Natl Acad Sci U S A 107(1):46–51
Culhane JC, Szewczuk LM, Liu X, Da G, Marmorstein R, Cole PA (2006) A mechanism-based inactivator for histone demethylase LSD1. J Am Chem Soc 128(14):4536–4537
Szewczuk LM, Culhane JC, Yang M, Majumdar A, Yu H, Cole PA (2007) Mechanistic analysis of a suicide inactivator of histone demethylase LSD1. Biochemistry 46(23): 6892–6902
Yang M, Culhane JC, Szewczuk LM, Gocke CB, Brautigam CA, Tomchick DR et al (2007) Structural basis of histone demethylation by LSD1 revealed by suicide inactivation. Nat Struct Mol Biol 14(6):535–539
Mimasu S, Sengoku T, Fukuzawa S, Umehara T, Yokoyama S (2008) Crystal structure of histone demethylase LSD1 and tranylcypromine at 2.25 A. Biochem Biophys Res Commun 366(1):15–22
Yang M, Culhane JC, Szewczuk LM, Jalili P, Ball HL, Machius M et al (2007) Structural basis for the inhibition of the LSD1 histone demethylase by the antidepressant trans-2-phenylcyclopropylamine. Biochemistry 46(27):8058–8065
Gooden DM, Schmidt DM, Pollock JA, Kabadi AM, McCafferty DG (2008) Facile synthesis of substituted trans-2-arylcyclopropylamine inhibitors of the human histone demethylase LSD1 and monoamine oxidases A and B. Bioorg Med Chem Lett 18(10):3047–3051
Ueda R, Suzuki T, Mino K, Tsumoto H, Nakagawa H, Hasegawa M et al (2009) Identification of cell-active lysine specific demethylase 1-selective inhibitors. J Am Chem Soc 131(48):17536–17537
Binda C, Valente S, Romanenghi M, Pilotto S, Cirilli R, Karytinos A et al (2010) Biochemical, structural, and biological evaluation of tranylcypromine derivatives as inhibitors of histone demethylases LSD1 and LSD2. J Am Chem Soc 132(19):6827–6833
Mimasu S, Umezawa N, Sato S, Higuchi T, Umehara T, Yokoyama S (2010) Structurally designed trans-2-phenylcyclopropylamine derivatives potently inhibit histone demethylase LSD1/KDM1. Biochemistry 49(30):6494–6503
Ogasawara D, Suzuki T, Mino K, Ueda R, Khan MN, Matsubara T et al (2011) Synthesis and biological activity of optically active NCL-1, a lysine-specific demethylase 1 selective inhibitor. Bioorg Med Chem 19(12):3702–3708
Huang Y, Greene E, Murray Stewart T, Goodwin AC, Baylin SB, Woster PM et al (2007) Inhibition of lysine-specific demethylase 1 by polyamine analogues results in reexpression of aberrantly silenced genes. Proc Natl Acad Sci U S A 104(19):8023–8028
Huang Y, Stewart TM, Wu Y, Baylin SB, Marton LJ, Perkins B et al (2009) Novel oligoamine analogues inhibit lysine-specific demethylase 1 and induce reexpression of epigenetically silenced genes. Clin Cancer Res 15(23):7217–7228
Sharma SK, Wu Y, Steinbergs N, Crowley ML, Hanson AS, Casero RA et al (2010) (Bis)urea and (bis)thiourea inhibitors of lysine-specific demethylase 1 as epigenetic modulators. J Med Chem 53(14):5197–5212
Wang J, Lu F, Ren Q, Sun H, Xu Z, Lan R et al (2011) Novel histone demethylase LSD1 inhibitors selectively target cancer cells with pluripotent stem cell properties. Cancer Res 71(23):7238–7249
Hamada S, Kim TD, Suzuki T, Itoh Y, Tsumoto H, Nakagawa H et al (2009) Synthesis and activity of N-oxalylglycine and its derivatives as Jumonji C-domain-containing histone lysine demethylase inhibitors. Bioorg Med Chem Lett 19(10):2852–2855
Hamada S, Suzuki T, Mino K, Koseki K, Oehme F, Flamme I et al (2010) Design, synthesis, enzyme-inhibitory activity, and effect on human cancer cells of a novel series of jumonji domain-containing protein 2 histone demethylase inhibitors. J Med Chem 53(15):5629–5638
Thalhammer A, Mecinovic J, Loenarz C, Tumber A, Rose NR, Heightman TD et al (2011) Inhibition of the histone demethylase JMJD2E by 3-substituted pyridine 2,4-dicarboxylates. Org Biomol Chem 9(1):127–135
King ON, Li XS, Sakurai M, Kawamura A, Rose NR, Ng SS et al (2010) Quantitative high-throughput screening identifies 8-hydroxyquinolines as cell-active histone demethylase inhibitors. PLoS One 5(11):e15535
Upadhyay AK, Rotili D, Han JW, Hu R, Chang Y, Labella D et al (2012) An analog of BIX-01294 selectively inhibits a family of histone H3 lysine 9 Jumonji demethylases. J Mol Biol 416(3):319–327
Kristensen LH, Nielsen AL, Helgstrand C, Lees M, Cloos P, Kastrup JS et al (2012) Studies of H3K4me3 demethylation by KDM5B/Jarid1B/PLU1 reveals strong substrate recognition in vitro and identifies 2,4-pyridine-dicarboxylic acid as an in vitro and in cell inhibitor. FEBS J 279(11):1905–1914
Nielsen AL, Kristensen LH, Stephansen KB, Kristensen JB, Helgstrand C, Lees M et al (2012) Identification of catechols as histone-lysine demethylase inhibitors. FEBS Lett 586(8):1190–1194
Kruidenier L, Chung CW, Cheng Z, Liddle J, Che K, Joberty G et al (2012) A selective jumonji H3K27 demethylase inhibitor modulates the proinflammatory macrophage response. Nature 488(7411):404–408
Metzger E, Wissmann M, Yin N, Muller JM, Schneider R, Peters AH et al (2005) LSD1 demethylates repressive histone marks to promote androgen-receptor-dependent transcription. Nature 437(7057):436–439
Metzger E, Imhof A, Patel D, Kahl P, Hoffmeyer K, Friedrichs N et al (2010) Phosphorylation of histone H3T6 by PKCbeta(I) controls demethylation at histone H3K4. Nature 464(7289):792–796
Kahl P, Gullotti L, Heukamp LC, Wolf S, Friedrichs N, Vorreuther R et al (2006) Androgen receptor coactivators lysine-specific histone demethylase 1 and four and a half LIM domain protein 2 predict risk of prostate cancer recurrence. Cancer Res 66(23):11341–11347
Wissmann M, Yin N, Muller JM, Greschik H, Fodor BD, Jenuwein T et al (2007) Cooperative demethylation by JMJD2C and LSD1 promotes androgen receptor-dependent gene expression. Nat Cell Biol 9(3):347–353
Garcia-Bassets I, Kwon YS, Telese F, Prefontaine GG, Hutt KR, Cheng CS et al (2007) Histone methylation-dependent mechanisms impose ligand dependency for gene activation by nuclear receptors. Cell 128(3):505–518
Yamane K, Toumazou C, Tsukada Y, Erdjument-Bromage H, Tempst P, Wong J et al (2006) JHDM2A, a JmjC-containing H3K9 demethylase, facilitates transcription activation by androgen receptor. Cell 125(3):483–495
Bjorkman M, Ostling P, Harma V, Virtanen J, Mpindi JP, Rantala J et al (2012) Systematic knockdown of epigenetic enzymes identifies a novel histone demethylase PHF8 overexpressed in prostate cancer with an impact on cell proliferation, migration and invasion. Oncogene 31(29):3444–3456
Stratmann A, Haendler B (2012) Histone demethylation and steroid receptor function in cancer. Mol Cell Endocrinol 348(1):12–20
Shin S, Janknecht R (2007) Activation of androgen receptor by histone demethylases JMJD2A and JMJD2D. Biochem Biophys Res Commun 359(3):742–746
Xiang Y, Zhu Z, Han G, Ye X, Xu B, Peng Z et al (2007) JARID1B is a histone H3 lysine 4 demethylase up-regulated in prostate cancer. Proc Natl Acad Sci U S A 104(49):19226–19231
Pinskaya M, Gourvennec S, Morillon A (2009) H3 lysine 4 di- and tri-methylation deposited by cryptic transcription attenuates promoter activation. EMBO J 28(12):1697–1707
Shi X, Hong T, Walter KL, Ewalt M, Michishita E, Hung T et al (2006) ING2 PHD domain links histone H3 lysine 4 methylation to active gene repression. Nature 442(7098):96–99
Chen Z, Zang J, Whetstine J, Hong X, Davrazou F, Kutateladze TG et al (2006) Structural insights into histone demethylation by JMJD2 family members. Cell 125(4):691–702
Hillringhaus L, Yue WW, Rose NR, Ng SS, Gileadi C, Loenarz C et al (2011) Structural and evolutionary basis for the dual substrate selectivity of human KDM4 histone demethylase family. J Biol Chem 286(48):41616–41625
Cai C, He HH, Chen S, Coleman I, Wang H, Fang Z et al (2011) Androgen receptor gene expression in prostate cancer is directly suppressed by the androgen receptor through recruitment of lysine-specific demethylase 1. Cancer Cell 20(4):457–471
Kim TD, Shin S, Janknecht R (2008) Repression of Smad3 activity by histone demethylase SMCX/JARID1C. Biochem Biophys Res Commun 366(2):563–567
Buijs JT, Henriquez NV, van Overveld PG, van der Horst G, ten Dijke P, van der Pluijm G (2007) TGF-beta and BMP7 interactions in tumour progression and bone metastasis. Clin Exp Metastasis 24(8):609–617
Seligson DB, Horvath S, Shi T, Yu H, Tze S, Grunstein M et al (2005) Global histone modification patterns predict risk of prostate cancer recurrence. Nature 435(7046):1262–1266
Yu J, Rhodes DR, Tomlins SA, Cao X, Chen G, Mehra R et al (2007) A polycomb repression signature in metastatic prostate cancer predicts cancer outcome. Cancer Res 67(22): 10657–10663
Zhou LX, Li T, Huang YR, Sha JJ, Sun P, Li D (2010) Application of histone modification in the risk prediction of the biochemical recurrence after radical prostatectomy. Asian J Androl 12(2):171–179
Ellinger J, Kahl P, von der Gathen J, Rogenhofer S, Heukamp LC, Gutgemann I et al (2010) Global levels of histone modifications predict prostate cancer recurrence. Prostate 70(1): 61–69
Wang Q, Li W, Zhang Y, Yuan X, Xu K, Yu J et al (2009) Androgen receptor regulates a distinct transcription program in androgen-independent prostate cancer. Cell 138(2): 245–256
Ke XS, Qu Y, Rostad K, Li WC, Lin B, Halvorsen OJ et al (2009) Genome-wide profiling of histone h3 lysine 4 and lysine 27 trimethylation reveals an epigenetic signature in prostate carcinogenesis. PLoS One 4(3):e4687
Kondo Y, Shen L, Cheng AS, Ahmed S, Boumber Y, Charo C et al (2008) Gene silencing in cancer by histone H3 lysine 27 trimethylation independent of promoter DNA methylation. Nat Genet 40(6):741–750
Harris WJ, Huang X, Lynch JT, Spencer GJ, Hitchin JR, Li Y et al (2012) The histone demethylase KDM1A sustains the oncogenic potential of MLL-AF9 leukemia stem cells. Cancer Cell 21(4):473–487
Hayami S, Kelly JD, Cho HS, Yoshimatsu M, Unoki M, Tsunoda T et al (2011) Overexpression of LSD1 contributes to human carcinogenesis through chromatin regulation in various cancers. Int J Cancer 128(3):574–586
Schulte JH, Lim S, Schramm A, Friedrichs N, Koster J, Versteeg R et al (2009) Lysine-specific demethylase 1 is strongly expressed in poorly differentiated neuroblastoma: implications for therapy. Cancer Res 69(5):2065–2071
Liao QL, Chen XD, Zhao L, Ding YQ (2008) [Comparative proteomics of the serum in patients with nasopharyngeal carcinoma: a study with two-dimensional electrophoresis and MALDI-TOF-MS]. Nan Fang Yi Ke Da Xue Xue Bao 28(2):154–8
Yamada D, Kobayashi S, Yamamoto H, Tomimaru Y, Noda T, Uemura M et al (2012) Role of the hypoxia-related gene, JMJD1A, in hepatocellular carcinoma: clinical impact on recurrence after hepatic resection. Ann Surg Oncol 19(Suppl 3):S355–S364
Guo X, Shi M, Sun L, Wang Y, Gui Y, Cai Z et al (2011) The expression of histone demethylase JMJD1A in renal cell carcinoma. Neoplasma 58(2):153–157
Kim JY, Kim KB, Eom GH, Choe N, Kee HJ, Son HJ et al (2012) KDM3B is the H3K9 demethylase involved in transcriptional activation of lmo2 in leukemia. Mol Cell Biol 32(14):2917–2933
Kauffman EC, Robinson BD, Downes MJ, Powell LG, Lee MM, Scherr DS et al (2011) Role of androgen receptor and associated lysine-demethylase coregulators, LSD1 and JMJD2A, in localized and advanced human bladder cancer. Mol Carcinog 50(12):931–944
Fu L, Chen L, Yang J, Ye T, Chen Y, Fang J (2012) HIF-1alpha-induced histone demethylase JMJD2B contributes to the malignant phenotype of colorectal cancer cells via an epigenetic mechanism. Carcinogenesis 33(9):1664–1673
Wu J, Liu S, Liu G, Dombkowski A, Abrams J, Martin-Trevino R et al (2012) Identification and functional analysis of 9p24 amplified genes in human breast cancer. Oncogene 31(3):333–341
Ehrbrecht A, Muller U, Wolter M, Hoischen A, Koch A, Radlwimmer B et al (2006) Comprehensive genomic analysis of desmoplastic medulloblastomas: identification of novel amplified genes and separate evaluation of the different histological components. J Pathol 208(4):554–563
Barrett A, Madsen B, Copier J, Lu PJ, Cooper L, Scibetta AG et al (2002) PLU-1 nuclear protein, which is upregulated in breast cancer, shows restricted expression in normal human adult tissues: a new cancer/testis antigen? Int J Cancer 101(6):581–588
Lu PJ, Sundquist K, Baeckstrom D, Poulsom R, Hanby A, Meier-Ewert S et al (1999) A novel gene (PLU-1) containing highly conserved putative DNA/chromatin binding motifs is specifically up-regulated in breast cancer. J Biol Chem 274(22):15633–15645
Anderton JA, Bose S, Vockerodt M, Vrzalikova K, Wei W, Kuo M et al (2011) The H3K27me3 demethylase, KDM6B, is induced by Epstein-Barr virus and over-expressed in Hodgkin’s lymphoma. Oncogene 30(17):2037–2043
Xiang Y, Zhu Z, Han G, Lin H, Xu L, Chen CD (2007) JMJD3 is a histone H3K27 demethylase. Cell Res 17(10):850–857
Acknowledgement
This work is supported by NIH and DOD grants to H.J.K. H.J.K. acknowledges the generous support from Auburn Community Cancer Endowment Fund.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer Science+Business Media New York
About this chapter
Cite this chapter
Wang, LY. et al. (2014). Histone Demethylases in Prostate Cancer. In: Kumar, R. (eds) Nuclear Signaling Pathways and Targeting Transcription in Cancer. Cancer Drug Discovery and Development. Humana Press, New York, NY. https://doi.org/10.1007/978-1-4614-8039-6_15
Download citation
DOI: https://doi.org/10.1007/978-1-4614-8039-6_15
Published:
Publisher Name: Humana Press, New York, NY
Print ISBN: 978-1-4614-8038-9
Online ISBN: 978-1-4614-8039-6
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)