
Abstract
Heart function vitally relies on precisely controlled intracellular Ca homeostasis during each cardiac cycle. Abnormalities in Ca regulation cause contractile dysfunction and arrhythmias under different pathological conditions including heart failure (HF). Playing a particularly important role in heart contraction is the sarcoplasmic reticulum (SR). In adult ventricular myocytes, the SR forms a highly interconnected network of tubules (free SR) and cisterns (junctional SR). Although the SR occupies only 2–4 % of the total cell volume [1], it provides the major portion of Ca that initiates contraction. The ability of the SR to accumulate large amounts of Ca is ensured by the SR-specific low affinity and high capacity Ca binding protein calsequestrin (CASQ) [2].
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Sarcoplasmic Reticulum
- Heart Failure Model
- SERCA Activity
- Junctional Sarcoplasmic Reticulum
- Sarcoplasmic Reticulum Depletion
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
SR Ca Regulation in Heart
Heart function vitally relies on precisely controlled intracellular Ca homeostasis during each cardiac cycle. Abnormalities in Ca regulation cause contractile dysfunction and arrhythmias under different pathological conditions including heart failure (HF). Playing a particularly important role in heart contraction is the sarcoplasmic reticulum (SR). In adult ventricular myocytes, the SR forms a highly interconnected network of tubules (free SR) and cisterns (junctional SR). Although the SR occupies only 2–4 % of the total cell volume [1], it provides the major portion of Ca that initiates contraction. The ability of the SR to accumulate large amounts of Ca is ensured by the SR-specific low affinity and high capacity Ca binding protein calsequestrin (CASQ) [2].
Ca is released from the SR as a result of activation of specialized Ca channels—ryanodine receptors (RyRs; type 2 isoform). These Ca channels are activated by a relatively small inward Ca current via L-type Ca channels (LTCCs) during an action potential (AP). This mechanism which mediates cardiac excitation–contraction coupling (ECC) is known as Ca-induced Ca release (CICR) [3]. In ventricular myocytes CICR occurs at specialized subcellular microdomains where a T-tubule of the sarcolemma is proximal to a junction of the SR forming a dyad (Fig. 1a). Dyadic junctional SR membrane contains clusters of ~100 RyRs [4] that are strategically aligned with LTCCs by junctophilins [5] which ensure that the dyadic cleft is narrow enough (~10–30 nm) to promote efficient signaling from the T-tubule network to the SR. Each of these microdomains constitutes a SR Ca release unit [4, 6] or couplon [7]. The simultaneous activation of RyRs within the release unit generates a locally restricted increase in cytosolic [Ca] ([Ca]i), or Ca spark [8, 9]. Ca release during a spark causes partial depletion of intra-SR [Ca] ([Ca]SR) limited to a single SR junction, also known as a Ca blink [10–13]. During ECC, the global SR Ca release that initiates contraction is the result of the spatio-temporal summation of Ca release from thousands of these individual release sites (Fig. 1b). Thus, the amplitude of the Ca transient is largely controlled at the local subcellular level by gradual recruitment of these Ca release units [14]. In addition to the RyRs organized in clusters, it has been proposed the existence of isolated “rogue” RyRs localized in free SR [15].

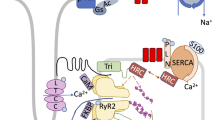
Structure and function of the SR Ca release unit in ventricular myocytes. (a) The organization of the main components of Ca regulation in ventricular myocytes at the subcellular level. The diagram illustrates L-type Ca channels (LTCCs) in the T-tubule (TT) and ryanodine receptor (RyR) clusters in the junctional sarcoplasmic reticulum (SR) form a Ca release unit. Ca-ATPase (SERCA) pumps cytosolic Ca back into the SR and the Na–Ca exchanger (NCX) removes Ca from the cell. The plasmalemmal Ca-ATPase (PMCA) and mitochondria play a minor role in cardiac relaxation. The mitochondrial Ca uniporter (MCU) and the mitochondrial Na–Ca exchanger (mtNCX) regulate mitochondrial Ca homeostasis. (b) A confocal image of diastolic Ca sparks and an action potential-induced Ca transient recorded from rabbit ventricular myocyte. Activation of a single Ca release unit generates a Ca spark (bottom inset), whereas simultaneous activation of thousands of these individual release units generates a global Ca transient
Because CICR by definition is a self-regenerative process, it would be expected to continue until SR Ca is fully exhausted. However, convincing results from different laboratories have shown that during a physiological stimulus (action potential or Ca current) [Ca]SR only partially depletes [10, 12, 16, 17]. Thus, a robust termination mechanism must exist to counteract the positive feedback of CICR and avoid “all-or-nothing” SR Ca release events [18]. Several possible mechanisms that control Ca release termination have been suggested including stochastic attrition, Ca-dependent inactivation, adaptation, and use-dependent inactivation of the RyR [14, 19–21]. However, none of these mechanisms can fully explain robust termination of Ca sparks in the intact cellular environment.
RyR gating is determined through complex regulation by both [Ca]i and [Ca]SR [22]. During Ca release, the [Ca] in the cytosol increases from ~100 nM to 1 μM reaching concentrations as high as 300 μM in the vicinity of the RyR [23], while free [Ca]SR reciprocally drops from 1–1.5 mM to 300–400 μM [24, 25]. While an increase of [Ca]i in the dyadic cleft is critical for triggering of CICR, partial depletion of [Ca]SR seems to be more important for termination of CICR. Direct experimental evidence demonstrates that Ca sparks cease when [Ca]SR falls to a certain critical level (Fig. 2 and also see [10, 12, 26]) supporting the functional link between partial [Ca]SR depletion and CICR termination [27, 28]. It has been proposed that luminal Ca can regulate RyR by passing through an opened channel and acting on the cytosolic Ca activation site of neighboring channels—a “feed-through” mechanism [29, 30]. Therefore, local [Ca]SR depletion could terminate Ca spark by reducing the unitary current of the RyR and thus breaking the positive feedback of local CICR within a cluster [31]. In addition, several independent groups found that the RyR can be directly activated by luminal [Ca] independent of Ca release flux [32, 33]. These findings suggest that the RyR complex contains low-affinity Ca binding site(s) accessible from the luminal side of the channel. Dissociation of Ca from these sites during [Ca]SR decline leads to RyR deactivation and termination of CICR [27]. It has been suggested that the SR Ca buffering protein CASQ together with auxiliary proteins triadin 1 and junctin form the luminal Ca sensor of RyR [34]. Additionally, purified and recombinant RyRs remain sensitive to [Ca]SR to some extent [35] suggesting that the RyR protein itself contains additional luminal Ca sensor(s). Thus, the sensitivity of the RyR to [Ca]SR is multifaceted, involving several Ca binding sites from both sides of the channel.

Elementary SR Ca release events. (a) Averaged images of Ca sparks (top) and Ca blinks (bottom) recorded in permeabilized ventricular myocytes. Ca sparks were measured using the high-affinity Ca indicator Rhod-2 loaded into the cytosol. Ca blinks were measured with the low-affinity Ca indicator Fluo-5N entrapper within the SR. For details see [11, 12]. Ca spark and blink profiles were obtained by averaging fluorescence from the 0.8 μm wide regions marked by black boxes. (b) Cartoon showing the relationship between the SR network and local SR Ca release. Elementary SR Ca release events (spark and blink) arise from the simultaneous opening of clustered RyRs in a single SR Ca release site. Ca blink recovery depended mainly on intra-SR Ca diffusion rather than SR Ca uptake [12]. (c) Distribution of the [Ca]SR termination level of Ca sparks. The spark termination level was measured as [Ca]SR at the nadir of blink
Besides safety mechanisms to control inherently unstable CICR, efficient regulatory mechanisms exist to allow CICR substantial flexibility, so that the heart can adequately respond to changes in the metabolic demands of the body during exercise or stress. The strength of cardiac contraction can be adjusted by at least two different mechanisms—by changing sensitivity of the myofilaments to Ca or by changing cytosolic Ca transient amplitude. β-Adrenergic receptor (β-AR) stimulation produces the most important positive inotropic effect on the heart. This effect is mainly mediated via activation of protein kinase A (PKA) [1, 36] which subsequently phosphorylates several key Ca regulatory systems, such as a SERCA regulatory protein phospholamban (PLB), LTCC, and RyR. This results in an increase of Ca flux through LTCC, elevation of SR Ca load, and greater synchronization of SR Ca release during systole [37–40]. These modifications of the Ca transport systems play an important role in increasing Ca transient amplitude and thereby cardiac contraction. We have recently described a novel inotropic mechanism which involves alteration of the CICR termination process [41]. During β-AR stimulation with isoproterenol SR Ca release can be increased at individual release junctions by lowering the [Ca]SR termination level (Fig. 3). Further studies are required to determine which post-translation modifications of RyR are involved in this effect. β-AR stimulation is also associated with increased heart rate (positive chronotropic effect) which by itself can increase Ca transient amplitude [42]. Among the many factors that contribute to the positive force–frequency relationship (FFR) [1], Ca/calmodulin-dependent kinase type II (CaMKII) seems to play a particularly important role by phosphorylation of PLB [43] and RyR [44]. Thus, the increased SR Ca release during β-AR stimulation is likely mediated by activation of two major protein kinases, PKA and CaMKII [25, 45–47].

β-Adrenergic receptor (β-AR) stimulation increases SR Ca release amplitude by decreasing the [Ca]SR termination level. Confocal images of [Ca]SR recorded in control conditions and during β-AR stimulation with isoproterenol (ISO). [Ca]SR profiles were obtained by averaging fluorescence from an individual SR Ca release junction (denoted by red bars to left of images). [Ca]SR depletions were induced by action potentials. ISO decreased [Ca]SR at which individual SR Ca release junctions terminate
During diastole, the heart relaxes in preparation for refilling with circulating blood. For relaxation to occur, CICR must terminate allowing the Ca-ATPase (SERCA) to pump cytosolic Ca back into the SR and the Na–Ca exchanger (NCX) to extrude Ca from the cell. Other Ca transport systems such as the plasmalemmal Ca-ATPase (PMCA) and mitochondria play only a minor role in cardiac relaxation [48] (Fig. 1a). Among many proteins (including histidine-rich Ca binding proteins (HRC), calreticulin, S100A, sarcolipin) that regulate SERCA activity, the small transmembrane protein PLB is particularly important [49]. Unphosphorylated PLB inhibits SERCA activity, whereas phosphorylated PLB (by PKA and CaMKII) relieves SERCA inhibition allowing Ca pumping into the SR. PLB phosphorylation by PKA plays a major role in acceleration of SR Ca uptake and myocyte relaxation (lusitropic effect) during β-AR stimulation.
After termination of systolic Ca release RyRs do not enter an absolute refractory state. Instead, the decrease in luminal [Ca] reduces the sensitivity of the RyR to CICR and, therefore, to the cytosolic Ca trigger [50]. This mechanism has been suggested to play a protective role in the healthy heart by limiting extrasystolic after contractions and preventing the occurrence of Ca-dependent arrhythmias [51]. Therefore, RyRs are not completely closed during diastole and spontaneous openings of RyRs can generate a substantial SR Ca efflux commonly termed SR Ca leak [52, 53]. The SERCA-mediated Ca uptake and diastolic Ca leak together determine SR Ca load and, therefore, the amplitude of Ca transients that initiate contraction. Because the fractional SR Ca release steeply depends on SR Ca load [54–56], a small shift in this balance can change SR Ca load and, therefore, Ca transient amplitude. At normal physiological conditions, SR Ca leak may also serve as an important protective mechanism against SR Ca overload [25]. However, in certain pathological conditions associated with Ca overload, SR Ca leak becomes exacerbated. The Ca released during a spontaneous spark diffuses to neighboring release units, triggers CICR, and generates diastolic Ca waves [8, 57]. Spontaneous Ca waves can generate delayed afterdepolarizations (DADs), an effective trigger of cardiac arrhythmias [58]. After the discovery of Ca sparks, it was proposed that the entire diastolic SR Ca leak can be mediated by spontaneous Ca sparks [8, 59]. A Ca spark was considered a release event when a cluster of RyRs is activated in “all-or-nothing” mode because Ca release flux through spontaneous opening of RyR is enough to simultaneously activate all neighboring channels (“cluster bomb effect”). However, this perception has been recently changed. It has been shown that in ventricular myocytes a significant portion of SR Ca leak occurs as undetectable openings of single RyRs or spark-independent Ca leak [53, 60]. The spark-independent Ca leak is particularly significant at low [Ca]SR [53]. The spark-independent leak can arise from the same RyR clusters responsible for Ca spark generation because at low [Ca]SR the RyR openings are insufficient to recruit neighboring RyRs to form a spark [61]. In addition, openings of unclustered RyRs can also contribute to SR Ca leak [15].
Although some important steps of SR Ca regulation are not well understood, it becomes increasingly clear that heart function highly relies on synchronized SR Ca release with robust termination, well-balanced diastolic SR Ca leak, and effective SR Ca uptake. In the following chapter, we will present an overview of the structural and functional alterations of SR Ca transport systems in the failing heart. We will also discuss how dysfunction of different components of the SR Ca release machinery (mainly RyR and SERCA) causes abnormalities in SR Ca regulation in failing heart.
Alteration of SR Ca Homeostasis in HF
Heart failure is a complex clinical syndrome which can be characterized in general as a decreased ability of the heart to provide sufficient cardiac output to meet the energy demand of the body. In addition to decreased pump function, HF is also associated with an increased rate of sudden cardiac death due to ventricular tachyarrhythmias [62]. The pioneering studies of intracellular Ca dynamics in multicellular myocardial preparations from patients with HF exhibited significant alteration of Ca homeostasis [63, 64]. Single cell studies of cardiomyocytes isolated from human HF revealed elevated diastolic [Ca] and reduced Ca transients with a slower decay kinetic [65, 66]. Henceforth, abnormal Ca handling has been considered a hallmark feature of myocytes from failing hearts that leads to contractile dysfunction and arrhythmias.
To maintain proper Ca homeostasis during periodic electrical pacing, the amount of Ca that enters the cell via LTCC during each cycle must be extruded by NCX and Ca released during CICR must be pumped back into the SR by SERCA [67, 68]. In the healthy human heart, the main portion of cytosolic Ca (~70 %) that activates contraction is released from the SR, whereas the rest of Ca (~30 %) enters the cell via LTCC [1]. During development of HF, however, this balance gradually changes and cardiomyocytes become more dependent on Ca influx from the extracellular space [1]. Although factors that cause this shift in the balance may vary (myocardial infarction, energetic stress, changes in neurohumoral tone and pacing rate), decreased ability of the SR to retain Ca seems to be a more consistent finding that can explain depressed contraction of the failing heart. This SR Ca mishandling occurs as a result of an alteration in expression and/or activity of several Ca transporters, including SERCA and RyR. Furthermore, the dependence of myocytes on the trans-sarcolemmal Ca fluxes has another important implication for HF pathology—increased propensity to ventricular arrhythmias. Compelling experimental evidence indicates that ventricular arrhythmias can be triggered by spontaneous SR Ca release events during diastole [58]. It has been suggested that in myocytes from failing hearts excessive SR Ca leak (particularly in the form of Ca waves) increases diastolic Ca extrusion via the electrogenic NCX causing the generation of DADs [69, 70].
Most studies showed that systolic SR Ca release is significantly depressed in HF. From the perspective of SR Ca regulation, Ca transient amplitude can be decreased in several ways (Fig. 4): (1) by diminishing SERCA pump function; (2) by increasing RyR-mediated Ca leak; (3) by decreasing intra-SR Ca buffer capacity; and (4) by decreasing the fidelity of the LTCC–RyR coupling resulting in asynchronous activation of SR Ca release units. Results from different HF models revealed that, with exception of the SR Ca buffer capacity, all these aforementioned mechanisms can contribute to abnormal Ca regulation. The total level of CASQ did not significantly change in HF [11, 24, 71]. However, abnormal distribution of CASQ within the endoplasmic reticulum (ER) of the failing heart has been reported previously [72]. Regarding the other explanations it is ambiguous as to which holds the most central role. Some studies indicate that downregulation of SERCA plays the essential role in depressing SR Ca release in HF [73–77], while other studies explain these abnormalities by increased activity of the RyR [11, 17, 70, 78]. There are also conflicting results about molecular mechanisms that lead to RyR overactivity in HF. Phosphorylation of RyR either by PKA [78] or by CaMKII [79] has been suggested to be critical in increasing the channel’s activity in HF. Furthermore, redox modification of the RyR is also an important factor during progression of HF [80, 81]. Other studies showed that HF is associated with remodeling of the T-tubular system leading to functional uncoupling of plasmalemmal LTCCs and RyRs on junctional SR [82–86]. Accordingly, the alteration of cell architecture has been suggested to play a critical role in the abnormality of SR Ca release in failing myocytes [84, 87]. The relative contributions of these different mechanisms may vary in different models of HF and with different degrees of severity of failure. Also, differences in Ca regulation between different species have to be taken into account [1].

Alteration of SR Ca regulation in failing heart. (a) In the healthy heart, SR Ca load is maintained by SERCA-mediated Ca uptake which effectively counterbalances a small RyR-mediated Ca leak. In failing heart, Ca transient amplitude can be decreased; (b) by diminishing SERCA pump function; (c) by increasing RyR-mediated Ca leak; (d) by decreasing intra-SR Ca buffer capacity; and (e) by decreasing the fidelity of the LTCC–RyR coupling resulting in asynchronous activation of SR Ca release units
Structure and Function of the RyR in HF
Properties of cardiac RyR (type 2) have been documented in several reviews [22, 88–90]. The RyR complex (~2,300 kDa) has an oligomeric structure formed by four identical subunits. The channel exhibits high conductance and low selectivity for Ca. The RyR is not only a Ca release channel but also a giant scaffolding protein on which several regulatory proteins and enzymes are concentrated [78]. On the cytosolic side, RyR interacts with calmodulin (CaM), FK-506 binding proteins (FKBP), sorcin, and Homer-1. It has been shown that FKBP12.6 can affect RyR by stabilizing the interaction between the channel subunits [91], whereas CaM and sorcin affect the channel activity via Ca-dependent mechanisms [92, 93]. The RyR also forms complex with two major protein kinases (PKA and CaMKII) and three phosphatases (PP1 and PP2A and PP2B) indicating the importance of RyR regulation by phosphorylation [88, 94]. The SR-membrane proteins, junctin and triadin, are believed to be crucial for the RyR’s ability to sense changes in luminal [Ca] via interactions with the major SR Ca-chelating protein CASQ [34] and HRC [95]. In addition, RyR has multiple sites for regulation by ions and small molecules, including Ca, Mg, and ATP. With so many associated proteins and regulators, a proper arrangement and stoichiometry of the RyR complex is critical for normal SR Ca regulation.
Structural Alteration of the RyR in HF
RyR expression. Numerous publications demonstrated a reduction of RyR expression on mRNA and protein level in human and animal HF preparations [11, 79, 96–101]. It has been shown that HF is associated with decreased binding of ryanodine to the receptor [102, 103] suggesting either reduction in the RyR expression level, open probability, or structural changes in the region of the protein responsible for ryanodine binding. Decrease in RyR expression level may be involved in the development of contractile dysfunction of the failing heart. The reduction in RyR level by itself would decrease SR Ca release. However, this effect can be partially compensated by increased RyR activity as it was shown by several studies (see below). Conversely, there are also several publications that did not find any significant difference in the RyR level between normal and HF [75, 87, 104].
Sub-domain and domain–domain interactions. Emerging evidence demonstrates significant rearrangements within the RyR macromolecular complex in HF [94]. Defective interactions between regulatory domains within the RyR or dissociation of key components from the RyR complex have been suggested to play a critical role in dysfunction of the channel contributing to abnormal SR Ca regulation. FKBP12.6 is one such component believed to be necessary for regulation of RyR at many levels. It has been proposed that FKBP12.6 binds to the cytosolic domain of RyR to stabilize the interaction among RyR subunits. This interaction functionally translates into coupled gating, meaning that all subunits of the RyR open and close simultaneously [96, 105]. FKBP12.6 may also be involved in physical and functional coupling between neighboring tetramers [91, 105]. It has been shown that during HF, chronic RyR hyperphosphorylation by PKA (Ser-2808) causes dissociation of FKBP12.6 from the channel. This alteration in RyR structure leads to increased SR Ca leak [99] as a result of prolonged subconductance openings of the channel [78]. A similar alteration in the RyR complex structure (but not in function) was reported by another group [79]. Furthermore, it has been suggested that FKBP12.6 is also responsible for stabilizing intra-domain interactions within the RyR subunit. In healthy hearts N-terminal and central domains of the RyR interact [106]. This zipping between domains keeps the RyR in closed state and prevents diastolic SR Ca leak. During HF, dissociation of FKBP12.6 from the channel (due to PKA phosphorylation or oxidative stress) causes domain unzipping and increases SR Ca leak [107, 108]. However, despite many years of research, there is still controversy about the functional role of FKBP12.6 and RyR phosphorylation. The role of FKBP12.6 in augmentation of SR Ca leak has been challenged by several different laboratories. It has been shown that level of FKBP12.6 associated with the RyR did not significantly change during RyR phosphorylation by PKA [109, 110] or during HF [75].
RyR phosphorylation. The processes of phosphorylation and dephosphorylation are posttranslational mechanisms utilized to rapidly change the function of many proteins including ion channels. Rapid and robust control of phosphorylation state of the SR Ca release channel is ensured by kinases PKA and CaMKII, phosphatases PP1, PP2A, and PP2B, and phosphodiesterase 4D3, all of which are bound to the RyR [88, 94, 111, 112]. In general, increased RyR phosphorylation in failing hearts can be explained either by increased localized activity of kinases or by diminished activity of phosphatases. In many animal HF models and also human HF, activity and expression of CaMKII are increased [79, 113]. However, this does not always translate into increased levels of CaMKII in the complex with the RyR [78, 114]. On the other hand, several independent groups have suggested that an increased level of RyR phosphorylation in HF can be explained by decreased levels of phosphatases PP1 and PP2A [78, 79, 114] and phosphodiesterase 4D3 [111] associated with the RyR.
It has been demonstrated that the RyR can be phosphorylated by serine–threonine kinases at multiple sites [115]. Thus far only three sites on cardiac RyR were suggested to be of functional relevance, namely PKA-specific sites Ser-2808 and Ser-2030 [78, 116, 117], and CaMKII-specific site Ser-2814 [118]. Functional consequences of RyR phosphorylation have been recently summarized in several reviews [119, 120]. Despite a major collective effort, the role of PKA-dependent phosphorylation in modulation of RyR function in normal and failing hearts remains a subject of heated debate [121, 122]. Studies from different laboratories have yielded conflicting results regarding the role of RyR phosphorylation at Ser-2808 in the progression of HF, which ranged from having an essential role [78, 107, 123, 124] to having only a very limited function [114, 125, 126]. On the contrary, increased RyR activity due to enhanced phosphorylation of RyR at CaMKII site Ser-2814 appears to be a more consistent finding. Hyperphosphorylation of RyR at the CaMKII site has been demonstrated in numerous (but not all [44]) HF models, and CaMKII inhibition was shown to attenuate abnormalities in Ca handling in myocytes from failing hearts [70, 79, 113, 127, 128].
RyR redox modifications. Alteration of RyR phosphorylation status is not the only type of posttranslational modification that could affect RyR function during development of HF. Changes in cellular metabolism that occur during the progression of HF can lead to depletion of the antioxidant defense and to an increase in reactive oxygen species (ROS) production [129]. Oxidation of the RyR by ROS can enhance the channel activity [130]. Accordingly, modulation of RyR activity by ROS in HF has been suggested as a major cause of the observed abnormality in Ca regulation [80, 81]. Cardiac RyR contains 89 reactive cysteine residues [131] and the number of free thiols has been shown to be dramatically decreased in HF. Incubation of myocytes isolated from canine failing hearts with ROS scavengers and antioxidants was able, to a large degree, to restore Ca transient amplitude and reduce RyR-mediated SR Ca leak [70]. ROS scavengers also normalized RyR sensitivity to luminal Ca in lipid bilayer experiments [81]. It has also been suggested that normalization of redox status of RyRs from failing hearts reverses inter-domain unzipping, thereby stabilizing RyR function [80]. The major sources of ROS in ventricular myocytes include NADPH oxidase (NOX), the mitochondria electron transport chain, xanthine oxidase/reductase (XOR), and uncoupled nitric oxide synthase (NOS). NOS1 and XOR were demonstrated to co-immunoprecipitate with the RyR, and their levels in HF are generally increased (for review, see [132, 133]). Activation of NOX2 residing in T-tubules was shown to increase Ca spark activity [134], while attenuation of ROS production by mitochondria stabilized SR Ca release during digitalis treatment [135] or β-AR stimulation [136].
Notably, no clinical studies have demonstrated beneficial effects of long-term treatment with antioxidants (for review see [129]), as well as treatment with a XOR inhibitor allopurinol [132, 133]. The failure of clinical trials can be a result of low scavenging efficacy of the antioxidant, but not the antioxidant therapy per se. Because ROS have very limited diffusion distance, they predominantly affect closely opposed targets. Therefore, it seems that a more beneficial strategy would be to design antioxidants that can prevent ROS production locally.
RyR luminal Ca sensor. Abnormally high activity of the RyR in HF has been attributed to increased sensitivity to luminal Ca [11]. This defect can be caused by chronic dissociation of several auxiliary proteins (junctin, triadin, and CASQ) that form the luminal Ca sensor of the channel [34, 137]. Although the total level of CASQ does not change in HF [11, 24, 71], it has been suggested that abnormal posttranslational processing of CASQ can cause its defective trafficking [72]. This can potentially lead to a local depletion of CASQ in the junctional SR. Additionally, it has been demonstrated that the level of another SR Ca buffer (HRC) that binds to the RyR complex is significantly lower in failing myocytes from human and animal models [138]. Finally, the levels of triadin and junctin were reported as being dramatically reduced in human end stage HF [139]. Future studies are needed to assess whether restoration of the levels of proteins that confer luminal Ca sensitivity to RyR in failing heart will lead to normalization of SR Ca release channel function.
Calmodulin. It has been reported that failing hearts have a reduced amount of CaM associated with the RyR complex [79, 140]. A molecular mechanism of CaM dissociation may be a defective interaction between N-terminal and central domains within the RyR [140]. However, it is not clear yet how CaM dissociation from the RyR affects SR Ca release. Several studies showed that CaM decreases single RyR channel activity and Ca spark frequency [92, 141], whereas other shows that CaM can increase the channel and spark activity [142]. Such discrepancies can be explained by the dual role of CaM: (1) it can serve as a stabilizer of RyR via direct binding to the channel and (2) it can be involved in regulation of phosphorylation–dephosphorylation balance of the RyR as an essential component for activation of CaMKII and phosphatase PP2B (also known as calcineurin or PP3).
Regardless of the molecular mechanisms, it is clear that during HF many important regulatory proteins detach from the RyR complex, and the channel simultaneously becomes more phosphorylated and oxidized (Fig. 5). These modifications of the RyR can cause SR Ca mishandling, contractile dysfunction, and arrhythmias.

Alteration of the RyR complex structure in failing heart. On the cytosolic side, RyR interacts with at least calmodulin (CaM), FKBP, nitric oxide synthase (NOS), two major protein kinases (PKA and CaMKII), and two phosphatases (PP1 and PP2A). Moreover, triadin and junction (Tr/Jn) form the luminal Ca sensor via interactions with the SR Ca-chelating protein calsequestrin (CASQ). RyR also contains ~100 reactive cysteine residues (SH–). During heart failure, CaM and phosphatases (PP1 and PP2A) dissociate from the RyR complex. The channel becomes more phosphorylated at the CaMKII and the PKA sites. Cysteine residues become oxidized forming the disulfide bonds. Dissociation of FKBP12.6 from the channel causes N-terminal and central domain unzipping. CaMKII and PKA become more active and NOS uncoupled
Functional Alteration of the RyR in HF
SR Ca release activation. Global SR Ca release is a summation of thousands of Ca sparks activated during an AP. Thus, a greater Ca transient can be achieved by more synchronized recruitment of individual SR Ca release units [39]. The degree of synchronization of SR Ca release depends on the strength of the trigger and the efficiency of coupling between the LTCC and RyR cluster. It has been suggested that decreased SR Ca release in HF is a result of decreased fidelity of LTCC–RyR coupling, leading to less organized and less synchronized SR Ca release [87]. HF myocytes from a myocardial infarction model showed diminished Ca transients, which are associated with slow rising and decay phases of the transient. It appears that on the subcellular level some RyR clusters have a delayed response to the stimulus. This desynchronization of SR Ca release in HF is due to downregulation of LTCC current [82, 143]. A similar effect can be achieved in healthy myocytes by partially inhibiting LTCC current [144]. However, in failing myocytes from spontaneously hypertensive rats, desynchronization of SR Ca release was a result of remodeling of the T-tubule system rather than a reduction in LTCC current [84, 87]. It appears that the reorganization of the T-tubular system leads to a substantial mismatch of the T-tubules with the junctional SR. Such spatial dissociation of triggering LTCCs and RyRs has been attributed to downregulation of junctophilins [145], proteins that tether T-tubules to the junctional SR [5]. It seems that this leaves some RyR clusters without local control by LTCC. At these “orphaned” clusters, Ca release is delayed because their activation depends on Ca diffusion from intact SR Ca release units instead of LTCC. Reports that the T-tubule system is significantly disorganized in cardiomyocytes from human [83, 85, 86] and animal HF models provide compelling evidence for this theory [82, 85, 104, 145]. However, other studies found that the T-tubule system remains relatively intact in HF [146].
Although the vast majority of SR Ca release during ECC is mediated by LTCC, it has also been proposed that Ca influx via NCX can trigger CICR [147]. It has been suggested that Ca entry via LTCC and NCX interacts synergistically to trigger greater SR Ca release during β-AR stimulation and PKA activation [148]. In HF induced either by chronic administration of β-adrenergic agonist isoproterenol or by volume overload, this mechanism has been found to be significantly compromised. This alteration in Ca regulation could account for the reduced SR Ca release and low response to β-AR stimulation in HF [148].
ECC gain. ECC gain quantifies the amplification strength of CICR. It is often calculated as SR Ca release divided by the amplitude of LTCC current [149]. Due to sensitivity of the RyR to luminal [Ca], ECC gain steeply depends on SR Ca load [55]. Thus, a small decrease in [Ca]SR would lead to significant decrease in Ca transient amplitude. Indeed, several studies of different HF models showed that the decrease in Ca transient amplitude was mainly due to depletion of the SR Ca load, but not to the LTCC current density [73–77]. Thus, it has been concluded that diminished SR Ca uptake, but not ECC gain, plays a crucial role in suppression of systolic SR Ca release in HF. However, experiments on HF myocytes from hypertensive rats showed that Ca transient amplitude was significantly reduced as a result of a decrease of ECC gain, not of SR Ca load [87]. The decreased ECC gain in this HF model was explained by inefficient coupling between LTCCs and RyRs, presumably due to loss of T-tubules. Additionally, a study that analyzed the changes of Ca handling during progression of HF found a significant increase in ECC gain at the early stage of HF, but diminished at advanced stage [70].
Fractional SR Ca release and Ca release termination. Recent studies suggest that depleted SR Ca load in HF would reduce Ca transient even more dramatically if it were not compensated for by an increase in RyR activity. Local depletion of [Ca]SR causes a decrease in the RyR open probability, and this mechanism is believed to be responsible for termination of Ca sparks and for refractoriness of SR Ca release [27,28]. As a result of the remodeling processes that occur in HF, the channel becomes more sensitive to [Ca]SR [11, 81]. It has been shown that under conditions of matched SR Ca load, the fraction of total [Ca]SR released to the cytosol (or fractional SR Ca release) was significantly larger in HF when compared to normal hearts [24, 41, 150]. This compensatory mechanism partially prevents reduction in Ca transient amplitude at a lower SR Ca load. Figure 6 shows examples of cytosolic Ca transients (red) and corresponding [Ca]SR depletions (green) recorded under different experimental conditions. Partial inhibition of SERCA in non-failing myocytes led to significant decrease of the Ca transient amplitude and fractional SR Ca release as a result of depletion of SR Ca load (Fig. 6, second images). Although SR Ca load was similarly decreased in myocytes from HF, the fractional SR Ca release and, consequently, Ca transient amplitude was significantly larger (Fig. 6, far right images). Similar to RyR activation with caffeine (Fig. 6, third images), the modifications of the RyR that occur in HF increase SR Ca release by decreasing the [Ca]SR level at which release terminates. It appears that during the early stages of HF, sensitization of the RyR allows myocytes to maintain Ca transients of nearly normal amplitude, despite the diminished SR Ca load [41, 70]. However as a drawback, these RyR modifications increase diastolic SR Ca leak (largely due to lowered SR [Ca] threshold for Ca spark activation and termination) and increase the propensity of cardiac arrhythmias [53, 107, 150–152]. At more advanced stages of HF, severe dysfunctions of the RyR cause a significant reduction of the Ca transient as a result of a massive loss of SR Ca content [70].

Cytosolic Ca transients, SR Ca load, and SR Ca release termination. From left to right: Cytosolic Ca transients induced by action potentials (top) and corresponding global [Ca]SR depletions (bottom) recorded in a non-failing myocyte at control conditions; in a non-failing myocyte after partial SERCA inhibition with thapsigargin (0.5 μM); in a non-failing myocyte after activation of the RyR with caffeine (200 μM); and in a myocyte from a failing heart. Cytosolic Ca transients were measured using the high-affinity Ca indicator Rhod-2 loaded into the cytosol. Global [Ca]SR depletions were measured with the low-affinity Ca indicator Fluo-5N loaded into the SR
SR Ca release refractoriness and RyR-mediated SR Ca leak. Abnormally high activity of RyRs due to increased sensitivity to luminal Ca has also been linked to shortened refractoriness of SR Ca release during diastole. Premature reopening of RyRs slows relaxation, exacerbates reduction of SR Ca content, and promotes generation of spontaneous Ca sparks which can ignite arrhythmogenic Ca waves [51, 70] contributing to diastolic and systolic dysfunction of HF. A growing body of evidence indicates that RyR-mediated Ca leak is significantly increased in the failing heart [53, 70, 78, 79, 81, 99, 107, 150, 152]. Augmentation of SR Ca leak is the one of the earliest alterations of Ca handling during the progression of HF that precedes changes in the amplitude of the Ca transient [70]. Increased RyR-mediated Ca leak can contribute to a reduction of SR Ca content and trigger Ca-dependent arrhythmias. This also implies that during diastole SERCA cannot achieve its maximal thermodynamic efficiency and more ATP must be consumed to balance the increased SR Ca leak. Thus, SR Ca leak is energetically costly, particularly in the metabolically compromised failing heart.
During diastole, Ca sparks serve as the main pathway for SR Ca leak [8, 53, 59]. Studies of Ca sparks in HF revealed varied results, ranging from decreased [41, 153] to increased spark frequency [11, 70, 85, 113]. Such diversity can arise from species- and model-specific differences in Ca regulation, and also from differences in experimental conditions. For example, a study of myocytes from human HF showed significantly lower Ca spark frequency compared to non-failing cells [153]. When SR Ca load was elevated in HF cells, spark frequency was increased to a level higher than control myocytes, suggesting that RyR activity is increased in human HF. Similar results were obtained in a study of rabbit HF induced by combined aortic insufficiency and stenosis [41]. In a rabbit pacing-induced HF model Ca spark frequency was similar to non-failing hearts, but the amplitude was significantly decreased [101]. Due to the observed decrease in RyR and SERCA expression levels, the authors concluded that alteration of spark properties was a result of combined defects in SR Ca release and uptake. In a study of a canine rapid pacing-induced HF model, Ca spark frequency was significantly higher in failing myocytes, despite a decreased SR Ca load [11]. These seemingly contradicting results were explained by increased sensitivity of the RyR to luminal [Ca] which was directly confirmed by single RyR measurements. It appears that increased RyR sensitivity to luminal [Ca] during HF contributes not only to the augmentation of spark frequency but also to the augmentation of non-spark-mediated Ca leak.
It has been shown recently that in ventricular myocytes a significant portion of SR Ca leak can occur as undetectable openings of the RyR, referred to as the spark-independent SR Ca leak [53]. This component of Ca leak becomes particularly predominant at low SR Ca loads. Because failing myocytes are characterized by depleted SR Ca content, this would imply that a significant portion of SR Ca leak in HF is mediated by spark-independent pathways. The specific role of increased non-spark-mediated Ca leak in HF is yet to be elucidated.
Other Pathways for Ca Release from the SR
Although RyR is the major Ca release channel on the SR of adult ventricular myocytes, contributions from other Ca pathways need to be considered. The existence of RyR-independent Ca leak in ventricular myocytes has been suggested before [53, 154], however the molecular mechanisms responsible for this Ca leak have not yet been identified.
IP 3 receptors. A potential candidate in this context is the inositol-1,4,5-trisphosphate receptor (IP3R) SR Ca release channel. There are three IP3R isoforms referred to as type 1, 2, and 3, encoded by three distinct genes. Type 2 is the primary isoform of the IP3R expressed in ventricular myocytes [155]. Cardiac IP3R is more sensitive to IP3 and almost insensitive to [Ca]i over the physiological range of [Ca]i [156] suggesting that the channel acts purely as a IP3 sensor. Although expressed at lower densities compared to RyRs, IP3R-dependent Ca release can facilitate CICR and even trigger arrhythmogenic Ca waves in cardiomyocytes [157, 158]. It has been shown that IP3Rs are upregulated during HF [79, 98]. Recently we have shown that activation of IP3Rs in normal myocytes nearly doubled RyR-independent Ca leak [53]. These results suggest that activation of the IP3R can contribute to the increased SR Ca leak during HF, particularly under conditions associated with elevated neurohumoral tone (e.g., endothelin-1) and stimulation of the phospholipase C-IP3 signaling pathway. Furthermore, it has been proposed that stimulation of IP3R-mediated Ca leak can regulate gene transcription during development of hypertrophy and HF [159]. Clearly more studies are needed to determine the pathological role of the IP3R in HF.
Phospholamban (PLB). PLB is a small transmembrane protein that regulates SERCA activity [49]. PLB exists in dynamic equilibrium between three different forms: in complex with SERCA, as a single molecule, and as a pentamer. Phosphorylation of PLB by PKA (Ser-16) shifts this balance toward formation of PLB pentamers [160]. It has been shown that PLB pentamers can function as Ca channels in lipid bilayers [161] and thereby can contribute to SR Ca leak. Thus, changes in PLB phosphorylation level that occur during HF [71, 79, 162, 163] can potentially affect diastolic SR Ca leak. However, Ca leak measured from SR vesicles isolated from wild-type and PLB-knockout mice was not significantly different [164]. Some studies have even reported that the PLB pentamer does not possess Ca channel activity [165]. Thus there is yet to be any conclusive evidence on this theory.
Other pathways. Cardiac SR, besides being the major organelle to store Ca, is responsible for other primary functions of endoplasmic reticulum as for any cell type (such as protein folding). Therefore the same may be said for other Ca leak pathways which exist in non-muscle cells. These pathways may include ATPase back-flux, Ca leak mediated by a small apoptosis-related protein BCL2, and the protein complex associated with the translocation of nascent polypeptides across membranes called translocon [166]. In addition, the transient receptor canonical type 1 (TRPC1) involved in capacitative Ca entry via sarcolemma was recently found to reside on the SR membrane of skeletal muscle and overexpression of this Ca channel led to accelerated loss of Ca from the SR [167]. Despite the potential importance of the aforementioned pathways in health and disease their role in cardiac Ca homeostasis has not been studied yet.
Structure and Function of SERCA in HF
Relaxation of cardiac muscle relies on removal of Ca from the cytosol. One mechanism by which this is achieved is through re-sequestration of Ca into the SR by SERCA. By hydrolyzing one ATP molecule, SERCA can transport two Ca ions from the cytosol into the SR lumen. SERCA activity also plays a critical role in setting of diastolic [Ca]i and SR Ca load. A specific SERCA isoform, SERCA2a, is one of the most abundant proteins of the cardiac SR. This Ca pump contains ten transmembrane domains (M1–M10) and several cytoplasmic domains, including a nucleotide binding (N) domain, an actuator (A) domain, and a phosphorylation (P) domain [168]. SERCA is freely mobile in the SR membrane [169], suggesting that the pump can be equally distributed throughout the SR network. Although SERCA interacts with a wide array of proteins (including HRC, PP1, calreticulin, S100A, and sarcolipin), PLB is the most important regulator of the pump activity [49]. Under the basal (unphosphorylated) state, PLB inhibits SERCA activity by lowering the apparent affinity of the pump for Ca. Phosphorylation of PLB at Ser-16 by PKA [170] largely relieves this inhibition, increasing the pumping rate by several fold. Stimulation of SR Ca uptake by PLB phosphorylation appears to be the major mechanism of acceleration of relaxation (lusitropic effect) during β-AR stimulation. SERCA activity can also be increased via phosphorylation of PLB by CaMKII at Thr-17 [170]. This mechanism has been suggested to be responsible for a stimulation frequency-dependent acceleration of SR Ca uptake. In addition to protein interactions, SERCA is highly sensitive to changes in the metabolic status of the cytosol, including the ATP/ADP ratio, pH, and the redox potential [1, 171].
Structural Alteration of the SERCA/PLB Complex in HF
SERCA and PLB expression. It is still a matter of debate whether the decreased SR Ca uptake in HF is due to a decrease in SERCA expression level or in SERCA activity. Downregulation of SERCA2a at the mRNA and protein levels has been shown in numerous studies of human and animal HF models [74, 75, 97, 101, 163, 172–176]. All these studies highlight the critical role of SERCA in abnormal Ca homeostasis and contractile dysfunction of the failing heart. For example, it has been shown that the transition from hypertrophy (induced by aortic banding in rats) to HF is associated with significant decrease in the SERCA mRNA level [172]. It has also been shown that increasing the level of SERCA protein via adenoviral infection can improve the function of hypertrophied hearts and can delay its transition to HF [177]. However, there are a significant number of publications that did not find any alteration in the SERCA expression level in human HF [162, 178–180] and animal HF models [11, 181].
It has been shown that HF is also associated with a decrease in the PLB expression level [74, 75, 97, 101], but to a lesser degree than SERCA [175, 176, 182]. It has been suggested that the decreased SERCA–PLB ratio can lead to inhibition of SR Ca uptake in HF, because an increased SERCA fraction would be inhibited by PLB binding. On the contrary, the level of PLB remains unchanged in HF models characterized by an unchanged SERCA level [11, 79, 162, 178, 179].
Posttranslational modifications of SERCA and PLB. In addition, the diminished SR Ca uptake in HF can be a result of posttranslational modifications of the SERCA/PLB complex. It has been shown that the responsiveness of SERCA to activation either by cAMP or by PKA was significantly decreased in human failing heart compared to non-failing myocardium [162]. Furthermore, the PLB phosphorylation level at the PKA-specific site was significantly decreased in HF. Another group showed that SERCA activity and its Ca sensitivity were significantly decreased in HF compared to normal heart [71, 180]. These changes in the pump’s activity were associated with decreased PLB phosphorylation at both the PKA and the CaMKII sites; however, the total level of SERCA and PLB remained unchanged [71, 180]. A decrease in PLB phosphorylation level can result from increased PP1 activity, observed in failing hearts [183]. In contrast, another study showed that the total level of PLB phosphorylation was higher in HF preparations [163]. Decreased SR Ca uptake in failing heart was explained by downregulation of the SERCA expression level. Another study showed that PLB phosphorylation level was increased at the CaMKII site but was decreased at the PKA site [79]. The authors suggested that the overall PLB phosphorylation level in HF would have only a minor effect on SERCA activity.
It has been recently established that several naturally occurring mutations of PLB can give rise to human dilated cardiomyopathy. For example, the missense mutation of Arg-9 to Cys (R9C) impairs SERCA regulation by preventing PKA-mediated phosphorylation of PLB [184]. Moreover, this mutation may also increase the sensitivity of PLB to oxidation, mediating disulfide bond formation between adjacent proteins in the PLB pentamer [185]. Such cross-linking prevents dissociation of PLB into the active monomers, and may increase association of protein into nonphosphorylatable aggregates. Thus, the R9C mutation may cause impairment of Ca handling leading to development of HF.
There is only a small amount of information regarding posttranslational modifications of the SERCA protein that affect Ca handling in HF. It has been reported that the contractile dysfunction in Gαq-induced cardiomyopathy was mediated by inhibition of SERCA activity as a result of oxidation of cysteine residue(s) on the pump [186]. Another study has shown that the depressed SR Ca uptake of failing myocytes is associated with dissociation of the small ubiquitin-related modifier 1 (SUMO1) from SERCA [187]. It has been suggested that restoring the sumoylation level of SERCA can stabilize the pump structure and improve SR Ca uptake in mouse and human failing hearts. However, these results are controversial, since a protein of the molecular weight corresponding to the sumoylated SERCA has never been detected in many previous immunoblotting studies of normal and failed human hearts [188].
Alteration of SERCA Activity in HF
In human and different animal HF models, evidence from SR Ca uptake studies indicates there is a reduction of SERCA activity [71, 75, 163, 178, 189, 190]. In many cases (but not all), this effect was linked to the downregulation of SERCA at the protein and mRNA levels. Furthermore, numerous studies of failing cardiomyocytes showed that the decrease in Ca transient amplitude was associated with a depletion of the SR Ca load [73–77]. Based on these results, it has been suggested that abnormal SR Ca uptake (rather than SR Ca release and Ca current) is one of the primary causes of depressed SR Ca release and contraction in failing heart. Decreased SR Ca uptake can also contribute to an increase in diastolic [Ca]i. Thus, reduction in SERCA activity can cause the impairment of both systolic and diastolic function in the failing myocardium. Studies by another group showed that the decrease in SR Ca load was a result of a significant increase in NCX activity and only a modest decrease in SERCA function [181, 191]. It has been proposed that upregulation of NCX together with downregulation of SERCA plays a key role in alteration of Ca regulation that leads to depletion of SR Ca load in the failing heart [182, 192]. Notably, several recent publications showed no significant alteration in SERCA-mediated Ca uptake during ECC in intact failing myocytes as well as during rest in permeabilized cells [70, 152]. To explain a similar HF phenotype, the authors reported significant augmentation of RyR-mediated Ca leak which causes depletion in SR Ca load.
A normal heart responds to increased stimulation frequency by generating larger force (positive FFR or the Bowditch effect). This mechanism allows increased cardiac output, thereby, providing a sufficient energy supply to the body during stresses. It has been shown that at low stimulation frequencies the failing myocardium has similar contractile characteristics as the non-failing heart. However, increasing the pacing rate causes contractility to decrease in HF [63]. The blunted or negative FFR is one of the prominent characteristics of failing hearts [193]. It has been suggested that the negative force–frequency response of the failing heart stems from an inability of the SR to increase Ca load at increased stimulation frequencies. This effect was explained by a decrease in SERCA activity and an enhancement of NCX extrusion of Ca [189, 190, 194]. Furthermore, the same mechanism was attributed to a decreased response to inotropic agents of human failing heart at the end stage [195]. Alternatively, it has been proposed that defective regulation of the RyR by CaMKII plays a critical role in the blunted FFR of the failing heart [44].
SERCA is regulated thermodynamically (i.e., by ΔG ATP) as well as kinetically (i.e., via changes in ATP hydrolysis products) (for review see [1, 171]). The dependence of cardiac SR Ca uptake on energy supply has been demonstrated in different experimental conditions [196–198]. Furthermore, SERCA activity also depends on the removal of ATP hydrolysis products, ADP and P i [199]. Since SR Ca uptake is highly dependent on the effective energy supply, SERCA activity can be reduced as a result of metabolic imbalance that commonly occurs during HF [200]. It has been demonstrated that a glycolytic metabolite pyruvate is able to improve SERCA activity and the contractile performance of isolated failing human myocardium [201]. It has also been proposed that an increase in phosphorylation potential (i.e., the ratio of [ATP] to [ADP] × [P i]) would improve the efficiency of ATP-dependent processes, especially those participating in the ECC cycle, such as the acto-myosin ATPase and the SR Ca-ATPase.
Normalization of SERCA and RyR Function as a Treatment Strategy in HF
Abnormalities in SR Ca regulation play a critical role in contractile dysfunction and arrhythmogenesis of the failing heart, making the RyR and SERCA clinically relevant sites for potential therapeutic interventions. Normalization of SR Ca regulation in HF can be achieved in two conceptually different ways—either by improving SR Ca uptake or by preventing loss of SR Ca during diastole.
An increase in SR Ca uptake can be achieved by overexpression of the SERCA pump, knock-out of PLB, or overexpression of phospho-mimetic PLB. These approaches have been demonstrated to improve contractile performance and delay progression of HF in several animal HF models (for review see [202]). It seems, however, that these approaches would be less beneficial for conditions of HF that are associated with increased RyR sensitivity to luminal [Ca] [11, 70, 150]. An attempt to increase SR Ca load in these HF models would also enhance SR Ca leak, thus increasing the likelihood of life-threatening arrhythmias. Moreover, increasing SERCA activity without reducing SR Ca leak would further increase energy consumption in already energy compromised tissue. In support of this reasoning, it has been shown that the loss-of-function PLB mutation has been associated with the development of cardiomyopathy in humans [203]. Interestingly, however, restoration of SERCA expression level in HF can stabilize SR Ca load and prevent arrhythmias by decreasing SR Ca leak and normalizing RyR phosphorylation [204]. It is possible that in this experimental setting the beneficial effect of SERCA overexpression primarily stems from increased [Ca]i buffering, as in the case of parvalbumin overexpression [205], and not due to increased SR Ca uptake.
The therapeutic approaches that target RyR-mediated Ca leak are considered to be another promising strategy to improve Ca homeostasis in the failing heart. Initially, it seems counterintuitive since systolic Ca release is already reduced in HF and inhibition of RyR may worsen systolic dysfunction. In practice, however, moderate inhibition of RyR activity in HF was proven to restore Ca cycling to a substantial degree. Partial RyR inhibition efficiently prevents diastolic loss of SR Ca, thus increasing SR Ca content. Subsequently, increased [Ca]SR helps to override RyR block during systole resulting in nearly normal Ca transients. Several groups showed potential benefit of using dantrolene, an anti-malignant hyperthermia drug, for normalization of cardiac RyR function in failing hearts. It has no effect on cardiac RyR from normal hearts. However, in HF it has been demonstrated to prevent arrhythmogenic Ca release and improve FFR and β-AR responsiveness [206, 207]. The proposed mechanism of these beneficial effects is that dantrolene stabilizes aberrant inter-domain interactions within the RyR from failing hearts, similarly to another pharmacological agent, a 1,4-benzothiazepine derivative JTV519 (K201) [208, 209]. It has been suggested that one of the beneficial effects of beta-blockers is to reduce phosphorylation of the RyR by PKA, restore FKBP12.6 binding, and normalize SR Ca leak [78, 99]. More recently, inhibition of CaMKII and restoration of the RyR redox status have emerged as promising strategies to normalize RyR function in diseased hearts [79–81, 113, 127]. The apparent disadvantage of these approaches is that available pharmacological agents cannot reduce phosphorylation or oxidation of the RyR specifically, without affecting other cellular systems and producing adverse effects.
Conclusion. Heart failure remains an incurable disease with the highest mortality rate. Over the last 2 decades, significant progress has been made in the understanding of mechanisms of SR Ca mishandling that leads to contractile dysfunction and arrhythmogenesis of the failing heart. It has become increasingly clear that functional impairment of two major Ca regulatory systems, the RyR and the SERCA/PLB complexes, contributes in great degree to aberrant SR Ca homeostasis. However, the previous studies indicate that the role of RyR or SERCA malfunction in SR Ca mishandling varies significantly, possibly depending on the stage and etiology of HF. Therefore, the development of individualized therapeutic strategies that specifically target the malfunctioning component of SR Ca machinery remains the highest priority.
References
Bers, D. M. (2001). Excitation-contraction coupling and cardiac contractile force. Dordrecht: Kluwer.
Cala, S. E., & Jones, L. R. (1983). Rapid purification of calsequestrin from cardiac and skeletal muscle sarcoplasmic reticulum vesicles by Ca2+-dependent elution from phenyl-sepharose. Journal of Biological Chemistry, 258, 11932–11936.
Fabiato, A. (1983). Calcium-induced release of calcium from the cardiac sarcoplasmic reticulum. American Journal of Physiology, 245, C1–C14.
Franzini-Armstrong, C., Protasi, F., & Ramesh, V. (1999). Shape, size, and distribution of Ca(2+) release units and couplons in skeletal and cardiac muscles. Biophysical Journal, 77, 1528–1539.
Garbino, A., van Oort, R. J., Dixit, S. S., Landstrom, A. P., Ackerman, M. J., & Wehrens, X. H. (2009). Molecular evolution of the junctophilin gene family. Physiological Genomics, 37, 175–186.
Cheng, H., & Lederer, W. J. (2008). Calcium sparks. Physiological Reviews, 88, 1491–1545.
Stern, M. D., Pizarro, G., & Rios, E. (1997). Local control model of excitation-contraction coupling in skeletal muscle. Journal of General Physiology, 110, 415–440.
Cheng, H., Lederer, W. J., & Cannell, M. B. (1993). Calcium sparks: Elementary events underlying excitation-contraction coupling in heart muscle. Science, 262, 740–744.
Lopez-Lopez, J. R., Shacklock, P. S., Balke, C. W., & Wier, W. G. (1995). Local calcium transients triggered by single L-type calcium channel currents in cardiac cells. Science, 268, 1042–1045.
Brochet, D. X., Yang, D., Di Maio, A., Lederer, W. J., Franzini-Armstrong, C., & Cheng, H. (2005). Ca2+ blinks: Rapid nanoscopic store calcium signaling. Proceedings of the National Academy of Sciences of the United States of America, 102, 3099–3104.
Kubalova, Z., Terentyev, D., Viatchenko-Karpinski, S., Nishijima, Y., Gyorke, I., Terentyeva, R., et al. (2005). Abnormal intrastore calcium signaling in chronic heart failure. Proceedings of the National Academy of Sciences of the United States of America, 102, 14104–14109.
Zima, A. V., Picht, E., Bers, D. M., & Blatter, L. A. (2008). Termination of cardiac Ca2+ sparks: Role of intra-SR [Ca2+], release flux, and intra-SR Ca2+ diffusion. Circulation Research, 103, e105–e115.
Picht, E., Zima, A. V., Shannon, T. R., Duncan, A. M., Blatter, L. A., & Bers, D. M. (2011). Dynamic calcium movement inside cardiac sarcoplasmic reticulum during release. Circulation Research, 108, 847–856.
Stern, M. D. (1992). Theory of excitation-contraction coupling in cardiac muscle. Biophysical Journal, 63, 497–517.
Sobie, E. A., Guatimosim, S., Gomez-Viquez, L., Song, L. S., Hartmann, H., Saleet, J. M., et al. (2006). The Ca 2+ leak paradox and rogue ryanodine receptors: SR Ca 2+ efflux theory and practice. Progress in Biophysics and Molecular Biology, 90, 172–185.
Shannon, T. R., Guo, T., & Bers, D. M. (2003). Ca2+ scraps: Local depletions of free [Ca2+] in cardiac sarcoplasmic reticulum during contractions leave substantial Ca2+ reserve. Circulation Research, 93, 40–45.
Belevych, A. E., Terentyev, D., Viatchenko-Karpinski, S., Terentyeva, R., Sridhar, A., Nishijima, Y., et al. (2009). Redox modification of ryanodine receptors underlies calcium alternans in a canine model of sudden cardiac death. Cardiovascular Research, 84, 387–395.
Stern, M. D., & Cheng, H. (2004). Putting out the fire: What terminates calcium-induced calcium release in cardiac muscle? Cell Calcium, 35, 591–601.
Gyorke, S., & Fill, M. (1993). Ryanodine receptor adaptation: Control mechanism of Ca(2+)-induced Ca2+ release in heart. Science, 260, 807–809.
Lukyanenko, V., Wiesner, T. F., & Gyorke, S. (1998). Termination of Ca2+ release during Ca2+ sparks in rat ventricular myocytes. The Journal of Physiology, 507(Pt 3), 667–677.
Sham, J. S., Song, L. S., Chen, Y., Deng, L. H., Stern, M. D., Lakatta, E. G., et al. (1998). Termination of Ca2+ release by a local inactivation of ryanodine receptors in cardiac myocytes. Proceedings of the National Academy of Sciences of the United States of America, 95, 15096–15101.
Fill, M., & Copello, J. A. (2002). Ryanodine receptor calcium release channels. Physiological Reviews, 82, 893–922.
Saucerman, J. J., & Bers, D. M. (2008). Calmodulin mediates differential sensitivity of CaMKII and calcineurin to local Ca2+ in cardiac myocytes. Biophysical Journal, 95, 4597–4612.
Guo, T., Ai, X., Shannon, T. R., Pogwizd, S. M., & Bers, D. M. (2007). Intra-sarcoplasmic reticulum free [Ca2+] and buffering in arrhythmogenic failing rabbit heart. Circulation Research, 101, 802–810.
Bovo, E., Mazurek, S. R., Blatter, L. A., & Zima, A. V. (2011). Regulation of sarcoplasmic reticulum Ca2+ leak by cytosolic Ca2+ in rabbit ventricular myocytes. The Journal of Physiology, 589, 6039–6050.
Terentyev, D., Kubalova, Z., Valle, G., Nori, A., Vedamoorthyrao, S., Terentyeva, R., et al. (2008). Modulation of SR Ca release by luminal Ca and calsequestrin in cardiac myocytes: Effects of CASQ2 mutations linked to sudden cardiac death. Biophysical Journal, 95, 2037–2048.
Terentyev, D., Viatchenko-Karpinski, S., Valdivia, H. H., Escobar, A. L., & Gyorke, S. (2002). Luminal Ca2+ controls termination and refractory behavior of Ca2+-induced Ca2+ release in cardiac myocytes. Circulation Research, 91, 414–420.
Sobie, E. A., Dilly, K. W., dos Santos, C. J., Lederer, W. J., & Jafri, M. S. (2002). Termination of cardiac Ca(2+) sparks: An investigative mathematical model of calcium-induced calcium release. Biophysical Journal, 83, 59–78.
Xu, L., & Meissner, G. (1998). Regulation of cardiac muscle Ca2+ release channel by sarcoplasmic reticulum lumenal Ca2+. Biophysical Journal, 75, 2302–2312.
Laver, D. R. (2007). Ca2+ stores regulate ryanodine receptor Ca2+ release channels via luminal and cytosolic Ca2+ sites. Biophysical Journal, 92, 3541–3555.
Guo, T., Gillespie, D., & Fill, M. (2012). Ryanodine receptor current amplitude controls Ca2+ sparks in cardiac muscle. Circulation Research, 111, 28–36.
Sitsapesan, R., & Williams, A. J. (1994). Regulation of the gating of the sheep cardiac sarcoplasmic reticulum Ca(2+)-release channel by luminal Ca2+. Journal of Membrane Biology, 137, 215–226.
Gyorke, I., & Gyorke, S. (1998). Regulation of the cardiac ryanodine receptor channel by luminal Ca2+ involves luminal Ca2+ sensing sites. Biophysical Journal, 75, 2801–2810.
Gyorke, I., Hester, N., Jones, L. R., & Gyorke, S. (2004). The role of calsequestrin, triadin, and junctin in conferring cardiac ryanodine receptor responsiveness to luminal calcium. Biophysical Journal, 86, 2121–2128.
Jiang, D., Chen, W., Wang, R., Zhang, L., & Chen, S. R. (2007). Loss of luminal Ca2+ activation in the cardiac ryanodine receptor is associated with ventricular fibrillation and sudden death. Proceedings of the National Academy of Sciences of the United States of America, 104, 18309–18314.
El-Armouche, A., & Eschenhagen, T. (2009). Beta-adrenergic stimulation and myocardial function in the failing heart. Heart Failure Reviews, 14, 225–241.
Hussain, M., & Orchard, C. H. (1997). Sarcoplasmic reticulum Ca2+ content, L-type Ca2+ current and the Ca2+ transient in rat myocytes during beta-adrenergic stimulation. The Journal of Physiology, 505(Pt 2), 385–402.
Callewaert, G., Cleemann, L., & Morad, M. (1988). Epinephrine enhances Ca2+ current-regulated Ca2+ release and Ca2+ reuptake in rat ventricular myocytes. Proceedings of the National Academy of Sciences of the United States of America, 85, 2009–2013.
Song, L. S., Wang, S. Q., Xiao, R. P., Spurgeon, H., Lakatta, E. G., & Cheng, H. (2001). Beta-adrenergic stimulation synchronizes intracellular Ca(2+) release during excitation-contraction coupling in cardiac myocytes. Circulation Research, 88, 794–801.
Ginsburg, K. S., & Bers, D. M. (2004). Modulation of excitation-contraction coupling by isoproterenol in cardiomyocytes with controlled SR Ca2+ load and Ca2+ current trigger. The Journal of Physiology, 556, 463–480.
Domeier, T. L., Blatter, L. A., & Zima, A. V. (2009). Alteration of sarcoplasmic reticulum Ca2+ release termination by ryanodine receptor sensitization and in heart failure. The Journal of Physiology, 587, 5197–5209.
Allen, D. G., & Blinks, J. R. (1978). Calcium transients in aequorin-injected frog cardiac muscle. Nature, 273, 509–513.
Zhao, W., Uehara, Y., Chu, G., Song, Q., Qian, J., Young, K., et al. (2004). Threonine-17 phosphorylation of phospholamban: A key determinant of frequency-dependent increase of cardiac contractility. Journal of Molecular and Cellular Cardiology, 37, 607–612.
Kushnir, A., Shan, J., Betzenhauser, M. J., Reiken, S., & Marks, A. R. (2010). Role of CaMKIIdelta phosphorylation of the cardiac ryanodine receptor in the force frequency relationship and heart failure. Proceedings of the National Academy of Sciences of the United States of America, 107, 10274–10279.
Wang, W., Zhu, W., Wang, S., Yang, D., Crow, M. T., Xiao, R. P., et al. (2004). Sustained beta1-adrenergic stimulation modulates cardiac contractility by Ca2+/calmodulin kinase signaling pathway. Circulation Research, 95, 798–806.
Curran, J., Hinton, M. J., Rios, E., Bers, D. M., & Shannon, T. R. (2007). Beta-adrenergic enhancement of sarcoplasmic reticulum calcium leak in cardiac myocytes is mediated by calcium/calmodulin-dependent protein kinase. Circulation Research, 100, 391–398.
Ferrero, P., Said, M., Sanchez, G., Vittone, L., Valverde, C., Donoso, P., et al. (2007). Ca2+/calmodulin kinase II increases ryanodine binding and Ca2+-induced sarcoplasmic reticulum Ca2+ release kinetics during beta-adrenergic stimulation. Journal of Molecular and Cellular Cardiology, 43, 281–291.
Bassani, J. W., Bassani, R. A., & Bers, D. M. (1994). Relaxation in rabbit and rat cardiac cells: Species-dependent differences in cellular mechanisms. The Journal of Physiology, 476, 279–293.
Kranias, E. G., & Hajjar, R. J. (2012). Modulation of cardiac contractility by the phospholamban/SERCA2a regulatome. Circulation Research, 110, 1646–1660.
Stevens, S. C., Terentyev, D., Kalyanasundaram, A., Periasamy, M., & Gyorke, S. (2009). Intra-sarcoplasmic reticulum Ca2+ oscillations are driven by dynamic regulation of ryanodine receptor function by luminal Ca2+ in cardiomyocytes. The Journal of Physiology, 587, 4863–4872.
Belevych, A. E., Terentyev, D., Terentyeva, R., Ho, H. T., Gyorke, I., Bonilla, I. M., et al. (2012). Shortened Ca2+ signaling refractoriness underlies cellular arrhythmogenesis in a postinfarction model of sudden cardiac death. Circulation Research, 110, 569–577.
Shannon, T. R., Ginsburg, K. S., & Bers, D. M. (2002). Quantitative assessment of the SR Ca2+ leak-load relationship. Circulation Research, 91, 594–600.
Zima, A. V., Bovo, E., Bers, D. M., & Blatter, L. A. (2010). Ca2+ spark-dependent and -independent sarcoplasmic reticulum Ca2+ leak in normal and failing rabbit ventricular myocytes. The Journal of Physiology, 588, 4743–4757.
Bassani, J. W., Yuan, W., & Bers, D. M. (1995). Fractional SR Ca release is regulated by trigger Ca and SR Ca content in cardiac myocytes. American Journal of Physiology, 268, C1313–C1319.
Shannon, T. R., Ginsburg, K. S., & Bers, D. M. (2000). Potentiation of fractional sarcoplasmic reticulum calcium release by total and free intra-sarcoplasmic reticulum calcium concentration. Biophysical Journal, 78, 334–343.
Venetucci, L. A., Trafford, A. W., O’Neill, S. C., & Eisner, D. A. (2008). The sarcoplasmic reticulum and arrhythmogenic calcium release. Cardiovascular Research, 77, 285–292.
Diaz, M. E., Trafford, A. W., O’Neill, S. C., & Eisner, D. A. (1997). Measurement of sarcoplasmic reticulum Ca2+ content and sarcolemmal Ca2+ fluxes in isolated rat ventricular myocytes during spontaneous Ca2+ release. The Journal of Physiology, 501(Pt 1), 3–16.
Schlotthauer, K., & Bers, D. M. (2000). Sarcoplasmic reticulum Ca(2+) release causes myocyte depolarization. Underlying mechanism and threshold for triggered action potentials. Circulation Research, 87, 774–780.
Bassani, R. A., & Bers, D. M. (1995). Rate of diastolic Ca release from the sarcoplasmic reticulum of intact rabbit and rat ventricular myocytes. Biophysical Journal, 68, 2015–2022.
Santiago, D. J., Curran, J. W., Bers, D. M., Lederer, W. J., Stern, M. D., Rios, E., et al. (2010). Ca sparks do not explain all ryanodine receptor-mediated SR Ca leak in mouse ventricular myocytes. Biophysical Journal, 98, 2111–2120.
Porta, M., Zima, A. V., Nani, A., Diaz-Sylvester, P. L., Copello, J. A., Ramos-Franco, J., et al. (2011). Single ryanodine receptor channel basis of caffeine’s action on Ca2+ sparks. Biophysical Journal, 100, 931–938.
Janse, M. J. (2004). Electrophysiological changes in heart failure and their relationship to arrhythmogenesis. Cardiovascular Research, 61, 208–217.
Gwathmey, J. K., Copelas, L., MacKinnon, R., Schoen, F. J., Feldman, M. D., Grossman, W., et al. (1987). Abnormal intracellular calcium handling in myocardium from patients with end-stage heart failure. Circulation Research, 61, 70–76.
Gwathmey, J. K., Slawsky, M. T., Hajjar, R. J., Briggs, G. M., & Morgan, J. P. (1990). Role of intracellular calcium handling in force-interval relationships of human ventricular myocardium. Journal of Clinical Investigation, 85, 1599–1613.
Beuckelmann, D. J., & Erdmann, E. (1992). Ca(2+)-currents and intracellular [Ca2+]i-transients in single ventricular myocytes isolated from terminally failing human myocardium. Basic Research in Cardiology, 87(Suppl. 1), 235–243.
Beuckelmann, D. J., Nabauer, M., & Erdmann, E. (1992). Intracellular calcium handling in isolated ventricular myocytes from patients with terminal heart failure. Circulation, 85, 1046–1055.
Eisner, D. A., Choi, H. S., Diaz, M. E., O’Neill, S. C., & Trafford, A. W. (2000). Integrative analysis of calcium cycling in cardiac muscle. Circulation Research, 87, 1087–1094.
Bers, D. M. (2002). Cardiac excitation-contraction coupling. Nature, 415, 198–205.
Curran, J., Brown, K. H., Santiago, D. J., Pogwizd, S., Bers, D. M., & Shannon, T. R. (2010). Spontaneous Ca waves in ventricular myocytes from failing hearts depend on Ca(2+)-calmodulin-dependent protein kinase II. Journal of Molecular and Cellular Cardiology, 49, 25–32.
Belevych, A. E., Terentyev, D., Terentyeva, R., Nishijima, Y., Sridhar, A., Hamlin, R. L., et al. (2011). The relationship between arrhythmogenesis and impaired contractility in heart failure: Role of altered ryanodine receptor function. Cardiovascular Research, 90, 493–502.
Schwinger, R. H., Munch, G., Bolck, B., Karczewski, P., Krause, E. G., & Erdmann, E. (1999). Reduced Ca(2+)-sensitivity of SERCA 2a in failing human myocardium due to reduced serin-16 phospholamban phosphorylation. Journal of Molecular and Cellular Cardiology, 31, 479–491.
Kiarash, A., Kelly, C. E., Phinney, B. S., Valdivia, H. H., Abrams, J., & Cala, S. E. (2004). Defective glycosylation of calsequestrin in heart failure. Cardiovascular Research, 63, 264–272.
Lindner, M., Erdmann, E., & Beuckelmann, D. J. (1998). Calcium content of the sarcoplasmic reticulum in isolated ventricular myocytes from patients with terminal heart failure. Journal of Molecular and Cellular Cardiology, 30, 743–749.
O’Rourke, B., Kass, D. A., Tomaselli, G. F., Kaab, S., Tunin, R., & Marban, E. (1999). Mechanisms of altered excitation-contraction coupling in canine tachycardia-induced heart failure, I: Experimental studies. Circulation Research, 84, 562–570.
Jiang, M. T., Lokuta, A. J., Farrell, E. F., Wolff, M. R., Haworth, R. A., & Valdivia, H. H. (2002). Abnormal Ca2+ release, but normal ryanodine receptors, in canine and human heart failure. Circulation Research, 91, 1015–1022.
Hobai, I. A., & O’Rourke, B. (2001). Decreased sarcoplasmic reticulum calcium content is responsible for defective excitation-contraction coupling in canine heart failure. Circulation, 103, 1577–1584.
Piacentino, V., III, Weber, C. R., Chen, X., Weisser-Thomas, J., Margulies, K. B., Bers, D. M., et al. (2003). Cellular basis of abnormal calcium transients of failing human ventricular myocytes. Circulation Research, 92, 651–658.
Marx, S. O., Reiken, S., Hisamatsu, Y., Jayaraman, T., Burkhoff, D., Rosemblit, N., et al. (2000). PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): Defective regulation in failing hearts. Cell, 101, 365–376.
Ai, X., Curran, J. W., Shannon, T. R., Bers, D. M., & Pogwizd, S. M. (2005). Ca2+/calmodulin-dependent protein kinase modulates cardiac ryanodine receptor phosphorylation and sarcoplasmic reticulum Ca2+ leak in heart failure. Circulation Research, 97, 1314–1322.
Mochizuki, M., Yano, M., Oda, T., Tateishi, H., Kobayashi, S., Yamamoto, T., et al. (2007). Scavenging free radicals by low-dose carvedilol prevents redox-dependent Ca2+ leak via stabilization of ryanodine receptor in heart failure. Journal of the American College of Cardiology, 49, 1722–1732.
Terentyev, D., Gyorke, I., Belevych, A. E., Terentyeva, R., Sridhar, A., Nishijima, Y., et al. (2008). Redox modification of ryanodine receptors contributes to sarcoplasmic reticulum Ca2+ leak in chronic heart failure. Circulation Research, 103, 1466–1472.
He, J., Conklin, M. W., Foell, J. D., Wolff, M. R., Haworth, R. A., Coronado, R., et al. (2001). Reduction in density of transverse tubules and L-type Ca(2+) channels in canine tachycardia-induced heart failure. Cardiovascular Research, 49, 298–307.
Louch, W. E., Bito, V., Heinzel, F. R., Macianskiene, R., Vanhaecke, J., Flameng, W., et al. (2004). Reduced synchrony of Ca2+ release with loss of T-tubules-a comparison to Ca2+ release in human failing cardiomyocytes. Cardiovascular Research, 62, 63–73.
Song, L. S., Sobie, E. A., McCulle, S., Lederer, W. J., Balke, C. W., & Cheng, H. (2006). Orphaned ryanodine receptors in the failing heart. Proceedings of the National Academy of Sciences of the United States of America, 103, 4305–4310.
Lyon, A. R., MacLeod, K. T., Zhang, Y., Garcia, E., Kanda, G. K., Lab, M. J., et al. (2009). Loss of T-tubules and other changes to surface topography in ventricular myocytes from failing human and rat heart. Proceedings of the National Academy of Sciences of the United States of America, 106, 6854–6859.
Crossman, D. J., Ruygrok, P. N., Soeller, C., & Cannell, M. B. (2011). Changes in the organization of excitation-contraction coupling structures in failing human heart. PLoS One, 6, e17901.
Gomez, A. M., Valdivia, H. H., Cheng, H., Lederer, M. R., Santana, L. F., Cannell, M. B., et al. (1997). Defective excitation-contraction coupling in experimental cardiac hypertrophy and heart failure. Science, 276, 800–806.
Bers, D. M. (2004). Macromolecular complexes regulating cardiac ryanodine receptor function. Journal of Molecular and Cellular Cardiology, 37, 417–429.
Meissner, G. (2004). Molecular regulation of cardiac ryanodine receptor ion channel. Cell Calcium, 35, 621–628.
Gyorke, S., & Carnes, C. (2008). Dysregulated sarcoplasmic reticulum calcium release: Potential pharmacological target in cardiac disease. Pharmacology and Therapeutics, 119, 340–354.
Marx, S. O., Gaburjakova, J., Gaburjakova, M., Henrikson, C., Ondrias, K., & Marks, A. R. (2001). Coupled gating between cardiac calcium release channels (ryanodine receptors). Circulation Research, 88, 1151–1158.
Balshaw, D. M., Xu, L., Yamaguchi, N., Pasek, D. A., & Meissner, G. (2001). Calmodulin binding and inhibition of cardiac muscle calcium release channel (ryanodine receptor). Journal of Biological Chemistry, 276, 20144–20153.
Farrell, E. F., Antaramian, A., Rueda, A., Gomez, A. M., & Valdivia, H. H. (2003). Sorcin inhibits calcium release and modulates excitation-contraction coupling in the heart. Journal of Biological Chemistry, 278, 34660–34666.
Marks, A. R. (2000). Cardiac intracellular calcium release channels: Role in heart failure. Circulation Research, 87, 8–11.
Pritchard, T. J., & Kranias, E. G. (2009). Junctin and the histidine-rich Ca2+ binding protein: Potential roles in heart failure and arrhythmogenesis. The Journal of Physiology, 587, 3125–3133.
Brillantes, A. M., Allen, P., Takahashi, T., Izumo, S., & Marks, A. R. (1992). Differences in cardiac calcium release channel (ryanodine receptor) expression in myocardium from patients with end-stage heart failure caused by ischemic versus dilated cardiomyopathy. Circulation Research, 71, 18–26.
Arai, M., Alpert, N. R., MacLennan, D. H., Barton, P., & Periasamy, M. (1993). Alterations in sarcoplasmic reticulum gene expression in human heart failure. A possible mechanism for alterations in systolic and diastolic properties of the failing myocardium. Circulation Research, 72, 463–469.
Go, L. O., Moschella, M. C., Watras, J., Handa, K. K., Fyfe, B. S., & Marks, A. R. (1995). Differential regulation of two types of intracellular calcium release channels during end-stage heart failure. Journal of Clinical Investigation, 95, 888–894.
Yano, M., Ono, K., Ohkusa, T., Suetsugu, M., Kohno, M., Hisaoka, T., et al. (2000). Altered stoichiometry of FKBP12.6 versus ryanodine receptor as a cause of abnormal Ca(2+) leak through ryanodine receptor in heart failure. Circulation, 102, 2131–2136.
Song, L. S., Pi, Y., Kim, S. J., Yatani, A., Guatimosim, S., Kudej, R. K., et al. (2005). Paradoxical cellular Ca2+ signaling in severe but compensated canine left ventricular hypertrophy. Circulation Research, 97, 457–464.
Armoundas, A. A., Rose, J., Aggarwal, R., Stuyvers, B. D., O’Rourke, B., Kass, D. A., et al. (2007). Cellular and molecular determinants of altered Ca2+ handling in the failing rabbit heart: Primary defects in SR Ca2+ uptake and release mechanisms. American Journal of Physiology. Heart and Circulatory Physiology, 292, H1607–H1618.
Vatner, D. E., Sato, N., Kiuchi, K., Shannon, R. P., & Vatner, S. F. (1994). Decrease in myocardial ryanodine receptors and altered excitation-contraction coupling early in the development of heart failure. Circulation, 90, 1423–1430.
Nimer, L. R., Needleman, D. H., Hamilton, S. L., Krall, J., & Movsesian, M. A. (1995). Effect of ryanodine on sarcoplasmic reticulum Ca2+ accumulation in nonfailing and failing human myocardium. Circulation, 92, 2504–2510.
Balijepalli, R. C., Lokuta, A. J., Maertz, N. A., Buck, J. M., Haworth, R. A., Valdivia, H. H., et al. (2003). Depletion of T-tubules and specific subcellular changes in sarcolemmal proteins in tachycardia-induced heart failure. Cardiovascular Research, 59, 67–77.
Marx, S. O., Ondrias, K., & Marks, A. R. (1998). Coupled gating between individual skeletal muscle Ca2+ release channels (ryanodine receptors). Science, 281, 818–821.
Ikemoto, N., & Yamamoto, T. (2002). Regulation of calcium release by interdomain interaction within ryanodine receptors. Frontiers in Bioscience, 7, d671–d683.
Oda, T., Yano, M., Yamamoto, T., Tokuhisa, T., Okuda, S., Doi, M., et al. (2005). Defective regulation of interdomain interactions within the ryanodine receptor plays a key role in the pathogenesis of heart failure. Circulation, 111, 3400–3410.
Yano, M., Okuda, S., Oda, T., Tokuhisa, T., Tateishi, H., Mochizuki, M., et al. (2005). Correction of defective interdomain interaction within ryanodine receptor by antioxidant is a new therapeutic strategy against heart failure. Circulation, 112, 3633–3643.
Xiao, B., Sutherland, C., Walsh, M. P., & Chen, S. R. (2004). Protein kinase A phosphorylation at serine-2808 of the cardiac Ca2+-release channel (ryanodine receptor) does not dissociate 12.6-kDa FK506-binding protein (FKBP12.6). Circulation Research, 94, 487–495.
Guo, T., Cornea, R. L., Huke, S., Camors, E., Yang, Y., Picht, E., et al. (2010). Kinetics of FKBP12.6 binding to ryanodine receptors in permeabilized cardiac myocytes and effects on Ca sparks. Circulation Research, 106, 1743–1752.
Lehnart, S. E., Wehrens, X. H., Reiken, S., Warrier, S., Belevych, A. E., Harvey, R. D., et al. (2005). Phosphodiesterase 4D deficiency in the ryanodine-receptor complex promotes heart failure and arrhythmias. Cell, 123, 25–35.
Bauman, A. L., Michel, J. J., Henson, E., Dodge-Kafka, K. L., & Kapiloff, M. S. (2007). The mAKAP signalosome and cardiac myocyte hypertrophy. IUBMB Life, 59, 163–169.
Sossalla, S., Fluschnik, N., Schotola, H., Ort, K. R., Neef, S., Schulte, T., et al. (2010). Inhibition of elevated Ca2+/calmodulin-dependent protein kinase II improves contractility in human failing myocardium. Circulation Research, 107, 1150–1161.
Belevych, A. E., Sansom, S. E., Terentyeva, R., Ho, H. T., Nishijima, Y., Martin, M. M., et al. (2011). MicroRNA-1 and -133 increase arrhythmogenesis in heart failure by dissociating phosphatase activity from RyR2 complex. PLoS One, 6, e28324.
Takasago, T., Imagawa, T., Furukawa, K., Ogurusu, T., & Shigekawa, M. (1991). Regulation of the cardiac ryanodine receptor by protein kinase-dependent phosphorylation. Journal of Biochemistry, 109, 163–170.
Xiao, B., Zhong, G., Obayashi, M., Yang, D., Chen, K., Walsh, M. P., et al. (2006). Ser-2030, but not Ser-2808, is the major phosphorylation site in cardiac ryanodine receptors responding to protein kinase A activation upon beta-adrenergic stimulation in normal and failing hearts. Biochemical Journal, 396, 7–16.
Huke, S., & Bers, D. M. (2008). Ryanodine receptor phosphorylation at serine 2030, 2808 and 2814 in rat cardiomyocytes. Biochemical and Biophysical Research Communications, 376, 80–85.
Wehrens, X. H., Lehnart, S. E., Reiken, S. R., & Marks, A. R. (2004). Ca2+/calmodulin-dependent protein kinase II phosphorylation regulates the cardiac ryanodine receptor. Circulation Research, 94, e61–e70.
George, C. H. (2008). Sarcoplasmic reticulum Ca2+ leak in heart failure: Mere observation or functional relevance? Cardiovascular Research, 77, 302–314.
Niggli, E., Ullrich, N. D., Gutierrez, D., Kyrychenko, S., Polakova, E., & Shirokova, N. (2013). Posttranslational modifications of cardiac ryanodine receptors: Ca(2+) signaling and EC-coupling. Biochimica et Biophysica Acta, 1833(4), 866–875.
Bers, D. M. (2012). Ryanodine receptor S2808 phosphorylation in heart failure: Smoking gun or red herring. Circulation Research, 110, 796–799.
Valdivia, H. H. (2012). Ryanodine receptor phosphorylation and heart failure: Phasing out S2808 and “criminalizing” S2814. Circulation Research, 110, 1398–1402.
Wehrens, X. H., Lehnart, S. E., Reiken, S., Vest, J. A., Wronska, A., & Marks, A. R. (2006). Ryanodine receptor/calcium release channel PKA phosphorylation: A critical mediator of heart failure progression. Proceedings of the National Academy of Sciences of the United States of America, 103, 511–518.
Shan, J., Betzenhauser, M. J., Kushnir, A., Reiken, S., Meli, A. C., Wronska, A., et al. (2010). Role of chronic ryanodine receptor phosphorylation in heart failure and beta-adrenergic receptor blockade in mice. Journal of Clinical Investigation, 120, 4375–4387.
Benkusky, N. A., Weber, C. S., Scherman, J. A., Farrell, E. F., Hacker, T. A., John, M. C., et al. (2007). Intact beta-adrenergic response and unmodified progression toward heart failure in mice with genetic ablation of a major protein kinase A phosphorylation site in the cardiac ryanodine receptor. Circulation Research, 101, 819–829.
Zhang, H., Makarewich, C. A., Kubo, H., Wang, W., Duran, J. M., Li, Y., et al. (2012). Hyperphosphorylation of the cardiac ryanodine receptor at serine 2808 is not involved in cardiac dysfunction after myocardial infarction. Circulation Research, 110, 831–840.
Zhang, R., Khoo, M. S., Wu, Y., Yang, Y., Grueter, C. E., Ni, G., et al. (2005). Calmodulin kinase II inhibition protects against structural heart disease. Nature Medicine, 11, 409–417.
Respress, J. L., van Oort, R. J., Li, N., Rolim, N., Dixit, S. S., deAlmeida, A., et al. (2012). Role of RyR2 phosphorylation at S2814 during heart failure progression. Circulation Research, 110, 1474–1483.
Mak, S., & Newton, G. E. (2001). The oxidative stress hypothesis of congestive heart failure: Radical thoughts. Chest, 120, 2035–2046.
Zima, A. V., & Blatter, L. A. (2006). Redox regulation of cardiac calcium channels and transporters. Cardiovascular Research, 71, 310–321.
Xu, L., Eu, J. P., Meissner, G., & Stamler, J. S. (1998). Activation of the cardiac calcium release channel (ryanodine receptor) by poly-S-nitrosylation. Science, 279, 234–237.
Tziomalos, K., & Hare, J. M. (2009). Role of xanthine oxidoreductase in cardiac nitroso-redox imbalance. Frontiers in Bioscience, 14, 237–262.
Kass, D. A., Carnicer, R., Crabtree, M. J., Sivakumaran, V., & Casadei, B. (2013). Nitric oxide synthases in heart failure. Antioxidants & Redox Signaling, 18(9), 1078–1099.
Prosser, B. L., Ward, C. W., & Lederer, W. J. (2011). X-ROS signaling: Rapid mechano-chemo transduction in heart. Science, 333, 1440–1445.
Ho, H. T., Stevens, S. C., Terentyeva, R., Carnes, C. A., Terentyev, D., & Gyorke, S. (2011). Arrhythmogenic adverse effects of cardiac glycosides are mediated by redox modification of ryanodine receptors. The Journal of Physiology, 589, 4697–4708.
Bovo, E., Lipsius, S. L., & Zima, A. V. (2012). Reactive oxygen species contribute to the development of arrhythmogenic Ca2+ waves during beta-adrenergic receptor stimulation in rabbit cardiomyocytes. The Journal of Physiology, 590, 3291–3304.
Gyorke, S., & Terentyev, D. (2008). Modulation of ryanodine receptor by luminal calcium and accessory proteins in health and cardiac disease. Cardiovascular Research, 77, 245–255.
Fan, G. C., Gregory, K. N., Zhao, W., Park, W. J., & Kranias, E. G. (2004). Regulation of myocardial function by histidine-rich, calcium-binding protein. American Journal of Physiology. Heart and Circulatory Physiology, 287, H1705–H1711.
Gergs, U., Berndt, T., Buskase, J., Jones, L. R., Kirchhefer, U., Muller, F. U., et al. (2007). On the role of junctin in cardiac Ca2+ handling, contractility, and heart failure. American Journal of Physiology. Heart and Circulatory Physiology, 293, H728–H734.
Ono, M., Yano, M., Hino, A., Suetomi, T., Xu, X., Susa, T., et al. (2010). Dissociation of calmodulin from cardiac ryanodine receptor causes aberrant Ca(2+) release in heart failure. Cardiovascular Research, 87, 609–617.
Guo, T., Zhang, T., Mestril, R., & Bers, D. M. (2006). Ca2+/calmodulin-dependent protein kinase II phosphorylation of ryanodine receptor does affect calcium sparks in mouse ventricular myocytes. Circulation Research, 99, 398–406.
Sigalas, C., Bent, S., Kitmitto, A., O’Neill, S., & Sitsapesan, R. (2009). Ca(2+)-calmodulin can activate and inactivate cardiac ryanodine receptors. British Journal of Pharmacology, 156, 794–806.
Litwin, S. E., Zhang, D., & Bridge, J. H. (2000). Dyssynchronous Ca(2+) sparks in myocytes from infarcted hearts. Circulation Research, 87, 1040–1047.
Diaz, M. E., O’Neill, S. C., & Eisner, D. A. (2004). Sarcoplasmic reticulum calcium content fluctuation is the key to cardiac alternans. Circulation Research, 94, 650–656.
Wei, S., Guo, A., Chen, B., Kutschke, W., Xie, Y. P., Zimmerman, K., et al. (2010). T-tubule remodeling during transition from hypertrophy to heart failure. Circulation Research, 107, 520–531.
Ohler, A., Weisser-Thomas, J., Piacentino, V., Houser, S. R., Tomaselli, G. F., & O’Rourke, B. (2009). Two-photon laser scanning microscopy of the transverse-axial tubule system in ventricular cardiomyocytes from failing and non-failing human hearts. Cardiology Research and Practice, 2009, 802373.
Litwin, S. E., Li, J., & Bridge, J. H. (1998). Na-Ca exchange and the trigger for sarcoplasmic reticulum Ca release: Studies in adult rabbit ventricular myocytes. Biophysical Journal, 75, 359–371.
Viatchenko-Karpinski, S., Terentyev, D., Jenkins, L. A., Lutherer, L. O., & Gyorke, S. (2005). Synergistic interactions between Ca2+ entries through L-type Ca2+ channels and Na+-Ca2+ exchanger in normal and failing rat heart. The Journal of Physiology, 567, 493–504.
Wier, W. G., Egan, T. M., Lopez-Lopez, J. R., & Balke, C. W. (1994). Local control of excitation-contraction coupling in rat heart cells. The Journal of Physiology, 474, 463–471.
Shannon, T. R., Pogwizd, S. M., & Bers, D. M. (2003). Elevated sarcoplasmic reticulum Ca2+ leak in intact ventricular myocytes from rabbits in heart failure. Circulation Research, 93, 592–594.
Wehrens, X. H., Lehnart, S. E., Reiken, S., van der Nagel, R., Morales, R., Sun, J., et al. (2005). Enhancing calstabin binding to ryanodine receptors improves cardiac and skeletal muscle function in heart failure. Proceedings of the National Academy of Sciences of the United States of America, 102, 9607–9612.
Belevych, A., Kubalova, Z., Terentyev, D., Hamlin, R. L., Carnes, C. A., & Gyorke, S. (2007). Enhanced ryanodine receptor-mediated calcium leak determines reduced sarcoplasmic reticulum calcium content in chronic canine heart failure. Biophysical Journal, 93, 4083–4092.
Lindner, M., Brandt, M. C., Sauer, H., Hescheler, J., Bohle, T., & Beuckelmann, D. J. (2002). Calcium sparks in human ventricular cardiomyocytes from patients with terminal heart failure. Cell Calcium, 31, 175–182.
Neary, P., Duncan, A. M., Cobbe, S. M., & Smith, G. L. (2002). Assessment of sarcoplasmic reticulum Ca(2+) flux pathways in cardiomyocytes from rabbits with infarct-induced left-ventricular dysfunction. Pflügers Archiv, 444, 360–371.
Perez, P. J., Ramos-Franco, J., Fill, M., & Mignery, G. A. (1997). Identification and functional reconstitution of the type 2 inositol 1,4,5-trisphosphate receptor from ventricular cardiac myocytes. Journal of Biological Chemistry, 272, 23961–23969.
Ramos-Franco, J., Fill, M., & Mignery, G. A. (1998). Isoform-specific function of single inositol 1,4,5-trisphosphate receptor channels. Biophysical Journal, 75, 834–839.
Proven, A., Roderick, H. L., Conway, S. J., Berridge, M. J., Horton, J. K., Capper, S. J., et al. (2006). Inositol 1,4,5-trisphosphate supports the arrhythmogenic action of endothelin-1 on ventricular cardiac myocytes. Journal of Cell Science, 119, 3363–3375.
Domeier, T. L., Zima, A. V., Maxwell, J. T., Huke, S., Mignery, G. A., & Blatter, L. A. (2008). IP3 receptor-dependent Ca2+ release modulates excitation-contraction coupling in rabbit ventricular myocytes. American Journal of Physiology. Heart and Circulatory Physiology, 294, H596–H604.
Wu, X., Zhang, T., Bossuyt, J., Li, X., McKinsey, T. A., Dedman, J. R., et al. (2006). Local InsP3-dependent perinuclear Ca2+ signaling in cardiac myocyte excitation-transcription coupling. Journal of Clinical Investigation, 116, 675–682.
Hou, Z., Kelly, E. M., & Robia, S. L. (2008). Phosphomimetic mutations increase phospholamban oligomerization and alter the structure of its regulatory complex. Journal of Biological Chemistry, 283, 28996–29003.
Kovacs, R. J., Nelson, M. T., Simmerman, H. K., & Jones, L. R. (1988). Phospholamban forms Ca2+-selective channels in lipid bilayers. Journal of Biological Chemistry, 263, 18364–18368.
Schmidt, U., Hajjar, R. J., Kim, C. S., Lebeche, D., Doye, A. A., & Gwathmey, J. K. (1999). Human heart failure: cAMP stimulation of SR Ca(2+)-ATPase activity and phosphorylation level of phospholamban. American Journal of Physiology, 277, H474–H480.
Currie, S., & Smith, G. L. (1999). Enhanced phosphorylation of phospholamban and downregulation of sarco/endoplasmic reticulum Ca2+ ATPase type 2 (SERCA 2) in cardiac sarcoplasmic reticulum from rabbits with heart failure. Cardiovascular Research, 41, 135–146.
Shannon, T. R., Chu, G., Kranias, E. G., & Bers, D. M. (2001). Phospholamban decreases the energetic efficiency of the sarcoplasmic reticulum Ca pump. Journal of Biological Chemistry, 276, 7195–7201.
Becucci, L., Cembran, A., Karim, C. B., Thomas, D. D., Guidelli, R., Gao, J., et al. (2009). On the function of pentameric phospholamban: Ion channel or storage form? Biophysical Journal, 96, L60–L62.
Camello, C., Lomax, R., Petersen, O. H., & Tepikin, A. V. (2002). Calcium leak from intracellular stores—The enigma of calcium signalling. Cell Calcium, 32, 355–361.
Berbey, C., Weiss, N., Legrand, C., & Allard, B. (2009). Transient receptor potential canonical type 1 (TRPC1) operates as a sarcoplasmic reticulum calcium leak channel in skeletal muscle. Journal of Biological Chemistry, 284, 36387–36394.
Moller, J. V., Olesen, C., Winther, A. M., & Nissen, P. (2010). The sarcoplasmic Ca2+-ATPase: Design of a perfect chemi-osmotic pump. Quarterly Reviews of Biophysics, 43, 501–566.
Bidwell, P., Blackwell, D. J., Hou, Z., Zima, A. V., & Robia, S. L. (2011). Phospholamban binds with differential affinity to calcium pump conformers. Journal of Biological Chemistry, 286, 35044–35050.
Simmerman, H. K., Collins, J. H., Theibert, J. L., Wegener, A. D., & Jones, L. R. (1986). Sequence analysis of phospholamban. Identification of phosphorylation sites and two major structural domains. Journal of Biological Chemistry, 261, 13333–13341.
Kodama, T. (1985). Thermodynamic analysis of muscle ATPase mechanisms. Physiological Reviews, 65, 467–551.
Feldman, A. M., Weinberg, E. O., Ray, P. E., & Lorell, B. H. (1993). Selective changes in cardiac gene expression during compensated hypertrophy and the transition to cardiac decompensation in rats with chronic aortic banding. Circulation Research, 73, 184–192.
Hasenfuss, G., Reinecke, H., Studer, R., Meyer, M., Pieske, B., Holtz, J., et al. (1994). Relation between myocardial function and expression of sarcoplasmic reticulum Ca(2+)-ATPase in failing and nonfailing human myocardium. Circulation Research, 75, 434–442.
Studer, R., Reinecke, H., Bilger, J., Eschenhagen, T., Bohm, M., Hasenfuss, G., et al. (1994). Gene expression of the cardiac Na(+)-Ca2+ exchanger in end-stage human heart failure. Circulation Research, 75, 443–453.
Meyer, M., Schillinger, W., Pieske, B., Holubarsch, C., Heilmann, C., Posival, H., et al. (1995). Alterations of sarcoplasmic reticulum proteins in failing human dilated cardiomyopathy. Circulation, 92, 778–784.
Kiss, E., Ball, N. A., Kranias, E. G., & Walsh, R. A. (1995). Differential changes in cardiac phospholamban and sarcoplasmic reticular Ca(2+)-ATPase protein levels. Effects on Ca2+ transport and mechanics in compensated pressure-overload hypertrophy and congestive heart failure. Circulation Research, 77, 759–764.
Miyamoto, M. I., del, M. F., Schmidt, U., DiSalvo, T. S., Kang, Z. B., Matsui, T., et al. (2000). Adenoviral gene transfer of SERCA2a improves left-ventricular function in aortic-banded rats in transition to heart failure. Proceedings of the National Academy of Sciences of the United States of America, 97, 793–798.
Schwinger, R. H., Bohm, M., Schmidt, U., Karczewski, P., Bavendiek, U., Flesch, M., et al. (1995). Unchanged protein levels of SERCA II and phospholamban but reduced Ca2+ uptake and Ca(2+)-ATPase activity of cardiac sarcoplasmic reticulum from dilated cardiomyopathy patients compared with patients with nonfailing hearts. Circulation, 92, 3220–3228.
Linck, B., Boknik, P., Eschenhagen, T., Muller, F. U., Neumann, J., Nose, M., et al. (1996). Messenger RNA expression and immunological quantification of phospholamban and SR-Ca(2+)-ATPase in failing and nonfailing human hearts. Cardiovascular Research, 31, 625–632.
Schwinger, R. H., Bolck, B., Munch, G., Brixius, K., Muller-Ehmsen, J., & Erdmann, E. (1998). cAMP-dependent protein kinase A-stimulated sarcoplasmic reticulum function in heart failure. Annals of the New York Academy of Sciences, 853, 240–250.
Pogwizd, S. M., Qi, M., Yuan, W., Samarel, A. M., & Bers, D. M. (1999). Upregulation of Na(+)/Ca(2+) exchanger expression and function in an arrhythmogenic rabbit model of heart failure. Circulation Research, 85, 1009–1019.
Hasenfuss, G., Meyer, M., Schillinger, W., Preuss, M., Pieske, B., & Just, H. (1997). Calcium handling proteins in the failing human heart. Basic Research in Cardiology, 92(Suppl. 1), 87–93.
Neumann, J., Eschenhagen, T., Jones, L. R., Linck, B., Schmitz, W., Scholz, H., et al. (1997). Increased expression of cardiac phosphatases in patients with end-stage heart failure. Journal of Molecular and Cellular Cardiology, 29, 265–272.
Schmitt, J. P., Kamisago, M., Asahi, M., Li, G. H., Ahmad, F., Mende, U., et al. (2003). Dilated cardiomyopathy and heart failure caused by a mutation in phospholamban. Science, 299, 1410–1413.
Ha, K. N., Masterson, L. R., Hou, Z., Verardi, R., Walsh, N., Veglia, G., et al. (2011). Lethal Arg9Cys phospholamban mutation hinders Ca2+-ATPase regulation and phosphorylation by protein kinase A. Proceedings of the National Academy of Sciences of the United States of America, 108, 2735–2740.
Lancel, S., Qin, F., Lennon, S. L., Zhang, J., Tong, X., Mazzini, M. J., et al. (2010). Oxidative posttranslational modifications mediate decreased SERCA activity and myocyte dysfunction in Galphaq-overexpressing mice. Circulation Research, 107, 228–232.
Kho, C., Lee, A., Jeong, D., Oh, J. G., Chaanine, A. H., Kizana, E., et al. (2011). SUMO1-dependent modulation of SERCA2a in heart failure. Nature, 477, 601–605.
Akin, B. L., & Jones, L. R. (2012). Characterizing phospholamban to sarco(endo)plasmic reticulum Ca2+-ATPase 2a (SERCA2a) protein binding interactions in human cardiac sarcoplasmic reticulum vesicles using chemical cross-linking. Journal of Biological Chemistry, 287, 7582–7593.
Pieske, B., Kretschmann, B., Meyer, M., Holubarsch, C., Weirich, J., Posival, H., et al. (1995). Alterations in intracellular calcium handling associated with the inverse force-frequency relation in human dilated cardiomyopathy. Circulation, 92, 1169–1178.
Schmidt, U., Hajjar, R. J., Helm, P. A., Kim, C. S., Doye, A. A., & Gwathmey, J. K. (1998). Contribution of abnormal sarcoplasmic reticulum ATPase activity to systolic and diastolic dysfunction in human heart failure. Journal of Molecular and Cellular Cardiology, 30, 1929–1937.
Pogwizd, S. M., Schlotthauer, K., Li, L., Yuan, W., & Bers, D. M. (2001). Arrhythmogenesis and contractile dysfunction in heart failure: Roles of sodium-calcium exchange, inward rectifier potassium current, and residual beta-adrenergic responsiveness. Circulation Research, 88, 1159–1167.
Bers, D. M., Eisner, D. A., & Valdivia, H. H. (2003). Sarcoplasmic reticulum Ca2+ and heart failure: Roles of diastolic leak and Ca2+ transport. Circulation Research, 93, 487–490.
Hasenfuss, G., Reinecke, H., Studer, R., Pieske, B., Meyer, M., Drexler, H., et al. (1996). Calcium cycling proteins and force-frequency relationship in heart failure. Basic Research in Cardiology, 91(Suppl. 2), 17–22.
Pieske, B., Maier, L. S., Bers, D. M., & Hasenfuss, G. (1999). Ca2+ handling and sarcoplasmic reticulum Ca2+ content in isolated failing and nonfailing human myocardium. Circulation Research, 85, 38–46.
Maier, L. S., Braunhalter, J., Horn, W., Weichert, S., & Pieske, B. (2002). The role of SR Ca(2+)-content in blunted inotropic responsiveness of failing human myocardium. Journal of Molecular and Cellular Cardiology, 34, 455–467.
Wimsatt, D. K., Hohl, C. M., Brierley, G. P., & Altschuld, R. A. (1990). Calcium accumulation and release by the sarcoplasmic reticulum of digitonin-lysed adult mammalian ventricular cardiomyocytes. Journal of Biological Chemistry, 265, 14849–14857.
Xiang, J. Z., & Kentish, J. C. (1995). Effects of inorganic phosphate and ADP on calcium handling by the sarcoplasmic reticulum in rat skinned cardiac muscles. Cardiovascular Research, 29, 391–400.
Shannon, T. R., & Bers, D. M. (1997). Assessment of intra-SR free [Ca] and buffering in rat heart. Biophysical Journal, 73, 1524–1531.
Inesi, G., & de Meis, L. (1989). Regulation of steady state filling in sarcoplasmic reticulum. Roles of back-inhibition, leakage, and slippage of the calcium pump. Journal of Biological Chemistry, 264, 5929–5936.
Ventura-Clapier, R., Garnier, A., & Veksler, V. (2004). Energy metabolism in heart failure. The Journal of Physiology, 555, 1–13.
Hasenfuss, G., Maier, L. S., Hermann, H. P., Luers, C., Hunlich, M., Zeitz, O., et al. (2002). Influence of pyruvate on contractile performance and Ca(2+) cycling in isolated failing human myocardium. Circulation, 105, 194–199.
Lompre, A. M., Hajjar, R. J., Harding, S. E., Kranias, E. G., Lohse, M. J., & Marks, A. R. (2010). Ca2+ cycling and new therapeutic approaches for heart failure. Circulation, 121, 822–830.
Haghighi, K., Kolokathis, F., Pater, L., Lynch, R. A., Asahi, M., Gramolini, A. O., et al. (2003). Human phospholamban null results in lethal dilated cardiomyopathy revealing a critical difference between mouse and human. Journal of Clinical Investigation, 111, 869–876.
Lyon, A. R., Bannister, M. L., Collins, T., Pearce, E., Sepehripour, A. H., Dubb, S. S., et al. (2011). SERCA2a gene transfer decreases sarcoplasmic reticulum calcium leak and reduces ventricular arrhythmias in a model of chronic heart failure. Circulation. Arrhythmia and Electrophysiology, 4, 362–372.
Sakata, S., Lebeche, D., Sakata, N., Sakata, Y., Chemaly, E. R., Liang, L. F., et al. (2007). Restoration of mechanical and energetic function in failing aortic-banded rat hearts by gene transfer of calcium cycling proteins. Journal of Molecular and Cellular Cardiology, 42, 852–861.
Kobayashi, S., Yano, M., Suetomi, T., Ono, M., Tateishi, H., Mochizuki, M., et al. (2009). Dantrolene, a therapeutic agent for malignant hyperthermia, markedly improves the function of failing cardiomyocytes by stabilizing interdomain interactions within the ryanodine receptor. Journal of the American College of Cardiology, 53, 1993–2005.
Maxwell, J. T., Domeier, T. L., & Blatter, L. A. (2012). Dantrolene prevents arrhythmogenic Ca2+ release in heart failure. American Journal of Physiology. Heart and Circulatory Physiology, 302, H953–H963.
Tateishi, H., Yano, M., Mochizuki, M., Suetomi, T., Ono, M., Xu, X., et al. (2009). Defective domain-domain interactions within the ryanodine receptor as a critical cause of diastolic Ca2+ leak in failing hearts. Cardiovascular Research, 81, 536–545.
Toischer, K., Lehnart, S. E., Tenderich, G., Milting, H., Korfer, R., Schmitto, J. D., et al. (2010). K201 improves aspects of the contractile performance of human failing myocardium via reduction in Ca2+ leak from the sarcoplasmic reticulum. Basic Research in Cardiology, 105, 279–287.
Acknowledgements
The authors also would like to thank Drs. Elisa Bovo, Seth L. Robia, Joshua Maxwell, and Ms. Yukiko Kunitomo for critical reading of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer Science+Business Media, LLC
About this chapter
Cite this chapter
Zima, A.V., Terentyev, D. (2013). Sarcoplasmic Reticulum Ca Homeostasis and Heart Failure. In: Solaro, R., Tardiff, J. (eds) Biophysics of the Failing Heart. Biological and Medical Physics, Biomedical Engineering. Springer, New York, NY. https://doi.org/10.1007/978-1-4614-7678-8_2
Download citation
DOI: https://doi.org/10.1007/978-1-4614-7678-8_2
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4614-7677-1
Online ISBN: 978-1-4614-7678-8
eBook Packages: Physics and AstronomyPhysics and Astronomy (R0)