
Abstract
In view of its ability to release and accumulate Ca2+, the sarcoplasmic reticulum (SR) plays a major role in cardiomyocyte excitation–contraction coupling. Since Ca2+ is important in the functions of both mitochondria and nucleus, it appears that these organelles are involved in excitation–metabolism coupling and excitation–transcription coupling. This brief article discusses some of the characteristics of the SR, mitochondria, and nucleus with respect to their Ca2+ transport systems in cardiomyocytes and the role of Ca2+ in different coupling processes. In addition, the influence of fluctuations in the cytoplasmic Ca2+ concentration on cardiomyocyte contractile activity, metabolism, and gene expression in heart disease is presented. The potential of the SR and the mitochondria and nucleus as therapeutic targets for the treatment of heart disease is also highlighted.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Heart
- Excitation–contraction coupling
- Excitation–metabolism coupling
- Excitation–transcription coupling
- Intracellular Ca2+ handling
- Mitochondria
- Nucleus
- Sarcoplasmic reticulum
- SERCA
- Ryanodine receptors
- Inositol trisphosphate receptor
1 Introduction
The precise control of intracellular Ca2+ cycling depends on the relationships among various channels and pumps that are involved in different membranous systems of cardiomyocytes. Accordingly, two significant aspects have been proposed: (a) structural coupling in which the transporters are organized within the dyad, linking the transverse tubule and sarcoplasmic reticulum and ensuring proximity of Ca2+ entry to sites of release and (b) functional coupling, where the fluxes across all membranes must be balanced such that, in the steady state, Ca2+ influx equals Ca2+ efflux on every beat [1]. Depolarization of the cardiomyocyte causes Ca2+ influx through the sarcolemmal membrane (SL) L-type Ca2+ channels (LTCCs), which subsequently elicits a greater release of Ca2+ upon activation of the sarcoplasmic reticulum (SR), ryanodine receptors (RyR), or Ca2+-release channels. This increase in Ca2+ in the cytosol is rapidly dissipated by four mechanisms: (a) Ca2+ binding by proteins such as calmodulin and troponin [2], (b) Ca2+ efflux through SL or plasma membrane Ca2+-pump ATPase (PMCA) and SL Na+–Ca2+ exchanger (NCX), (c) storage in the SR by SR Ca2+-pump ATPase (SERCA), and (d) Ca2+ buffering by intracellular organelles such as mitochondria and nucleus [3]. In this regard, the uptake of Ca2+ by the mitochondria occurs mainly through the mitochondrial Ca2+ uniporter (mCUP), while the nucleoplasmic reticulum serves as a Ca2+ storing site in the nucleus [4].
It is now well known that the process of cardiac contraction is initiated by the release of Ca2+ from the SR, whereas the process of relaxation is associated with Ca2+ uptake by the SR in cardiomyocytes. Furthermore, SR is also known to possess inositol trisphosphate receptors (IP3R), the activation of which is considered to release Ca2+ in the cardiomyocyte [5, 6]. Fluctuations in the cytosolic levels of Ca2+ affect mitochondrial metabolism that may contribute to the pathogenesis of congestive heart failure (CHF) and ischemic heart disease (IHD) [5]. Cytosolic Ca2+ is also considered to regulate myocardial gene expression during subcellular remodeling in ischemia-reperfusion (I-R) injury, cardiac hypertrophy, and CHF [6, 7]. Accordingly, this chapter is intended to discuss the major characteristics of the Ca2+-handling and regulatory proteins such as SR RyR, SR Ca2+-pump ATPase, and IP3R. Furthermore, the coupling of Ca2+ to the cardiac contraction–relaxation cycle, Ca2+ to mitochondrial metabolism, and Ca2+ to nuclear transcription is described following the excitation of cardiomyocytes. In addition, their role in the pathophysiology of different myocardial diseases will be briefly discussed, and the potential of the SR, mitochondria, and nucleus as therapeutic targets for heart disease will be highlighted.
2 Excitation–Contraction Coupling
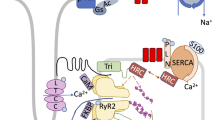
The excitation–contraction (E–C) unit comprises synergistic lines of communication between the SL and SR [8]. Since the major functions of the SR are the release of Ca2+ for cardiac contraction upon excitation and uptake of Ca2+ for cardiac relaxation, it is planned to describe the characteristics as well as the role of SR Ca2+-release channel, SR Ca2+-pump ATPase and SR IP3R (Fig. 21.1) in Ca2+ handling and regulation of the intracellular concentration of Ca2+ ([Ca2+]i) in cardiomyocytes.

Schematic representation of SR Ca2+-handling proteins involved in the regulation of the intracellular concentration of Ca2+ in cardiomyocytes. RyR ryanodine receptor, PLB phospholamban, SERCA sarcoplasmic reticulum Ca2+- pump ATPase, IP3R inositol trisphoshate receptor
2.1 SR Ca2 + -release Channel
The SR RyR or Ca2+-release channel is a 564 kDa tetrameric protein consisting of at least four transmembrane segments and a large cytoplasmic domain. Three isoforms have been identified, namely, RyR1 and RyR3 in skeletal muscle and RyR2 in cardiac muscle; these isoforms share up to 60% sequence homology. RyR in cardiac muscle is organized in groups of approximately 20 receptors each. It has been suggested that the close approximation of these receptors is required for orchestrated opening and closing of the Ca2+-release channels [9]. Ca2+ has a biphasic effect on RyR; submicromolar Ca2+ concentrations activate RyR, whereas higher Ca2+ concentrations (>1 μM) inhibit the channel. RyR is inhibited by cytosolic Mg2+ but an increase in cytosolic Ca2+ relieves the Mg2+ inhibition [10]. RyR is also modulated by several cytosolic proteins; however, the most thoroughly studied modulators are calmodulin (CaM), calmodulin kinase (CaMKII), protein kinase A (PKA), and protein phosphatases. CaM binding to RyR is Ca2+ dependent, where Ca2+ binding increases the sensitivity of RyR to CaM. CaM binding inhibits the cardiac RyR in the presence of high concentration of Ca2+ (10 μM). Several kinases have been shown to affect the activity of cardiac RyR. These include protein kinase A, C, and G in addition to the CaM-activated CaMKII [6, 10]. The RyR phosphorylation is of great importance in the pathogenesis of heart failure and arrhythmias [11]. Hyperphosphorylation of RyR in heart failure leads to the dissociation of “calstabin”, an accessory protein that stabilizes the closed conformation of the channel [12]. Phosphorylation of RyR cannot be interpreted in isolation from other SR proteins such as the Ca2+-pump ATPase (SERCA) and phospholamban (PLB). PLB phosphorylation activates SERCA and increases the SR Ca2+ load, which then stimulates RyR [13]. While the overall effect of phosphorylation is stimulatory, protein phosphatases are inhibitory [10]. In addition to kinases and phosphatases, several other cytoplasmic proteins are considered to modulate the activity of cardiac RyR. These include the transmembrane proteins, junctin and triadin, and the cytosolic proteins, S100, sorcin, glutathione transferase as well as intracellular chloride ion channels, glycolytic enzymes, and the accessory protein, calstabin [14].
Mutations in cardiac RyR have been linked to the development of premature heartbeats. A “leaky” RyR promotes the buildup of Ca2+ in the cytosol and subsequently triggers the SL NCX to remove the excess Ca2+ in exchange for Na+; this then depolarizes the cell membrane leading to extrasystolic depolarizations and premature beats. On the other hand, adrenergic stimulation leading to the phosphorylation of RyR can cause fatal arrhythmias such as catecholaminergic polymorphic ventricular tachycardia (CPVT) in susceptible individuals. Susceptibility to CPVT was linked mainly to autosomal dominant mutations of RyR2; these mutations destabilize the channel or impair the binding to calstabin that stabilizes RyR [15, 16]. Recently, a mutation linked to the development of CPVT (R2474S) was found to produce “leaky” RyR in the heart and to the development of CPVT in transgenic mice. It has been shown that these RyRs could be stabilized by a novel compound, S107, for averting fatal arrhythmias [17]. Interestingly, the expression of RyR is reduced in heart failure due to the development of myocardial infarction (MI); this change is believed to be mediated through angiotensin receptors because it is inhibited by angiotensin-converting enzymes (ACE) inhibitors such as imidapril and in fact, close involvement of the SR RyR in the cellular remodeling process has been suggested [18]. In addition to suppressing cardiac hypertrophy, ACE inhibitor therapy has been shown to decrease the hyperadrenergic state associated with heart failure, resulting in a reduction of PKA activity, which abolishes the phosphorylation of RyR. Furthermore, ACE inhibitors have been reported to promote the interaction of RyR with the accessory protein calstabin, which stabilizes the channel and reduces the RyR leak [19].
2.2 SR Ca2+-Pump ATPase
Most of the Ca2+ released from SR stores is rapidly taken up again via the SR Ca2+-pump ATPase (SERCA), while the rest is extruded from the cell by the PMCA and SL NCX. The SERCA transports two Ca2+ ions for the hydrolysis of a single ATP molecule [19]. SERCA is organized into three interacting domains: the cytosolic nucleotide-binding domain, the phosphorylation domain containing Asp351, and the transmembrane translocation domain. The cytosolic and transmembrane domains consist of 10 transmembrane segments, which are connected by the stalk domain [20]. At least six isoforms of SERCA have been identified belonging to three different gene families, namely, SERCA1, 2, and 3; cardiomyocytes express the SERCA2a isoform [21]. The SERCA activity is regulated by SR-associated phospholamban (PLB). In this regard, the dephosphorylated form of PLB inhibits the pump by interacting with the enzyme phosphorylation site, which is needed for ATP binding, whereas phosphorylation of PLB by PKA or CaMK relieves this inhibition [22]. During adrenergic stimulation, PKA reduces the affinity of Ca2+-pump ATPase for Ca2+ and thus enhances cardiac muscle relaxation. The adrenergic stimulation of the heart enhances the SERCA activity and such a regulatory action has been suggested to be a compensatory mechanism to improve cardiac performance.
Sarcolipin is another modulator of SERCA activity [23]. Sarcolipin is a shorter homologue (31 amino acids) of PLB, which inhibits SERCA. However, unlike PLB, the level of expression of sarcolipin determines its interaction with SERCA rather than its phosphorylation level. It has been found that sarcolipin modulates SERCA in a Ca2+-dependent manner; at low Ca2+ levels, sarcolipin reduces its affinity for Ca2+ and thus inhibits its activity, whereas at high Ca2+ levels, it increases its affinity for Ca2+ by increasing the maximum turnover rate [24]. Sarcolipin and PLB both synergistically inhibit SERCA; coexpression of sarcolipin in HEK cells leads to a very strong inhibition of the SERCA activity, whereas a sarcolipin knockout leads to enhanced SERCA activity [25]. On the other hand, the S100 protein, which is abundantly expressed in the heart and skeletal muscle, is believed to stimulate Ca2+ uptake by SERCA2a through a direct protein–protein interaction [26].
In cardiac hypertrophy and heart failure, the levels of SERCA expression as well as the SERCA activity have been found to be reduced, leading to compromised SR function [27]. In fact, overexpression of SERCA can improve cardiac functional defects [28]. In heart failure, a reduction in the SERCA activity by about 50% was evident without any changes in the creatine kinase activity and mitochondrial functions [29]. While several studies have indicated a role for SERCA in heart function, heterozygous loss of function of the SERCA2a and 2b in Darier disease was not associated with any defects in cardiac performance [30, 31].
2.3 SR Inositol Trisphosphate Receptors
Although IP3R plays a minor role in EC coupling compared to the RyR in ventricular cardiomyocytes [32, 33], the higher (3.5–10 fold) IP3R expression in atrial myocytes vs. ventricular myocytes, is suggestive of a greater role of IP3R in atrial contraction [34]. There are several isoforms of the IP3R in both excitable and non-excitable tissues; the heart expresses the 300 KDa IP3R2 isoform that co-assembles to form a tetrameric channel. The channel is predicted to have six transmembrane segments and a large regulatory cytoplasmic domain [35]. IP3R1 is regulated by Ca2+ in a biphasic pattern similar to the RyR. In fact, it is activated by submicromolar concentration of Ca2+ (<300 nM) and inhibited by micromolar concentrations of Ca2+ (>1 μM). The cardiac IP3R2 is resistant to inhibition by high Ca2+, as it remains active in the presence of high Ca2+ concentration (>100 μM) [36, 37]. The IP3R is modulated by cytosolic proteins such as calstabin, PKA, and CaM [38]. The most significant interaction of IP3R is with CaM, because Ca2+-free CaM inhibits the cardiac IP3R2 in a Ca2+-independent manner, indicating a permanent inhibition of these receptors by CaM in the absence of Ca2+ [39]. The development and progression of cardiac hypertrophy have been linked to increases in the phospholipase C-IP3R expression levels leading to a stimulation of hypertrophic gene transcription. IP3R2 has been shown to be upregulated, while, in contrast, a reduction in RyR2 expression and activity has been reported in heart failure [34]. In addition, the relative abundance of IP3R in Purkinje fibers suggests its potential participation in ventricular arrhythmias [40], however; the precise role of IP3R in the generation of ventricular arrhythmias remains to be fully elucidated.
In addition to the role of SR in Ca2+-handling in cardiomyocytes, other cellular structures can also be seen to participate in the process of excitation–contraction–relaxation cycle. It should be mentioned that induced pluripotent stem cell-derived cardiomyocytes (iPSC-CMs) hold enormous potential in many fields of cardiovascular research. Overcoming many of the limitations of their embryonic counterparts, the application of iPSC-CMs ranges from facilitating the investigation of familial cardiac disease and pharmacological toxicity screening to personalized medicine and autologous cardiac cell therapies. The main factor preventing the full realization of this potential is the limited maturity of iPSC-CMs, which display a number of substantial differences in comparison to adult cardiomyocytes. E–C coupling, a fundamental property of cardiomyocytes, is often described in iPSC-CMs as being more analogous to neonatal than adult cardiomyocytes. With Ca2+-handling linked, directly or indirectly to almost all other properties of cardiomyocytes, a solid understanding of this process will be crucial to fully realizing the potential of human iPSC-CMs [41].
3 Excitation–Metabolism Coupling
E–C coupling consumes a large amount of energy through myosin ATPase, the SL Na+-K+ ATPase, and SERCA activities [6]. On the other hand, the mitochondria are the main source of energy production. Mitochondria take up to 30% of the myocyte volume and are present in close proximity to the contractile machinery where energy is most required [42]. Indeed, [Ca2+]i concentration in different cellular compartments is intimately linked to cell metabolism, because (a) ATP production requires low Ca2+, (b) Ca2+ homeostatic systems consume ATP, and (c) Ca2+ signals in mitochondria stimulate ATP synthesis, being an essential part of excitation-metabolism (E-M) coupling [43]. The regulation of mitochondrial metabolism by Ca2+ is attributed to three key enzymes in the citric acid cycle, namely pyruvate dehydrogenase, isocitrate dehydrogenase, and α-ketoglutarate dehydrogenase, which are activated by low concentrations of Ca2+ [44]; in addition, Ca2+ also activates the mitochondrial F1/F0 ATPase [45].
Mitochondria in cardiac muscles also possess the molecular components for efficient uptake and release of Ca2+. In this regard, Ca2+ enters the mitochondrial membrane through the mitochondrial Ca2+ uniporter (mCUP), a low affinity (10–20 μM Ca2+) and highly selective ion channel, which accumulates Ca2+ in the mitochondria using the potential difference across the mitochondrial membrane; the mCUP is Ca2+-gated and requires calmodulin for its activation [46]. Although mitochondria are generally considered to serve as a Ca2+ sink, mitochondrial Ca2+ uptake is inhibited by high concentrations of cytosolic Ca2+ [47]. Ca2+ is extruded from mitochondria through the activity of mitochondrial Na+-Ca2+ exchanger (mNCX), which is believed to possess a stoichiometry similar to that of the SL NCX of three Na+ to one Ca2+ [48]. Since the Ca2+ extrusion mechanism is slower relative to the rate of Ca2+ entry, this leads to the accumulation of Ca2+ in the mitochondria [49]. Ca2+ and mitochondria may also be intimately linked to cardiac function as Ca2+ is central to cardiac excitation–contraction coupling and stimulates mitochondrial energy production to fuel contraction. It should be mentioned that under conditions of dysregulated Ca2+ cycling, mitochondrial Ca2+ overload activates cell death pathways [50]. Indeed, excessive accumulation of Ca2+ in the mitochondria may induce the translocation of cytochrome C as well as other pro-apoptotic factors [51]. Thus, taken together, the mitochondrial Ca2+ microdomain is where contraction, energy, and cardiomyocyte death converge [50, 52].
Studies have shown that increases in mitochondrial [Ca2+] occur simultaneously with the increase in cytosolic [Ca2+] in response to an increase in cardiomyocyte pacing or β-adrenergic stimulation [53, 54]. The rise of mitochondrial [Ca2+] was shown to be dependent on the rise of cytosolic Ca2+ in the proximity of the SR, suggesting that mitochondria may also accumulate some of the Ca2+ released by the SR [55]. The ability of mitochondria to sense Ca2+ in a microdomain rather than the whole cytosol is predicted in modeling studies [56] and can explain the high threshold for Ca2+ uptake by the mCUP (1–3 μM). Indeed, localized Ca2+ sparks (synchronous coordinated activity of 30–100 RyR) elicit miniature mitochondrial matrix Ca2+ signals that last less than 500 ms [55]. The rapid buffering of cytosolic Ca2+ by mitochondria may be due to mCUP activity [57] or may be due to some other mechanism, which remains to be investigated.
E-M coupling is modulated by two messengers, ADP and NADH. The ADP production activates the mitochondrial F1/F0 ATPase to generate more ATP, which consequently leads to the reduction of inner membrane potential. This drop in potential activates NADH production (the second messenger) to supply electrons to compensate for the electron loss and maintain the inner mitochondrial membrane potential [58]. The other modulatory control of mitochondrial metabolism occurs through the mNCX, which is needed to maintain the intramitochondrial Ca2+ concentration. A study examining the influence of cytoplasmic Na+ and mitochondrial potential on mNCX activity has confirmed the role of mNCX as a Ca2+ extrusion mechanism for the mitochondria [59]. In addition, it was demonstrated that mNCX is electrogenic and that depolarization of the mitochondrial membrane activates the mNCX to shuffle Ca2+ into the mitochondria rather than eliminating Ca2+; this seems to be a potential protective mechanism against loss of mitochondrial potential that follows ATP depletion [58]. A third potential mechanism modulating E–M coupling is mediated by ATP-sensitive potassium (KATP) channels. These channels are activated by ADP and inhibited by ATP. It has been shown that KATP channels can be imported into the mitochondria and localize to the inner mitochondrial membrane following phosphorylation by protein kinase C. KATP are believed to have a protective effect against I-R-induced increase in cellular Ca2+ [60].
A rise in mitochondrial Ca2+ is associated with an increase in metabolism [61]. Cardiac pathologies, including CHF, IHD, or potential arrhythmias, lead to an increase in cytosolic Ca2+ and have a deleterious effect on mitochondrial metabolism [5]. In fact, a higher Na+ concentration in the cytosol stimulates the mitochondrial NCX (mNCX), leading to reduced Ca2+ accumulation and reduced activity of mitochondrial function [49]. In cardiomyopathies, the reduced cytoplasmic Ca2+ negatively affects the mitochondrial Ca2+ transient, which is expected to inhibit mitochondrial function. Furthermore, a reduced rate of glycolysis, resulting in reduced availability of pyruvate, contributes to energy starvation in cardiomyopathy [62]. Thus, the mitochondria appear to be a therapeutic target in I-R injury and cardiomyopathies. It has been suggested that inhibition of the mNCX could be beneficial in cases of heart failure as well as during I-R injury.
In heart failure, the increase in cytosolic Na+ activates the forward mode of mNCX to release mitochondrial Ca2+. On the other hand, an increase in the cytosolic Ca2+ will trigger the reverse mode of mNCX and promote the accumulation of excess cytosolic Ca2+ in the mitochondria. As a consequence of its effect, the inhibition of mNCX has been shown to improve Ca2+ accumulation and mitochondrial energetics in isolated cardiomyocytes [62, 63]. Maintaining a delicate balance in mitochondrial Ca2+ content depends on the activities of mCUP and mNCX, where the balance between Ca2+ uptake and Ca2+ extrusion directly affects mitochondrial metabolism. Thus, inhibition of mNCX may be beneficial in preventing I-R injury and heart failure [5]. In addition, various interventions such as trimetazidine, ranolazine, dichloroacetate, carnitine palmitoyl transferase, and coenzyme Q10 have been proposed to prevent ischemic injury via mitochondrial modulation [63].
It is pointed out that current therapies for patients with stable systolic heart failure are largely limited to treatments that interfere with neurohormonal activation. Critical pathophysiological hallmarks of heart failure are an energetic deficit and oxidative stress, and both may be the result of mitochondrial dysfunction. This dysfunction is not only the result of a defect within mitochondria per se, but is in particular related to defects in intermediary metabolism. The regulatory interplay between excitation–contraction coupling and mitochondrial energetics, where defects of cytosolic Ca2+ and Na+ handling in failing hearts may play important roles [64]. The contribution and mechanisms of cardiomyocyte mitochondrial Ca2+ handling in E-C/E-M coupling as well as how mitochondrial Ca2+ regulates physiological mitochondrial and cellular functions in cardiac muscles in health and disease remain to be fully understood [65]. Figure 21.2 summarizes the major components of the E-M coupling process.

Scheme depicting the role of mitochondria in the excitation-metabolism coupling in cardiomyocytes. mCUP mitochondrial Ca2+ uniporter, CaM calmodulin, mNCX mitochondrial Na+-Ca2+ exchanger, RyR ryanodine receptor, SL Sarcolemma, SR sarcoplasmic reticulum
4 Excitation–Transcription Coupling
Ca2+ is a universal regulator of various cellular functions. In cardiomyocytes, Ca2+ is the central element of E–C coupling, but it also exerts a great impact on diverse signaling cascades and influences the regulation of gene expression, referred to as excitation–transcription (E–T) coupling. Disturbances in cellular Ca2+ handling and alterations in Ca2+-dependent gene expression patterns are pivotal characteristics of failing cardiomyocytes, with several -E–T coupling pathways shown to be critically involved in structural and functional remodeling [66]. Electrical activity initiates a program of selective gene expression in excitable cells. Although such transcriptional activation is commonly attributed to depolarization-induced changes in intracellular Ca2+ [67], it should be noted that the specific and localized elevations of Ca2+ that are converted into changes in gene expression can be taken as long-term effects on the adaptation of the heart to a sustained stimulus [68].
The role of Ca2+ in controlling nuclear signaling has been established in some studies [69, 70]. In cardiomyocytes, nuclei are closely associated with SR; the nucleus of adult cardiomyocytes is reported to possess a nucleoplasmic reticulum, a nuclear Ca2+ store that is continuous with SR and the nuclear envelope [33]. The nucleoplasmic reticulum is loaded from cytosolic Ca2+ microdomains [71,72,73,74]. This organelle expresses functional IP3R and RyR. There is evidence that the nucleus contains key components of the phosphoinositide-PLC signaling cascade, where the production of IP3 has been speculated. It is suggested that the nucleus is able to control the effect of Ca2+ on gene expression, allowing nuclear Ca2+ to regulate cellular functions independently of the cytosolic Ca2+ increase [75]. Likewise, IP3 can trigger the release of Ca2+ directly into the nucleoplasm, which may have an important impact on the excitation–transcription process [75]. An increase in nuclear Ca2+ concentration is reported to control the Ca2+-activated gene expression mediated by the cAMP response element [76]. Moreover, cytoplasmic and nuclear Ca2+ signals activate transcription through different pathways. Cytoplasmic Ca2+ signal activates transcription through the serum response element (SRE) transcription factor and does not require an increase in nuclear Ca2+ [77]. Similar to SR, the nucleoplasmic reticulum seems to possess counter-ion channels such as K+ channels [78]; it is plausible that channels control the change in the potential across the nucleoplasm. This would be particularly important as the nucleoplasmic reticulum contains the voltage-gated R-type Ca2+ channels [78]. In addition, there is evidence that the nuclear membrane contains both NCX and NHE that may contribute to nuclear potential and cellular homeostasis.
CaMKII signaling regulates diverse cellular processes in a spatiotemporal manner including excitation–contraction and excitation–transcription coupling, mechanics and energetics in cardiac myocytes. Chronic activation of CaMKII results in cellular remodeling and ultimately arrhythmogenic alterations in Ca2+ handling, ion channels, cell-to-cell coupling, and metabolism [79]. Specific alterations in nuclear Ca2+ handling via altered excitation–transcription coupling contribute to the development and progression of heart failure. During cardiac remodeling, early changes of cardiomyocyte nuclei cause altered nuclear Ca2+ signaling implicated in hypertrophic gene program activation. Normalization of nuclear Ca2+ regulation may therefore be a novel therapeutic approach to prevent adverse cardiac remodeling [80].
The increase in nuclear Ca2+ signal has been closely associated with cardiac hypertrophy. The most well-characterized mechanism is via Ca2+–CaM. This pathway is under the influence of Ca2+–CaM–CaMK on histone deactylase (HDAC) or the effect of Ca2+–CAM on the protein phosphatase calcineurin (CaN); the two ways may act in parallel, contributing to cardiac hypertrophy [51, 81, 82]. It has been shown that depolarization-mediated Ca2+ influx occurs through CaMKII to inhibit the HDAC5, thereby sustaining high activity of the cerebellar granule neuron maintained under myocyte enhancer factor 2 (MEF2) depolarizing conditions. In adult rabbit and mouse cardiomyocytes, phenylephrine and endothelin-1-induced nuclear export of HDAC5 depends not only on CaMK II but also on protein kinase D (PKD) [82]. The nuclear export required type II IP3R, Ca2+ release from stores, CaM, HDAC5 phosphorylation but was completely insensitive to Ca2+ transients associated with both nuclear and cytosolic Ca2+ and PKC inhibition [51, 82]. HDAC class II is expressed in the heart and possesses a unique extension to bind MEF2, repressing its transcription activation; the relief of MEF2 repression by HDAC comes mainly through phosphorylation of HDAC by CaMK [83]. The latter argues for local control of Ca2+ release in the nuclear region, where local activation of nuclear IP3R releases Ca2+ locally that activates CaMKII to phosphorylate HDAC and relieves transcription inhibition. This novel local Ca2+ signaling in excitation–transcription coupling is analogous to, but separate from, that involved in excitation–contraction coupling [51]. It is pointed out that MEF2 activation is strongly implicated in cardiac hypertrophy; this has been shown in transgenic animals overexpressing CaMKII and IV [83]. The link between MEF2 and CaMK is through HDAC [84].
Interestingly, acute activation of exchange protein activated by cyclic-AMP (Epac) has been shown to increase Ca2+ sparks and diastolic [Ca2+]i but decrease systolic [Ca2+]i. Epac preferentially increases intranuclear Ca2+ concentration ([Ca2+]n) during both diastole and systole, correlating with the perinuclear expression pattern of Epac. Moreover, Epac activation induces HDAC5 nuclear export, with consequent activation of the prohypertrophic transcription factor MEF2. The cAMP-binding protein Epac modulates cardiac nuclear Ca2+ signaling by increasing [Ca2+]n through PLC, IP3R, and CaMKII activation and initiates a prohypertrophic program via HDAC5 nuclear export and subsequent activation of the transcription factor MEF2 [85].
Other transcription factors that are regulated by CaMK include activating protein (AP1), activating transcription factor (ATF-1), serum response factor (SRF), cyclic AMP response element (CREB), and the myocyte enhancing factor (MEF-2) [86]. While CREB can be phosphorylated by CaMKII, transgenic mice overexpressing CaMKII or CaMKIV that develop hypertrophy did not show an enhanced level of CREB phosphorylation [84, 87]. MEF2 activation is strongly implicated in hypertrophy; this has been shown in transgenic animals overexpressing CaMKII and IV [88]. The other mechanism by which Ca2+–CaM controls transcription is through CaN, a Ca2+-CaM binding phosphatase (500 times more sensitive to Ca2+ than CaM), allowing CaN to be more sensitive to small sustained Ca2+ transients [89]. CaN dephosphorylates NFAT, which leads to its translocation into the nucleus, where it binds to the transcription factor GATA4 to activate hypertrophic gene transcription [90]. In some studies, a sustained global rise in Ca2+ is needed to activate NFAT [91], whereas in others, Ca2+ oscillations were more efficient NFAT activators [92]. In neurons, the C-terminal fragment of LTCCs was found to be proteolytically cleaved as it translocated into the nucleus; this is then bound to a transcriptional regulator, Ca2+ channel associated transcription (CCAT) factor, leading to an increase in the length of neurites [93, 94]. The latter leads to an increase in some genes such as connexins, regulators of G-protein and catenin, while other proteins such as K+ channel (Kcn3), NCX, myosin, NMDA receptor, serine-threonine kinase, and glucokinase are down-regulated.
While the role of this catalytic fragment in cardiomyocytes remains to be examined, the potential for developing specific inhibitors of nuclear Ca2+ signaling is unlimited. The control of such a mechanism could provide treatments for heart failure and cardiac hypertrophy as well as other conditions where Ca2+ oscillations affect gene expression and consequently the expression of signaling molecules that modulate Ca2+ cycling. It is now well recognized that CaMKII and a Ca-calmodulin-dependent phosphatase calcineurin are major Ca2+-dependent mediators of transcriptional regulation. Moreover, these pathways contribute to changes in the gene expression of proteins involved in the HF phenotype, including some of the ion channels and Ca2+ transporters that are acutely involved in systolic dysfunction and arrhythmias [95]. In fact, the development of intracellular Ca2+ overload in the heart has been demonstrated to depress cardiac gene expression for SL Na+–K+ ATPase isoforms, SR RyR and SERCA2 proteins as well as α- and β-myosin [96] Fig. 21.3 summarizes the role of Ca2+ in E–T coupling in the cardiomyocyte.

Scheme showing the role of Ca2+ in excitation-transcription coupling in the cardiomyocyte. NR nucleoplasmic reticulum, RyR ryanodine receptor, IP3R inositol trisphosphate receptor, SL sarcolemma, LTCC L-type Ca2+-channel, SR sarcoplasmic reticulum, PIP2 phosphatidylinositol-bisphosphate, PLC phospholipase C, DAG diacylglycerol, IP3 inositol trisphosphate, PKC protein kinase C, Raf Rapidly accelerated fibrosarcoma, MEK Mitogen-activated protein kinase kinase, ERK1/2 extracellular signal-regulated kinase 1/2
5 Conclusions
From the aforementioned, it is evident that novel therapeutic interventions will continue to target ion channels; however, the focus may now also be on mitochondrial channels and exchangers in order to influence cellular metabolism and nuclear ion channels to control cell proliferation and growth. Targeting intracellular ion channels for the control of E-C, E-M and E-T coupling has emerged as an attractive new area of cardiovascular research that constitutes novel and exciting approaches for the treatment of IHD, cardiac hypertrophy, and heart failure.
References
Eisner DA, Caldwell JL, Kistamás K, Trafford AW. Calcium and excitation-contraction coupling in the heart. Circ Res. 2017;121:181–95.
Berlin JR, Bassani JW, Bers DM. Intrinsic cytosolic calcium buffering properties of single rat cardiac myocytes. Biophys J. 1994;67:1775–87.
Dhalla NS. Excitation-contraction coupling in heart. I. Comparison of calcium uptake by the sarcoplasmic reticulum and mitochondria of the rat heart. Arch Int Physiol Biochim. 1969;77:916–34.
Alonso MT, Villalobos C, Chamero P, Alvarez J, Garcia-Sancho J. Calcium microdomains in mitochondria and nucleus. Cell Calcium. 2006;40:513–25.
Dhalla NS, Saini-Chohan HK, Rodriguez-Leyva D, Elimban V, Dent MR, Tappia PS. Subcellular remodeling may induce cardiac dysfunction in congestive heart failure. Cardiovasc Res. 2009;81:429–38.
Bers DM. Calcium cycling and signaling in cardiac myocytes. Annu Rev Physiol. 2008;70:23–49.
Dhalla NS, Saini HK, Tappia PS, Sethi R, Mengi SA, Gupta SK. Potential role and mechanisms of subcellular remodeling in cardiac dysfunction due to ischemic heart disease. J Cardiovasc Med. 2007;8:238–50.
Nader M. The SLMAP/Striatin complex: an emerging regulator of normal and abnormal cardiac excitation-contraction coupling. Eur J Pharmacol. 2009;5:858.
Lukyanenko V, Ziman A, Lukyanenko A, Salnikov V, Lederer WJ. Functional groups of ryanodine receptors in rat ventricular cells. J Physiol. 2007;583:251–69.
Meissner G. Molecular regulation of cardiac ryanodine receptor ion channel. Cell Calcium. 2004;35:621–8.
Antos CL, Frey N, Marx SO, Reiken S, Gaburjakova M, Richardson JA, et al. Dilated cardiomyopathy and sudden death resulting from constitutive activation of protein kinase A. Circ Res. 2001;89:997–1004.
Marx SO, Reiken S, Hisamatsu Y, Jayaraman T, Burkhoff D, Rosemblit N, et al. PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell. 2000;101:365–76.
Diaz ME, Graham HK, O’neill SC, Trafford AW, Eisner DA. The control of sarcoplasmic reticulum Ca2+ content in cardiac muscle. Cell Calcium. 2005;38:391–6.
Zalk R, Lehnart SE, Marks AR. Modulation of the ryanodine receptor and intracellular calcium. Annu Rev Biochem. 2007;76:367–85.
Yamamoto T, Yano M, Xu X, Uchinoumi H, Tateishi H, Mochizuki M, et al. Identification of target domains of the cardiac ryanodine receptor to correct channel disorder in failing hearts. Circulation. 2008;117:762–72.
Yano M. Ryanodine receptor as a new therapeutic target of heart failure and lethal arrhythmia. Circ J. 2008;72:509–14.
Lehnart SE, Mongillo M, Bellinger A, Lindegger N, Chen BX, Hsueh W, et al. Leaky Ca2+ release channel/ryanodine receptor 2 causes seizures and sudden cardiac death in mice. J Clin Invest. 2008;118:2230–45.
Dhalla NS, Dent MR, Tappia PS, Sethi R, Barta J, Goyal RK. Subcellular remodeling as a viable target for the treatment of congestive heart failure. J Cardiovasc Pharmacol Therapeut. 2006;11:31–45.
Tada M, Yamada M, Kadoma M, Inui M, Ohmori F. Calcium transport by cardiac sarcoplasmic reticulum and phosphorylation of phospholamban. Mol Cell Biochem. 1982;46:73–95.
Toyoshima C, Nakasako M, Nomura H, Ogawa H. Crystal structure of the calcium pump of sarcoplasmic reticulum at 2.6 Å resolution. Nature. 2000;405:647–55.
Tada M, Yamada M, Inui M, Ohmori F. Regulation of Ca2+-dependent ATPase of cardiac sarcoplasmic reticulum by cAMP- and calmodulin-dependent phosphorylation of phospholamban. Tanpakushitsu Kakusan Koso. 1982;27:2350–64.
James P, Inui M, Tada M, Chiesi M, Carafoli E. Nature and site of phospholamban regulation of the Ca2+ pump of sarcoplasmic reticulum. Nature. 1989;342:90–2.
Asahi M, Nakayama H, Tada M, Otsu K. Regulation of sarco(endo)plasmic reticulum Ca2+ adenosine triphosphatase by phospholamban and sarcolipin: implication for cardiac hypertrophy and failure. Trends Cardiovasc Med. 2003;13:152–7.
Babu GJ, Bhupathy P, Timofeyev V, Petrashevskaya NN, Reiser PJ, Chiamvimonvat N, et al. Ablation of sarcolipin enhances sarcoplasmic reticulum calcium transport and atrial contractility. Proc Natl Acad Sci U S A. 2007;104:17867–72.
Kiewitz R, Acklin C, Schafer BW, Maco B, Uhrik B, Wuytack F, et al. Ca2+ −dependent interaction of S100A1 with the sarcoplasmic reticulum Ca2+-ATPase2a and phospholamban in the human heart. Biochem Biophys Res Commun. 2003;306:550–7.
Remppis A, Most P, Löffler E, Ehlermann P, Bernotat J, Pleger S, et al. The small EF-hand Ca2+ binding protein S100A1 increases contractility and Ca2+ cycling in rat cardiac myocytes. Basic Res Cardiol. 2002;97:I56–162.
Mercadier JJ, Lompre AM, Duc P, Boheler KR, Fraysse JB, Wisnewsky C, et al. Altered sarcoplasmic reticulum Ca2+-ATPase gene expression in the human ventricle during end-stage heart failure. J Clin Invest. 1990;85:305–9.
del Monte F, Williams E, Lebeche D, Schmidt U, Rosenzweig A, Gwathmey JK, et al. Improvement in survival and cardiac metabolism after gene transfer of sarcoplasmic reticulum Ca2+-ATPase in a rat model of heart failure. Circulation. 2001;104:1424–9.
Joubert F, Wilding JR, Fortin D, Domergue-Dupont V, Novotova M, Ventura-Clapier R, et al. Local energetic regulation of sarcoplasmic and myosin ATPase is differently impaired in rats with heart failure. J Physiol. 2008;586:5181–92.
Prasad V, Okunade GW, Miller ML, Shull GE. Phenotypes of SERCA and PMCA knockout mice. Biochem Biophys Res Commun. 2004;322:1192–203.
Mayosi BM, Kardos A, Davies CH, Gumedze F, Hovnanian A, Burge S, et al. Heterozygous disruption of SERCA2a is not associated with impairment of cardiac performance in humans: implications for SERCA2a as a therapeutic target in heart failure. Heart. 2006;92:105–9.
Moschella MC, Marks AR. Inositol 1,4,5-trisphosphate receptor expression in cardiac myocytes. J Cell Biol. 1993;120:1137–46.
Kockskamper J, Zima AV, Roderick HL, Pieske B, Blatter LA, Bootman MD. Emerging roles of inositol 1,4,5-trisphosphate signaling in cardiac myocytes. J Mol Cell Cardiol. 2008;45:128–47.
Hund TJ, Ziman AP, Lederer WJ, Mohler PJ. The cardiac IP3 receptor: uncovering the role of "the other" calcium-release channel. J Mol Cell Cardiol. 2008;45:159–61.
Lakatta EG, Vinogradova TM, Maltsev VA. The missing link in the mystery of normal automaticity of cardiac pacemaker cells. Ann N Y Acad Sci. 2008;1123:41–57.
Jiang QX, Thrower EC, Chester DW, Ehrlich BE, Sigworth FJ. Three-dimensional structure of the type 1 inositol 1,4,5-trisphosphate receptor at 24 a resolution. EMBO J. 2002;21:3575–81.
Ramos-Franco J, Fill M, Mignery GA. Isoform-specific function of single inositol 1,4,5-trisphosphate receptor channels. Biophys J. 1998;75:834–9.
Swatton JE, Morris SA, Cardy TJ, Taylor CW. Type 3 inositol trisphosphate receptors in RINm5F cells are biphasically regulated by cytosolic Ca2+ and mediate quantal Ca2+ mobilization. Biochem J. 1999;344:55–60.
Adkins CE, Morris SA, De SH, Sienaert I, Torok K, Taylor CW. Ca2+-calmodulin inhibits Ca2+ release mediated by type-1, 2 and 3 inositol trisphosphate receptors. Biochem J. 2000;345:357–63.
Bultynck G, Sienaert I, Parys JB, Callewaert G, De SH, Boens N, et al. Pharmacology of inositol trisphosphate receptors. Pflugers Arch. 2003;445:629–42.
Kane C, Couch L, Terracciano CM. Excitation-contraction coupling of human induced pluripotent stem cell-derived cardiomyocytes. Front Cell Dev Biol. 2015;3:59.
Yoshikane H, Nihei T, Moriyama K. Three-dimensional observation of intracellular membranous structures in dog heart muscle cells by scanning electron microscopy. J Submicrosc Cytol. 1986;18:629–36.
Guerrero-Hernandez A, Verkhratsky A. Calcium signalling in diabetes. Cell Calcium. 2014;56:297–301.
Denton RM, McCormack JG. Ca2+ as a second messenger within mitochondria of the heart and other tissues. Annu Rev Physiol. 1990;52:451–66.
Territo PR, French SA, Dunleavy MC, Evans FJ, Balaban RS. Calcium activation of heart mitochondrial oxidative phosphorylation: rapid kinetics of mVO2, NADH AND light scattering. J Biol Chem. 2001;276:2586–99.
Kirichok Y, Krapivinsky G, Clapham DE. The mitochondrial calcium uniporter is a highly selective ion channel. Nature. 2004;427:360–4.
Moreau B, Nelson C, Parekh AB. Biphasic regulation of mitochondrial Ca2+ uptake by cytosolic Ca2+ concentration. Curr Biol. 2006;16:1672–7.
Gunter KK, Gunter TE. Transport of calcium by mitochondria. J Bioenerg Biomembr. 1994;26:471–85.
Maack C, Cortassa S, Aon MA, Ganesan AN, Liu T, O'Rourke B. Elevated cytosolic Na+ decreases mitochondrial Ca2+ uptake during excitation-contraction coupling and impairs energetic adaptation in cardiac myocytes. Circ Res. 2006;99:172–82.
Kwong JQ. The mitochondrial calcium uniporter in the heart: energetics and beyond. J Physiol. 2017;595:3743–51.
Wu X, Zhang T, Bossuyt J, Li X, McKinsey TA, Dedman JR, et al. Local InsP3-dependent perinuclear Ca2+ signaling in cardiac myocyte excitation-transcription coupling. J Clin Invest. 2006;116:675–82.
Gustafsson AB, Gottlieb RA. Heart mitochondria: gates of life and death. Cardiovasc Res. 2008;77:334–43.
Maack C, O'Rourke B. Excitation-contraction coupling and mitochondrial energetics. Basic Res Cardiol. 2007;102:369–92.
Ohata H, Chacon E, Tesfai SA, Harper IS, Herman B, Lemasters JJ. Mitochondrial Ca2+ transients in cardiac myocytes during the excitation-contraction cycle: effects of pacing and hormonal stimulation. J Bioenerg Biomembr. 1998;30:207–22.
Pacher P, Csordas P, Schneider T, Hajnoczky G. Quantification of calcium signal transmission from sarco-endoplasmic reticulum to the mitochondria. J Physiol. 2000;529:553–64.
Cortassa S, Aon MA, Marbán E, Winslow RL, O'Rourke B. An integrated model of cardiac mitochondrial energy metabolism and calcium dynamics. Biophys J. 2003;84:2734–55.
Beutner G, Sharma VK, Lin L, Ryu SY, Dirksen RT, Sheu SS. Type 1 ryanodine receptor in cardiac mitochondria: transducer of excitation-metabolism coupling. Biochim Biophys Acta. 2005;1717:1–10.
Stanley WC, Chandler MP. Energy metabolism in the normal and failing heart: potential for therapeutic interventions. Heart Fail Rev. 2002;7:115–30.
Kim B, Matsuoka S. Cytoplasmic Na+-dependent modulation of mitochondrial Ca2+ via electrogenic mitochondrial Na+-Ca2+ exchange. J Physiol. 2008;586:1683–97.
Light PE, Kanji HD, Fox JE, French RJ. Distinct myoprotective roles of cardiac sarcolemmal and mitochondrial KATP channels during metabolic inhibition and recovery. FASEB J. 2001;15:2586–94.
Brandes R, Bers DM. Simultaneous measurements of mitochondrial NADH and Ca2+ during increased work in intact rat heart trabeculae. Biophys J. 2002;83:587–604.
Kuo TH, Zhu L, Golden K, Marsh JD, Bhattacharya SK, Liu BF. Altered Ca2+ homeostasis and impaired mitochondrial function in cardiomyopathy. Mol Cell Biochem. 2002;238:119–27.
O’Rourke B, Cortassa S, Akar F, Aon M. Mitochondrial ion channels in cardiac function and dysfunction. Novartis Found Symp. 2007;287:140–51.
von Hardenberg A, Maack C. Mitochondrial therapies in heart failure. Handb Exp Pharmacol. 2017;243:491–514.
Cao JL, Adaniya SM, Cypress MW, Suzuki Y, Kusakari Y, Jhun BS, et al. Role of mitochondrial Ca2+ homeostasis in cardiac muscles. Arch Biochem Biophys. 2019;663:276–87.
Dewenter M, von der Lieth A, Katus HA, Backs J. Calcium signaling and transcriptional regulation in cardiomyocytes. Circ Res. 2017;121:1000–20.
Atar D, Backx PH, Appel MM, Gao WD, Marban E. Excitation-transcription coupling mediated by zinc influx through voltage-dependent calcium channels. J Biol Chem. 1995;270:2473–7.
Benitah JP, Gomez AM, Virsolvy A, Richard S. New perspectives on the key role of calcium in the progression of heart disease. J Muscle Res Cell Motil. 2003;24:275–83.
Hamdan M, Urien S, Le LH, Tillement JP, Morin D. Inhibition of mitochondrial carnitine palmitoyltransferase-1 by a trimetazidine derivative, S-15176. Pharmacol Res. 2001;44:99–104.
Mellstrom B, Savignac M, Gomez-Villafuertes R, Naranjo JR. Ca2+-operated transcriptional networks: molecular mechanisms and in vivo models. Physiol Rev. 2008;88:421–49.
Abrenica B, Gilchrist JS. Nucleoplasmic Ca2+loading is regulated by mobilization of perinuclear Ca2+. Cell Calcium. 2000;28:127–36.
Abrenica B, Pierce GN, Gilchrist JS. Nucleoplasmic calcium regulation in rabbit aortic vascular smooth muscle cells. Can J Physiol Pharmacol. 2003;81:301–10.
Echevarria W, Leite MF, Guerra MT, Zipfel WR, Nathanson MH. Regulation of calcium signals in the nucleus by a nucleoplasmic reticulum. Nat Cell Biol. 2003;5:440–6.
Chi TH, Crabtree GR. Perspectives: signal transduction. Inositol phosphates in the nucleus. Science. 2000;287:1937–9.
Zima AV, Bare DJ, Mignery GA, Blatter LA. IP3-dependent nuclear Ca2+ signalling in the mammalian heart. J Physiol. 2007;584:601–11.
Hardingham GE, Chawla S, Johnson CM, Bading H. Distinct functions of nuclear and cytoplasmic calcium in the control of gene expression. Nature. 1997;385:260–5.
Dolmetsch RE, Xu K, Lewis RS. Calcium oscillations increase the efficiency and specificity of gene expression. Nature. 1998;392:933–6.
Bkaily G, Avedanian L, Jacques D. Nuclear membrane receptors and channels as targets for drug development in cardiovascular diseases. Can J Physiol Pharmacol. 2009;87:108–19.
Hegyi B, Bers DM, Bossuyt J. CaMKII signaling in heart diseases: emerging role in diabetic cardiomyopathy. J Mol Cell Cardiol. 2019;127:246–59.
Ljubojevic S, Radulovic S, Leitinger G, Sedej S, Sacherer M, Holzer M, et al. Early remodeling of perinuclear Ca2+ stores and nucleoplasmic Ca2+ signaling during the development of hypertrophy and heart failure. Circulation. 2014;130:244–55.
Bkaily G, Nader M, Avedanian L, Choufani S, Jacques D, D’Orléans-Juste P. G-protein-coupled receptors, channels, and Na+-H+ exchanger in nuclear membranes of heart, hepatic, vascular endothelial, and smooth muscle cells. Can J Physiol Pharmacol. 2006;84:431–41.
Bossuyt J, Helmstadter K, Wu X, Clements-Jewery H, Haworth RS, Avkiran M, et al. Ca2+ /calmodulin-dependent protein kinase II delta and protein kinase D overexpression reinforce the histone deacetylase 5 redistribution in heart failure. Circ Res. 2008;102:695–702.
Wu H, Naya FJ, McKinsey TA, Mercer B, Shelton JM, Chin ER, et al. MEF2 responds to multiple calcium-regulated signals in the control of skeletal muscle fiber type. EMBO J. 2000;19:1963–73.
Zhang T, Kohlhaas M, Backs J, Mishra S, Phillips W, Dybkova N, et al. CaMKIIdelta isoforms differentially affect calcium handling but similarly regulate HDAC/MEF2 transcriptional responses. J Biol Chem. 2007;282:35078–87.
Pereira L, Ruiz-Hurtado G, Morel E, Laurent AC, Métrich M, Domínguez-Rodríguez A, et al. Epac enhances excitation-transcription coupling in cardiac myocytes. J Mol Cell Cardiol. 2012;52:283–91.
Backs J, Song K, Bezprozvannaya S, Chang S, Olson EN. CaM kinase II selectively signals to histone deacetylase 4 during cardiomyocyte hypertrophy. J Clin Invest. 2006;116:1853–64.
McKinsey TA, Zhang CL, Olson EN. Activation of the myocyte enhancer factor-2 transcription factor by Ca2+ /calmodulin-dependent protein kinase-stimulated binding of 14-3-3 to histone deacetylase 5. Proc Natl Acad Sci U S A. 2000;97:14400–5.
Colella M, Grisan F, Robert V, Turner JD, Thomas AP, Pozzan T. Ca2+ oscillation frequency decoding in cardiac cell hypertrophy: role of calcineurin/NFAT as Ca2+ signal integrators. Proc Natl Acad Sci U S A. 2008;105:2859–64.
Molkentin JD. Calcineurin-NFAT signaling regulates the cardiac hypertrophic response in coordination with the MAPKs. Cardiovasc Res. 2004;63:467–75.
Timmerman LA, Clipstone NA, Ho SN, Northrop JP, Crabtree GR. Rapid shuttling of NF-AT in discrimination of Ca2+ signals and immunosuppression. Nature. 1996;383:837–40.
Hallhuber M, Burkard N, Wu R, Buch MH, Engelhardt S, Hein L, et al. Inhibition of nuclear import of calcineurin prevents myocardial hypertrophy. Circ Res. 2006;99:626–35.
Dolmetsch RE, Lewis RS, Goodnow CC, Healy JI. Differential activation of transcription factors induced by Ca2+ response amplitude and duration. Nature. 1997;386:855–8.
Gomez-Ospina N, Tsuruta F, Barreto-Chang O, Hu L, Dolmetsch R. The C terminus of the L-type voltage-gated calcium channel Ca(V)1.2 encodes a transcription factor. Cell. 2006;127:591–606.
Chung HJ, Jan LY. Channeling to the nucleus. Neuron. 2006;52:937–40.
Bers DM. Ca2+-calmodulin-dependent protein kinase II regulation of cardiac excitation- transcription coupling. Heart Rhythm. 2011;8:1101–4.
Ozcelikay AT, Chapman D, Elimban V, Dhalla NS. Role of intracellular Ca2+-overload in inducing changes in cardiac gene expression. Curr Res Cardiol. 2014;1:13–6.
Acknowledgements
The infrastructural support for the work presented in this article was provided by the St. Boniface Hospital Research Foundation. Dr. Nusier is a Visiting Professor from Jordan University of Science and Technology, School of Medicine, Department of Physiology and Biochemistry, Jordan.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2023 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Dhalla, N.S., Nusier, M., Shah, A.K., Tappia, P.S. (2023). Characteristics of Intracellular Ca2+ Handling Proteins in Heart Function in Health and Disease. In: Tripathi, O.N., Quinn, T.A., Ravens, U. (eds) Heart Rate and Rhythm. Springer, Cham. https://doi.org/10.1007/978-3-031-33588-4_21
Download citation
DOI: https://doi.org/10.1007/978-3-031-33588-4_21
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-031-33587-7
Online ISBN: 978-3-031-33588-4
eBook Packages: MedicineMedicine (R0)