
Abstract
Apoptosis is conventionally regarded as an anti-cancer mechanism that eliminates or prevents mutant cell expansion necessary for tumor development and progression. However, evidence for the paradoxical role of apoptosis in tumor progression is accumulating. In this chapter, we describe the mechanisms by which apoptosis serves as a vehicle for accumulating genomic instability to promote malignant progression of tumors, and show a direct association between apoptosis and tumor progression in clinical settings. The negative therapeutic implications of increased apoptosis on clinical outcome and the need to inhibit apoptosis or disable proliferation in apoptotic tumors are discussed.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Genomic instability
- Horizontal transfer
- Cell–cell fusion
- Cancer stem cell
- Comedo-ductal carcinoma in situ
Introduction
Apoptosis represents a major mechanism by which tissue maintains homeostasis. Apoptosis also plays an important role in protecting tissues against tumorigenesis and malignant conversion. Cell loss occurs once tumors have grown more than a few million cells [1, 2]. Intracellular stresses such as hypoxia, nutrient deprivation, telomere shortening, and cell elimination by the host immune system are thought to contribute to steady attrition of a portion of cancer cells in solid tumors [3–5]. A hypothetical link between apoptosis and tumorigenesis has been described by Hanahan and Weinberg [6]. According to their model, the process of tumorigenesis and malignant conversion forces expression of proapoptotic factors that assist with elimination of ‘mutant’ cells and protection from transformation. Thus, mechanisms contributing to acquired resistance to apoptosis serve as a potential mechanism for cell survival and neoplastic conversion.
For cells to acquire the ability for limitless replicative potential or resistance to apoptosis, cells need to uncouple their growth/proliferation programs from environmental signals. It is, however, becoming abundantly clear that there are situations in which tumor cells undergo programmed cell death under conditions of optimal growth stimulation and in the absence of environmental stress. In these tumors, the apoptosis program is wired independently of cell–cell and cell-environmental communications. Interestingly, despite the proficiency for spontaneous cell death, the surviving tumor cells exhibit increased ability for malignant progression. This suggests that the very same process used for protecting cells against malignant conversion can contribute to greater vulnerability to malignancy. In metastatic melanoma, spontaneous regression of individual metastatic deposits is seen throughout the life history of the disease, yet the final outcome is usually death [7]. Similarly, spontaneous regression has been reported in metastatic renal carcinoma, neuroblastoma, colorectal carcinoma, and a wide range of solid tumors [8], suggesting that spontaneous cell loss is not an infrequent event. Yet, despite the loss of tumor cells, the final outcome is not often favorable. The fact that cell loss can occur in vitro under optimal growth and normoxic conditions suggests that cell loss seen in vivo is not necessarily due to nutrient deprivation or hypoxia but rather these tumor cells are intrinsically programmed to undergo apoptosis.
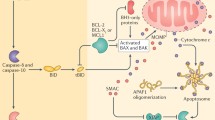
Causes of spontaneous death. Spontaneous loss of cells occurring in the absence of extracellular factors suggests that the fully transformed cells may be intrinsically unstable due to specific genetic abnormalities, and that in the process of achieving stability they undergo apoptotic cell death. Factors responsible for spontaneous apoptosis in tumors are diverse. In some tumors apoptotic cells are found near foci of confluent necrosis suggesting that mild ischemia may contribute to the initiation of apoptosis. The release of cytokines such as TNF-α by infiltrating macrophages is regarded as one of the contributing factors. The extrinsic or death receptor pathway of apoptosis involves stimulation of external surface receptors by FasL and TRAIL leading to activation of caspase-8. In the intrinsic or mitochondrial pathway, proapoptotic signals meet at the mitochondria with resultant loss of the mitochondrial membrane potential, release of cytochrome c and activation of the caspase-9 signaling cascade [9, 10]. The intrinsic pathway is regulated by members of the Bcl-2 family that have either antiapoptotic (Bcl-2, Bcl-xL, Bcl-W, Mcl-1) or proapoptotic (Bax, Bim, Bak, Bid) function. The p53 tumor suppressor protein functions in apoptosis by transcriptional upregulation of genes (e.g., Bax, PUMA) that are directly involved in apoptosis [11, 12]. Both apoptotic pathways converge on the same execution pathway resulting in the activation of caspase-3, DNA fragmentation and formation of apoptotic bodies. Tumor cells exhibit resistance to apoptosis through overexpression of FLIP, reduced expression of CD95, TRAIL DR4 or DR5 receptors, or by overexpression of IAPs or altered expression of the Bcl-2 family members [10].
Paradoxical role of Bcl-2 and Bax in tumor progression. Upregulation of Bcl-2, Bcl-xL, and Mcl-1 are associated with inhibition of apoptosis; however, their expressions do not often correlate strictly with poor clinical prognosis [13, 14]. In breast cancer, Bcl-2 overexpression is associated with normal ploidy, estrogen receptor positivity, and absence of metastasis; all characteristics associated with better clinical outcome and a more favorable prognosis that is contradictory to its predicted role in apoptosis resistance [15, 16]. In colorectal adenoma, Bcl-2 levels are decreased compared to the adjacent normal tissue [17]. Similar data are reported in cervical, prostate, and endometrial cancers [18]. Overexpression of Mcl-1 is associated with poor prognosis for lung, and head and neck cancers [17–20]. Thus, there are malignancies in which Bcl-2 family member overexpression correlates with favorable prognosis and those in which it is correlative of high tumor grade. Paradoxically, elevated expressions of the proapoptotic proteins Bax and Bak have been associated with poor prognosis in esophageal carcinoma and bladder cancer, respectively [21–23]. In breast tumors, an increase in the proportion of apoptotic cells was observed in recurrent tumors as compared with primary lesions, and patients with tumors with higher apoptotic indices were associated with shorter survival [24]. Several examples of a positive correlation between the apoptotic index and the tumor grade have been reported [25–27]. The role of apoptosis suppression as a vehicle for enhancing genomic instability was tested by measuring the effects of Bcl-2 overexpression on the frequency of CAD gene amplification in cells exposed to the CAD inhibitor PALA. Bcl-2 overexpression failed to increase the frequency of CAD gene amplification. However, similar analysis on cells expressing mutant p53 showed enhanced frequency of CAD gene amplification. These data suggest that although both p53 inactivation and Bcl-2 overexpression suppress apoptosis, apoptosis inhibition by Bcl-2 overexpression does not make the cells genetically unstable [28]. These data are consistent with low grade tumors produced by Bcl-2 overexpressing cells, further confirming that apoptosis inhibition by Bcl-2 overexpression does not favor selection of tumor cell variants to increase genomic instability [28].
Apoptosis as a mechanism for driving genomic instability. Reduced rates of apoptosis correlate with fewer cell divisions and, hence, the possibility of generating fewer mutant clones. On the other hand, higher rates of apoptosis would require increased rates of cell division to compensate for cell loss, and consequently lay the foundation for accruing genomic instability [29, 30]. As discussed above, Bcl-2 overexpression and p53 loss both act by blocking apoptosis; however, only the latter enhances accumulation of genomic instability. In this context, it is interesting to note that Bcl-2 overexpression and mutant p53 are rarely coexpressed in a tumor. Bcl-2 overexpression was found in breast and head and neck cancers expressing wild type p53 [13], [31–34], whereas breast tumors with p53 accumulation (indicative of mutant p53 expression) showed low Bcl-2 expression [31, 35]. p53 is inactivated late in most cancers, suggesting that loss of p53 function in later stages of tumorigenesis may translate into higher rates of apoptosis and consequently higher rates of proliferation during the course of tumor development that would enable expansion and accumulation of mutant clones favoring malignant progression. Clinical data show that noncomedo-ductal carcinoma in situ (DCIS) breast cancers express normal levels of Bcl-2 compared to comedo-DCIS that express weak or negligible Bcl-2, strong Bax and mutant p53. Accordingly, comedo-DCIS lesions are characterized by high apoptotic and mitotic indices [36], and are at greater risk for recurrence and malignant progression compared to noncomedo-DCIS. Evidence for a direct link between increased apoptosis and tumorigenesis was recently demonstrated with a mouse model harboring the loxP-targeted allele of Mcl-1 and albumin promoter-driven Cre-recombinase. Hepatocytes of Mcl-1 fl/fl-AlbCre mice lacking Mcl-1 exhibited increased apoptosis and spontaneously developed hepatocellular carcinoma-like lesions at >50 % higher incidence [37]. These data suggest that apoptosis and proliferation work in concert to drive malignant progression.
Mechanisms contributing to tumor progression in apoptotic tumors. Apoptosis allows elimination of unwanted or unstable cells by processing them into an array of smaller bodies called apoptotic bodies by cellular blebbing and fragmentation. Apoptotic bodies have intact plasma membrane and adequate energy supply to maintain their membrane integrity. Cells undergoing apoptosis and apoptotic bodies expose phosphatidylserine on their surface that promote recognition by professional (macrophages, dendritic cells, B-lymphocytes) and nonprofessional (epithelial cells and fibroblasts) phagocytes, resulting in quick ingestion and lysosomal degradation [38]. It is generally believed that apoptosis does not induce inflammation because the apoptotic bodies are rapidly cleared to prevent release of harmful immunogenic materials from the dying cells [39]. We posit that the clearance rates of apoptotic bodies and the presence of associated inflammation will depend upon the extent and rate of apoptosis. Tumor tissues undergoing massive apoptotic cell loss such as comedo-DCIS breast cancers are generally associated with a strong inflammatory response. It is possible that these cells are recruited to mop-up the overwhelming amounts of released apoptotic bodies, or that the slow clearing of apoptotic bodies induces the inflammatory response. Either way, a tumor microenvironment rich in inflammatory cells would promote tumor progression. Additionally, slow or incomplete removal of apoptotic bodies would result in the accumulation of released genetic material into the cellular milieu and, thus, trigger the horizontal transfer or uptake of the DNA by neighboring tumor or stromal cells.
Horizontal transfer of genetic material via cell–cell fusion. Horizontal transfer of genes has been reported in bacteria and fungi and plays an important role in the generation of antibiotic resistance and the adaptation to new environments [40]. Thus, horizontal transfer of genes represents a powerful mechanism for bacterial diversification. Cell–cell fusion is considered to play an important role in horizontal transmission of genes and malignant transformation [41–43]. Transfer of genetic information could occur via fusions between tumor and stromal cells or between tumor cells. Human glioblastoma grafted to hamster cheek pouches produced hybrid human/hamster tumors in which human chromosome segregation occurred within the first transplant generation and showed widespread metastasis. At least seven genes from the six human chromosomes were retained of which three genes implicated in oncogenesis (CD7, CXCR4 and PLAGL2) showed continued expression [44]. CXCR4 (also called fusin), a G-protein coupled chemokine receptor for SDF-1 has been implicated in proliferation, motility, homing and metastasis of cancer cells and is associated with regions of cell death and angiogenesis. These data suggest that in vivo stability of the resulting hybrids depends upon the selective growth advantage provided by the DNA taken up.
Horizontal transfer of DNA via apoptotic conversion. There is increasing evidence that even cell-free cancer DNA can be transferred to induce malignancy. Holmgren et al. [45] demonstrated that DNA can be transferred from apoptotic cells to recipient neighboring cells by phagocytosis. Cocultivation of cell lines containing integrated copies of the Epstein-Barr virus (EBV) resulted in rapid a uptake of EBV DNA to the nucleus of the phagocytosing cell. Once transferred, the expression of EBV encoded genes was detected at both the mRNA and protein levels. Similarly, apoptotic bodies derived from c-Ha-ras Val [12] and c-myc transformed rat embryo fibroblasts (REFs) were able to transform p53-/- or p21-/- mouse embryo fibroblasts (MEFs) but not wild type MEFs [46, 47]. FISH analysis confirmed that entire chromosomes are transferred from the apoptotic bodies of REFs and become integrated into the mouse host genome. However, the stability of the integrated DNA can be maintained only if it confers a selective growth advantage to the recipient cell. Following uptake of the apoptotic genetic material, normal recipient cells with the activated Chk2/p53/p21 DNA damage response pathway block replication of the transferred DNA, thereby protecting them from the potentially harmful effects of the apoptotic DNA [48]. Similar uptake of apoptotic bodies with resultant acquisition of and propagation of drug resistance genes has been demonstrated in prostate cancer cells [49]. Since p53 is lost in most cancers, horizontal transfer of genetic material from the dying tumor cells to recipient tumor cells may serve as a driving force for accumulation of genomic instability and high mutability of tumor cells. Apoptotic DNA from the dying tumor cells can also be transferred to recipient stromal cells (fibroblasts, endothelial cells, macrophages). Such transfers could actively modify the structure and behavior of the surrounding microenvironment, and potentially provide an explanation for pro-tumorigenic and pro-metastatic properties of the tumor microenvironment. Apoptotic DNA was detected in the nuclei of ~15 % of the phagocytosing cells [45], suggesting that horizontal DNA transfer is an efficient mode of enhancing genomic diversity of tumor cells that is dictated by the rate and extent of apoptosis in the tumor. In contrast, mutation of specific genes is an inefficient process that requires amplification of the mutated cells and is limited by activities of surveillance mechanisms that monitor and maintain the genomic integrity. It is possible that the fluidity of the cell membrane is increased in cells sensitive to apoptosis, making it more receptive for cell fusion and DNA transfer. Given the manner in which even highly differentiated epithelial cells are stimulated to become phagocytic by the proximity of an apoptotic body, it is one of the less studied but more remarkable components of apoptosis [48, 50]. Horizontal DNA transfer or apoptotic conversion may be clinically important particularly in tumors characterized by high spontaneous apoptosis such as comedo-DCIS breast cancer since these tumor cells also express mutant p53.
Genetic exchanges occurring either via cell–cell fusions or by apoptotic conversion can provide the residual tumor with new attributes for survival, growth, progression and metastasis. It is possible that the phenotypic and genotypic diversity (or heterogeneity) observed in cells within a tumor, between primary and recurrent tumors, primary and metastatic tumors, and/or between metastases of the same tumor arise at least in part by horizontal transmission of genes and gene products by cell–cell fusions or direct incorporation of apoptotic DNA into cancer or stromal cells.
Cancer stem cell activation. Recent studies have invoked the role of cancer stem cells to explain the relationship between enhanced apoptosis and tumor progression. It has been proposed that in tumors with high apoptotic rates, the dying cells may free up space for cancer stem cells to proliferate into and populate the tumor [51, 52]. To determine the association between apoptosis and increased tumor growth/progression, Enderling et al. [53] simulated tumor development for different spontaneous cell death rates, and initialized each simulation with one cancer stem cell, stopping the simulation after 35 months or when the tumor reached confluence. When random cell death among tumor cells was increased, an increase in the number of stem cells was observed [53]. While spontaneous cell death can reduce the number of tumor cells in the short run, they can facilitate sufficient symmetric stem cell divisions to enrich the stem cell pool and ultimately promote malignant expansion [54]. These findings have therapeutic implications since conventional anti-cancer therapies are directed towards eradicating apoptosis-sensitive tumor bulk populations while sparing the therapy of resistant cancer stem cells. Thus, the accelerated tumor recurrence following therapy may be explained by the opportunistic proliferation of quiescent tumor cells into the space made available by the initial killing. This treatment recovery cycle could favor the creation of new stem cells by symmetrical division of previously quiescent stem cells as the latter are considered to be more resistant to radiation [55] and chemotherapy [56] compared to their nonstem counterparts [53]. As the tumor returns to its pretreatment size, the tumor could become more refractory to treatment. This notion is supported by observations that high rates of apoptosis in cancer cells correlate with tumor progression [30], whereas upregulation of anti-apoptotic factors suppresses tumor progression and improves prognosis [18, 28]. Alternatively, apoptosis may accelerate tumor generation or progression by preferentially eliminating cells that retain normal apoptosis sensitivity while sparing apoptosis-resistant mutant cells for expansion of mutant clones [57, 58].
A prototype clinical disease depicting pertinence of apoptosis adverse effects. The relevance of detrimental effects of apoptosis to clinical settings is best illustrated with comedo-type ductal carcinoma in situ. Among the several ductal carcinoma in situ (DCIS) subtypes of preinvasive breast cancer, comedo type DCIS or comedo-DCIS accounts for ~10 % of all DCIS and confers the greatest risk for progression and post-operative recurrence [59, 60]. Comedo-DCIS tumors are easily distinguished from other DCIS by the characteristic central comedo-necrosis [61] that results from extensive spontaneous apoptosis [62]. Yet despite abundant cell loss, comedo-DCIS often demonstrates microinvasion, chromosome aneuploidy, and higher proliferation and recurrence rates compared to non-comedo DCIS tumors [63–65]. MCF10DCIS.com human breast cancer cells produce tumors that resemble clinical comedo-DCIS and recapitulate the temporal sequence of progression from in situ to invasive cancer [62, 66, 67]. Using the MCF10DCIS.com model, we have demonstrated that spontaneous apoptosis contributes to the etiology and progression of comedo-DCIS [62] (Fig. 1). Spontaneous MCF10DCIS.com cell loss is activated by the mitochondrial pathway with upregulation of Bax, decreases in Bcl-2 and loss of p53 [62]. Clinical comedo-DCIS, like MCF10DCIS.com cells, show a significant drop in Bcl-2 expression combined with an increase in mutant p53 levels [36]. MCF10DCIS.com cells undergo spontaneous apoptosis in vitro under optimal growth conditions indicating that cell loss in vivo is not due to extraneous factors, but rather they are preprogrammed to undergo apoptosis [62]. The high rates of apoptosis in comedo-DCIS are accompanied by compensatory increases in PCNA-positive cells that are enriched for the CD44+/CD24− phenotype (Fig. 2). CD44+/CD24− cells have tumor-initiating properties in breast cancer [68]. The CD44+/CD24− phenotype is associated with stem cell-like characteristics [69], enhanced potential for invasion [70], radiation resistance [71] and with distinct genetic profiles that correlate with adverse prognosis [72]. These data corroborate that abundant cell loss in comedo-DCIS provides increased opportunities for selecting cell variants with greater malignancy potential. The presence of an intact myoepithelial layer is indicative of DCIS lacking invasive potential. Interestingly, apoptosis is also implicated in the initiation of progression of clinical and MCF10DCIS.com-derived comedo-DCIS tumors as both luminal epithelial and myoepithelial cells are concurrently eliminated by apoptosis [62] (Fig. 2). These data provide clinical support for the adverse effects of apoptosis in neoplastic progression and show that apoptosis is tightly linked with the coordinated and concerted events that lead up to malignant progression (summarized in Fig. 3).

Progression of MCF10DCIS.com to comedo-DCIS occurs by high rates of spontaneous apoptosis of luminal epithelial and myoepithelial cells. Panels a, a′: early DCIS lesions; b, b′: comedo-DCIS lesions; c, c′: MCF10DCIS.com multicellular tumor spheroids undergoing spontaneous apoptosis in vitro. Panels a and c, H&E staining; b, silver staining; a′–c′, TUNEL staining. Thin and thick arrows in b′ show apoptotic luminal epithelial and myoepithelial cells, respectively

Progression of MCF10DCIS.com-derived comedo-DCIS lesions is accompanied by increase in PCNA-positive and CD44+ (CD24−negative, not shown) cells. Panels a, a′ PCNA expression in early and comedo-DCIS lesions, respectively. Note the presence of cells with uniform nuclei in early lesions (panel a) compared to cells with larger or pleiomorphic nuclei in the vicinity of the apoptotic core (indicated by arrow) of advanced lesions (panel a′). Panels b, b′ CD44 expression. Note low CD44 expression in early lesion (thin arrow in b) and strong CD44 expression in advanced comedo-DCIS (thick arrow in b and b′). Panels c, c′ H&E and PCNA staining, respectively, of MCF10DCIS.com multicellular tumor spheroids generated in vitro. Note the appearance of large nucleated cells in the apoptotic core (arrow in c) with PCNA expression (c′). Panels d–e cell–cell fusion propensity. MCF10DCIS.com cells were prelabeled with DiI (red) or DiO (green) and cocultured. Note the formation of fused cells in panel d′ and e

Schematic model depicting the role of high apoptosis in malignant progression using the comedo-DCIS model of breast cancer
Therapeutic implications. The majority of anti-cancer therapies work by inducing apoptosis in tumors. In tumors undergoing high rates of natural (spontaneous) or therapy-induced apoptosis, increased horizontal transfer of genetic material by cell–cell fusions or apoptotic conversion, cancer stem cell activation, and/or associated inflammatory response may all contribute to the adverse effects of chemo- and radio-therapy, viz., initial tumor regression that is accompanied by quicker relapse, a greater tumor burden and therapy resistance [73]. Thus, it would seem that using therapies that induce apoptosis in tumors experiencing high rates of spontaneous apoptosis is counterintuitive. Inhibiting apoptosis in these tumor tissues may be beneficial as it could decrease compensatory proliferation of surviving tumor cells, decrease activation and expansion of initiated or cancer stem cells, decrease acquisition of genetic instability by inhibiting horizontal DNA transfer and reduce inflammation. Taking into account the adverse effects of apoptosis in malignant transformation and progression, it would seem that disabling proliferation of stem and nonstem cells in “apoptotic tumors” may be necessary to achieve a longer lasting clinical response.
Conclusion
Contrary to the popular belief that apoptosis acts to safeguard cells from neoplastic conversion and progression, apoptosis must also be viewed as a “not so innocent” participant that actively promotes tumor progression. The latter paradoxical role becomes relevant to tumors experiencing high rates of natural (spontaneous) apoptosis or therapy-induced apoptosis, and must be taken into account when therapy decisions are made.
Abbreviations
- CAD gene:
-
Trifunctional protein with carbamoyl-phosphate synthetase 2, aspartate transcarbamylase, and dihydroorotase activities
- DCIS:
-
Ductal carcinoma in situ
- EBV:
-
Epstein-Barr virus
- REF:
-
Rat embryo fibroblast
- MEF:
-
Mouse embryo fibroblast
- IAP:
-
Inhibitors of apoptosis
- PUMA:
-
P53 upregulated modulator of apoptosis
- FLIP:
-
FADD-like IL-1β-converting enzyme)-inhibitory protein
- TRAIL:
-
TNF-related apoptosis-inducing ligand
- DR4, DR5:
-
Death receptors
References
Steel GG. Cell loss from experimental tumors. Cell Tissue Kinet. 1968;1:193–207.
Bertuzzi A, Gandolfi A, Sinisgalli C, Starce G, Ubezio P. Cell loss and the concept of potential doubling time. Cytometry. 1997;29:34–40.
Kerr J, Winterford C, Harmon B. Apoptosis, Its significance in cancer and cancer therapy. Cancer. 1994;73:2013–23.
Elston D. Mechanisms of regression. Clin Med Res. 2004;2:85–8.
Smyth M, Kershaw M. Discovery of an innate cancer resistance gene. Mol Intervent. 2003;3:186–9.
Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70.
McGovern V. Spontaneous regression of melanoma. Pathology. 1975;2:91–9.
Chang W. Complete spontaneous regression of cancer: four case reports, review of literature and discussion of possible mechanisms involved. Haw Med J. 2000;59:379–87.
Green DR, Reed JC. Mitochondria and apoptosis. Science. 1998;281:1309–12.
Fulda S. Inhibitor of apoptosis proteins as targets for anticancer therapy. Expert Rev Anticancer Ther. 2007;7:1255–64.
Miyashita T, Reed JC. Tumor suppressor p53 is a direct transcriptional activator of the bax gene. Cell. 1995;80:293–9.
Nakano K, Vousden KH. PUMA, a novel proapoptotic gene, is induced by p53. Mol Cell. 2001;7:683–94.
Joensuu H, Pylkkanen L, Toikkanen S. Bcl-2 protein expression and long term survival in breast cancer. Am J Pathol. 1994;145:1191–8.
Hamilton A, Piccart M. The contribution of molecular markers to the prediction of response in the treatment of breast cancer: a review of the literature on Her-2, p53, and BCL-2. Ann Oncol. 2000;11:647–63.
Kobayashi S, Iwase H, Ito Y, Yamashita H, Iwata H, Yamashita T, Ito K, Toyama T, Nakamura T, Masaoka A. Clinical significance of bcl-2 gene expression in human breast cancer tissues. Breast Cancer Res Treat. 1997;42:173–81.
Inada T, Kikuyama S, Ichikawa A, Igarashi S, Ogata Y. Bcl-2 expression as a prognostic factor of survival of gastric carcinoma. Anticancer Res. 1998;18:2003–10.
Krajewska M, Moss SF, Krajevski S, Song K, Holt PR, Reed JC. Elevated expression of Bcl-X and reduced Bak in primary colorectal adenocarcinomas. Cancer Res. 1996;56:2422–7.
Gurova KV, Gudkov AV. Paradoxical role of apoptosis in tumor progression. J Cell Biochem. 2003;88:128–37.
Hotz MA, Bosq J, Zbaeren P, Reed J, Schwab G, Krajewski S, Brousset P, Borner MM. Spontaneous apoptosis and the expression of p53 and Bcl-2 family proteins in locally advanced head and neck cancer. Arch Otolaryngol Head Neck Surg. 1999;125:417–22.
Eerola AK, Ruokolainen H, Soini Y, Raunio H, Paakko P. Accelerated apoptosis and low bcl-2 expression associated with neuroendocrine differentiation predict shortened survival in operated large cell carcinoma of the lung. Pathol Oncol Res. 1999;5:179–86.
Haitek A, Posch B, El-Baz M, Mokhtar AA, Susani M, Ghoneim MA, Marberger M. Bilharzial related, organ confined, muscle invasive bladder cancer: Prognostic value of apoptotic markers, p53, E-cadherin, epidermal growth factor receptor and c-erbB-2. J Urol. 2001;165:1481–7.
Kurabayashi A, Furihata M, Matsumoto M, Ohtsuki Y, Sasaguri S, Ogoshi S. Expression of Bax and apoptosis-related proteins in esophageal squamous cell carcinoma including dysplasia. Mod Pathol. 2001;14:741–7.
Takayama T, Nagao M, Sawada H, Yamada Y, Emoto K, Fujimoto H, Ueno M, Hirao S, Nakajima Y. Bcl-X expression in esophageal squamous cell carcinoma: association with tumor progression and prognosis. J Surg Oncol. 2001;78:116–23.
Vakkala M, Lahteenmaki K, Raunio H, Paakko P, Soini Y. Apoptosis during breast carcinoma progression. Clin Cancer Res. 1999;5:319–24.
Tanji N, Yokoyama M, Sugamoto T, Takeuchi M, Terada N. Apoptosis in prostatic adenocarcinomas: a study of relationship to Ki-67 and Bcl-2 protein expression. Anticancer Res. 1998;18:1111–6.
Li L, Yan L, Wang Z, Liu Z, Wei Y, Huang G. Change of apoptotic status in human colorectal adenoma-carcinoma stage sequences and its correlation with carcinogenesis and prognosis. Clin Med J. 2000;113:886–8.
Sjostrom J, Bergh J. How apoptosis is regulated, and what goes wrong in cancer. BMJ. 2001;322:1538–9.
Gurova KV, Kwek SS, Koman IE, Kamarov AP, Kandel E, Nikiforov MA, Gudkov AV. Apoptosis inhibitor as a suppressor of tumor progression: expression of Bcl-2 eliminates selective advantages for p53-deficient cells in the tumor. Cancer Biol Ther. 2002;1:39–44.
Nowell PC. The clonal evolution of tumor cell populations. Science. 1976;194:23–8.
Wodarz D, Komarova N. Can loss of apoptosis protect against cancer? Trends Genet. 2007;23:232–7.
Berardo MD, Elledge RM, de Moor C, Clark GM, Osborn CK, Allred C. Bcl-2 and apoptosis in lymph node positive breast carcinoma. Cancer. 1998;82:1296–302.
Castiglione F, Sarotto I, Fontana V, Destifanis M, Venturino A, Ferro S, Cardaropoli S, Orengo MA, Porcile G. Bcl-2, p53 and clinical outcome in series of 138 operable breast cancer patients. Anticancer Res. 1999;19:4555–63.
Tete S, Pappalardo S, Fioroni M, Salini L, Imperatrice AM, Perfetti G. Bcl-2, p53, Ki-67 and apoptotic index in cancerous and precancerous lesions of the oral mucosa. Minerva Stomatol. 1999;48:419–25.
Lazaris AC, Lenardi I, Kavantzas N, Kandiloros D, Adamapoulos G, Davaris P. Correlation of tumor markers p53, bcl-2 and cathepsin-D with clinicopathological features and disease-free survival in laryngeal squamous cell carcinoma. Pathol Int. 2000;50:717–24.
van Slooten HJ, van de Vijver MJ, van de Velde CJ, van Dierendonck JH. Loss of bcl-2 in invasive breast cancer is associated with high rates of cell death, but also with increased proliferative activity. Br J Cancer. 1998;77:789–96.
Megha T, Ferrari F, Arcuri F, Lalinga AV, Lazzi S, Cardone C, Cevenini G, Leoncini L, Tosi P. Cellular kinetics and expression of bcl-2 and p53 in ductal carcinoma of the breast. Oncol Rep. 2000;7:473–8.
Weber A, Boger R, Vick B, Urbanik T, Haybaeck J, Zoller S, Teufel A, Krammer PH, Opferman JT, Galle PR, Schuchmann M, Heikenwalder M, Schulze-Bergkamen H. Hepatocyte-specific deletion of the anti-apoptotic protein Mcl-1 triggers proliferation and hepatocarcinogenesis in mice. Hepatology. 2010;51:1226–36.
Grimsley C, Ravichandran KS. Cues for apoptotic cell engulfment: eat-me, don’t eat-me and come-get-me signals. Trends Cell Biol. 2003;13:648–56.
Ren Y, Savill J. Apoptosis: the importance of being eaten. Cell Death Differ. 1998;5:563–8.
Akiba T, Koyama K, Ishiki Y, Kimura S, Fukushima T. On the mechanism of the development of multiple-drug-resistant clones of Shigella. Jpn J Microbiol. 1960;4:219–27.
Goldenberg DM. Ü ber die Progression der Malignita ät: Eine Hypothese [on the progression of malignancy: a hypothesis]. Klin Wochenschr. 1968;46:898–9.
Goldenberg DM, Pavia RA, Tsao MC. In vivo hybridization of human tumour and normal hamster cells. Nature. 2012;250:649–51.
Goldenberg DM, Pavia RA. Horizontal transmission of malignant conditions rediscovered. New Engl J Med. 1981;305:283–4.
Goldenberg DM, Zagzag D, Heselmeyer-Haddad KM, Berroa Garcia LY, Ried T, Loo M, Chang CH, Gold DV. Horizontal transmission and retention of malignancy, as well as functional human genes, after spontaneous fusion of human glioblastoma and hamster host cells in vivo. Int J Cancer. 2012;131:49–58.
Holmgren L, Szeles A, Rajnavölgyi E, Folkman J, Klein G, Ernberg I, Falk KI. Horizontal transfer of DNA by the uptake of apoptotic bodies. Blood. 1999;93:3956–63.
Bergsmedh A, Szeles A, Henriksson M, Bratt A, Folkman MJ, Spetz AL, Holmgren L. Horizontal transfer of oncogenes by uptake of apoptotic bodies. Proc Natl Acad Sci. 2001;98:6407–11.
Bergsmedh A, Szeles A, Spetz AL, Holmgren L. Loss of the p21(Cip1/Waf1)cyclin kinase inhibitor results in propagation of horizontally transferred DNA. Cancer Res. 2002;62:575–9.
Bergsmedh A, Ehnfors J, Kawane K, Motoyama N, Nagata S, Holmgren L. DNase II and the chk2 DNA damage pathway form a genetic barrier blocking replication of horizontally transferred DNA. Mol Cancer Res. 2006;4:187–95.
de la Taille A, Chen MW, Burchardt M, Chopin DK, Buttyan R. Apoptotic conversion: evidence for exchange of genetic information between prostate cancer cells mediated by apoptosis. Cancer Res. 1999;59:5461–3.
Chakraborty AK, Sodi S, Rachkovsky M, Kolesnikova N, Platt JT, Bolognia JL, Pawelek JM. A spontaneous murine melanoma lung metastasis comprised of host x tumor hybrids. Cancer Res. 2000;60:2512–9.
Ehemann V, Sykora J, Vera-Delgado J, Lange A, Otto HF. Flow cytometric detection of spontaneous apoptosis in human breast cancer using the TUNEL-technique. Cancer Lett. 2003;194:125–31.
Meggiato T, Calabrese F, Valente M, Favaretto E, Baliello E, Del Favero G. Spontaneous apoptosis and proliferation in human pancreatic cancer. Pancreas. 2000;20:117–22.
Enderling H, Anderson ARA, Chaplain MAJ, Behesti A, Hlatky L, Hahnfeldt P. Paradoxical dependencies of tumor dormancy and progression on basic cell kinetics. Cancer Res. 2009;69:8814–21.
Lynch MD. The role of cellular senescence may be to prevent proliferation of neighboring cells within stem cell niches. Ann NY Acad Sci. 2004;1019:191–4.
Diehn M, Cho RW, Lobo NA, Kalisky T, Dorie MJ, Kulp AN, Qian D, Lam JS, Ailles LE, Wong M, Joshua B, Kaplan MJ, Wapnir I, Dirbas F, Somlo G, Garberoglio C, Paz B, Shen J, Lau SK, Quake SR, Brown JM, Weissman IL, Clarke MF. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature. 2009;458:780–3.
Gangemi R, Paleari L, Orengo AM, Cesario A, Chessa L, Ferrini S, Russo P. Cancer stem cells: a new paradigm for understanding tumor growth and progression and drug resistance. Curr Med Chem. 2009;16:1688–703.
Ziegler A, Jonason AS, Leffell DJ, Simon JA, Sharma HW, Kimmelman J, Remington L, Jacks T, Brash DE. Sunburn and p53 in the onset of skin cancer. Nature. 1994;372:773–6.
Graeber TG, Osmanian C, Jacks T, Housman DE, Koch CJ, Lowe SW, Giaccia AJ. Hypoxia-mediated selection of cells with diminished apoptotic potential in solid tumors. Nature. 1996;379:88–91.
Fisher ER, Land SR, Saad RS, Fisher B, Wickerham DL, Wang M, et al. Pathologic variables predictive of breast events in patients with ductal carcinoma in situ. Am J Clin Pathol. 2007;128:86.
Yang M, Moriya T, Oguma M, Cruz CDL, Endoh M, Ishida T, Hirakawa H, Orita Y, Ohuchi N, Sasano H. Microinvasive ductal carcinoma (T1mic) of the breast. The clinicopathological profile and immunohistochemical features of 28 cases. Pathol Int. 2003;53:422–8.
Moinfar F, Mannion C, Man YG, Tavassoli FA. Mammary “comedo”-DCIS: apoptosis, oncosis, and necrosis: an electron microscopic examination of 8 cases. Ultrastruct Pathol. 2000;24:135.
Shekhar MP, Tait L, Pauley RJ, Wu GS, Santner SJ, Nangia-Makker P, Shekhar V, Nassar H, Visscher DW, Heppner GH, Miller FR. Comedo-ductal carcinoma in situ: a paradoxical role for programmed cell death. Cancer Biol Ther. 2008;7:1774.
Jaffer S, Bleiweiss IJ. Histologic classification of ductal carcinoma in situ. Microsc Res Tech. 2002;59:92.
Aasmundstad T, Haugen O. DNA ploidy in intraductal breast carcinomas. Eur J Cancer. 1992;26:956.
Meyer J. Cell kinetics of histologic variants of in situ breast carcinoma. Breast Cancer Res Treat. 1986;7:171.
Miller FR, Santner SJ, Tait L, Dawson PJ. MCF10DCIS. com xenograft model of human comedo ductal carcinoma in situ. J Nat Cancer Inst. 2000;92:1185.
Tait LR, Pauley RJ, Santner SJ, Heppner GH, Heng HH, Rak JW, et al. Dynamic stromal-epithelial interactions during progression of MCF10DCIS.com xenografts. Int J Cancer. 2007;120:2127.
Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci USA. 2003;100:3983–8.
Ponti D, Costa A, Zaffaroni N, Pratesi G, Petrangolini G, Coradini D, Pilotti S, Pierotti MA, Daidone MG. Isolation and in vitro propagation of tumorigenic breast cancer cells with stem/progenitor cell properties. Cancer Res. 2005;65:5506–11.
Sheridan C, Kishimoto H, Fuchs RK, Mehrotra S, Bhat-Nakshatri P, Turner CH, Goulet R Jr, Turner CH, Badve S, Nakshatri H. CD44+/CD24− breast cancer cells exhibit enhanced invasive properties: an early step necessary for metastasis. Breast Cancer Res. 2006;8:R59.
Phillips TM, McBride WH, Pajonk F. The response of CD24(-/low)/CD44+ breast cancer-initiating cells to radiation. J Natl Cancer Inst. 2006;98:1777–85.
Shipitsin M, Campbell LL, Argani P, Weremowicz S, Bloushtain-Qimron N, Yao J, Nikolskaya T, Serebryiskaya T, Beroukhim R, Hu M, Halushka MK, Sukumar S, Parker LM, Anderson KS, Harris LN, Garber JE, Richardson AL, Schnitt SJ, Nikolsky Y, Gelman RS, Polyak K. Molecular definition of breast tumor heterogeneity. Cancer Cell. 2007;11:259–73.
Michor F, Hughes TP, Iwasa Y, Branford S, Shah NP, Sawyers CL, Nowak MA. Dynamics of chronic myeloid leukemia. Nature. 2005;435:1267–70.
Acknowledgments
Supported by U.S. Army Medical Research and Materiel Command DAMD17-02-1-0618.
Conflicts of Interest
No potential conflicts of interest were disclosed.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer Science+Business Media New York
About this chapter
Cite this chapter
Shekhar, M.P.V. (2013). The Dark Side of Apoptosis. In: Bonavida, B. (eds) Molecular Mechanisms of Tumor Cell Resistance to Chemotherapy. Resistance to Targeted Anti-Cancer Therapeutics, vol 1. Springer, New York, NY. https://doi.org/10.1007/978-1-4614-7070-0_12
Download citation
DOI: https://doi.org/10.1007/978-1-4614-7070-0_12
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4614-7069-4
Online ISBN: 978-1-4614-7070-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)