
Abstract
-
Hepatocellular carcinoma (HCC) is a common malignancy worldwide that is increasing in incidence in the United States.
-
Viral and alcohol-related liver diseases account for most cases; however, a significant number of patients with HCC do not have a known underlying chronic liver disease.
-
New evidence suggests that obesity and type 2 diabetes may play a significant role in the development of HCC.
-
Nonalcoholic fatty liver disease (NAFLD) is very common in patients with obesity and diabetes and may progress to cirrhosis and HCC.
-
Insulin resistance, adipose tissue inflammation, adipokines, and inflammatory cytokines comprise mechanistic pathways that link obesity with NAFLD and HCC.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Alcoholic Liver Disease
- Nonalcoholic Fatty Liver Disease
- Primary Liver Cancer
- NAFLD Patient
- NASH Patient
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
9.1 Introduction: The Changing Epidemiologic Pattern of HCC
Primary liver cancer is the sixth most common liver cancer worldwide and the third most common cause of cancer-related death [1]. Hepatocellular carcinoma (HCC) accounts for approximately 90 % of all primary liver cancers, and these two terms are frequently used interchangeably [2]. Each year, 20,000 new cases of HCC are diagnosed in the United States with an incidence that has tripled over the past 2 decades making HCC the fastest growing cause of cancer death in the male population [3]. Liver cirrhosis is present in approximately 70–90 % of HCC cases. Although chronic hepatitis B virus (HBV) infection is the major risk factor for HCC worldwide, infection with hepatitis C virus (HCV) is the causal factor in the majority of cases in the United States and Japan [4]. Other etiologies of chronic liver disease/cirrhosis associated with HCC include alcoholic liver disease, exposure to aflatoxin, hereditary hemochromatosis, α1-antitrypsin deficiency, primary biliary cirrhosis, and autoimmune hepatitis. However, a significant number of patients with HCC (15–50 %) do not have a known underlying chronic liver disease (cryptogenic) [5].
New evidence suggests that obesity and obesity-related complications, such as type 2 diabetes (DM2), metabolic syndrome (MetS), and nonalcoholic fatty liver disease (NAFLD), play a significant role in the development of HCC in susceptible patients.
In fact, epidemiological data have demonstrated a parallel increase in prevalence of HCC and obesity. In multiple population-based cohort studies from the USA and Europe [6–8], HCC was approximately twice as likely to develop in obese individuals as in those who were not obese, and similar results were found in patients with DM2 compared to nondiabetic subjects [9, 10]. NAFLD is the hepatic manifestation of MetS, and its prevalence (33 % of the adult US population) has been increasing with the growing epidemics of obesity and DM2. In addition, NAFLD appears to be a significant factor in the tumorigenesis of HCC in the setting of obesity [11].
9.2 Mechanistic Pathways Liking Obesity and Inflammation to Chronic Liver Disease and HCC
It has become clear that a state of low-grade chronic inflammation is typically associated with obesity and plays a crucial role in the development of insulin resistance (IR) [12, 13]. An important initiator of this inflammatory response is the adipose tissue, which actively secretes a variety of products such as cytokines, adipokines, and fatty acids into the circulation [14]. Abnormal adipose tissue metabolism has also been identified as a critical mechanistic link between obesity and NAFLD [15, 16]. As shown in Fig. 9.1, the increased release of free fatty acids (FFA) from adipose tissue due to a state of insulin resistance, which is characteristic of obesity, represents the main source of FFA during development of hepatic steatosis and progressive liver injury in NAFLD [17]. A surplus of FFA in non-adipose cells (ectopic fat deposition) may enter deleterious pathways leading to hepatocyte dysfunction (lipotoxicity) and apoptotic cell death (lipoapoptosis) [18, 19] which can occur through death receptors, the mitochondrial-lysosomal pathway, and endoplasmic reticulum (ER) stress [17]. The ensuing responses of cell repair, inflammation, regeneration, and fibrosis may all be triggered by apoptosis of adjacent cells [20]. Of these processes in the liver, hepatic fibrosis has the potential to be the most deleterious, as progressive fibrosis may result in cirrhosis and end-stage liver disease [21, 22].

The role of insulin resistance, free fatty acids, obesity, and diabetes in the development and progression of nonalcoholic fatty liver disease (NAFLD)
Liver fibrosis is strongly associated with the development of HCC; in fact, up to 90 % of HCC cases arise in cirrhotic livers [23]. As shown in Fig. 9.2, excess extracellular matrix and stiffness in cirrhotic livers provides a reservoir for bound growth factors, promotes angiogenesis, enhances survival of pre-neoplastic hepatocytes and activated hepatic stellate cells (HSCs), and reduces the activity of natural killer and natural killer T cells that have critical roles in tumor surveillance [24].

The importance of fibrosis and the immune system for creating an environment in the liver conducive for the development of hepatocellular carcinoma (HCC)
9.3 Obesity and Insulin Resistance in the Pathogenesis of HCC
9.3.1 Insulin Resistance/IGF Axis and HCC
IR is frequently present in obese individuals and can lead to a state of compensatory hyperinsulinemia in order to maintain normal metabolic functions. As shown in Fig. 9.3, hyperinsulinemia activates downstream pathways through the insulin receptor substrate-1 (IRS-1), which inhibit apoptosis and increase mitogenesis promoting tumorigenesis. IRS-1 plays an important role in cytokine signaling pathways and is upregulated in HCC [25]. The c-Jun amino-terminal kinase 1 (JNK1) has emerged as a key link between obesity and IR [26]. Obesity is associated with increased release of FFA, pro-inflammatory cytokines, and reactive oxygen species, all potent activators of JNK, which lead to phosphorylation of IRS-1 and IR. JNK1 plays a significant role in the development of nonalcoholic steatohepatitis (NASH) [27] and was found recently to be associated with the development of HCC as more than 50 % of human HCC samples were observed to have increased activation of JNK1 [11, 28, 29].

The transcriptional factors and adipokines currently considered the most important carcinogenic in NAFLD
The liver synthesizes and secretes insulin-like growth factors (IGFs), in response to growth hormone. Insulin upregulates hepatic growth hormone receptors, which in turn increase the activation of IGFs. Accumulating nascent data suggest that dysregulation of the IGF axis, which consists of IGF-1 and IGF-2, their receptors IGF1R and IGF2R, and their binding proteins (IGFBP1–6), plays a role in the development of HCC in animal models and human HCC cell lines [30, 31]. Binding of IGF-1 and IGF-2 to their cell surface receptor results in the activation of the Ras-mitogen-activated protein kinase (MAPK)-ERK pathway and the phosphoinositide 3-kinase (PI3K)/Akt/mTOR pathway, which are major signaling pathways in cellular proliferation and apoptosis [32–34]. Hyperinsulinemia decreases liver production and blood levels of IGFBPs leading to increased bioavailability of IGFs [35]. Indeed, decreased expression of IGFBP-3 in HCC was found to be significantly associated with portal vein invasion and poor prognosis [36], while the treatment of HCC cells with IGFBP-3 in vitro controlled cell proliferation [37].
9.3.2 Inflammatory Cytokines and HCC
Obesity is associated with the upregulation of pro-inflammatory cytokines such as tumor necrosis factor-α (TNF-α) and interleukin 6 (IL-6), both of which can stimulate normal and malignant hepatocyte proliferation [38, 39]. The binding of TNF-α to its receptor activates the nuclear factor-κB pathway (NF-κB), which is considered a major factor in determining the ability of pre-neoplastic and malignant cells to avoid apoptosis and therefore providing a link between inflammation and cancer [40]. After activation, NF-κB translocate dimers to the nucleus where they affect transcriptional activation of hundreds of target genes. By using the Mdr-2 knockout mouse model, which spontaneously develops cholestatic hepatitis leading to dysplasia and eventually HCC, Pikarsky et al. demonstrated that TNF-α expression was upregulated in adjacent inflammatory and endothelial cells, triggering NF-κB signaling in hepatocytes, thereby promoting survival and the ultimate formation of HCC [41]. Furthermore, anti-TNF treatment suppressed NF-κB activation resulting in apoptosis of transformed hepatocytes and failure to progress to HCC. In contrast with the tumor-promoting role of NF-κB pathway in the animal model described above, the inactivation of the pathway at an early stage in other models can also promote carcinogenesis because of increased hepatocyte apoptosis which triggers compensatory hyperplasia, regeneration, and fibrosis leading to malignant transformation [42].
Another important inflammation-associated pathway is the signal transducer and activator of transcription 3 (STAT3) which is triggered by IL-6 and inhibited by SOCS3 (suppressor of cytokine signaling 3) [43]. In a study by He et al., hepatocyte-specific STAT3-deficient mice (Stat3 Δhep) exhibit more than sixfold reduction in HCC load relative to Stat3 F/F in response to the chemical procarcinogen diethylnitrosamine (DEN)-induced liver tumorigenesis [44]. The authors also examined the status of STAT3 activation in a large number of human HCC specimens and found that approximately 60 % exhibited activated nuclear STAT3, with STAT3-positive tumors being more aggressive. Furthermore, hepatocyte-specific SOCS3 deletion in mice with DEN-induced HCC leads to larger and more numerous tumors [45].
9.3.3 Adipokines and HCC
Leptin is a specialized peptide hormone produced mainly by adipocytes (adipokine) and is the product of the obese (ob) gene. Leptin regulates energy intake and expenditure through binding to its receptors in the central nervous system [46, 47]. In overweight and obese subjects, leptin levels are elevated indicating a state of leptin resistance. Using HCC cell lines, Saxena et al. demonstrated that leptin promoted HCC through concomitant activation of the STAT3 and PI3K/Akt pathways and that blocking these signaling pathways can effectively block leptin-mediated migration and invasion of HCC cells [48]. Moreover, leptin can promote tumorigenesis by protecting HCC cells from apoptosis induced by transforming growth factor-β (TGF-β), which occurs by downregulating the pro-apoptotic Bax gene [49].
Adiponectin is another adipokine mainly secreted by visceral adipose tissue. Adiponectin has three receptors, AdipoR1, AdipoR2, and T-cadherin, with AdipoR2 being the one predominantly expressed in the liver [50]. Unlike leptin, adiponectin levels are markedly decreased in obesity, DM2, NAFLD, and atherosclerosis indicating a protective effect of adiponectin against obesity-related disorders [51–53]. Similarly, reduced plasma or hepatic adiponectin correlates closely with the development of HCC with an inverse relationship between adiponectin expression and tumor size [54, 55]. Interestingly, adiponectin may inhibit tumor growth induced by leptin by reducing STAT3 and PI3K/Akt activity and increasing SOCS3 which is a negative regulator of leptin signaling [56].
9.4 HCC and Obesity
9.4.1 Obesity and HCC in the General Population
Obesity has been established as a risk factor for HCC by multiple large cohort studies [57]. This increased risk may be related to the increased prevalence of NAFLD and NASH in obese individuals, and the carcinogenic potential exerted by obesity alone, as discussed above. In a large Danish study of nearly 44,000 obese individuals, Moller et al. found an overall 16 % increased incidence of cancers and a relative risk of 1.9 for liver cancer [7]. Wolk et al. found that obesity was associated with a threefold increase in HCC risk in a large Swedish study that included more than 28,000 obese patients [8]. A Korean study that followed 781,283 males without a prior diagnosis of cancer for over a 10-year period found a relative risk for HCC of 1.53 in obese patients compared to normal controls after controlling for HBV infection (the most common cause of HCC in Korea) [58]. A mortality study conducted by the American Cancer Society that followed more than 900,000 US adults initially free from cancer for 16 years demonstrated that the relative risk of dying from liver cancer was 1.68 times higher in women and 4.52 times higher in men who had a BMI ≥ 35 kg/m2 compared to lean controls with normal BMI (18.5–24.9 kg/m2) [6]. Of note, the relative risk of liver cancer mortality among males was the highest of all the cancers studied. A meta-analysis that included 11 cohort studies found that compared to normal-weight individuals, the relative risk of liver cancer was 1.17 (95 % CI, 1.02–1.34) for overweight patients and 1.89 (95 % CI, 1.51–2.36) for obese patients [57].
9.4.2 Obesity and HCC in Patients with Cirrhosis
In addition to the association between obesity and HCC in the general population, obesity is an independent risk factor for HCC in patients with cirrhosis of different etiologies. A review of the United Network of Organ Sharing (UNOS) database that included 19,271 cirrhotic patients who had liver transplantation between 1991 and 2000 revealed that the overall incidence of HCC was higher in obese patients compared to non-obese patients (4.0 % vs. 3.0 %, p = 0.013) [59]. In the multivariate analysis of this study, obesity was an independent predictor of HCC in cryptogenic cirrhosis (OR = 11.1, 95 % CI, 1.5–87.4) and alcoholic cirrhosis (OR = 3.2, 95 % CI, 1.5–6.6) but not in cirrhosis due to viral hepatitis or autoimmune liver disease. A prospective 7-year study in France that followed 771 patients with alcohol- or HCV-related cirrhosis demonstrated that a BMI ≥ 30 kg/m2 was associated with a hazard ratio for HCC of 2.8 [60].
Beyond the effect of obesity (assessed by BMI) on the risk of HCC, it appears that visceral fat accumulation per se may play a role in tumor initiation and progression. Visceral adipose tissue is a highly active endocrine organ that produces multiple pro-inflammatory cytokines (such as TNF-α and IL-6), promotes insulin resistance, and causes hepatocyte fat accumulation with ensuing hepatocyte injury and possibly carcinogenesis through FFA toxicity [31]. Ohki et al. studied the effect of visceral fat area, as measured by contrast-enhanced dynamic CT, on the recurrence of HCC after curative ablation [61]. They found that patients with high visceral fat area (defined as >130 cm2 in male and >90 cm2 in female) had a cumulative recurrence rates at 1 and 3 years of 15.9 % and 75.1 %, respectively, compared to 9.7 and 43.1 % in the control group (p = 0.018).
9.5 HCC and Type 2 Diabetes Mellitus (DM2)
Establishing the causal relationship between diabetes and HCC has proven difficult for multiple reasons. First, cirrhosis and end-stage liver disease can cause impaired glucose tolerance and overt diabetes [62] introducing potential bias in case–control studies. Second, some etiologies of chronic liver disease such as HCV and hemochromatosis are associated with increased diabetes risk [63]. Finally, diabetes is a strong risk factor for NAFLD and NASH which can progress to cirrhosis and HCC [64]. However, several large cohort studies have established the presence of DM2 as an independent risk factor for the development of HCC. Earlier population-based studies from Sweden and Denmark [65, 66] have shown a significant increase in HCC risk among patients with diabetes alone and in the presence of viral hepatitis, alcoholic liver disease, and cirrhosis. A large longitudinal study from the USA followed 173,643 diabetic patients and 650,620 patients without diabetes for 10–15 years and found that diabetes was associated with a hazard ratio for HCC of 2.16 (95 % confidence interval [CI], 1.86–1.09; p < 0.0001) even after excluding patients with HCV, HBV, and alcoholic liver disease [67]. Importantly, this study provided evidence for a causal association between DM2 and HCC by demonstrating that diabetes preceded the development of chronic liver disease, as well as an increased risk for HCC in patients with longer duration of diabetes. Similar findings were reported from a hospital-based case–control study with diabetes being present in 87 % of cases before the diagnosis of HCC and patients with diabetes duration >10 years having an adjusted hazard ratio for HCC of 2.2 (95 % CI, 1.2–4.8) compared to those with a diabetes duration of 2–5 years. More robust support for diabetes as a risk factor for HCC came from a meta-analysis of 13 cohort studies that aimed to assess the association between diabetes and HCC. The analysis revealed a statistically significant 2.5-fold increase in HCC incidence among diabetic patients (95 % CI, 1.9–3.2; p < 0.01) [9]. A more recent meta-analysis that included a total of 17 case–control studies and 32 cohort studies confirmed the association between diabetes and risk of HCC with a pooled relative risk of 2.31 for HCC among diabetic patients (95 % CI, 1.87–2.84) [68].
9.5.1 Association of Diabetes Treatment with HCC Risk
Several lines of evidence suggest that the type of antidiabetic therapy may increase or decrease HCC risk contingent upon mechanism of action [68].
On one hand, insulin sensitizers such as metformin can improve insulin sensitivity and suppress cell growth by activating the AMP-activated protein kinase yielding a protective effect on cancer risk [69]. A retrospective study from Italy that included 465 patients with HCC, 618 cirrhotics, and 490 controls found a statistically significant reduction in HCC risk for diabetics receiving metformin (OR = 0.33; 95 % CI, 0.1–0.61) [70]. Another study from the University of Texas reported a similar decrease in the risk for HCC with metformin (OR = 0.3; 95 % CI, 0.2–0.6) [71]. Moreover, a recent study demonstrated that metformin decreased the risk of HCC in diabetic patients in a dose-dependent manner with a 7 % risk reduction of HCC for each incremental year increase in metformin use [72]. Interestingly, metformin inhibited hepatoma cell line proliferation by inducing cell cycle arrest at the G0/G1 phase via AMP-activated protein kinase. Furthermore, metformin had chemosensitizing effect in combination with doxorubicin to accelerate HCC regression in a mouse xenograft model.
On the other hand, exogenous insulin and sulfonylureas lead to an increase in circulating insulin levels promoting carcinogenesis and increasing the risk for HCC. Donadon et al. found a statistically significant increase in HCC risk for diabetics receiving sulfonylureas or insulin (OR = 2.99; 95 % CI, 1.34–6.65), and similar findings were also reported by Hassan et al. (insulin use was associated with HCC OR of 1.9; 95 % CI, 0.8–4.6, and sulfonylurea use was associated with HCC OR of 7.1; 95 % CI, 2.9–16.9) [70, 71].
Overall, in the meta-analysis by Wang et al., the pooled risk estimates for developing HCC were 0.31 (95 % CI, 0.19–0.49) for diabetics treated with metformin and 4.0 (95 % CI, 1.94–8.24) for those treated with sulfonylurea or exogenous insulin [68].
In addition to the effects of antidiabetic therapy on the risk for liver cancer, statin use in diabetics appears to have a protective effect against HCC. Proposed mechanisms include the inhibition of downstream products of the mevalonate pathway which are important for the growth of malignant cells [73] and the inhibition of HSC proliferation and their production of collagen [74]. In a nested, matched, case–control study in diabetics, El-Serag et al. examined 1,303 cases and 5,212 controls and found a risk reduction for the development of HCC that ranged between 25 and 40 % providing evidence of the cancer-preventive effect of statins specific to HCC [75].
9.6 HCC and Nonalcoholic Fatty Liver Disease
9.6.1 The Epidemic of NAFLD
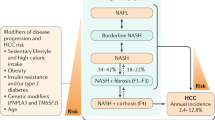
NAFLD is considered the hepatic manifestation of MetS and as such has become the most common form of chronic liver disease in the world [76–78]. NAFLD encompasses a wide histological spectrum of disease ranging from simple steatosis characterized by lipid accumulation in the liver in the form of triglyceride (TG) to NASH characterized by the association of lipid accumulation with evidence of hepatocyte injury, inflammation, and various degrees of fibrosis [79]. NASH is a serious condition that can progress to cirrhosis and its feared complications of portal hypertension and end-stage liver disease requiring liver transplantation. It is estimated that in the United States, NAFLD affects 30–45 % of the general population and as high as 90 % of the morbidly obese [80, 81]. Furthermore, the aggressive form of NASH affects between 5 and 13 % of Americans, making this entity a rising cause of liver-related morbidity and mortality and potentially the main indication for liver transplantation in the next decade [82]. A recent study found that among diabetic patients, NAFLD was present in 74 % and NASH in 22.2 % [81].
9.6.2 HCC and Cryptogenic Cirrhosis
When NASH progresses to cirrhosis, most of the classic histological features of the disease disappear, making the diagnosis of the underlying etiology difficult [83]. It has been proposed that NASH accounts for the majority of cases of cryptogenic cirrhosis (CC) because these patients have a significantly higher prevalence of conditions associated with NASH including obesity and diabetes compared to patients with cirrhosis of well-defined etiology [84, 85]. Three landmark studies published in 2002 expanded the spectrum of NAFLD from CC to HCC. In the first study, Bugianesi et al. retrospectively identified 646 Italian patients with cirrhosis-associated HCC and found that the prevalence of CC was 6.9 % compared to 55 % for HCV-cirrhosis, 16 % for HBV-cirrhosis, and 13 % for alcoholic cirrhosis [86]. Interestingly, the prevalence of pre-cirrhotic obesity, DM2, and dyslipidemia were more than twice as prevalent in patients who had CC as in the control group (p value <0.05 for all metabolic factors). The second study by Marrero et al. demonstrated that CC was the second most common etiology of underlying liver disease (29 %) after HCV (51 %) in a group of 105 patients with HCC from Michigan likely related to the high prevalence of obesity in the United States [87]. The third study by Ratziu et al. in France found CC in 27 % of HCC patients and corroborated the association between DM2, IR, and dyslipidemia with both CC and HCC [88].
9.6.3 HCC and NASH-Cirrhosis
Several studies have directly examined the incidence of HCC in patients with NAFLD or NASH-cirrhosis (Table 9.1). In a large cohort study that included 7,326 patients discharged with a diagnosis of fatty liver from Danish hospitals over a 16-year period, Sorensen et al. found that the risk for primary liver cancer was significantly elevated in NAFLD patients compared to the Danish general population with a standardized incidence ratio of 4.4 (95 % CI, 1.2–11.4) [89]. A large prospective US study compared 152 patients with NASH-cirrhosis (median age of 55 years) with 150 matched patients with HCV-cirrhosis. Those with NASH-cirrhosis had significantly lower risk of developing HCC over a 10-year follow-up period (6.7 % vs. 17 %, respectively, p < 0.01) [90]. Hashimoto et al. conducted a case-controlled study that included 34 NASH patients with HCC and 348 patients NASH patients without HCC [91]. Risk factors for HCC in this study included older age, low AST level, low NAFLD activity score, and advanced fibrosis stage. A prospective cohort study of 137 NASH patients with advanced fibrosis was included in that same study; this prospective cohort demonstrated that the 5-year cumulative incidence of HCC was 7.6 % [91]. Finally, a study from the Cleveland Clinic compared 195 patients with NASH-cirrhosis to 315 patients with HCV-cirrhosis that were evaluated for liver transplantation between 2003 and 2007 [92]. The yearly cumulative incidence of HCC in cirrhotic patients with NASH was 2.6 % compared to 4.0 % for those with hepatitis C infection. Interestingly, older age at the time of cirrhosis diagnosis and any alcohol consumption were independently associated with the development of HCC in the multivariate analysis (hazard ratio of 1.08 and 3.8, respectively; p < 0.005), supporting the notion that alcohol intake even in small quantities may increase the risk of HCC development in the setting of cirrhosis.
9.6.4 HCC and Non-cirrhotic NAFLD
New data indicate that HCC can arise in steatotic livers in the absence of cirrhosis. In fact, several case series have described the occurrence of HCC in non-cirrhotic NAFLD patients. In a Japanese study that included 9 patients with HCC in the setting of NAFLD, one third of patients only had mild hepatic fibrosis and no evidence of cirrhosis [93]. Hashimoto et al. confirmed these findings by demonstrating that 12 % of their patients with NASH-HCC had mild fibrosis (fibrosis stage 1–2) [91].
In a large nationwide survey of 14,530 HCC patients in Japan, NAFLD-HCC and unknown HCC accounted for 7.1 % of all cases, but only 62 % of NAFLD-HCC had cirrhosis [94]. A European study by Ertle et al. that enrolled 150 patients with HCC (including 36 with NAFLD) intriguingly found that only 58 % of patients with NAFLD-HCC had evidence of cirrhosis [95]. These studies underscore the importance of HCC screening in high-risk NAFLD patients such as those with family history of HCC.
9.7 Summary
NAFLD has emerged as the most prevalent chronic liver disease. Population-based studies in the United States estimate that the prevalence of NAFLD is at least 30 % in adults and as high as 80 % in obese patients and in patients with type 2 diabetes. Of additional concern is the fact that the incidence of NAFLD has increased from 4.5/100,000 to 38/100,000 over the epoch of 1980–1999. Consistent with these data is the recognition that obesity and diabetes are risk factors for both NAFLD and HCC. Although viral and alcohol-related liver disease account for the majority of HCC, the increased incidence of HCC has occurred in parallel with incidences of NAFLD, obesity, and diabetes, with the latter two being risk factors for both the development and progression of NAFLD. This increased incidence of HCC in NAFLD and the fact that there is a predilection for HCC to develop in NAFLD even in the absence of cirrhosis has important clinical implications that may change the current paradigms for cancer surveillance in these patients. Finally, our increased knowledge of the mechanistic pathways that link obesity and diabetes to the pathogenesis of HCC in NAFLD (discussed above) provide strategies for both the clinical management of NAFLD and the development of targeted treatments for HCC associated with NAFLD.
References
Ferlay J, Shin HR, Bray F et al (2010) Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer 127:2893–2917
Nordenstedt H, White DL, El-Serag HB (2010) The changing pattern of epidemiology in hepatocellular carcinoma. Dig Liver Dis 42(suppl 3):S206–S214
El-Serag HB (2011) Hepatocellular carcinoma. N Engl J Med 365:1118–1127
McGlynn KA, London WT (2011) The global epidemiology of hepatocellular carcinoma: present and future. Clin Liver Dis 15:223–243, vii–x
Page JM, Harrison SA (2009) NASH and HCC. Clin Liver Dis 13:631–647
Calle EE, Rodriguez C, Walker-Thurmond K et al (2003) Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. N Engl J Med 348:1625–1638
Moller H, Mellemgaard A, Lindvig K et al (1994) Obesity and cancer risk: a Danish record-linkage study. Eur J Cancer 30A:344–350
Wolk A, Gridley G, Svensson M et al (2001) A prospective study of obesity and cancer risk (Sweden). Cancer Causes Control 12:13–21
El-Serag HB, Hampel H, Javadi F (2006) The association between diabetes and hepatocellular carcinoma: a systematic review of epidemiologic evidence. Clin Gastroenterol Hepatol 4:369–380
El-Serag HB, Tran T, Everhart JE (2004) Diabetes increases the risk of chronic liver disease and hepatocellular carcinoma. Gastroenterology 126:460–468
Starley BQ, Calcagno CJ, Harrison SA (2010) Nonalcoholic fatty liver disease and hepatocellular carcinoma: a weighty connection. Hepatology 51:1820–1832
Wellen KE, Hotamisligil GS (2005) Inflammation, stress, and diabetes. J Clin Invest 115:1111–1119
Hotamisligil GS (2006) Inflammation and metabolic disorders. Nature 444:860–867
Weisberg SP, McCann D, Desai M et al (2003) Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest 112:1796–1808
Jou J, Choi SS, Diehl AM (2008) Mechanisms of disease progression in nonalcoholic fatty liver disease. Semin Liver Dis 28:370–379
Wree A, Kahraman A, Gerken G et al (2010) Obesity affects the liver—the link between adipocytes and hepatocytes. Digestion 83:124–133
Alkhouri N, Dixon LJ, Feldstein AE (2009) Lipotoxicity in nonalcoholic fatty liver disease: not all lipids are created equal. Expert Rev Gastroenterol Hepatol 3:445–451
Listenberger LL, Han X, Lewis SE et al (2003) Triglyceride accumulation protects against fatty acid-induced lipotoxicity. Proc Natl Acad Sci U S A 100:3077–3082
Unger RH (2002) Lipotoxic diseases. Annu Rev Med 53:319–336
Guicciardi ME, Gores GJ (2005) Apoptosis: a mechanism of acute and chronic liver injury. Gut 54:1024–1033
Bataller R, Brenner DA (2005) Liver fibrosis. J Clin Invest 115:209–218
Friedman SL (2003) Liver fibrosis—from bench to bedside. J Hepatol 38(suppl 1):S38–S53
Seitz HK, Stickel F (2006) Risk factors and mechanisms of hepatocarcinogenesis with special emphasis on alcohol and oxidative stress. Biol Chem 387:349–360
Zhang DY, Friedman SL (2012) Fibrosis-dependent mechanisms of hepatocarcinogenesis. Hepatology 56:769–775
Tanaka S, Mohr L, Schmidt EV et al (1997) Biological effects of human insulin receptor substrate-1 overexpression in hepatocytes. Hepatology 26:598–604
Hirosumi J, Tuncman G, Chang L et al (2002) A central role for JNK in obesity and insulin resistance. Nature 420:333–336
Puri P, Mirshahi F, Cheung O et al (2008) Activation and dysregulation of the unfolded protein response in nonalcoholic fatty liver disease. Gastroenterology 134:568–576
Chang Q, Zhang Y, Beezhold KJ et al (2009) Sustained JNK1 activation is associated with altered histone H3 methylations in human liver cancer. J Hepatol 50:323–333
Hui L, Zatloukal K, Scheuch H et al (2008) Proliferation of human HCC cells and chemically induced mouse liver cancers requires JNK1-dependent p21 downregulation. J Clin Invest 118:3943–3953
Shen C, Zhao CY, Zhang R et al (2012) Obesity-related hepatocellular carcinoma: roles of risk factors altered in obesity. Front Biosci 17:2356–2370
Siddique A, Kowdley KV (2011) Insulin resistance and other metabolic risk factors in the pathogenesis of hepatocellular carcinoma. Clin Liver Dis 15:281–296, vii–x
Kurmasheva RT, Houghton PJ (2006) IGF-I mediated survival pathways in normal and malignant cells. Biochim Biophys Acta 1766:1–22
Chen JS, Wang Q, Fu XH et al (2009) Involvement of PI3K/PTEN/AKT/mTOR pathway in invasion and metastasis in hepatocellular carcinoma: association with MMP-9. Hepatol Res 39:177–186
Lawlor MA, Alessi DR (2001) PKB/Akt: a key mediator of cell proliferation, survival and insulin responses? J Cell Sci 114:2903–2910
Nam SY, Lee EJ, Kim KR et al (1997) Effect of obesity on total and free insulin-like growth factor (IGF)-1, and their relationship to IGF-binding protein (BP)-1, IGFBP-2, IGFBP-3, insulin, and growth hormone. Int J Obes Relat Metab Disord 21:355–359
Aishima S, Basaki Y, Oda Y et al (2006) High expression of insulin-like growth factor binding protein-3 is correlated with lower portal invasion and better prognosis in human hepatocellular carcinoma. Cancer Sci 97:1182–1190
Huynh H, Chow PK, Ooi LL et al (2002) A possible role for insulin-like growth factor-binding protein-3 autocrine/paracrine loops in controlling hepatocellular carcinoma cell proliferation. Cell Growth Differ 13:115–122
Alison MR, Nicholson LJ, Lin WR (2011) Chronic inflammation and hepatocellular carcinoma. Recent results in cancer research. Fortschr Krebsforsch 185:135–148
Park EJ, Lee JH, Yu GY et al (2010) Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell 140:197–208
Ben-Neriah Y, Karin M (2011) Inflammation meets cancer, with NF-kappaB as the matchmaker. Nat Immunol 12:715–723
Pikarsky E, Porat RM, Stein I et al (2004) NF-kappaB functions as a tumour promoter in inflammation-associated cancer. Nature 431:461–466
Luedde T, Beraza N, Kotsikoris V et al (2007) Deletion of NEMO/IKKgamma in liver parenchymal cells causes steatohepatitis and hepatocellular carcinoma. Cancer Cell 11:119–132
He G, Karin M (2011) NF-kappaB and STAT3—key players in liver inflammation and cancer. Cell Res 21:159–168
He G, Yu GY, Temkin V et al (2010) Hepatocyte IKKbeta/NF-kappaB inhibits tumor promotion and progression by preventing oxidative stress-driven STAT3 activation. Cancer Cell 17:286–297
Ogata H, Kobayashi T, Chinen T et al (2006) Deletion of the SOCS3 gene in liver parenchymal cells promotes hepatitis-induced hepatocarcinogenesis. Gastroenterology 131:179–193
Jequier E (2002) Leptin signaling, adiposity, and energy balance. Ann N Y Acad Sci 967:379–388
Sun B, Karin M (2012) Obesity, inflammation, and liver cancer. J Hepatol 56:704–713
Saxena NK, Sharma D, Ding X et al (2007) Concomitant activation of the JAK/STAT, PI3K/AKT, and ERK signaling is involved in leptin-mediated promotion of invasion and migration of hepatocellular carcinoma cells. Cancer Res 67:2497–2507
Chen C, Chang YC, Liu CL et al (2007) Leptin induces proliferation and anti-apoptosis in human hepatocarcinoma cells by up-regulating cyclin D1 and down-regulating Bax via a Janus kinase 2-linked pathway. Endocr Relat Cancer 14:513–529
Kadowaki T, Yamauchi T (2005) Adiponectin and adiponectin receptors. Endocr Rev 26:439–451
Hotta K, Funahashi T, Arita Y et al (2000) Plasma concentrations of a novel, adipose-specific protein, adiponectin, in type 2 diabetic patients. Arterioscler Thromb Vasc Biol 20:1595–1599
Jiang LL, Li L, Hong XF et al (2009) Patients with nonalcoholic fatty liver disease display increased serum resistin levels and decreased adiponectin levels. Eur J Gastroenterol Hepatol 21:662–666
Skrabal CA, Czaja J, Honz K et al (2011) Adiponectin—its potential to predict and prevent coronary artery disease. Thorac Cardiovasc Surg 59:201–206
Man K, Ng KT, Xu A et al (2010) Suppression of liver tumor growth and metastasis by adiponectin in nude mice through inhibition of tumor angiogenesis and downregulation of Rho kinase/IFN-inducible protein 10/matrix metalloproteinase 9 signaling. Clin Cancer Res 16:967–977
Saxena NK, Fu PP, Nagalingam A et al (2010) Adiponectin modulates C-jun N-terminal kinase and mammalian target of rapamycin and inhibits hepatocellular carcinoma. Gastroenterology 139:1762–1773, 1773e1–5
Sharma D, Wang J, Fu PP et al (2010) Adiponectin antagonizes the oncogenic actions of leptin in hepatocellular carcinogenesis. Hepatology 52:1713–1722
Larsson SC, Wolk A (2007) Overweight, obesity and risk of liver cancer: a meta-analysis of cohort studies. Br J Cancer 97:1005–1008
Oh SW, Yoon YS, Shin SA (2005) Effects of excess weight on cancer incidences depending on cancer sites and histologic findings among men: Korea National Health Insurance Corporation Study. J Clin Oncol 23:4742–4754
Nair S, Mason A, Eason J et al (2002) Is obesity an independent risk factor for hepatocellular carcinoma in cirrhosis? Hepatology 36:150–155
N’Kontchou G, Paries J, Htar MT et al (2006) Risk factors for hepatocellular carcinoma in patients with alcoholic or viral C cirrhosis. Clin Gastroenterol Hepatol 4:1062–1068
Ohki T, Tateishi R, Shiina S et al (2009) Visceral fat accumulation is an independent risk factor for hepatocellular carcinoma recurrence after curative treatment in patients with suspected NASH. Gut 58:839–844
Petrides AS, Vogt C, Schulze-Berge D et al (1994) Pathogenesis of glucose intolerance and diabetes mellitus in cirrhosis. Hepatology 19:616–627
Zein CO, Levy C, Basu A et al (2005) Chronic hepatitis C and type II diabetes mellitus: a prospective cross-sectional study. Am J Gastroenterol 100:48–55
Angulo P, Keach JC, Batts KP et al (1999) Independent predictors of liver fibrosis in patients with nonalcoholic steatohepatitis. Hepatology 30:1356–1362
Adami HO, Chow WH, Nyren O et al (1996) Excess risk of primary liver cancer in patients with diabetes mellitus. J Natl Cancer Inst 88:1472–1477
Wideroff L, Gridley G, Mellemkjaer L et al (1997) Cancer incidence in a population-based cohort of patients hospitalized with diabetes mellitus in Denmark. J Natl Cancer Inst 89:1360–1365
El-Serag HB (2004) Hepatocellular carcinoma: recent trends in the United States. Gastroenterology 127:S27–S34
Wang P, Kang D, Cao W et al (2012) Diabetes mellitus and risk of hepatocellular carcinoma: a systematic review and meta-analysis. Diabetes Metab Res Rev 28:109–122
Hardie DG (2007) AMP-activated protein kinase as a drug target. Annu Rev Pharmacol Toxicol 47:185–210
Donadon V, Balbi M, Ghersetti M et al (2009) Antidiabetic therapy and increased risk of hepatocellular carcinoma in chronic liver disease. World J Gastroenterol 15:2506–2511
Hassan MM, Curley SA, Li D et al (2010) Association of diabetes duration and diabetes treatment with the risk of hepatocellular carcinoma. Cancer 116:1938–1946
Chen HP, Shieh JJ, Chang CC et al (2013) Metformin decreases hepatocellular carcinoma risk in a dose-dependent manner: population-based and in vitro studies. Gut 62(4):606–615
Blanco-Colio LM, Villa A, Ortego M et al (2002) 3-Hydroxy-3-methyl-glutaryl coenzyme A reductase inhibitors, atorvastatin and simvastatin, induce apoptosis of vascular smooth muscle cells by downregulation of Bcl-2 expression and Rho A prenylation. Atherosclerosis 161:17–26
Rombouts K, Kisanga E, Hellemans K et al (2003) Effect of HMG-CoA reductase inhibitors on proliferation and protein synthesis by rat hepatic stellate cells. J Hepatol 38:564–572
El-Serag HB, Johnson ML, Hachem C et al (2009) Statins are associated with a reduced risk of hepatocellular carcinoma in a large cohort of patients with diabetes. Gastroenterology 136:1601–1608
Angulo P (2002) Nonalcoholic fatty liver disease. N Engl J Med 346:1221–1231
Browning JD, Szczepaniak LS, Dobbins R et al (2004) Prevalence of hepatic steatosis in an urban population in the United States: impact of ethnicity. Hepatology 40:1387–1395
Fan JG, Zhu J, Li XJ et al (2005) Prevalence of and risk factors for fatty liver in a general population of Shanghai, China. J Hepatol 43:508–514
Brunt EM (2005) Pathology of nonalcoholic steatohepatitis. Hepatol Res 33:68–71
Torres DM, Harrison SA (2008) Diagnosis and therapy of nonalcoholic steatohepatitis. Gastroenterology 134:1682–1698
Williams CD, Stengel J, Asike MI et al (2011) Prevalence of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis among a largely middle-aged population utilizing ultrasound and liver biopsy: a prospective study. Gastroenterology 140:124–131
Charlton MR, Burns JM, Pedersen RA, Watt KD, Heimbach JK, Dierkhising RA (2011) Frequency and outcomes of Liver transplantation for nonalcoholic streatohepatitis in the United States. Gastroenterology 141:1249–1253
Falck-Ytter Y, Younossi ZM, Marchesini G et al (2001) Clinical features and natural history of nonalcoholic steatosis syndromes. Semin Liver Dis 21:17–26
Caldwell SH, Oelsner DH, Iezzoni JC et al (1999) Cryptogenic cirrhosis: clinical characterization and risk factors for underlying disease. Hepatology 29:664–669
Poonawala A, Nair SP, Thuluvath PJ (2000) Prevalence of obesity and diabetes in patients with cryptogenic cirrhosis: a case–control study. Hepatology 32:689–692
Bugianesi E, Leone N, Vanni E et al (2002) Expanding the natural history of nonalcoholic steatohepatitis: from cryptogenic cirrhosis to hepatocellular carcinoma. Gastroenterology 123:134–140
Marrero JA, Fontana RJ, Su GL et al (2002) NAFLD may be a common underlying liver disease in patients with hepatocellular carcinoma in the United States. Hepatology 36:1349–1354
Ratziu V, Bonyhay L, Di Martino V et al (2002) Survival, liver failure, and hepatocellular carcinoma in obesity-related cryptogenic cirrhosis. Hepatology 35:1485–1493
Sorensen HT, Mellemkjaer L, Jepsen P et al (2003) Risk of cancer in patients hospitalized with fatty liver: a Danish cohort study. J Clin Gastroenterol 36:356–359
Sanyal AJ, Banas C, Sargeant C et al (2006) Similarities and differences in outcomes of cirrhosis due to nonalcoholic steatohepatitis and hepatitis C. Hepatology 43:682–689
Hashimoto E, Yatsuji S, Tobari M et al (2009) Hepatocellular carcinoma in patients with nonalcoholic steatohepatitis. J Gastroenterol 44(suppl 19):89–95
Ascha MS, Hanouneh IA, Lopez R et al (2010) The incidence and risk factors of hepatocellular carcinoma in patients with nonalcoholic steatohepatitis. Hepatology 51:1972–1978
Hashizume H, Sato K, Takagi H et al (2007) Primary liver cancers with nonalcoholic steatohepatitis. Eur J Gastroenterol Hepatol 19:827–834
Tokushige K, Hashimoto E, Horie Y et al (2011) Hepatocellular carcinoma in Japanese patients with nonalcoholic fatty liver disease, alcoholic liver disease, and chronic liver disease of unknown etiology: report of the nationwide survey. J Gastroenterol 46:1230–1237
Ertle J, Dechene A, Sowa JP et al (2011) Non-alcoholic fatty liver disease progresses to hepatocellular carcinoma in the absence of apparent cirrhosis. Int J Cancer 128:2436–2443
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer Science+Business Media New York
About this chapter
Cite this chapter
Alkhouri, N., McCullough, A. (2013). Obesity, Inflammation, Nonalcoholic Fatty Liver Disease, and Hepatocellular Carcinoma. In: Dannenberg, A., Berger, N. (eds) Obesity, Inflammation and Cancer. Energy Balance and Cancer, vol 7. Springer, New York, NY. https://doi.org/10.1007/978-1-4614-6819-6_9
Download citation
DOI: https://doi.org/10.1007/978-1-4614-6819-6_9
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4614-6818-9
Online ISBN: 978-1-4614-6819-6
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)