
Abstract
The family of innate lymphoid cells (ILCs) comprises of natural killer (NK) cells, Rorγt-dependent ILCs (lymphoid tissue inducer (LTi) cells, ILC22, and ILC17), and type 2 ILCs. Apart from a common requirement for inhibitor of DNA binding 2 (Id2) expression and common γ-chain (γc) signaling, the differentiation of ILC populations is regulated by distinct transcription factors. ILCs play fundamental roles in processes such as cytotoxicity, lymphoid organogenesis, intestinal homeostasis, immunity against infections, and wound healing. However, the dysregulation of ILCs has been implicated in autoimmune and inflammatory diseases. Here, we will review the recent advances in ILC development and their roles in immunity and disease, with a primary focus on type 2 ILCs.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Type 2
- Innate lymphoid cells
- ILC
- Id2
- Rorγt
- Rorα
- LTi
- ILC22
- ILC17
- Nuocyte
- NHC
- Ih2
- Immunity
- Disease
- Anti-helminth
- Inflammatory bowel disease
- IBD
- Fibrosis
- Allergy
- Airway hyperreactivity
- AHR
2.1 Introduction
The immune system has evolved as protection against a wide range of infectious agents ranging from simple pathogens such as viruses, bacteria, and fungi to multicellular parasites such as helminths. In vertebrates, the immune system can be broadly divided into two interdependent effector arms, the adaptive and innate immune responses. In the adaptive immune response, lymphocytes are activated to generate potent pathogen-specific responses (e.g., antibodies and cytotoxic T cells) via VDJ recombination and generation of memory T and B cells. The innate immune response is evolutionarily older, and pathogen recognition does not adapt to the infection. The innate immune system serves as the “first line of defense” in organisms by providing an immediate protective response against infection and helping to initiate the adaptive immune response.
The innate immune system comprises of leukocytes such as mast cells, eosinophils, basophils, macrophages, neutrophils, dendritic cells (DCs), and natural killer cells (NK cells). These leukocytes act together to combat infectious agents by secreting cytokines, chemokines, and antimicrobials. This leads to inflammation, phagocytosis of microorganisms and infected cells, antigen processing and presentation, and activation of the adaptive immune response. Up until the last decade, NK cells were unique in being the only identified innate cell derived from a lymphoid progenitor. Recent developments have now classified NK cells as the earliest identified member of a family of hematopoietic effector cells termed innate lymphoid cells (ILCs) that are dependent on the transcription factor Id2. Currently, ILCs can be broadly classified into three groups: (a) NK cells, (b) the retinoic acid receptor-related orphan receptor γ t (Rorγt)-dependent ILCs (lymphoid tissue inducer (LTi) cells, ILC17, ILC22), and (c) type 2 ILCs. These groups have recently been named ILC1, ILC3 and ILC2 respectively. These various ILCs have now been implicated in protection against infectious organisms, organogenesis of lymphoid tissue, tissue remodeling during wound healing and homeostasis in tissue stromal cells.
Because the key cytokines secreted by some ILCs mirror those of various T helper cell populations, it has been proposed that ILCs may represent the innate counterparts of T helper lymphocytes, at least in terms of cytokine production [1, 2] (Table 2.1). In this review we will provide a general overview of the ILC family, focusing on the recent advances with regard to type 2 ILCs in immunity.
2.2 Phenotype of ILCs
2.2.1 NK Cell Phenotype
NK cells were first described in 1975 and later defined as an innate effector lymphocyte [15]. They are mostly differentiated in the bone marrow and are widely distributed in many tissues such as the lungs, liver, spleen, and lymph nodes [16, 17]. In humans, a majority of NK cells (approximately 90%) are CD56dim and CD16hi and the minority (10%) are CD56hi and CD16dim/− [18]. A population of thymic derived NK cells has been described in mice that may be similar to human CD56hiCD16dim/− NK cells. These thymic NK cells express CD127, high levels of Gata-3 and are Notch independent [3].
2.2.2 Ror γ t-Dependent ILC Phenotype
In mice, LTi cells are characterized by a lack of T, B, and myeloid cell markers, but express integrin α4β7, CD45, CD4, lymphotoxin-α (LT-α), and LT-β, as well as multiple chemokine and cytokine receptors (CD127, CD117, c-Kit)[19]. Adult LTi cells differ from their fetal counterpart due to expression of the T cell costimulatory molecule ligand (OX40-L) and CD30L [20]. Human LTi cells have been described in fetal mesentery and adult lymph node, spleen, gut, and tonsils [21]. They are similar to mouse LTi cells except that human LTi cells are all CD4- while a proportion of mouse LTi cells are CD4+ [22].
ILC22 were first identified as an IL-22 producing NK cell subset [9, 10, 23, 24]. The reason for this classification was due to surface expression of NK cell markers such as CD56, NKp44 and low NKp46 expression (in humans), and NKp46 and some low NK1.1 expression (in mice). However, ILC22 differ from conventional NK cells because they lack cytolytic properties, lack killer inhibitory receptors (in humans), lack Ly49 (in mice) and do not produce IFN-γ. They also share similarities with LTi cells by expressing Rorγt and IL-22. In both human and mice, ILC22 are found mainly in the small intestine, colon, mesenteric lymph nodes and liver [9]. ILC22 (ILC3) have been termed NK22, NCR22, NKR+LTi, LTi-like NK cells and NKp46+Rorγt+ ILCs. We have adopted the nomenclature proposed by Spits and Di Santo and use ILC22 to describe this Rorγt+IL-22+NK receptor+ LTi-like ILC population [1].
IL-1β and IL-23 upregulate the production of IL-22 from ILC22 in both mice and humans [10], while IL-12 and IL-18 induce IL-22 production from mouse ILC22 [25]. It has also been shown that IL-25 produced by intestinal epithelial cells negatively regulates IL-22 production by Rorγt+ ILCs [26]. The common γ-chain (γc) cytokines (for example, IL-2, IL-7, and IL-15) can also activate proliferation and cytokine production of human ILC22 [27]. Depending on culture conditions, human ILC22 can be induced to secrete a spectrum of cytokines, including IL-2, IL-5, IL-8, IL-13, IL-17, TNF, IFN-γ, and B cell activation factor [27, 28]. Whether this is because human ILC22 possess cytokine plasticity, or because it is a heterogeneous population of cells, has yet to be determined.
Another mouse non-LTi population has been described, which is specialized to produce IL-17. These cells, termed ILC17, are Rorγt dependent and are CD4-CD117-NKp46-, which separates them from both LTi and ILC22 [29]. In humans, an IL-17-producing ILC population has been described; however its expression of cell surface markers is different from mouse ILC17 [7]. Analysis of lineage−CD127+CD117+ adult tonsil cells identified that a proportion of them produce IL-17, but not IL-22 [7, 30]. However, these lineage−CD127+CD117+ cells appear to be heterogeneous because an IL-17+IL-22+ subpopulation was also identified [30].
2.2.3 Type 2 ILC Phenotype
The type 2 ILCs were independently discovered in 2010 by three separate groups, and were called nuocytes, natural helper cells (NHCs), and innate type 2 helper (Ih2) cells [13, 14, 31]. Using a combination of flow cytometry and microarray analyses, type 2 ILCs were shown to lack the expression of lineage defining surface markers for T cells, B cells, NKT cells, DCs, macrophages, neutrophils, eosinophils, mast cells, basophils, and LTi cells. Type 2 ILCs share a number of surface and functional similarities [32] (Table 2.2). Variability of surface expression markers may be attributed to the different tissues these type 2 ILCs were taken from, indicate a different activation state of the cell, or identify different type 2 ILC subsets. Certainly, activated nuocytes isolated from lung tissue showed a lower expression of Sca-1 and CCR9 compared to those isolated from the MLN [33]. Multiple other groups have since described type 2 ILC-like populations in the liver, bone marrow, lungs, and intestine [34–44]. All identified type 2 ILCs are lineage negative, respond to treatment with either IL-25 and/or IL-33, and can produce type 2 cytokines (IL-5 and/or IL-13). A report of particular interest by Mjösberg et al. characterized a possible human equivalent of mouse type 2 ILCs [36]. These human type 2 ILC cells share a similar phenotype and function with mouse type 2 ILCs and are found in the fetal and adult lung and gut tissues (Table 2.2). They also found these cells in the peripheral blood, but these cells express the chemokine receptor CCR6 and did not produce type 2 cytokines. This suggests that human type 2 ILCs may initially be released into the bloodstream in an inactivate form after which they home into the lung and gut tissue. There, they may mature, becoming activated in situ to start producing type 2 cytokines. Further research is required to determine if the cells identified by Mjösberg et al. are truly the human equivalent of type 2 ILCs.
Although multipotent progenitor type 2 (MPPtype2) cells also respond to IL-25 treatment, they can differentiate into myeloid cells following treatment with SCF and IL-3 [45]. This suggests that MPPtype2 cells may represent a heterogeneous population which includes precursor cells which are not terminally differentiated. This differs from the other members of the type 2 ILC family and they are not included herein.
2.3 Development of ILCs
2.3.1 Inhibitor of DNA Binding 2 (Id2): An Early Common “Switch” for ILCs
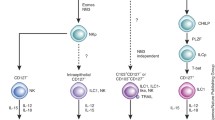
The development of T cells, B cells, and DCs from progenitor cells is dependent on a group of basic helix-turn-helix (bHLH) proteins termed E proteins, which include the E2a isotypes (E12 and E47), E2-2, and human bHLH factor (HEB) [46]. Conversely, the E proteins inhibit the development of several ILC populations [47]. The transcription factor function of E proteins is neutralized by Id (inhibitor of DNA binding) proteins, by forming a heterodimer with each other [48]. Of the 4 Id protein members, Id2 has been shown to be important for the development of NK cells and LTi cells by blocking the transcriptional activity of E47 [47]. Id2 also promotes development of ILC22 and type 2 ILCs [14, 49]. It has been demonstrated in Rorγt+ ILCs that Id2 is upregulated prior to Rorγt expression [50]. Taken together, the evidence suggests that the progenitors of different ILC populations share an early expression of Id2 protein, which acts as a developmental block against differentiation down the T cell and B cell pathway (Fig. 2.1).

An overview of known ILC developmental requirements. All ILC members are thought to derive from an Id2 expressing progenitor. Differentiation into NK cells, type 2 ILCs, or Rorγt-dependent ILCs is dictated by various cytokines, signals (above arrow), and transcription factors (below arrow). A brief summary of where these subsets are thought to differentiate as well as functions is included
2.3.2 NK Cells Development
NK cell development in the bone marrow is dependent on early IL-15 and Flt3 ligand signaling [51, 52]. Other transcription factors affecting NK cell differentiation and maturation include transcription factors such as Ets-1, Id2, Ikaros, PU.1, T-bet, Gata-3, NFIL3 (E4BP4), and Eomesodermin, as well as the Tox nuclear factor [51, 53]. The development of mouse thymic NK cells is dependent on IL-15, IL-7, and Gata-3 [4] (Fig. 2.1).
2.3.3 Rorgt-Dependent ILCs Development
As their name suggests, Rorγt-dependent ILCs express Rorγt, which is important for their development and function [2]. The retinoic acid receptor-related orphan receptors (Rorα, Rorβ, and Rorγ) are a family of DNA-binding transcription factors which are nuclear receptors. Cholesterol and its derivatives have been identified as natural ligands for Rorα while hydroxycholesterols have been proposed as a natural ligand for Rorγ [54, 55]. Rorγt is a short Rorγ isoform that is specifically expressed in cells of the immune system, and Rorγt-deficient mice lack lymph nodes and Peyer’s patches [56]. All the Rorγt-dependent ILCs express the IL-7 receptor CD127, and IL-7 has been shown to be important for homeostasis of these ILCs [57, 58] (Fig. 2.1).
Fetal LTi cells differentiate from fetal liver CLPs by first upregulating Id2 which results in upregulation of α4β7, with the loss of B cell potential. This is followed by upregulation of the chemokine marker CXCR6, extinguishing their T cell potential, before final expression of Rorγt. An early pulse of Notch signaling has been reported to maximize the efficiency of LTi cell differentiation [50]. However the necessity of Notch signaling is still contentious as results show fetal liver CLPs can still generate LTi cells in the absence of Notch [59]. Postnatal LTi cells are derived from bone marrow CLPs, which enter the periphery as α4β7+ cells to colonize the spleen and lamina propria before completing differentiation in situ into Rorγt+ cells via a Notch-dependent pathway [59]. Additionally, LTi cell differentiation has also been shown to depend on the transcriptions factors Runx1 and Tox [60, 61].
Fate-mapping experiments and genome-wide microarray profiling have demonstrated that mouse ILC22 derive from Rorγt+ precursors that are α4β7- [62, 63]. Although ILC22 development does not require the expression of α4β7, Id2-deficient mice do not possess ILC22 [49], suggesting that another function of Id2, apart from inducing α4β7 expression, is required for the differentiation of ILC22 [50]. ILC22 can develop from a CXCR6+ CLP population in adult lamina propria but not in spleen, showing a compartmental specificity for the chemokine receptor [59]. These Rorγt+ precursors in the small intestine, colon, and secondary lymphoid organs require Notch signaling to stably upregulate NK receptors in vivo and will express IL-22 as they mature into ILC22 [59]. ILC22 retain some degree of plasticity, for example, ILC22 in the small intestine remain Rorγt+, while those in the colon and secondary lymphoid organs become RORγt-IL-22- IFN-γ+ cells, gaining NK cell markers but lacking cytolytic ability, thus differentiating them from true NK cells [63, 64]. In vitro, IL-7 has been implicated in maintaining Rorγt expression and IL-22 production, while IL-2 and IL-15 promote the loss of Rorγt and the gain of IFN-γ expression [64]. Vornarboug et al. showed that the presence of commensal microflora also plays a role in maintaining Rorγt expression and subsequent induction of IL-22. However, the microflora is not essential for ILC22 development since they continue to develop in the small intestine of germfree mice [23, 24, 26, 62, 64].
Another factor implicated in Rorγt-dependent ILC development and function is the ligand-dependent transcription factor aryl hydrocarbon receptor (AhR) (Fig. 2.1) [65–67]. AhR is expressed by ILC22 in both mice and humans, and AhR-deficient mice have fewer ILC22 that have impaired IL-22 production [67, 68]. These mice also show defects in cryptopatch clusters and isolated lymphoid follicles (ILFs), suggesting that AhR is also important for postnatal LTi function [66]. The natural ligands for AhR are flavanoids and glucosinolates, which are dietary compounds commonly found in vegetables such as of the family Brassicaceae [66]. This suggests that balanced nutrition plays a part in priming the innate immune system in the gut, and maybe mother was right to make you eat your greens [65].
Some key questions about the Rorγt-dependent ILCs (LTi, ILC22, and ILC17) still remain to be answered. These include the developmental relationships between these subsets; should they be classified as distinct subsets or differently activated cells, and do they possess a degree of plasticity to change from one subset to another? Previous attempts to differentiate fetal mouse LTi cells into ILC22 have been unsuccessful, but human LTi cells from fetal lymph nodes and adult tonsils can become ILC22, suggesting that some degree of trans-differentiation is possible [62]. Future research will hopefully better define the development and function of the Rorγt-dependent ILCs in both humans and mice.
2.3.4 Type 2 ILCs Development
Type 2 ILCs are derived from the lymphoid lineage [69]. Collectively, they express a combination of hematopoietic markers such as CD45, c-Kit, and Sca-1, as well as lymphoid markers such as IL-7Rα, ICOS, Thy1.2, and CD44 [13, 14, 31, 36]. The development of NHCs and Ih2 cells is dependent on expression of the γc surface receptor, suggesting the developmental importance of γc-dependent cytokines (IL-2, IL-4, IL-7, IL-9, IL-15, or IL-21), some of which are central regulators of lymphocyte homeostasis [14, 31, 58]. Type 2 ILCs do not differentiate into other lineage cell types in a cytokine cocktail with SCF and IL-3, suggesting that they are terminally differentiated [13]. However, it is currently unknown if they can trans-differentiate under specific stimulus.
Yang et al. proposed that NHCs are derived from bone marrow lymphoid progenitor cells. They found that most NHCs expressed Rag1 at some point in their development and they differentiate in vivo from lymphoid progenitor cells [69]. Their findings were corroborated by Wong et al., wherein they showed that functional nuocytes differentiated from bone marrow CLP in both in vivo and in vitro models when treated with IL-7 and IL-33 [34]. Both studies agreed that NHCs and nuocytes required IL-7 receptor for in vivo development. In addition, Wong et al. demonstrated the requirement for Notch signaling for in vitro differentiation of nuocytes. Notch signaling is important for hematopoiesis, especially for T cell commitment of progenitor cells and T cell maturation in the thymus [70]. T cell precursors in the double negative stage 1 and stage 2 thymocytes also retain nuocyte differentiation potential when treated with IL-7 and IL-33 [34]. However, further differentiation down the T cell pathway has to be inhibited. The absence of NHCs in Id2-deficient mice suggests that Id2 is important for type 2 ILCs development, and could be responsible for the inhibition of T cell differentiation [14].
-
1
Significantly, Wong et al. also reported the requirement for Rorα for nuocyte differentiation [34]. Rorα is expressed in a wide variety of tissues, but is especially important for neuronal development [71, 72]. Mice deficient for Rorα exhibit ataxia, cerebellar atrophy, and a significantly diminished life span [73, 74]. Rorα had also been loosely linked to immunity because Rorα-deficient mice were reported to have reduced T cell/B cell numbers in the spleen and thymus, as well as reduced OVA-induced airway hyperreactivity [71, 75]. Rorα has also been implicated in Th17 differentiation [76]. Microarray data for both nuocytes and NHCs show that they express Rorα mRNA and not Rorγt mRNA, further differentiating them from the Rorγt-dependent ILCs [13, 14]. A natural knockout of Rorα occurs in staggerer (Rora sg/sg) mice [73]. Reconstituting lethally irradiated mice with staggerer mouse bone marrow followed by infection with Nippostrongylus brasiliensis showed that the nuocyte population did not expand in the reconstituted mice and these animals displayed impaired worm expulsion. The adaptive immune response remained normal based on normal T cell development and numbers [34]. An OVA-induced asthma model using staggerer mice demonstrated that these mice developed less airway inflammation, goblet cell hyperplasia, eosinophilia, and production of type 2 cytokines [75]. These results, together with those from a study by Halim et al. [160], support a model for Rorα as an important transcription factor for type 2 ILC development in the bone marrow. Interestingly, human type 2 ILCs express Rorγt, albeit at lower levels than the Rorγt-dependent ILCs [36]. It remains to be proved if Rorα is also important for the development of human type 2 ILCs.
Thus, type 2 ILCs differentiate from CLPs, and this is dependent on Notch and γc-dependent cytokine (IL-7) signaling [77]. A possible source of Notch ligands and IL-7 is the stromal cells in the bone marrow [78, 79], although this requires further investigation. Notch signaling encourages CLPs towards a T cell fate [70], but it appears that the expression of Id2 (by signals as yet unknown) inhibits the T cell commitment of these progenitor cells [2, 14]. Expression of Rorα correlates with the further differentiation of mouse type 2 ILCs [34] (Fig. 2.1), but the factors that regulate Rorα expression remain to be identified. Recently, Gata-3 expression has been demonstrated to license type 2 ILCs for IL-13 expression, and that Gata-3 and STAT6 both contribute to type 2 ILC development [80, 161].
2.4 ILC Roles in the Host Organism
2.4.1 NK Cells: Cytolytic Activity and Cytokine Production
The CD56dim NK cell population is biased towards rapidly initiating a cytolytic response against virus-infected host organism cells or tumor cells without the need for pre-sensitization or activation via the major histocompatibility (MHC) molecules [81]. There are two mechanisms for this cytolytic activity. The first is granule-dependent cytotoxicity, where NK cells are activated to release perforin and granzymes in proximity to an infected cell to kill it. The other triggers the apoptosis pathway in target cells via NK cell-secreted tumor necrosis factor-α (TNF-α) that binds to the target cell, or via direct cell contact with NK cells leading to signaling through TNF-related apoptosis-inducing ligand (TRAIL) and/or Fas ligand (FasL) [16, 82].
CD56hi NK cells and mouse thymic NK cells lack cytolytic activity and are primed towards producing cytokines such as IFN-γ, TNF-α, IL-10, and other growth factors [3, 81] (Fig. 2.1).
Apart from being effector cells, NK cells have a regulatory role during an immune response by affecting DCs, macrophages, and mast cells [83]. Recently, the notion that NK cells are truly innate cells has been called into question because specific subsets of mouse liver NK cells have been described to have the adaptive immunity property of lasting memory against specific viral antigens [84, 85].
2.4.2 LTi Cells and Organogenesis of Lymphoid Structures
LTi cells induce the formation of lymph tissues such as lymph nodes and Peyer’s patches during embryogenesis in both humans and mice [86–88]. During mouse embryogenesis, fetal liver-derived LT-α1LT-β2 +LTi cells colonize developing lymph tissues and interact with mesenchymal-derived stromal organizer cells called lymphoid tissue organizer (LTo), which express vascular cell adhesion molecule (VCAM-1) and LT-β receptor. Signaling through the LT-β receptor induces the upregulation of various cell adhesion molecules, production of IL-7 and TNF-related activation-induced cytokine (TRANCE), and secretion of lymphoid chemokines such as CXCL13, CCL19, and CCL21. These factors recruit additional LTi precursors as well as other hematopoietic cells, including B and T cells, and DCs, to the developing lymph node [89, 90].
Postnatal LTi cells have further developmental roles in secondary lymphoid tissues [7]. They are important for the formation of ILFs in the gut after recognition of pathogen-associated patterns (PAMPs) on commensal bacteria in order to maintain intestinal homeostasis [91, 92]. Additionally, it has been reported that postnatal LTi cells are involved in the repair of damaged lymph nodes after acute viral infections that destroy the T cell zone stromal cells [93]. They have also been shown to be involved in the segregation of B and T cell zones in spleen architecture, as well as in memory CD4 T cell generation [94, 95].
2.4.3 Ror γ t-Dependent ILCs: IL-17 and IL-22 Producers for Intestinal Homeostasis
IL-17 is a pro-inflammatory cytokine that recruits neutrophils and promotes cytokine and antimicrobial peptide production from a variety of cells such as bronchial epithelial cells [96]. IL-17 has also been shown to have a role in the formation of germinal centers and neutrophilia in allergic asthma [97, 98]. IL-22 is a member of the IL-10 family and binds to its receptor, which is found exclusively on epithelial cells to induce the production of cytokines, microbial peptides, and mucins [99]. It can act as either a pro-inflammatory or an anti-inflammatory cytokine depending on the cellular and cytokine environment. It acts as a pro-inflammatory cytokine in diseases such as psoriasis and multiple sclerosis [100], but limits damage caused by the immune system in hepatitis and helps maintain mucosal immunity and integrity in eosinophilic airway inflammation and inflammatory bowel disease [101].
LTi cells are producers of the cytokines IL-17 and/or IL-22 after stimulation with IL-23 [21, 102]. Therefore, LTi cells may be involved with the early protection against microbial infections and maintaining the mucosal barrier in the host organism. ILC22 and ILC17 act as specialized producers of IL-17 and IL22, to support the protective responses in the gut during microbial infections.
Both ILC22 and ILC17 are recruited to the intestine under inflammatory conditions, and are involved in a protective role during intestinal infection and inflammation. IL-23 induces ILC22 and ILC17 to produce their respective cytokine [99]. ILC22 serve as a critical early source of IL-22 to protect against colitis-inducing Citrobacter rodentium infections [103], as well as other colitis models such as inflammatory bowel disease (IBD) and dextran sulfate sodium (DSS)-induced colitis [104]. Although ILC22 are not required to control a Listeria monocytogenes infection, the oral introduction of the pathogen still enhances IL-22 production from ILC22 [63]. It is unknown if these human-specialized IL-17 producers are present in the intestine during an infection. The different effector functions of ILC17 and ILC22 might explain the presence of specialized subsets of IL-22- and IL-17-producing ILCs, which would tailor the innate immune response to infections and maintain intestinal homeostasis.
2.4.4 Type 2 ILCs: Protective Response Against Helminths
Type 2 ILCs were initially described as important innate cells responsible for anti-helminth protection [105]. IL-25 and IL-33 have been shown to be important to activate type 2 ILCs to produce effector cytokines.
IL-25 (IL-17E) is a member of the IL-17 family that is associated with Th2-like inflammation and disease [106–108]. IL-25 mRNA transcripts are produced in Th2 cells and lung epithelial cells while the protein has been reported to be produced by alveolar macrophages, mast cells, eosinophils, and basophils [109–111]. IL-25 upregulates the production of type 2 cytokines by eosinophils, mast cells, type 2 ILCs, and Th2 cells [33, 112]. IL-25 signaling acts via the signaling molecule Act1 to increase expression of Gata-3 and subsequent production of type 2 cytokines [109, 113, 114].
IL-33 (IL-1F11) is a member of the IL-1 family that binds to the ST2 receptor (Il1lr1) in complex with IL1RAP [115]. ST2 is primarily expressed on mast cells, Th2 cells, and type 2 ILCs [33, 116, 117]. IL-33 mRNA is expressed in epithelial cells, endothelial cells, lung fibroblasts, DCs, and alveolar macrophages [118], and plays roles in disease symptoms such as fibrosis and airway hyperreactivity, as well as in autoimmune diseases such as arthritis [119–122]. The mechanism by which IL-33 is released by cells is unclear; it is thought that IL-33 acts as an “alarmin” during necrosis and initiates inflammatory signaling [123]. By contrast, if the cell undergoes programmed cell death, i.e., apoptosis, then caspase-1 cleaves the cytokine domain of IL-33 into a nonfunctional form that fails to initiate the inflammatory response [124].
Infection with helminths, such as N. brasiliensis, breaches and irritates the epithelial barrier of the lung and gut. TFF2 signaling and other unknown signals induce the production and release of IL-25 and IL-33 from epithelial cells and other cells such as alveolar macrophages [125]. Since IL-25-responsive epithelial cells are important for downstream IL-5 and IL-13 production in the lung, this suggests that epithelial cells could self-upregulate factors in a positive feedback loop that amplifies the downstream type 2 response [113, 114].
-
2
Type 2 ILCs activated by IL-25 or IL-33 are important for N. brasiliensis expulsion. Neill et al. demonstrated that mice lacking either one or both cytokine receptors have very few nuocytes and are unable to effectively clear helminth infections. However, the adoptive transfer of activated type 2 ILCs was able to rescue this defect. Furthermore, worm expulsion was dependent on IL-13 since transferring IL-13-deficient nuocytes into IL-13-deficient mice failed to mediate worm expulsion [13]. IL-13 is indispensible for the efficient expulsion of N. brasiliensis [126] because it induces a range of type 2 immune physiological responses such as smooth muscle contraction, goblet cell hyperplasia, and mucus hypersecretion, thus activating a “weep and sweep” mechanism which traps and expels the worms [127] (Fig. 2.2). IL-13 is partially involved during the infection with other parasite species such as Trichuris muris [128]. Therefore, type 2 ILCs are the early trigger of type 2 protective responses. An increased number of circulating Ih2 cells in the blood after IL-25 treatment suggests that additional type 2 ILCs could be recruited from the blood to enhance a local type 2 response and also suggests that a localized infection could initiate a systemic type 2 response via these circulating type 2 ILCs [31].
Fig. 2.2 
Schematic of type 2 ILC function. Allergens, chemicals, irritants, or parasites induce lung or intestinal epithelial cells to release the type 2 ILC-activating cytokines, IL-25, and IL-33. IL-25 may act in a positive feedback loop on epithelial cells to amplify the activation of type 2 ILCs. Activated type 2 ILCs rapidly produce amphiregulin, IL-5, and IL-13. Amphiregulin promotes epithelial cell proliferation, while IL-5 promotes eosinophilia into the lung or gut tissues. IL-13 promotes smooth muscle contraction, goblet cell hyperplasia, and mucus hypersecretion. It also encourages deposition of extracellular matrix (ECM) by directly inducing collagen production from fibroblasts, and indirectly by inducing fibrotic factor production from alternatively activated macrophages (dashed arrow). Professional antigen-presenting cells (APC) activate adaptive Th2 cells in order to support the proliferation and function of type 2 ILCs. Th2 cells produce IL-5, IL-13, IL-9 (not shown), and IL-4, which perform functions specific to Th2 cells such as IL-4-driven class switching and antibody production

Apart from the innate immune system, the adaptive immune system is also activated following infection. Alarmins released by damaged epithelial cells, and parasite-derived antigens, promote Th2 cell differentiation via professional antigen-presenting cells [129]. This secondary wave of type 2 cytokines amplifies the effects of type 2 ILCs as well as initiating other physiological responses such as promoting Th2 cell differentiation, activating B cells to produce antibodies, inducing IgE class switching and upregulating mast cells [130–132].
Even though type 2 ILC-derived cytokines are sufficient to resolve a helminth infection, the presence of Th2 cells (even if IL-4 and IL13 deficient) is still essential for effective helminth expulsion [133]. This could be explained by the observation that Th2 cells are required to maintain type 2 ILCs numbers during an infection [13]. Nuocytes are present in Rag2 knockout mice (which lack B cells and T cells) and are responsive to IL-25 and IL-33, but their population numbers decrease soon after induction, and these mice are unable to expel the worm burden effectively [13]. This suggests that T cells play a role in nuocyte maintenance, and in addition, boost the type 2 immune response by producing more type 2 cytokines. An area for further investigation is the interrelationship of innate type 2 ILCs and adaptive Th2 cells.
2.4.5 Wound Healing
Evidence suggests that type 2 ILCs are involved in wound healing and fibrotic processes. Type 2 ILCs directly produce amphiregulin, which promotes the proliferation of epithelial cells [38, 134]. Type 2 ILCs can also indirectly promote tissue remodeling via IL-13 and IL-5. In vitro studies show that IL-13 can directly induce the proliferation of myofibroblasts and collagen production from fibroblasts [135, 136]. IL-13 indirectly promotes fibrosis via the induction of fibrotic factors such as arginase, TFG-β, and fibronectin from fibroblasts and alternatively activated macrophages [137–140]. In vivo, IL-13 has been shown to mediate Schistosoma mansoni-induced liver fibrosis in a TGF-β-independent pathway [141–143]. IL-5 promotes eosinophil recruitment and activation, which is thought to play a role in airway remodeling in chronic airway diseases [144].
Therefore, activation of type 2 ILCs may contribute to tissue repair following infection and injury to minimize the disease pathology (Fig. 2.2) [145].
Other ILC populations may also be involved in healing injuries sustained during infection. As mentioned previously, LTi cells can restore damaged lymph nodes after particularly severe viral infections. Other Rorγt-dependent ILCs may also promote healing via production of IL-22, which has been implicated in tissue repair after injury or alcohol-induced damage [146].
2.4.6 Dysregulation of ILCs: Autoimmunity, Allergy, and Fibrosis
The dysregulation of either IL-17 or IL-22 has been linked to autoimmune diseases such as psoriasis, rheumatoid arthritis, and IBD [147]. Therefore, if the activation of Rorγt-dependent ILCs (and production of IL-17 and IL-22) is not tightly regulated, they may contribute to these diseases. For example, Buonocore et al. have demonstrated that a Rorγt+ILC population is stimulated by IL-23 in the colon to produce IL-17 and induces intestinal colitis [29].
Chronic activation of the type 2 response can cause allergic airway diseases (such as asthma), inflammatory gut diseases, as well as excessive fibrosis and tissue remodeling [148–150]. As potent type 2 cytokine producers, type 2 ILCs would be expected to play a part in these diseases. Research has shown that the activator (IL-25, IL-33) and effector (IL-5, IL-13) cytokines of type 2 ILCs are involved in allergic diseases.
IL-25 and IL-33 expression correlates with allergic airway diseases [118, 151], and IL-33 has been identified as an asthma-related gene based on a genome-wide study [152]. In asthmatic lung tissue, increased production of IL-25 and IL-33 bring about the same physiological changes in the lungs as during a helminth infection, such as a rapid type 2 response, increased production of IL-5 and IL-13, and increased mucus production, eosinophilia, and airway hyperreactivity [118, 151]. Blocking either IL-25 or IL-33 signaling in the airways can reduce eosinophilia and inflammation in an ovalbumin (OVA)-driven model of allergic airway disease [43, 151]. Overexpression or ablation of IL-13 within the lungs has underlined its role in inducing asthma-like phenotypes, such as nonspecific airway hyperreactivity and mucus hyperproduction [153, 154]. As mentioned above, IL-13 also contributes to tissue remodeling and fibrosis, and thus may contribute to fibrosis in diseases dominated by a type 2 immune response [155]. IL-5 promotes eosinophil infiltration into the lungs [156, 157].
Recently, multiple teams have identified type 2 ILCs in the lungs and their role in airway allergy has been investigated [37–44, 137]. They have shown that when challenged with IL-25, IL-33, papain, allergens (Alternaria alternata, OVA, house dust mite, glycolipid antigen), parasites, or viruses, type 2 ILCs proliferated and were activated to produce a rapid type 2 response characterized by increased production of IL-5 and IL-13, increased mucus production, eosinophilia, and airway hyperreactivity, reminiscent of the response during an allergic asthma.
When mice were treated with OVA (as per an OVA-induced asthma model), IL-25, or IL-33, nuocytes were induced in the lung tissue and bronchoalveolar lavage (BAL). These nuocytes represent a major source of IL-13 in the lung, explaining why IL-13 from T cells is partially dispensable for the allergic inflammation during an airway hyperreactivity response. Adoptive transfer of nuocytes into IL-13-deficient mice (which do not respond to IL-25 treatment) restores both AHR and eosinophilia, indicating that nuocytes have the capacity to upregulate asthma even in the absence of T cell-derived IL-13. However, infiltration of neutrophils into the lung during challenge with IL-25 was not restored, indicating that other cells and cytokines are responsible for other aspects of the allergic response [41]. Kim et al. also showed the importance of type 2 ILCs in response to glycolipid antigens [40]. Halim et al. corroborated these earlier studies using the adoptive transfer of type 2 ILCs into Rag2 and γc double knockout mice, which restored the allergic phenotype [44]. Respiratory infections with rhinovirus or respiratory syncytial virus are known to promote type 2 responses, and exacerbate allergic asthma. Chang et al. demonstrated that influenza virus-induced asthma is not mediated by adaptive immunity, but by IL-33-dependent type 2 ILCs [39].
Significantly, Mjösberg et al. demonstrated that human type 2 ILCs are enriched in chronically inflamed airway tissues, such as the nasal polyps of patients suffering from chronic rhinosinusitis. These patients exhibited higher levels of IL-5 and IL-13 transcripts within the polyp tissue, which in turn contributes to eosinophil enrichment within the nasal polyps [36]. This may be attributable to the increased human type 2 ILC population.
The allergic response is not limited to only the lungs. As Camelo et al. demonstrate, activation of type 2 ILC leads to an overexpression of IL-13 in the gut, which then leads to chronic inflammation and ulcerative colitis [158]. It is also possible that type 2 ILCs represent a potent source of IL-13 in patients suffering from chronic asthma, which may contribute to the remodeling of lung tissue and lung fibrosis [159].
2.5 Conclusion
As we begin to understand the complexities of these newly identified ILC populations, it is apparent that the innate lymphoid cell compartment plays an important role for the host. It drives lymphoid tissue development, maintains tissue and barrier homeostasis, provides a rapid protective response against infectious agents, and promotes wound healing. In this way, they precede and also support the adaptive immune response.
Dysregulation of ILCs is also associated with disease. Rorγt-dependent cells are involved with colitis and IBD, while type 2 ILCs are associated with allergy in the gut and lungs. As we learn more about the innate lymphoid cells, they may come to represent viable therapeutic targets to combat such diseases.
References
Spits H, Di Santo JP. The expanding family of innate lymphoid cells: regulators and effectors of immunity and tissue remodeling. Nat Immunol. 2011;12:21–27.
Cherrier M, Ohnmacht C, Cording S, Eberl G. Development and function of intestinal innate lymphoid cells. Curr Opin Immunol. 2012.
Vosshenrich CA, Garcia-Ojeda ME, Samson-Villeger SI et al. A thymic pathway of mouse natural killer cell development characterized by expression of GATA-3 and CD127. Nat Immunol. 2006;7:1217–1224.
Ribeiro VS, Hasan M, Wilson A et al. Cutting edge: Thymic NK cells develop independently from T cell precursors. J Immunol. 2010;185:4993–4997.
Schoenborn JR, Wilson CB. Regulation of interferon-gamma during innate and adaptive immune responses. Adv Immunol. 2007;96:41–101.
Harrington LE, Hatton RD, Mangan PR et al. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol. 2005;6:1123–1132.
Cupedo T, Crellin NK, Papazian N et al. Human fetal lymphoid tissue-inducer cells are interleukin 17-producing precursors to RORC+ CD127+ natural killer-like cells. Nat Immunol. 2009;10:66–74.
Eyerich S, Eyerich K, Pennino D et al. Th22 cells represent a distinct human T cell subset involved in epidermal immunity and remodeling. J Clin Invest. 2009;119:3573–3585.
Cella M, Fuchs A, Vermi W et al. A human natural killer cell subset provides an innate source of IL-22 for mucosal immunity. Nature. 2009;457:722–725.
Luci C, Reynders A, Ivanov II et al. Influence of the transcription factor RORgammat on the development of NKp46+ cell populations in gut and skin. Nat Immunol. 2009;10:75–82.
Takatori H, Kanno Y, Watford WT et al. Lymphoid tissue inducer-like cells are an innate source of IL-17 and IL-22. J Exp Med. 2009;206:35–41.
Brinkmann V, Kristofic C. TCR-stimulated naive human CD4+ 45RO- T cells develop into effector cells that secrete IL-13, IL-5, and IFN-gamma, but no IL-4, and help efficient IgE production by B cells. J Immunol. 1995;154:3078–3087.
Neill DR, Wong SH, Bellosi A et al. Nuocytes represent a new innate effector leukocyte that mediates type-2 immunity. Nature. 2010;464:1367–1370.
Moro K, Yamada T, Tanabe M et al. Innate production of T(H)2 cytokines by adipose tissue-associated c-Kit(+)Sca-1(+) lymphoid cells. Nature. 2010;463:540–544.
Herberman RB, Nunn ME, Holden HT, Lavrin DH. Natural cytotoxic reactivity of mouse lymphoid cells against syngeneic and allogeneic tumors. II. Characterization of effector cells. Int J Cancer. 1975;16:230–239.
Di Santo JP. Natural killer cell developmental pathways: a question of balance. Annu Rev Immunol. 2006;24:257–286.
van den Brink MR, Palomba ML, Basse PH, Hiserodt JC. In situ localization of 3.2.3+ natural killer cells in tissues from normal and tumor-bearing rats. Cancer Res. 1991;51:4931–4936.
Lanier LL, Le AM, Civin CI, Loken MR, Phillips JH. The relationship of CD16 (Leu-11) and Leu-19 (NKH-1) antigen expression on human peripheral blood NK cells and cytotoxic T lymphocytes. J Immunol. 1986;136:4480–4486.
Mebius RE, Rennert P, Weissman IL. Developing lymph nodes collect CD4+CD3- LTbeta+ cells that can differentiate to APC, NK cells, and follicular cells but not T or B cells. Immunity. 1997;7:493–504.
Kim MY, Anderson G, White A et al. OX40 ligand and CD30 ligand are expressed on adult but not neonatal CD4+CD3- inducer cells: evidence that IL-7 signals regulate CD30 ligand but not OX40 ligand expression. J Immunol. 2005;174:6686–6691.
Kim MY, Kim KS, McConnell F, Lane P. Lymphoid tissue inducer cells: architects of CD4 immune responses in mice and men. Clin Exp Immunol. 2009;157:20–26.
Kim MY, Toellner KM, White A et al. Neonatal and adult CD4+ CD3- cells share similar gene expression profile, and neonatal cells up-regulate OX40 ligand in response to TL1A (TNFSF15). J Immunol. 2006;177:3074–3081.
Sanos SL, Bui VL, Mortha A et al. RORgammat and commensal microflora are required for the differentiation of mucosal interleukin 22-producing NKp46+ cells. Nat Immunol. 2009;10:83–91.
Satoh-Takayama N, Vosshenrich CA, Lesjean-Pottier S et al. Microbial flora drives interleukin 22 production in intestinal NKp46+ cells that provide innate mucosal immune defense. Immunity. 2008;29:958–970.
Satoh-Takayama N, Dumoutier L, Lesjean-Pottier S et al. The natural cytotoxicity receptor NKp46 is dispensable for IL-22-mediated innate intestinal immune defense against Citrobacter rodentium. J Immunol. 2009;183:6579–6587.
Sawa S, Lochner M, Satoh-Takayama N et al. RORgammat+ innate lymphoid cells regulate intestinal homeostasis by integrating negative signals from the symbiotic microbiota. Nat Immunol. 2011;12:320–326.
Crellin NK, Trifari S, Kaplan CD, Satoh-Takayama N, Di Santo JP, Spits H. Regulation of cytokine secretion in human CD127(+) LTi-like innate lymphoid cells by Toll-like receptor 2. Immunity. 2010;33:752–764.
Cella M, Otero K, Colonna M. Expansion of human NK-22 cells with IL-7, IL-2, and IL-1beta reveals intrinsic functional plasticity. Proc Natl Acad Sci USA. 2010;107:10961–10966.
Buonocore S, Ahern PP, Uhlig HH et al. Innate lymphoid cells drive interleukin-23-dependent innate intestinal pathology. Nature. 2010;464:1371–1375.
Crellin NK, Trifari S, Kaplan CD, Cupedo T, Spits H. Human NKp44+IL-22+ cells and LTi-like cells constitute a stable RORC+ lineage distinct from conventional natural killer cells. J Exp Med. 2010;207:281–290.
Price AE, Liang HE, Sullivan BM et al. Systemically dispersed innate IL-13-expressing cells in type 2 immunity. Proc Natl Acad Sci USA. 2010;107:11489–11494.
Saenz SA, Noti M, Artis D. Innate immune cell populations function as initiators and effectors in Th2 cytokine responses. Trends Immunol. 2010;31:407–413.
Neill DR, McKenzie AN. Nuocytes and beyond: new insights into helminth expulsion. Trends Parasitol. 2011;27:214–221.
Wong SH, Walker JA, Jolin HE et al. Transcription factor RORalpha is critical for nuocyte development. Nat Immunol. 2012;13:229–236.
Brickshawana A, Shapiro VS, Kita H, Pease LR. Lineage(-)Sca1+c-Kit(-)CD25+ cells are IL-33-responsive type 2 innate cells in the mouse bone marrow. J Immunol. 2011;187:5795–5804.
Mjosberg JM, Trifari S, Crellin NK et al. Human IL-25- and IL-33-responsive type 2 innate lymphoid cells are defined by expression of CRTH2 and CD161. Nat Immunol. 2011;12:1055–1062.
Yasuda K, Muto T, Kawagoe T et al. Contribution of IL-33-activated type II innate lymphoid cells to pulmonary eosinophilia in intestinal nematode-infected mice. Proc Natl Acad Sci USA. 2012;109:3451–3456.
Monticelli LA, Sonnenberg GF, Abt MC et al. Innate lymphoid cells promote lung-tissue homeostasis after infection with influenza virus. Nat Immunol. 2011;12:1045–1054.
Chang YJ, Kim HY, Albacker LA et al. Innate lymphoid cells mediate influenza-induced airway hyper-reactivity independently of adaptive immunity. Nat Immunol. 2011;12:631–638.
Kim HY, Chang YJ, Subramanian S et al. Innate lymphoid cells responding to IL-33 mediate airway hyperreactivity independently of adaptive immunity. J Allergy Clin Immunol. 2012;129:216–27. e1–6.
Barlow JL, Bellosi A, Hardman CS et al. Innate IL-13-producing nuocytes arise during allergic lung inflammation and contribute to airways hyperreactivity. J Allergy Clin Immunol. 2012;129:191–8. e1–4.
Wolterink RG, Kleinjan A, van Nimwegen M et al. Pulmonary innate lymphoid cells are major producers of IL-5 and IL-13 in murine models of allergic asthma. Eur J Immunol. 2012;42:1106–1116.
Bartemes KR, Iijima K, Kobayashi T, Kephart GM, McKenzie AN, Kita H. IL-33-responsive lineage- CD25+ CD44(hi) lymphoid cells mediate innate type 2 immunity and allergic inflammation in the lungs. J Immunol. 2012;188:1503–1513.
Halim TY, Krauss RH, Sun AC, Takei F. Lung natural helper cells are a critical source of Th2 cell-type cytokines in protease allergen-induced airway inflammation. Immunity. 2012;36:451–463.
Saenz SA, Siracusa MC, Perrigoue JG et al. IL25 elicits a multipotent progenitor cell population that promotes T(H)2 cytokine responses. Nature. 2010;464:1362–1366.
Kee BL. E and ID proteins branch out. Nat Rev Immunol. 2009;9:175–184.
Yokota Y, Mansouri A, Mori S et al. Development of peripheral lymphoid organs and natural killer cells depends on the helix-loop-helix inhibitor Id2. Nature. 1999;397:702–706.
Engel I, Murre C. The function of E- and Id proteins in lymphocyte development. Nat Rev Immunol. 2001;1:193–199.
Satoh-Takayama N, Lesjean-Pottier S, Vieira P et al. IL-7 and IL-15 independently program the differentiation of intestinal CD3-NKp46+ cell subsets from Id2-dependent precursors. J Exp Med. 2010;207:273–280.
Cherrier M, Sawa S, Eberl G. Notch, Id2, and RORgammat sequentially orchestrate the fetal development of lymphoid tissue inducer cells. J Exp Med. 2012;209:729–740.
Gascoyne DM, Long E, Veiga-Fernandes H et al. The basic leucine zipper transcription factor E4BP4 is essential for natural killer cell development. Nat Immunol. 2009;10:1118–1124.
Nakamori Y, Liu B, Ohishi K et al. Human bone marrow stromal cells simultaneously support B and T/NK lineage development from human haematopoietic progenitors: a principal role for flt3 ligand in lymphopoiesis. Br J Haematol. 2012
Martin-Fontecha A, Lord GM, Brady HJ. Transcriptional control of natural killer cell differentiation and function. Cell Mol Life Sci. 2011;68:3495–3503.
Kallen J, Schlaeppi JM, Bitsch F, Delhon I, Fournier B. Crystal structure of the human RORalpha Ligand binding domain in complex with cholesterol sulfate at 2.2 A. J Biol Chem. 2004;279:14033–14038.
Jin L, Martynowski D, Zheng S, Wada T, Xie W, Li Y. Structural basis for hydroxycholesterols as natural ligands of orphan nuclear receptor RORgamma. Mol Endocrinol. 2010;24:923–929.
Sun Z, Unutmaz D, Zou YR et al. Requirement for RORgamma in thymocyte survival and lymphoid organ development. Science. 2000;288:2369–2373.
Schmutz S, Bosco N, Chappaz S et al. Cutting edge: IL-7 regulates the peripheral pool of adult ROR gamma+ lymphoid tissue inducer cells. J Immunol. 2009;183:2217–2221.
Kang J, Coles M. IL-7: The global builder of the innate lymphoid network and beyond, one niche at a time. Semin Immunol. 2012
Possot C, Schmutz S, Chea S et al. Notch signaling is necessary for adult, but not fetal, development of RORgammat(+) innate lymphoid cells. Nat Immunol. 2011;12:949–958.
Tachibana M, Tenno M, Tezuka C, Sugiyama M, Yoshida H, Taniuchi I. Runx1/Cbfbeta2 complexes are required for lymphoid tissue inducer cell differentiation at two developmental stages. J Immunol. 2011;186:1450–1457.
Aliahmad P, de la Torre B, Kaye J. Shared dependence on the DNA-binding factor TOX for the development of lymphoid tissue-inducer cell and NK cell lineages. Nat Immunol. 2010;11:945–952.
Sawa S, Cherrier M, Lochner M et al. Lineage relationship analysis of RORgammat+ innate lymphoid cells. Science. 2010;330:665–669.
Reynders A, Yessaad N, Vu Manh TP et al. Identity, regulation and in vivo function of gut NKp46+RORgammat+ and NKp46+RORgammat- lymphoid cells. EMBO J. 2011;30:2934–2947.
Vonarbourg C, Mortha A, Bui VL et al. Regulated expression of nuclear receptor RORgammat confers distinct functional fates to NK cell receptor-expressing RORgammat(+) innate lymphocytes. Immunity. 2010;33:736–751.
Qiu J, Heller JJ, Guo X et al. The aryl hydrocarbon receptor regulates gut immunity through modulation of innate lymphoid cells. Immunity. 2012;36:92–104.
Kiss EA, Vonarbourg C, Kopfmann S et al. Natural aryl hydrocarbon receptor ligands control organogenesis of intestinal lymphoid follicles. Science. 2011;334:1561–1565.
Lee JS, Cella M, McDonald KG et al. AHR drives the development of gut ILC22 cells and postnatal lymphoid tissues via pathways dependent on and independent of Notch. Nat Immunol. 2012;13:144–151.
Veldhoen M, Hirota K, Westendorf AM et al. The aryl hydrocarbon receptor links TH17-cell-mediated autoimmunity to environmental toxins. Nature. 2008;453:106–109.
Yang Q, Saenz SA, Zlotoff DA, Artis D, Bhandoola A. Cutting edge: Natural helper cells derive from lymphoid progenitors. J Immunol. 2011;187:5505–5509.
Sandy AR, Jones M, Maillard I. Notch signaling and development of the hematopoietic system. Adv Exp Med Biol. 2012;727:71–88.
Dzhagalov I, Zhang N, He YW. The roles of orphan nuclear receptors in the development and function of the immune system. Cell Mol Immunol. 2004;1:401–407.
Jetten AM. Retinoid-related orphan receptors (RORs): critical roles in development, immunity, circadian rhythm, and cellular metabolism. Nucl Recept Signal. 2009;7:e003.
SIDMAN RL, LANE PW, DICKIE MM. Staggerer, a new mutation in the mouse affecting the cerebellum. Science. 1962;137:610–612.
Dussault I, Fawcett D, Matthyssen A, Bader JA, Giguere V. Orphan nuclear receptor ROR alpha-deficient mice display the cerebellar defects of staggerer. Mech Dev. 1998;70:147–153.
Jaradat M, Stapleton C, Tilley SL et al. Modulatory role for retinoid-related orphan receptor alpha in allergen-induced lung inflammation. Am J Respir Crit Care Med. 2006;174:1299–1309.
Yang XO, Pappu BP, Nurieva R et al. T helper 17 lineage differentiation is programmed by orphan nuclear receptors ROR alpha and ROR gamma. Immunity. 2008;28:29–39.
Vonarbourg C, Diefenbach A. Multifaceted roles of interleukin-7 signaling for the development and function of innate lymphoid cells. Semin Immunol. 2012
Weber JM, Calvi LM. Notch signaling and the bone marrow hematopoietic stem cell niche. Bone. 2010;46:281–285.
Welch PA, Burrows PD, Namen A, Gillis S, Cooper MD. Bone marrow stromal cells and interleukin-7 induce coordinate expression of the BP-1/6C3 antigen and pre-B cell growth. Int Immunol. 1990;2:697–705.
Liang HE, Reinhardt RL, Bando JK, Sullivan BM, Ho IC, Locksley RM. Divergent expression patterns of IL-4 and IL-13 define unique functions in allergic immunity. Nat Immunol. 2012;13:58–66.
Cooper MA, Fehniger TA, Caligiuri MA. The biology of human natural killer-cell subsets. Trends Immunol. 2001;22:633–640.
Smyth MJ, Cretney E, Kelly JM et al. Activation of NK cell cytotoxicity. Mol Immunol. 2005;42:501–510.
Moretta A, Marcenaro E, Sivori S, Della Chiesa M, Vitale M, Moretta L. Early liaisons between cells of the innate immune system in inflamed peripheral tissues. Trends Immunol. 2005;26:668–675.
Gillard GO, Bivas-Benita M, Hovav AH et al. Thy1+ NK [corrected] cells from vaccinia virus-primed mice confer protection against vaccinia virus challenge in the absence of adaptive lymphocytes. PLoS Pathog. 2011;7:e1002141.
Vivier E, Raulet DH, Moretta A et al. Innate or adaptive immunity? The example of natural killer cells. Science. 2011;331:44–49.
Kelly KA, Scollay R. Seeding of neonatal lymph nodes by T cells and identification of a novel population of CD3-CD4+ cells. Eur J Immunol. 1992;22:329–334.
Eberl G, Marmon S, Sunshine MJ, Rennert PD, Choi Y, Littman DR. An essential function for the nuclear receptor RORgamma(t) in the generation of fetal lymphoid tissue inducer cells. Nat Immunol. 2004;5:64–73.
Eberl G, Littman DR. The role of the nuclear hormone receptor RORgammat in the development of lymph nodes and Peyer’s patches. Immunol Rev. 2003;195:81–90.
Eberl G, Lochner M. The development of intestinal lymphoid tissues at the interface of self and microbiota. Mucosal Immunol. 2009;2:478–485.
van de Pavert SA, Mebius RE. New insights into the development of lymphoid tissues. Nat Rev Immunol. 2010;10:664–674.
Hamada H, Hiroi T, Nishiyama Y et al. Identification of multiple isolated lymphoid follicles on the antimesenteric wall of the mouse small intestine. J Immunol. 2002;168:57–64.
Bouskra D, Brezillon C, Berard M et al. Lymphoid tissue genesis induced by commensals through NOD1 regulates intestinal homeostasis. Nature. 2008;456:507–510.
Scandella E, Bolinger B, Lattmann E et al. Restoration of lymphoid organ integrity through the interaction of lymphoid tissue-inducer cells with stroma of the T cell zone. Nat Immunol. 2008;9:667–675.
Kim MY, McConnell FM, Gaspal FM et al. Function of CD4+CD3- cells in relation to B- and T-zone stroma in spleen. Blood. 2007;109:1602–1610.
Lane P, Kim MY, Withers D et al. Lymphoid tissue inducer cells in adaptive CD4 T cell dependent responses. Semin Immunol. 2008;20:159–163.
Onishi RM, Gaffen SL. Interleukin-17 and its target genes: mechanisms of interleukin-17 function in disease. Immunology. 2010;129:311–321.
Hsu HC, Yang P, Wang J et al. Interleukin 17-producing T helper cells and interleukin 17 orchestrate autoreactive germinal center development in autoimmune BXD2 mice. Nat Immunol. 2008;9:166–175.
Mizutani N, Goshima H, Nabe T, Yoshino S. Complement C3a-Induced IL-17 Plays a Critical Role in an IgE-Mediated Late-Phase Asthmatic Response and Airway Hyperresponsiveness via Neutrophilic Inflammation in Mice. J Immunol. 2012.
Zenewicz LA, Flavell RA. Recent advances in IL-22 biology. Int Immunol. 2011;23:159–163.
Zenewicz LA, Yancopoulos GD, Valenzuela DM, Murphy AJ, Stevens S, Flavell RA. Innate and adaptive interleukin-22 protects mice from inflammatory bowel disease. Immunity. 2008;29:947–957.
Takahashi K, Hirose K, Kawashima S et al. IL-22 attenuates IL-25 production by lung epithelial cells and inhibits antigen-induced eosinophilic airway inflammation. J Allergy Clin Immunol. 2011;128:1067–1076.
Kim S, Han S, Withers DR et al. CD117 CD3 CD56 OX40Lhigh cells express IL-22 and display an LTi phenotype in human secondary lymphoid tissues. Eur J Immunol. 2011;41:1563–1572.
Zheng Y, Valdez PA, Danilenko DM et al. Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nat Med. 2008;14:282–289.
Cox JH, Kljavin NM, Ota N et al. Opposing consequences of IL-23 signaling mediated by innate and adaptive cells in chemically induced colitis in mice. Mucosal Immunol. 2012;5:99–109.
Fallon PG, Ballantyne SJ, Mangan NE et al. Identification of an interleukin (IL)-25-dependent cell population that provides IL-4, IL-5, and IL-13 at the onset of helminth expulsion. J Exp Med. 2006;203:1105–1116.
Fort MM, Cheung J, Yen D et al. IL-25 induces IL-4, IL-5, and IL-13 and Th2-associated pathologies in vivo. Immunity. 2001;15:985–995.
Lee J, Ho WH, Maruoka M et al. IL-17E, a novel proinflammatory ligand for the IL-17 receptor homolog IL-17Rh1. J Biol Chem. 2001;276:1660–1664.
Saadoun D, Terrier B, Cacoub P. Interleukin-25: key regulator of inflammatory and autoimmune diseases. Curr Pharm Des. 2011;17:3781–3785.
Angkasekwinai P, Park H, Wang YH et al. Interleukin 25 promotes the initiation of proallergic type 2 responses. J Exp Med. 2007;204:1509–1517.
Ikeda K, Nakajima H, Suzuki K et al. Mast cells produce interleukin-25 upon Fc epsilon RI-mediated activation. Blood. 2003;101:3594–3596.
Kang CM, Jang AS, Ahn MH et al. Interleukin-25 and interleukin-13 production by alveolar macrophages in response to particles. Am J Respir Cell Mol Biol. 2005;33:290–296.
Corrigan CJ, Wang W, Meng Q et al. Allergen-induced expression of IL-25 and IL-25 receptor in atopic asthmatic airways and late-phase cutaneous responses. J Allergy Clin Immunol. 2011;128:116–124.
Claudio E, Sonder SU, Saret S et al. The adaptor protein CIKS/Act1 is essential for IL-25-mediated allergic airway inflammation. J Immunol. 2009;182:1617–1630.
Swaidani S, Bulek K, Kang Z et al. The critical role of epithelial-derived Act1 in IL-17- and IL-25-mediated pulmonary inflammation. J Immunol. 2009;182:1631–1640.
Townsend MJ, Fallon PG, Matthews DJ, Jolin HE, McKenzie AN. T1/ST2-deficient mice demonstrate the importance of T1/ST2 in developing primary T helper cell type 2 responses. J Exp Med. 2000;191:1069–1076.
Lohning M, Stroehmann A, Coyle AJ et al. T1/ST2 is preferentially expressed on murine Th2 cells, independent of interleukin 4, interleukin 5, and interleukin 10, and important for Th2 effector function. Proc Natl Acad Sci USA. 1998;95:6930–6935.
Yanagisawa K, Naito Y, Kuroiwa K et al. The expression of ST2 gene in helper T cells and the binding of ST2 protein to myeloma-derived RPMI8226 cells. J Biochem. 1997;121:95–103.
Schmitz J, Owyang A, Oldham E et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity. 2005;23:479–490.
Hong YS, Moon SJ, Joo YB et al. Measurement of interleukin-33 (IL-33) and IL-33 receptors (sST2 and ST2L) in patients with rheumatoid arthritis. J Korean Med Sci. 2011;26:1132–1139.
Kearley J, Buckland KF, Mathie SA, Lloyd CM. Resolution of allergic inflammation and airway hyperreactivity is dependent upon disruption of the T1/ST2-IL-33 pathway. Am J Respir Crit Care Med. 2009;179:772–781.
Kondo Y, Yoshimoto T, Yasuda K et al. Administration of IL-33 induces airway hyperresponsiveness and goblet cell hyperplasia in the lungs in the absence of adaptive immune system. Int Immunol. 2008;20:791–800.
Marvie P, Lisbonne M, L’helgoualc’h A et al. Interleukin-33 overexpression is associated with liver fibrosis in mice and humans. J Cell Mol Med. 2010;14:1726–1739.
Moussion C, Ortega N, Girard JP. The IL-1-like cytokine IL-33 is constitutively expressed in the nucleus of endothelial cells and epithelial cells in vivo: a novel “alarmin”? PLoS One. 2008;3:e3331.
Liew FY, Pitman NI, McInnes IB. Disease-associated functions of IL-33: the new kid in the IL-1 family. Nat Rev Immunol. 2010;10:103–110.
Wills-Karp M, Rani R, Dienger K et al. Trefoil factor 2 rapidly induces interleukin 33 to promote type 2 immunity during allergic asthma and hookworm infection. J Exp Med. 2012;209:607–622.
McKenzie GJ, Bancroft A, Grencis RK, McKenzie AN. A distinct role for interleukin-13 in Th2-cell-mediated immune responses. Curr Biol. 1998;8:339–342.
Anthony RM, Rutitzky LI, Urban JFJ, Stadecker MJ, Gause WC. Protective immune mechanisms in helminth infection. Nat Rev Immunol. 2007;7:975–987.
Bancroft AJ, McKenzie AN, Grencis RK. A critical role for IL-13 in resistance to intestinal nematode infection. J Immunol. 1998;160:3453–3461.
Oliphant CJ, Barlow JL, McKenzie AN. Insights into the initiation of type 2 immune responses. Immunology. 2011;134:378–385.
Dancescu M, Rubio-Trujillo M, Biron G, Bron D, Delespesse G, Sarfati M. Interleukin 4 protects chronic lymphocytic leukemic B cells from death by apoptosis and upregulates Bcl-2 expression. J Exp Med. 1992;176:1319–1326.
Goswami R, Kaplan MH. A brief history of IL-9. J Immunol. 2011;186:3283–3288.
Zhu J, Yamane H, Cote-Sierra J, Guo L, Paul WE. GATA-3 promotes Th2 responses through three different mechanisms: induction of Th2 cytokine production, selective growth of Th2 cells and inhibition of Th1 cell-specific factors. Cell Res. 2006;16:3–10.
Voehringer D, Reese TA, Huang X, Shinkai K, Locksley RM. Type 2 immunity is controlled by IL-4/IL-13 expression in hematopoietic non-eosinophil cells of the innate immune system. J Exp Med. 2006;203:1435–1446.
Enomoto Y, Orihara K, Takamasu T et al. Tissue remodeling induced by hypersecreted epidermal growth factor and amphiregulin in the airway after an acute asthma attack. J Allergy Clin Immunol. 2009;124:913–920. e1–7.
Ingram JL, Rice A, Geisenhoffer K, Madtes DK, Bonner JC. Interleukin-13 stimulates the proliferation of lung myofibroblasts via a signal transducer and activator of transcription-6-dependent mechanism: a possible mechanism for the development of airway fibrosis in asthma. Chest. 2003;123:422S–424S.
Oriente A, Fedarko NS, Pacocha SE, Huang SK, Lichtenstein LM, Essayan DM. Interleukin-13 modulates collagen homeostasis in human skin and keloid fibroblasts. J Pharmacol Exp Ther. 2000;292:988–994.
Monticelli LA, Sonnenberg GF, Artis D. Innate lymphoid cells: critical regulators of allergic inflammation and tissue repair in the lung. Curr Opin Immunol. 2012
Fichtner-Feigl S, Strober W, Kawakami K, Puri RK, Kitani A. IL-13 signaling through the IL-13alpha2 receptor is involved in induction of TGF-beta1 production and fibrosis. Nat Med. 2006;12:99–106.
Lee CG, Homer RJ, Zhu Z et al. Interleukin-13 induces tissue fibrosis by selectively stimulating and activating transforming growth factor beta(1). J Exp Med. 2001;194:809–821.
Lindemann D, Racke K. Glucocorticoid inhibition of interleukin-4 (IL-4) and interleukin-13 (IL-13) induced up-regulation of arginase in rat airway fibroblasts. Naunyn Schmiedebergs Arch Pharmacol. 2003;368:546–550.
Chiaramonte MG, Donaldson DD, Cheever AW, Wynn TA. An IL-13 inhibitor blocks the development of hepatic fibrosis during a T-helper type 2-dominated inflammatory response. J Clin Invest. 1999;104:777–785.
Chiaramonte MG, Cheever AW, Malley JD, Donaldson DD, Wynn TA. Studies of murine schistosomiasis reveal interleukin-13 blockade as a treatment for established and progressive liver fibrosis. Hepatology. 2001;34:273–282.
Kaviratne M, Hesse M, Leusink M et al. IL-13 activates a mechanism of tissue fibrosis that is completely TGF-beta independent. J Immunol. 2004;173:4020–4029.
Ohnishi T, Sur S, Collins DS, Fish JE, Gleich GJ, Peters SP. Eosinophil survival activity identified as interleukin-5 is associated with eosinophil recruitment and degranulation and lung injury twenty-four hours after segmental antigen lung challenge. J Allergy Clin Immunol. 1993;92:607–615.
Chen F, Liu Z, Wu W et al. An essential role for TH2-type responses in limiting acute tissue damage during experimental helminth infection. Nat Med. 2012;18:260–266.
Xing WW, Zou MJ, Liu S, Xu T, Wang JX, Xu DG. Interleukin-22 protects against acute alcohol-induced hepatotoxicity in mice. Biosci Biotechnol Biochem. 2011;75:1290–1294.
Ouyang W, Kolls JK, Zheng Y. The biological functions of T helper 17 cell effector cytokines in inflammation. Immunity. 2008;28:454–467.
Lloyd CM, Hessel EM. Functions of T cells in asthma: more than just T(H)2 cells. Nat Rev Immunol. 2010;10:838–848.
Shale M, Ghosh S. Beyond TNF, Th1 and Th2 in inflammatory bowel disease. Gut. 2008;57:1349–1351.
O’Reilly S, Hugle T, van Laar JM. T cells in systemic sclerosis: a reappraisal. Rheumatology (Oxford). 2012.
Ballantyne SJ, Barlow JL, Jolin HE et al. Blocking IL-25 prevents airway hyperresponsiveness in allergic asthma. J Allergy Clin Immunol. 2007;120:1324–1331.
Moffatt MF, Gut IG, Demenais F et al. A large-scale, consortium-based genomewide association study of asthma. N Engl J Med. 2010;363:1211–1221.
Wills-Karp M. Interleukin-13 in asthma pathogenesis. Immunol Rev. 2004;202:175–190.
Wynn TA. IL-13 effector functions. Annu Rev Immunol. 2003;21:425–456.
Fattouh R, Jordana M. TGF-beta, eosinophils and IL-13 in allergic airway remodeling: a critical appraisal with therapeutic considerations. Inflamm Allergy Drug Targets. 2008;7:224–236.
Warringa RA, Schweizer RC, Maikoe T, Kuijper PH, Bruijnzeel PL, Koendermann L. Modulation of eosinophil chemotaxis by interleukin-5. Am J Respir Cell Mol Biol. 1992;7:631–636.
Shi HZ, Xiao CQ, Zhong D et al. Effect of inhaled interleukin-5 on airway hyperreactivity and eosinophilia in asthmatics. Am J Respir Crit Care Med. 1998;157:204–209.
Camelo A, Barlow JL, Drynan LF et al. Blocking IL-25 signalling protects against gut inflammation in a type-2 model of colitis by suppressing nuocyte and NKT derived IL-13. J Gastroenterol. 2012.
Olman MA. Epithelial cell modulation of airway fibrosis in asthma. Am J Respir Cell Mol Biol. 2003;28:125–128.
Halim TY, MacLaren A, Romanish MT, Gold MJ, McNagny KM, Takei F. Retinoic-acid-receptor-related orphan nuclear receptor alpha is required for natural helper cell development and allergic inflammation. Immunity. 2012;37:463-474.
Hoyler T, Klose CS, Souabni A et al. The transcription factor GATA-3 controls cell fate and maintenance of type 2 innate lymphoid cells. Immunity. 2012;37:634-648.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer Science+Business Media New York
About this chapter
Cite this chapter
Hwang, Y.Y., McKenzie, A.N.J. (2013). Innate Lymphoid Cells in Immunity and Disease. In: Katsikis, P., Schoenberger, S., Pulendran, B. (eds) Crossroads Between Innate and Adaptive Immunity IV. Advances in Experimental Medicine and Biology, vol 785. Springer, New York, NY. https://doi.org/10.1007/978-1-4614-6217-0_2
Download citation
DOI: https://doi.org/10.1007/978-1-4614-6217-0_2
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4614-6216-3
Online ISBN: 978-1-4614-6217-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)