
Abstract
Over the past decade, mesenchymal stromal/stem cells (MSCs) have evolved into an important cell therapy demonstrating potential utility in a range of clinical applications, including bone and cartilage repair, cardiac repair, and immune disorders. MSCs can be isolated from a variety of tissue sources, including bone marrow, adipose tissue, dental pulp, and placenta. Groups have developed different manufacturing processes with a goal of improving the quality of clinical-grade cells and the overall efficiency of the manufacturing process. Variations in cell source and manufacturing process may have a significant impact on the efficacy of the final MSC product. Moreover, this variability in cell source and manufacturing processes has made it challenging to compare the resulting MSC products and associated results from clinical trials that have been conducted to date. The development of consistent, well-controlled manufacturing processes along with the implementation of thorough quality control testing, including rigorous potency assays, will insure high quality and may help to clarify the impact of cell source and manufacturing process on the resulting MSC product. In addition to providing an overview of the current good manufacturing practice (cGMP) methods for MSC production, this chapter summarizes key FDA regulatory requirements, including those related to cell source, raw materials, and quality control testing.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Umbilical Cord Blood
- Bovine Spongiform Encephalopathy
- Platelet Lysate
- Quality Control Testing
- Potency Assay
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
Introduction
Mesenchymal stromal/stem cells (MSCs) are adherent, fibroblast-like cells that are characterized by the expression of certain cell surface markers and the potential to differentiate into bone, fat, and cartilage [1, 2]. Although bone marrow (BM) is the most common source of starting material, cells with characteristics similar to BM-derived MSCs have been isolated from other tissue sources, including adipose, umbilical cord blood, placenta, and dental pulp [3–6]. Given the ability of MSCs to differentiate into adipocytes, osteoblasts, and chondrocytes, initial clinical applications focused on the use of MSCs to regenerate tissues using engineered bone constructs [7]. However, MSCs are excellent candidates for other applications due to several characteristics, including their ability to migrate to the site of injury/inflammation, the potential to stimulate proliferation and differentiation of resident progenitor cells, and the propensity to promote recovery of injured cells and/or modulate the immune system through secretion of growth factors [8–15]. Recent clinical applications have focused on utilizing the immunomodulatory properties and paracrine effects of MSCs in cardiovascular disease, neurological disorders, and immune dysregulation disorders. MSCs have demonstrated encouraging clinical results in Crohn’s disease [16] and graft-versus-host disease (GVHD) after allogeneic hematopoietic stem cell (HSC) transplantation, and these studies have now advanced to phase III clinical trials [17].
Many of these initial clinical trials have demonstrated significant promise in using MSCs as a therapeutic. However, efforts to repeat clinical observations have resulted in variable success. One major hurdle in comparing results from clinical studies is the potential variability in cell quality and characteristics between clinical sites. Due to the complex nature of cell therapeutics, it is important to recognize that the manufacturing process will likely have a significant impact on important cell properties that impact in vivo efficacy. In addition to variability in starting cell source, there is a wide range of cell culture media and culture practices that are currently employed in producing MSCs for clinical applications. It is therefore critical to establish a panel of quality control (QC) test methods that can be used to assess the impact of these variables on the safety and potency of the final MSC product. This chapter provides an overview of important considerations when producing MSCs for clinical applications. In addition to a brief overview of regulations for clinical production of MSCs, this chapter provides an overview of a number of manufacturing and testing considerations.
FDA Regulations and cGMP Compliance
A thorough understanding of applicable regulations and industry standards are essential when developing biotherapeutics. Regulatory requirements will often drive key decisions for manufacturing process development, selection of raw materials, and development of QC testing plans for raw materials and final product. This section provides a brief overview of regulations that are applicable to MSC-based therapies in the USA.
In the USA, cell therapies are regulated by the Center for Biologics Evaluation and Research (CBER) division of the Food and Drug Administration (FDA). Although the regulatory requirements for cell therapies are expected to evolve as new therapies move through human clinical trials toward approval, the FDA has provided guidance documents and regulations covering several key areas of production and testing.
In May 2005, Part 1271 of Chapter 21 of the US Code of Federal Regulations became effective. Part 1271, Human Cells, Tissues, and Cellular and Tissue-Based Products provides the basis for regulation of human cellular and tissue-based products (HCT/Ps). In addition to providing regulations for Donor Eligibility (Subpart C), Subpart D outlines Current Good Tissue Practices (cGTP). The regulations in 21 CFR 1271 Subpart D cover a broad range of requirements, including quality system, personnel, procedures, facilities, environmental monitoring and control, equipment, supplies and reagents, process changes/validation, and product labeling/storage/tracking [18]. The HCT/P regulations outlined in 21 CFR 1271 and cGMP regulations (21 CFR 210, 211, 610) are intended to be applied in a progressively more strict manner as therapeutics move toward the eventual filing of a Biologics License Application (BLA) [19]. However, the FDA expects that certain key requirements of the cGTP/cGMP regulations even will be met during early-stage human clinical trials [20].
In addition to the regulations outlined above, the FDA has issued several guidance documents that are applicable to HCT/Ps. The FDA issued a guidance in March 1998 that provides an overview of manufacturing and testing requirements for human somatic cell therapy and gene therapy products including procedures for cell collection, cell culture, cell banking systems, and release testing requirements for cellular therapy products [21]. The International Conference on Harmonization (ICH) has also issued several guidance documents that provide further details on testing requirements for cell therapeutics [22, 23]. Guidance documents are also available for issues related to the sourcing and testing of the initial cell material including donor eligibility determination and addressing xenotransplantation issues for cell therapeutics that were previously cultured ex vivo with live nonhuman animal feeder cells [24, 25]. Since HCT/Ps typically cannot undergo a terminal sterilization step, HCT/Ps must be manufactured following aseptic processing methods. Several documents are available providing general guidance for validation and cGMP compliance for aseptic processes [26]. In addition to guidance from the FDA, AABB (formerly the American Association of Blood Banks) and the Foundation for the Accreditation of Cellular Therapy, or FACT, have established standards to assist with meeting regulatory requirements [27, 28]. Several groups from academia and industry have published documents that provide guidelines for moving HCT/Ps into human clinical trials [29–31].
Cell Source
MSCs were originally isolated as an adherent cell population derived from bone marrow (BM) [1]. Subsequent studies have found that similar populations can be isolated from other adult and perinatal tissues, including adipose tissue (AT) [6], skeletal muscle [32], synovium [33], dental pulp [34], placenta [35], amniotic fluid [36], and umbilical cord blood (UCB) [37]. Several studies that have compared the properties of the cells derived from these diverse sources have found that the cells demonstrate very similar characteristics including cell marker expression, differentiation potential, and immunological properties [38–40]. However, a study that compared the gene expression profiles of MSCs derived from BM, AT, and UCB found that while MSCs derived from different donors using the same source material and expansion protocol exhibited consistent and reproducible profiles, MSCs from AT, BM, and UCB display differences in the transcriptome [41]. The impact of these differences on in vivo efficacy remains unclear. However, the results serve to highlight potentially important differences between MSCs derived from different sources. This section provides a brief overview of MSCs derived from BM, AT, and UCB. In addition, information is provided on donor screening and eligibility requirements that apply to all sources of starting cell material.
Bone Marrow
The starting BM for MSC production is typically obtained from a 25–100-mL BM aspirate from the posterior superior iliac crest of the donor. The procedure is performed in a clinical setting allowing for sterile harvest of the BM aspirate. In addition, donors typically go through a full medical screening process (see Donor Screening below) and a rigorous informed consent procedure, very similar to that of a blood donor.
Several important factors regarding the BM donation may have a significant impact on the quantity and quality of MSCs derived from the BM. The age, sex, and health of the donor, including factors such as smoking, may impact the quality of the BM harvest [42, 43]. Donor-to-donor variation has also been observed in the profile of cytokines and chemokines that are secreted by MSCs in response to stimulation with proinflammatory cytokines [44]. Freezing of BM prior to MSC isolation was also reported to have a negative impact on both MSC yield and immunosuppressive properties of the MSC in mixed lymphocyte cultures [45]. Finally, as discussed in section “MSC Isolation from Bone Marrow,” the method that is used for isolating the mononuclear cell fraction from the BM may have a signification impact on the resulting MSCs.
Adipose Tissue
Although the bulk of the published literature concerns BM-derived MSCs, AT is also considered to be an easily obtainable source of starting cells for MSC production. AT-derived MSCs have been used in a few small clinical trials for Crohn’s disease [46], steroid-refractory acute GVHD [47], enhancement of HSC engraftment [48], and as salvage therapy for refractory pure red cell aplasia after major ABO-incompatible HSC transplantation [49]. The procedure for producing MSCs from AT involves red blood cell (RBC) washing steps similar to BM processing with the density-gradient step essentially replaced by a collagenase digestion step. A number of factors including donor characteristics and anatomical location of AT harvest can impact the characteristics of the resulting MSCs [50].
Umbilical Cord Blood
UCB is the most recently established source of hematopoietic stem cells for clinical utility [51]. Although some investigators have had limited success [52, 53], it is also now generally accepted that UCB is a suitable starting material for MSC isolation and expansion [3, 54]. With efficiency of isolation varying among research groups with success rates in the range of 24–63% [3, 55], an effort has been made to optimize cell processing [55]. In general, the approach is very similar to that of marrow-derived MSCs. The mononuclear cell (MNC) fraction is isolated using a density-gradient centrifugation and then seeded into culture flasks (e.g., 1 × 106 MNC/cm2). Within 24 h the non-adherent cells are removed, and the remaining adherent cells are carried through culture much like MSCs from other sources. Interestingly, one group demonstrated UCB-derived MSCs to have a greater proliferation capacity, becoming senescent later than adipose- and marrow-derived MSCs [56]. The same group was unable to show adipogenic potential of UCB-derived MSCs, though others have been able to demonstrate in vitro differentiation to fat cells [3, 54]. In fact, some researchers have isolated MSC-like cells from UCB and succeeded in coaxing to cell types representative of all three embryonic lineages [57–59]. The potential value of UCB-derived MSCs over other types remains to be determined, though their unique qualities suggest there may be some advantages [56].
Donor Evaluation
Donor evaluation is an important requirement for cell therapeutics derived from human tissue sources. Requirements for donor evaluation are outlined in the HCT/P regulations (21 CFR 1271 Subpart C) and FDA guidance documents on donor eligibility [25]. A comprehensive donor evaluation is typically performed by a physician with expertise in the collection procedure at the time of initial evaluation. The donor evaluation typically consists of three components: donor questionnaire, medical examination, and testing for infectious disease markers. All potential donors fill out a questionnaire that screens donors for transmissible diseases on the basis of history [60]. The donor’s medical history is reviewed including information on transfusion history, surgical history, pregnancies, vaccination history, family history, social history, and health habits including smoking, alcohol, and recreational drug use. A general medical examination is performed prior to donation and typically includes routine laboratory testing (CBC with differential and platelet count, PT/INR, standard blood chemistry panel, and ABO/Rh type).
A blood sample is also taken from the donor at the time of initial donor assessment and, if needed, at the time of collection for infectious disease testing as required in 21 CFR 1271 Subpart C FDA Donor Eligibility. Infectious disease testing is performed using FDA-licensed test kits as summarized in Table 16.1. The results from donor testing, donor eligibility assessment, and the informed consent for tissue donation are typically retained in a file that is coded to protect patient confidentiality while maintaining traceability of the final MSC product back to the original tissue source.
MSC Production Methods
Along with advances in clinical applications for MSC-based therapies, strides have been made in several key technical areas related to production, testing, and banking of MSCs. Producing MSCs for clinical applications requires addressing several key issues [61, 62]. In addition to addressing regulatory compliance issues, manufacturers of MSC for clinical applications must address issues related to source material, cell culture conditions, and media source/quality. Several studies have been performed to determine the optimal conditions for culturing MSC for clinical applications [63–66]. New media formulations that avoid the use of FBS have been described recently [67, 68]. Efficient procedures for MSC cryopreservation and conditions for transporting and holding cells for transplantation have also been evaluated [66, 69].
In addition to developing well-defined and reproducible manufacturing procedures, quality control (QC) test methods must be established to characterize and evaluate the final cell product. Characterization assays are especially critical for MSC products given the diversity of starting material, isolation methods, and culture methods [70]. Several groups have published reports on QC test methods that are currently used for both in-process testing and testing MSC products intended for human clinical trials [71, 72]. This section provides a brief overview of a typical manufacturing process for BM-derived MSCs including discussions regarding key process steps and parameters that potentially impact the quality and efficacy of the final MSC product.
Overview of MSC Manufacturing Process
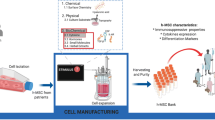
A typical MSC manufacturing process consists of the following steps: isolation of MNC fraction from BM, MSC seed/master cell bank (MCB) production (optional), MSC expansion, and cryopreservation. Final formulation may take place prior to or after cryopreservation following the thaw. The overall process is depicted in the process flow diagram presented in Fig. 16.1. Different seeding and passaging strategies can be used in the MSC production process. For example, a low seeding density of MSCs may be subjected to a single expansion step without production of an intermediate cell bank. Seeding density and passaging schedule have an impact on the final MSC population and this is discussed in section “MSC Culture Method.”

Process flow diagram for MSC production from bone marrow
Raw Materials
Raw materials that are used in cGMP manufacturing processes should be sourced from vendors that have been audited for compliance with cGMPs or other appropriate quality standards. QC testing and documentation should be maintained for each raw material, and traceability from final MSC product back to all raw materials should be maintained for each production lot. Raw materials should be reviewed to identify potential risks that may be introduced, for example, through the use of animal-derived raw materials. Table 16.2 provides an overview of common raw materials used in MSC production with recommendations for QC testing and documentation requirements.
MSC Isolation from Bone Marrow
MSCs are present in the mononuclear cell (MNC) fraction of the BM. The MNC fraction is enriched from the BM using density-gradient centrifugation. This step is typically performed using Ficoll-Hypaque density-gradient medium. cGMP-grade versions of density-gradient medium are commercially available. Following enrichment, the cells are washed with PBS or Hank’s Balanced Salts Solution (no phenol red, calcium, or magnesium) prior to initial plating. Studies have demonstrated that modifications to the MNC isolation step can have a significant impact on the yield and quality of the resulting MSC product. For example, MNC isolation using 1.073 g/mL Ficoll produced an MNC fraction that was lower in CD45+ cells resulting in about a twofold increase in MSC yield after four passages with higher expression of CD90, CD146, and GD2 [73].
MSC Seed Bank Production
Following enrichment of the MNC fraction by density-gradient centrifugation, the washed cells are typically plated (passage 0) in cell culture flasks and incubated at 37°C with 5% humidified CO2 using the selected MSC culture media (see section “Media Selection”). Twenty-four to forty-eight hours later, the non-adherent cells are removed (suctioned out) and the adherent cells are expanded in culture with media changed every 3–4 days.
At this point in the manufacturing process, the MSCs may either be expanded directly to final product or expanded to an intermediate stage (e.g., P = 2) where they are harvested and cryopreserved to create a seed bank for future production trials. The creation of MSC seed banks allows future production campaigns to be performed with a starting cell source that has undergone testing for key attributes such as growth characteristics and biological activity. This allows for more uniform production campaigns and can be used to address key issues such as donor-to-donor variability in MSC properties. Cells from the seed bank (P = 2) are typically expanded through several additional passages to generate the final MSC product (P = 4–6) to be used in clinical trials. It should be noted that this product will be several passages older than MSCs that are expanded directly without creating an intermediate seed bank. While there are advantages to such an expansion strategy from a time/yield and logistics perspective, the overall impact of time in culture and passaging on cell quality and potency remains to be established. Limited studies suggest that there is an impact of time in culture on MSCs possibly related to the age of the donor [74, 75]. Other studies have shown that moderate time in culture (4–7 passages) does not affect the immunosuppressive activity of MSCs [45]. It is advisable, however, that investigators qualify their chosen MSC manufacturing approach for the intended clinical use (see section “Potency Assays”).
Media Selection
Currently there is no standard method of culturing MSCs from any source/starting material, and there is no consensus among the investigators on the most efficient approach to producing MSCs. This is important since proliferation rate, differentiation potential, and immunophenotype of cells could change depending on the culture method. Nevertheless, clinical trials based on the use of MSCs generated at different academic centers have all showed that infusions of these cells are safe and potentially efficacious. The most commonly used media for MSC production appears to be fetal bovine serum (FBS)/alpha-minimum essential medium (αMEM). Considerations for the use of FBS in MSC culture along with several media options are presented below.
Fetal Bovine Serum
FBS has traditionally been utilized to expand human MSCs for both research and clinical applications. FBS is often added to alpha-MEM base media supplemented with glutamine with the FBS concentration ranging typically from 5 to 17%. Lot-to-lot variability is typically observed in the ability of FBS to support MSC expansion requiring screening of FBS lots and highlighting the potential impact on MSC quality and potency. Interestingly, the concentration of FBS can affect the subpopulation of MSCs that grow out in culture with serum deprivation resulted in selection of an Oct-4-positive early progenitor population [76]
The use of FBS in the production of cellular therapies generates several potential concerns including the introduction of the risk of transmission of zoonotic agents, bovine spongiform encephalopathy (BSE), and the introduction of antigens of animal origin that may be incorporated into the cell therapeutic (e.g., Neu5Gc) or present from residual contaminating FBS [77, 78]. The risk associated with BSE transmission may be reduced by selecting a FBS source from countries classified by the World Organization for Animal Health [Office International des Epizooties (OIE)] as negligible BSE risk or Geographical BSE-Risk (GBR) I, as classified by the European Food Safety Authority (EFSA) [79]. The potential risk of bovine pathogen transmission may also be mitigated by using only FBS that has undergone screening for bovine pathogens (9 CFR 113) and that has additionally undergone a viral inactivation step such as gamma irradiation.
The potential risks associated with BSE, pathogens, animal antigens, and variability drive the desire to identify other potential media for MSC clinical production. Alternatives to FBS that have been investigated include serum-free media, autologous serum, fresh-frozen plasma, and human platelet lysates [65, 68, 80–82].
Platelet Lysates
Among the current alternatives for FBS, media based on human platelet lysate have been studied the most extensively, including evaluation in human clinical trials [83]. One advantage of platelet lysate media is that it can be sourced from normal healthy donors that have passed all infectious disease testing. Platelet lysate media is typically produced using platelet concentrates collected from single donors by apheresis. The platelets are frozen, thawed, and then heat inactivated at 56°C for 30 min. After removal of the remaining platelets by centrifugation, the resulting platelet lysate is frozen in aliquots for future use in MSC culture. Despite its clear advantages, preparation of platelet lysate media requires additional time, and it may result in donor-to-donor (i.e., lot-to-lot) variability in MSC growth characteristics and potentially cell quality due to variability in growth factor content (e.g., platelet-derived growth factor – PDGF) [84].
Serum-Free Media
Several groups have developed serum-free media formulations that have demonstrated promise in MSC production. Meuleman et al. found that commercially available medium supplemented with a serum substitute demonstrated a significant increase in MSC yield compared to standard FBS/αMEM. In addition, the resulting MSCs were similar with respect to cell marker expression, differentiation potential, and the ability to support the growth of hematopoietic progenitors [85]. Chase et al. described development of a proprietary serum-free media that also demonstrated enhanced MSC growth over FBS/αMEM when the medium was supplemented with fibroblast growth factor-2 (FGF-2), transforming growth factor-beta (TGF-β) and PDGF [86]. MSCs produced using this medium showed similar cell surface marker expression by flow cytometry, differentiation potential, and gene expression profile relative to MSCs produced using standard FBS/αMEM. Although this initial version of media contained animal-derived components, a new xeno-free version is now commercially available [87, 88]. Additional in vitro potency studies and animal studies are needed to demonstrate whether the use of these serum-free media will have a significant impact on the in vivo efficacy of the MSCs. In addition, these media are proprietary formulations that contain undisclosed components. Care should be taken to identify potential risks from media components such as growth factors or other animal-derived components. For example, some growth factors may be produced using mammalian cell lines such as rodent cell lines (e.g., CHO, NS0) that inherently introduce the risk of retrovirus and retrovirus-like particle contamination [89]. Growth factors that are derived using such mammalian expression systems should utilize tested cell lines and have purification processes that have been validated for clearance of viral pathogens.
MSC Culture Method
MSCs are typically grown as adherent cells using standard tissue culture plasticware. Initial cultures of MSCs from the enriched MNC fraction or seed bank are typically expanded in T-flasks. Cells from T-flasks are then used to seed large-scale cell culture devices such as Cell Factories (Nunc) or CellSTACK (Corning). Cell Factories have demonstrated utility in producing MSCs for clinical applications [90]. Cell Factories/CellSTACK provide a convenient format for large-scale culture of adherent cells. Media may be prepared in disposable bioprocess containers, and bags and tubing sets can be used to allow the entire feeding and harvesting steps to be performed in a single-use, disposable, closed system. This format, therefore, provides the added benefits of decreased contamination risk and elimination of the need to perform cleaning validation as would be required for multiuse bioreactors.
Beyond the impact of donor characteristics, MNC isolation method, and media selection discussed above, a number of factors in MSC culture can impact the final MSC characteristics. Seeding density is one important major factor that has a significant impact on the MSCs. Low seeding densities (10–50 cells/cm2) have been shown to promote the growth of a subpopulation of MSCs that appears to represent early progenitors [91]. The resulting MSCs have an increased growth rate, thin spindle-shaped morphology, and have increased adipogenic potential relative to the later developing MSCs that have a wider morphology and greater chondrogenic potential.
Although several scalable formats including Cell Factories (Nunc) and CellSTACK have been used for MSC production, the relatively large doses (0.4–9 × 106 cells/kg) [92] of MSCs that are required for many indications suggest that other scalable production methods may be needed for future applications. Bioreactors offer a potential solution for large-scale production of cell therapeutics with the opportunity to provide greater control over cell growth conditions and potentially over cell quality. Most of the work aimed at growing MSCs in bioreactors is recent with a focus on growing MSCs on novel and commercially available microcarriers [93–96]. Although results to date have demonstrated modest levels of expansion, further optimization of seeding parameters, media formulation, feeding strategies, and bioreactor conditions will likely lead to further improvements in cell yield and manufacturing efficiencies.
Final Formulation and Cryopreservation
Following the final harvest, the MSCs are typically centrifuged, washed, and changed over to a formulation that is compatible with cryopreservation and administration to the patient. One formulation that has been used in previous clinical trials is PlasmaLyte A (Baxter, Deerfield, IL, USA) containing 5–10% human serum albumin and 10% DMSO. Alternative cryoprotectants have been evaluated with some success in reducing the required levels of DMSO by utilizing PEG and albumin [97]. The dose range for MSCs is typically 2–8 × 106 cells/kg or 1–6 × 108 MSCs/dose that is formulated as 25–100 mL of cells in a bag that is suitable for low-temperature storage. Bags of cells are typically frozen using a controlled rate freezer (−1°C/min) and stored in liquid nitrogen freezers in liquid or vapor phase at temperatures below −150°C.
MSCs have been thawed and immediately infused; however, they are often thawed and washed or diluted with an appropriate solution (e.g., Dextran 40, 5% human serum albumin) and then infused. Studies should be conducted to ensure that time limits are established for holding the final thawed product under defined conditions prior to administration. Previous studies have demonstrated a range of acceptable hold times depending on the formulation and hold temperature [66].
Manufacturing Controls
Cleanroom Environment
A key aspect of manufacturing cell therapeutics for clinical applications is the inability to perform a terminal sterilization step. This necessitates that the product be manufactured under strict aseptic conditions through the entire production process. The FDA has issued a guidance document that outlines key issues for aseptic manufacturing processes [98]. Key areas of focus that should be addressed for an aseptic manufacturing process include: clean room design, clean room cleaning practices, environmental monitoring practices, personnel gowning, personnel monitoring, and validation of aseptic processing methods. For cell therapy production, the clean room environment should, at minimum, meet class 10,000 (ISO class 7) clean room rating with a biosafety cabinet or other class 100 (ISO class 5) zone for performing open manipulations. Strict gowning practices, cleaning practices, and environmental monitoring (viable and nonviable) are critical for ensuring that the manufacturing environment is maintained in a controlled state during clinical production.
Process Qualification
Process validation is defined by the FDA as the “collection and evaluation of data, from the process design stage throughout production, which establishes scientific evidence that a process is capable of consistently delivering quality products” [99]. Initial process (performance) qualification (PQ) trials are typically conducted at the end of the initial process development studies and prior to initiating clinical production campaigns. Preclinical PQ trials typically consist of performing trials (3–5 runs) of the cGMP manufacturing process with full documentation and testing, including in-process testing. These trials allow final details to be worked out for manufacturing procedures and documentation and demonstrate that the manufacturing process is capable of producing material that will meet release testing requirements for clinical trials. Material from these initial PQ trials can typically be used as reference standard for future QC testing or for use in preclinical animal studies.
Process validation typically occurs throughout the product life cycle with data collected during production runs and process design experiments. The goal of this stage is to identify key process parameters and material attributes (e.g., donor variability) that impact process variability and product quality. Studies are then performed to demonstrate that the manufacturing process is capable of producing acceptable product within the limits established for these key operating parameters. Comprehensive process validation studies are required to be completed prior to commencing commercial distribution of the therapeutic [99].
Aseptic Processing Qualification
As discussed above, maintaining aseptic conditions during manufacturing is a critical aspect of clinical production for cell therapeutics. Demonstrating the ability to maintain aseptic conditions during the manufacturing process, especially during critical steps such as open manipulations, is therefore a critical component of process qualification. Media simulation studies are typically performed to validate aseptic processes [98]. These studies are performed using microbial growth media (e.g., soybean casein digest (SCD) medium) in place of cell culture medium with simulation of a full production run. Critical steps in cell production including seeding, feeding, harvest, and dispensing of the product into the final container should be included in the simulation runs. The final product containers containing SCD medium are incubated for 14 days, typically at two temperatures, with observation for any signs of microbial growth.
Quality Control Testing
Quality control testing is a critical component of the clinical production program. QC testing is typically performed at multiple points in the manufacturing process, prior to production (i.e., including donor material, raw material), cell (seed) bank, in-process samples, and final product release testing. Specifications are typically set for donor, raw materials, and final product based on key safety and performance requirements. In addition, data from PQ trials are used to establish process capabilities and set specifications for both in-process testing and final product release testing. Specifications are expected to address key attributes including identity, strength, quality, purity, and potency. Typical testing for donor tissue (see Donor Evalution) and raw materials (see Raw Materials) is discussed above. This section will cover QC testing requirements for the final MSC product.
Release Testing
Each lot of final MSC product will undergo testing to demonstrate that it meets preestablished specifications prior to release for clinical trials. Quality assurance is responsible for reviewing all production records, including QC testing, prior to release of final product. A summary of the typical final QC release testing performed on each lot of MSCs is provided in Table 16.3.
Identity Testing
Identity testing is typically performed using either short tandem repeat (STR) testing or human leukocyte antigen (HLA) testing. The identity tests create a genetic fingerprint that can be used to relate the cell source back to the original donor. This is especially important if multiple cell lines are being produced in the same facility. STR testing is typically performed using commercially available kits [100]. HLA testing is performed by high-resolution sequencing of the HLA-A, HLA-B, HLA-C, and HLA-DRB1 loci. This technique is becoming more efficient as techniques utilizing next-generation sequencing methods are developed [101].
Viable Cell Count
Viable cell counts are typically performed by staining cells with reagents such as Trypan Blue or acridine orange (AO)/propidium iodide (PI) and performing manual counts with a hemacytometer or using an automated cell counter. Alternatively, viable counts can be performed using 7-AAD or PI staining in conjunction with flow cytometry analysis of MSC cell maker expression [102].
Microbial and Fungal Contamination
Sterility testing is typically conducted using the direct transfer method in accordance with 21 CFR 610.12. The test article is inoculated into SCD and FTM and incubated at 20–25 and 30–35°C, respectively, for 14 days. Alternative strategies such as use of automated testing systems (e.g., BACTEC, BD, Franklin Lakes, NJ, USA) commonly used in the clinical setting may be employed if agreeable by FDA. Bacteriostasis and fungistasis testing described in United States Pharmacopeia (USP) <71> is also performed on the product at a minimum with the PQ to insure that the product components, or residual antibiotics if used in initial isolation, do not interfere with sterility testing.
Mycoplasma
Mycoplasma testing is conducted on both the cells and supernatant from the final product as well as the master cell bank, if that manufacturing approach is taken. Although PCR or chemical testing can be used as a screening assay for mycoplasma, the Points to Consider (PTC) culture method is preferred for release testing. The PTC method, which takes 28 days for completion, includes both a direct assay and an indirect assay [103]. The direct assay involves cultivation of the test article in agar and broth media under conditions suitable for growth of cultivatable mycoplasmas. The indirect method involves culturing the test article in Vero indicator cells followed by staining with a DNA-binding fluorochrome (Hoechst stain) to detect nuclear and extranuclear fluorescence. Appropriate positive controls are included in each arm of the assay.
Endotoxin
Endotoxin testing that is performed on the final production should conform with USP <85> Bacterial Endotoxins Tests. Testing is typically based on the Limulus amebocyte lysate (LAL) assay utilizing commercially available reagents and test kits (e.g., Endosafe, Charles River). Testing should include inhibition and enhancement test controls. A typical recommended specification for endotoxin is <5.0 EU/kg/dose.
MSC Antigen Expression
Flow cytometry is performed on the MSC seed bank and final product to verify appropriate expression of MSC markers. Most groups use the guidelines as proposed by the Mesenchymal and Tissue Stem Cell Committee of the ISCT [2]. This group defined criteria for MSC identification to include presence of CD105, CD73, and CD90 as positive markers (≥95%), and absence of CD45, CD34, CD14/CD11b, CD19/CD79α, and HLA-DR as negative markers (≤2%).
Karyotype
Karyotyping is typically performed using standard Giemsa/Trypsin/Leishman (GTL) banding (or simply G-banding) on 20 metaphase spreads. Analysis is performed in compliance with the Clinical Cytogenetics Standards and Guidelines published by the American College of Medical Genetics. Chromosome counts are performed on 20 cells with full band analysis performed on 5–10 cells [104].
Residual FBS
Levels of residual FBS in the final product are typically determined based on the level of residual bovine serum albumin (BSA). Levels of BSA can be measured using a commercially available ELISA kit. A typical target of reduction for therapeutics is <1 ppm residual FBS. However, acceptable specifications should be based on process capabilities and potential risk to the patient population. Cross-reactivity of the ELISA with human serum albumin will be a primary consideration if the final product is formulated with HSA. In that case, other components of FBS can be exploited to determine residual amounts of FBS in the final product (e.g., bovine transferrin) [105].
Potency Assays
The FDA requires that biological products meet requirements of safety, purity, and potency for biologics license application approval. A potency assay must be established prior to initiating phase III trials, and it must be validated before BLA submission. Potency is defined by FDA as “the specific ability or capacity of the product, as indicated by appropriate laboratory tests or by adequately controlled clinical data obtained through the administration of the product in the manner intended, to effect a given result” [21 CFR 600.3(s)]. The regulations allow potency assays to be in vitro, in vivo, or both as long as the assay(s) is designed specifically for the given product to assess potency as described above [106].
Since MSCs are used for a variety of clinical applications, the intended effect will undoubtedly vary. MSCs may be administered for an immunomodulatory effect (e.g., graft-versus-host disease), tissue or organ repair (e.g., meniscal repair), enhancement of engraftment following blood/marrow transplant, etc. Table 16.4 lists a few resources for potency testing of MSCs for various medical applications. Some approaches are more developed than others. Cytokine-based analysis is listed below as a possible potency assay, and this approach is expected to grow given the expansion of research in this area [116].
There are several advantages to establishing a potency assay as early in the developmental pathway as possible. These include evaluating multiple candidate assays, evaluating the impact of media and production methods, generating data to support lot release specifications, and establishing a stability program. In the 2008 guidance, the FDA provides more practical benefits to early work on potency testing, as well as direction toward relevant biologics and cGMP regulations for consideration of potency assays [106].
Conclusion
MSCs can be produced from a variety of different cell sources with many variations in the initial isolation, cell expansion, and formulation/cryopreservation procedures. In addition, a variety of different test methods are used by groups to assess the quality of MSCs. This chapter provides a brief overview of some of the more common methods that are used for producing and testing MSCs for clinical applications. Clearly one of the major challenges facing the field of MSC-based therapeutics is the need to develop better analytical methods, including potency assays, to better assess how differences in production methods impact cell quality and in vivo potency.
References
Friedenstein AJ, Petrakova KV, Kurolesova AI, Frolova GP (1968) Heterotopic of bone marrow. Analysis of precursor cells for osteogenic and hematopoietic tissues. Transplantation 6(2):230–247
Dominici M, Le BK, Mueller I, Slaper-Cortenbach I, Marini F, Krause D et al (2006) Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 8(4):315–317
Erices A, Conget P, Minguell JJ (2000) Mesenchymal progenitor cells in human umbilical cord blood. Br J Haematol 109(1):235–242
Huang GTJ, Gronthos S, Shi S (2009) Mesenchymal stem cells derived from dental tissues vs. those from other sources: their biology and role in regenerative medicine. J Dent Res 88(9):792–806
Parolini O, Alviano F, Bagnara GP, Bilic G, Bühring H Jr, Evangelista M et al (2008) Concise review: isolation and characterization of cells from human term placenta: outcome of the first international workshop on placenta derived stem cells. Stem Cells 26(2):300–311
Zuk PA, Zhu M, Mizuno H, Huang J, Futrell JW, Katz AJ et al (2001) Multilineage cells from human adipose tissue: implications for cell-based therapies. Tissue Eng 7(2):211–228
Lee K, Chan CK, Patil N, Goodman SB (2009) Cell therapy for bone regenerationGÇöBench to bedside. J Biomed Mater Res 89B(1):252–263
Phinney DG, Prockop DJ (2007) Concise review: mesenchymal stem/multipotent stromal cells: the state of transdifferentiation and modes of tissue repair – current views. Stem Cells (Dayton, Ohio) 25(11):2896–2902
Chamberlain G, Fox J, Ashton B, Middleton J (2007) Concise review: mesenchymal stem cells: their phenotype, differentiation capacity, immunological features, and potential for homing. Stem Cells (Dayton, Ohio) 25(11):2739–2749
Caplan AI (2007) Adult mesenchymal stem cells for tissue engineering versus regenerative medicine. J Cell Physiol 213(2):341–347
Uccelli A, Pistoia V, Moretta L (2007) Mesenchymal stem cells: a new strategy for immunosuppression? Trends Immunol 28(5):219–226
Prockop DJ (2007) “Stemness” does not explain the repair of many tissues by mesenchymal stem/multipotent stromal cells (MSCs). Clin Pharmacol Ther 82(3):241–243
Deans RJ, Moseley AB (2000) Mesenchymal stem cells: biology and potential clinical uses. Exp Hematol 28(8):875–884
Dazzi F, Horwood NJ (2007) Potential of mesenchymal stem cell therapy. Curr Opin Oncol 19(6):650–655
Brooke G, Cook M, Blair C, Han R, Heazlewood C, Jones B et al (2007) Therapeutic applications of mesenchymal stromal cells. Semin Cell Dev Biol 18(6):846–858
Lanzoni G, Roda G, Belluzzi A, Roda E, Bagnara GP (2008) Inflammatory bowel disease: moving toward a stem cell-based therapy. World J Gastroenterol 14(29):4616–4626
Le Blanc K, Frassoni F, Ball L, Locatelli F, Roelofs H, Lewis I et al (2008) Mesenchymal stem cells for treatment of steroid-resistant, severe, acute graft-versus-host disease: a phase II study. Lancet 371(9624):1579–1586
FDA (2005) Human cells, tissues, and cellular and tissue-based products. Report No: 21
FDA (2007) Guidance for industry – regulation of human cells, tissues, and cellular and tissue-based products HCT/Ps – small entity compliance guide U.S. Department of Health and Human Services Food and Drug Administration Center for Biologies Evaluation and Research Rockville, MD
FDA (2008) Guidance for industry – CGMP for phase 1 investigational drugs U.S. Department of Health and Human Services Food and Drug Administration Center for Biologies Evaluation and Research Rockville, MD
FDA (1998) Guidance for industry – guidance for human somatic cell therapy and gene therapy. U.S. Department of Health and Human Services, Food and Drug Administration. Report No.: March 1998
ICH (2008) ICH harmonized tripartite guideline Q5D – derivation and characterization of cell substrates used for production of biotechnological/biological products
FDA (1998) International conference on harmonization; Guidance on viral safety evaluation of biotechnology products derived from cell lines of human or animal origin. Report No.: 63
FDA (2003) Guidance for industry – source animal, product, preclinical, and clinical issues concerning the use of xenotransplantation products in human U.S. Department of Health and Human Services Food and Drug Administration Center for Biologies Evaluation and Research Rockville, MD
FDA (2007) Guidance for industry – eligibility determination for donors of human cells, tissues, and cellular and tissue-based products (HCT/Ps) U.S. Department of Health and Human Services Food and Drug Administration Center for Biologies Evaluation and Research Rockville, MD
FDA (2004) Guidance for industry – sterile drug products produced by aseptic processing – current good manufacturing practice U.S. Department of Health and Human Services Food and Drug Administration Center for Biologies Evaluation and Research Rockville, MD
FACT-JACIE (2008) International standard for cellular therapy product collection, processing, and administration
AABB (2009) Standards for cellular therapy product services, 4th edn. AABB, Bethesda
International Society for Stem Cell Research (2008) Guidelines for the clinical translation of stem cells Dec 3, 2008 ISSCR, Skokle, IL. United States Pharmacopeial Convention, Inc. Rockville, MD
U.S.Pharmacopeia (2009) Cell and gene therapy products, USP 32 p 436–466. United States Pharmacopeial Convention, Inc. Rockville, MD
U.S.Pharmacopeia (2009) Ancillary materials for cell, gene, and tissue-engineered products, USP 32 p 420–426. United States Pharmacopeial Convention, Inc. Rockville, MD
Williams JT, Southerland SS, Souza J, Calcutt AF, Cartledge RG (1999) Cells isolated from adult human skeletal muscle capable of differentiating into multiple mesodermal phenotypes. Am Surg 65(1):22–26
De Bari C, Dell’Accio F, Tylzanowski P, Luyten FP (2001) Multipotent mesenchymal stem cells from adult human synovial membrane. Arthritis Rheum 44(8):1928–1942
Gronthos S, Mankani M, Brahim J, Robey PG, Shi S (2000) Postnatal human dental pulp stem cells (DPSCs) in vitro and in vivo. Proc Natl Acad Sci USA 97(25):13625–13630
In’t Anker PS, Scherjon SA, Kleijburg-van der Keur C, de Groot-Swings GM, Claas FH, Fibbe WE et al (2004) Isolation of mesenchymal stem cells of fetal or maternal origin from human placenta. Stem Cells (Dayton, Ohio) 22(7):1338–1345
In’t Anker PS, Scherjon SA, Kleijburg-van der Keur C, Noort WA, Claas FH, Willemze R et al (2003) Amniotic fluid as a novel source of mesenchymal stem cells for therapeutic transplantation. Blood 102(4):1548–1549
Bieback K, Kern S, Kluter H, Eichler H (2004) Critical parameters for the isolation of mesenchymal stem cells from umbilical cord blood. Stem Cells (Dayton, Ohio) 22(4):625–634
Hoogduijn MJ, Crop MJ, Peeters AM, Van Osch GJ, Balk AH, Ijzermans JN et al (2007) Human heart, spleen, and perirenal fat-derived mesenchymal stem cells have immunomodulatory capacities. Stem Cells Dev 16(4):597–604
Puissant B, Barreau C, Bourin P, Clavel C, Corre J, Bousquet C et al (2005) Immunomodulatory effect of human adipose tissue-derived adult stem cells: comparison with bone marrow mesenchymal stem cells. Br J Haematol 129(1):118–129
Gotherstrom C, Ringden O, Westgren M, Tammik C, Le Blanc K (2003) Immunomodulatory effects of human foetal liver-derived mesenchymal stem cells. Bone Marrow Transplant 32(3):265–272
Wagner W, Wein F, Seckinger A, Frankhauser M, Wirkner U, Krause U et al (2005) Comparative characteristics of mesenchymal stem cells from human bone marrow, adipose tissue, and umbilical cord blood. Exp Hematol 33(11):1402–1416
Bouwmeester W, Fechter MM, Heymans MW, Twisk JWR, Ebeling LJ, Brand A (2010) Prediction of nucleated cells in bone marrow stem cell products by donor characteristics: a retrospective single centre analysis. Vox Sang 98(3):e276–e283
Crisostomo PR, Markel TA, Wang MJ, Lahm T, Lillemoe KD, Meldrum DR (2007) In the adult mesenchymal stem cell population, source gender is a biologically relevant aspect of protective power. Surgery 142(2):215–221
Zhukareva V, Obrocka M, Houle JD, Fischer I, Neuhuber B (2010) Secretion profile of human bone marrow stromal cells: donor variability and response to inflammatory stimuli. Cytokine 50(3):317–321
Samuelsson H, Ringden O, Lonnies H, Le Blanc K (2009) Optimizing in vitro conditions for immunomodulation and expansion of mesenchymal stromal cells. Cytotherapy 11(2):129–136
Garcia-Olmo D, Garcia-Arranz M, Herreros D, Pascual I, Peiro C, Rodriguez-Montes JA (2005) A phase I clinical trial of the treatment of Crohn’s fistula by adipose mesenchymal stem cell transplantation. Dis Colon Rectum 48(7):1416–1423
Fang B, Song Y, Lin Q, Zhang Y, Cao Y, Zhao RC et al (2007) Human adipose tissue-derived mesenchymal stromal cells as salvage therapy for treatment of severe refractory acute graft-vs.-host disease in two children. Pediatr Transplant 11(7):814–817
Fang B, Li N, Song Y, Li J, Zhao RC, Ma Y (2009) Cotransplantation of haploidentical mesenchymal stem cells to enhance engraftment of hematopoietic stem cells and to reduce the risk of graft failure in two children with severe aplastic anemia. Pediatr Transplant 13(4):499–502
Fang B, Song Y, Li N, Li J, Han Q, Zhao RC (2009) Mesenchymal stem cells for the treatment of refractory pure red cell aplasia after major ABO-incompatible hematopoietic stem cell transplantation. Ann Hematol 88(3):261–266
Schaffler A, Buchler C (2007) Concise review: adipose tissue-derived stromal cells – basic and clinical implications for novel cell-based therapies. Stem Cells 25(4):818–827
Wagner JE, Gluckman E (2010) Umbilical cord blood transplantation: the first 20 years. Semin Hematol 47(1):3–12
Mareschi K, Biasin E, Piacibello W, Aglietta M, Madon E, Fagioli F (2001) Isolation of human mesenchymal stem cells: bone marrow versus umbilical cord blood. Haematologica 86(10):1099–1100
Wexler SA, Donaldson C, Denning-Kendall P, Rice C, Bradley B, Hows JM (2003) Adult bone marrow is a rich source of human mesenchymal ‘stem’ cells but umbilical cord and mobilized adult blood are not. Br J Haematol 121(2):368–374
Goodwin HS, Bicknese AR, Chien SN, Bogucki BD, Oliver DA, Quinn CO et al (2001) Multilineage differentiation activity by cells isolated from umbilical cord blood: expression of bone, fat, and neural markers. Biol Blood Marrow Transplant 7(11):581–588
Bieback K, Kern S, Kluter H, Eichler H (2004) Critical parameters for the isolation of mesenchymal stem cells from umbilical cord blood. Stem Cells 22(4):625–634
Kern S, Eichler H, Stoeve J, Kluter H, Bieback K (2006) Comparative analysis of mesenchymal stem cells from bone marrow, umbilical cord blood, or adipose tissue. Stem Cells 24(5):1294–1301
Berger MJ, Adams SD, Tigges BM, Sprague SL, Wang XJ, Collins DP et al (2006) Differentiation of umbilical cord blood-derived multilineage progenitor cells into respiratory epithelial cells. Cytotherapy 8(5):480–487
Kogler G, Sensken S, Airey JA, Trapp T, Muschen M, Feldhahn N et al (2004) A new human somatic stem cell from placental cord blood with intrinsic pluripotent differentiation potential. J Exp Med 200(2):123–135
Lee OK, Kuo TK, Chen WM, Lee KD, Hsieh SL, Chen TH (2004) Isolation of multipotent mesenchymal stem cells from umbilical cord blood. Blood 103(5):1669–1675
FDA (2006) Guidance for industry – implementation of acceptable full-length donor history questionnaire and accompanying materials for use in screeing donors of blood and blood components U.S. Department of Health and Human Services Food and Drug Administration Center for Biologies Evaluation and Research Rockville, MD
Sensebe L, Bourin P (2008) Producing MSC according GMP: process and controls. Biomed Mater Eng 18(4–5):173–177
Sensebe L (2008) Clinical grade production of mesenchymal stem cells. Biomed Mater Eng 18:S3–S10
Sotiropoulou PA, Perez SA, Salagianni M, Baxevanis CN, Papamichail M (2006) Cell culture medium composition and translational adult bone marrow-derived stem cell research. Stem Cells 24(5):1409–1410
Sotiropoulou PA, Perez SA, Salagianni M, Baxevanis CN, Papamichail M (2006) Characterization of the optimal culture conditions for clinical scale production of human mesenchymal stem cells. Stem Cells 24(2):462–471
Haack-Sorensen M, Friis T, Bindslev L, Mortensen S, Johnsen HE, Kastrup J (2008) Comparison of different culture conditions for human mesenchymal stromal cells for clinical stem cell therapy. Scand J Clin Lab Invest 68(3):192–203
Pal R, Hanwate M, Totey SM (2008) Effect of holding time, temperature and different parenteral solutions on viability and functionality of adult bone marrow-derived mesenchymal stem cells before transplantation. J Tissue Eng Regen Med 2(7):436–444
Meuleman N, Tondreau T, Delforge A, Dejeneffe M, Massy M, Libertalis M et al (2006) Human marrow mesenchymal stem cell culture: serum-free medium allows better expansion than classical alpha-MEM medium. Eur J Haematol 76(4):309–316
Lange C, Cakiroglu F, Spiess AN, Cappallo-Obermann H, Dierlamm J, Zander AR (2007) Accelerated and safe expansion of human mesenchymal stromal cells in animal serum-free medium for transplantation and regenerative medicine. J Cell Physiol 213(1):18–26
Haack-Sorensen M, Bindslev L, Mortensen S, Friis T, Kastrup J (2007) The influence of freezing and storage on the characteristics and functions of human mesenchymal stromal cells isolated for clinical use. Cytotherapy 9(4):328–337
Wagner W, Ho AD (2007) Mesenchymal stem cell preparations – comparing apples and oranges. Stem Cell Rev 3(4):239–248
Burunova VV, Suzdaltseva YG, Voronov AV, Cheglakov IB, Vakhrushev IV, Yarygin KN et al (2008) Development and introduction of production standards for cell products of mesenchymal origin. Bull Exp Biol Med 145(4):526–530
Veyrat-Masson R, Boiret-Dupre N, Rapatel C, Descamps S, Guillouard L, Guerin JJ et al (2007) Mesenchymal content of fresh bone marrow: a proposed quality control method for cell therapy. Br J Haematol 139(2):312–320
Grisendi G, Anneren C, Cafarelli L, Sternieri R, Veronesi E, Cervo GL et al (2010) GMP-manufactured density gradient media for optimized mesenchymal stromal/stem cell isolation and expansion. Cytotherapy 12(4):466–477
Crisostomo PR, Wang MJ, Wairiuko GM, Morrell ED, Terrell AM, Seshadri P et al (2006) High passage number of stem cells adversely affects stem cell activation and myocardial protection. Shock 26(6):575–580
Bonab MM, Alimoghaddam K, Talebian F, Ghaffari SH, Ghavamzadeh A, Nikbin B (2006) Aging of mesenchymal stem cell in vitro. BMC Cell Biol 10:7
Pochampally RR, Smith JR, Ylostalo J, Prockop DJ (2004) Serum deprivation of human marrow stromal cells (hMSCs) selects for a subpopulation of early progenitor cells with enhanced expression of OCT-4 and other embryonic genes. Blood 103(5):1647–1652
Heiskanen A, Satomaa T, Tiitinen S, Laitinen A, Mannelin S, Impola U et al (2007) N-glycolylneuraminic acid xenoantigen contamination of human embryonic and mesenchymal stem cells is substantially reversible. Stem Cells 25(1):197–202
Sundin M, Ringden O, Sundberg B, Nava S, Gotherstrom C, Le Blanc K (2007) No alloantibodies against mesenchymal stromal cells, but presence of anti-fetal calf serum antibodies, after transplantation in allogeneic hematopoietic stem cell recipients. Haematologica 92(9):1208–1215
WHO Expert Committee on Biological Standardization (2010) Recommendations for the evaluation of animal cell cultures as substrates for the manufacture of biological medicinal products and for the characterization of cell banks. Report No.: WHO/BS/10.2132
Stute N, Holtz K, Bubenheim M, Lange C, Blake F, Zander AR (2004) Autologous serum for isolation and expansion of human mesenchymal stem cells for clinical use. Exp Hematol 32(12):1212–1225
Muller I, Kordowich S, Holzwarth C, Spano C, Isensee G, Staiber A et al (2006) Animal serum-free culture conditions for isolation and expansion of multipotent mesenchymal stromal cells from human BM. Cytotherapy 8(5):437–444
Le Blanc K, Samuelsson H, Lonnies L, Sundin M, Ringden O (2007) Generation of immunosuppressive mesenchymal stem cells in allogeneic human serum. Transplantation 84(8):1055–1059
von Bonin M, Stolzel F, Goedecke A, Richter K, Wuschek N, Holig K et al (2009) Treatment of refractory acute GVHD with third-party MSC expanded in platelet lysate-containing medium. Bone Marrow Transplant 43(3):245–251
Horn P, Bokermann G, Cholewa D, Bork S, Walenda T, Koch C et al (2010) Impact of individual platelet lysates on isolation and growth of human mesenchymal stromal cells. Cytotherapy 12(7):888–898
Meuleman N, Tondreau T, Bron D, Lagneaux L (2007) Human marrow mesenchymal stem cell culture: serum-free medium allows better expansion than classical alpha-minimal essential medium (MEM). Eur J Haematol 78(2):168
Chase L, Lakshmipathy U, Solchaga L, Rao M, Verfaillie CM (2010) A novel serum-free medium for the expansion of human mesenchymal stem cells. Stem Cell Res Ther 1(8):1–8
Chase L, Boucher S, Vemuri M (2010) Serum-free and xeno-free culture medium for the expansion of human mesenchymal stem cells. Hum Gene Ther 21(6):785–786
Lindroos B, Boucher S, Chase L, Kuokkanen H, Huhtala H, Haataja R et al (2009) Serum-free, xeno-free culture media maintain the proliferation rate and multipotentiality of adipose stem cells in vitro. Cytotherapy 11(7):958–972
Shepherd AJ, Wilson NJ, Smith KT (2003) Characterisation of endogenous retrovirus in rodent cell lines used for production of biologicals. Biologicals 31(4):251–260
Wolfe M, Pochampally R, Swaney W, Reger R (2008) Isolation and culture of bone marrow-derived human multipotent stromal cells (hMSCs). Methods Mol Biol 449:3–25
Sekiya I, Larson BL, Smith JR, Pochampally R, Cui JG, Prockop DJ (2002) Expansion of human adult stem cells from bone marrow stroma: conditions that maximize the yields of early progenitors and evaluate their quality. Stem Cells 20(6):530–541
LeBlanc K, Frassoni F, Ball L, Locatelli F, Roelofs H, Lewis I et al (2008) Mesenchymal stem cells for treatment of steroid-resistant, severe, acute graft-versus-host disease: a phase II study. Lancet 371(9624):1579–1586
Yang HS, Jeon O, Bhang SH, Lee SH, Kim BS (2010) Suspension culture of mammalian cells using thermosensitive microcarrier that allows cell detachment without proteolytic enzyme treatment. Cell Transplant 19:1123–1132
Eibes G, dos Santos F, Andrade PZ, Boura JS, Abecasis MMA, da Silva CL et al (2010) Maximizing the ex vivo expansion of human mesenchymal stem cells using a microcarrier-based stirred culture system. J Biotechnol 146(4):194–197
Frauenschuh S, Reichmann E, Ibold Y, Goetz PM, Sittinger M, Ringe J (2007) A microcarrier-based cultivation system for expansion of primary mesenchymal stem cells. Biotechnol Prog 23(1):187–193
Schop D, Janssen FW, Borgart E, de Bruijn JD, Dijkhuizen-Radersma R (2008) Expansion of mesenchymal stem cells using a microcarrier-based cultivation system: growth and metabolism. J Tissue Eng Regen Med 2(2–3):126–135
Liu Y, Xu X, Ma X, Martin-Rendon E, Watt S, Cui Z (2010) Cryopreservation of human bone marrow-derived mesenchymal stem cells with reduced dimethylsulfoxide and well-defined freezing solutions. Biotechnol Prog 26:1635–1643
Center for Biologics Evaluation and Research (2004) Guidance for industry – sterile drug products produced by aseptic processing – current good manufacturing practice. p 1–59 U.S. Department of Health and Human Services Food and Drug Administration Center for Biologies Evaluation and Research Rockville, MD
FDA (2008) Guidance for industry – process validation: general principals and practices (draft guidance) U.S. Department of Health and Human Services Food and Drug Administration Center for Biologies Evaluation and Research Rockville, MD
Butler JM (2006) Genetics and genomics of core short tandem repeat loci used in human identity testing. J Forensic Sci 51(2):253–265
Bentley G, Higuchi R, Hoglund B, Goodridge D, Sayer D, Trachtenberg EA et al (2009) High-resolution, high-throughput HLA genotyping by next-generation sequencing. Tissue Antigens 74(5):393–403
Schmid I, Krall WJ, Uittenbogaart CH, Braun J, Giorgi JV (1992) Dead cell discrimination with 7-amino-actinomycin-D in combination with dual color immunofluorescence in single laser flow-cytometry. Cytometry 13(2):204–208
FDA (1993) Points to consider in the characterization of cell lines used to produce biologics U.S. Department of Health and Human Services Food and Drug Administration Center for Biologies Evaluation and Research Rockville, MD
Catalina P, Cobo F, Cortes JL, Nieto AI, Cabrera C, Montes R et al (2007) Conventional and molecular cytogenetic diagnostic methods in stem cell research: a concise review. Cell Biol Int 31(9):861–869
Sumstad D, Carlson M, Adams S, Kadidlo D, Bostrom N, Wagner J et al (2009) Reduction of non-clinical-/non-cGMP-grade culture reagents and measurement of residual ingredients in final early phase cellular therapy products. Presented at the International Society for Cellular Therapy 2009 Annual Meeting, San Diego, CA
FDA (2008) Guidance for industry: potency tests for cellular and gene therapy products (draft guidance) U.S. Department of Health and Human Services Food and Drug Administration Center for Biologies Evaluation and Research Rockville, MD
Le Blanc K, Rasmusson I, Gotherstrom C, Seidel C, Sundberg B, Sundin M et al (2004) Mesenchymal stem cells inhibit the expression of CD25 (interleukin-2 receptor) and CD38 on phytohaemagglutinin-activated lymphocytes. Scand J Immunol 60(3):307–315
Di Nicola M, Carlo-Stella C, Magni M, Milanesi M, Longoni PD, Matteucci P et al (2002) Human bone marrow stromal cells suppress T-lymphocyte proliferation induced by cellular or nonspecific mitogenic stimuli. Blood 99(10):3838–3843
Matthay MA, Thompson BT, Read EJ, McKenna DH, Liu KD, Calfee CS et al (2010) Therapeutic potential of mesenchymal stem cells for severe acute lung injury. Chest 138(4):965–972
Bernardo ME, Avanzini MA, Ciccocioppo R, Perotti C, Cometa AM, Moretta A et al (2008) Phenotypical and functional characterization of in vitro expanded bone marrow-derived mesenchymal stromal cells from patients with Crohn’s disease. Blood 112(11):888
De Bari C, Dell’Accio F, Karystinou A, Guillot PV, Fisk NM, Jones EA et al (2008) A biomarker-based mathematical model to predict bone-forming potency of human synovial and periosteal mesenchymal stem cells. Arthritis Rheum 58(1):240–250
Pittenger MF, Mackay AM, Beck SC, Jaiswal RK, Douglas R, Mosca JD et al (1999) Multilineage potential of adult human mesenchymal stem cells. Science 284(5411):143–147
Wagner W, Wein F, Roderburg C, Saffrich R, Diehlmann A, Eckstein V et al (2008) Adhesion of human hematopoietic progenitor cells to mesenchymal stromal cells involves CD44. Cells Tissues Organs 188(1–2):160–169
Kadereit S, Deeds LS, Haynesworth SE, Koc ON, Kozik MM, Szekely E et al (2002) Expansion of LSTC-ICs and maintenance of p21 and BCL-2 expression in cord blood CD34(+)/CD38(−) early progenitors cultured over human MSCs as a feeder layer. Stem Cells 20(6):573–582
Hatzistergos KE, Quevedo H, Oskouei BN, Hu QH, Feigenbaum GS, Margitich IS et al (2010) Bone marrow mesenchymal stem cells stimulate cardiac stem cell proliferation and differentiation. Circ Res 107(7):913
Ghannam S, Bouffi C, Djouad F, Jorgensen C, Noel D (2010) Immunosuppression by mesenchymal stem cells: mechanisms and clinical applications. Stem Cell Res Ther 1(2):1–7
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer Science+Business Media New York
About this chapter
Cite this chapter
Hei, D.J., McKenna, D.H. (2013). cGMP Production of MSCs. In: Hematti, P., Keating, A. (eds) Mesenchymal Stromal Cells. Stem Cell Biology and Regenerative Medicine. Humana Press, New York, NY. https://doi.org/10.1007/978-1-4614-5711-4_16
Download citation
DOI: https://doi.org/10.1007/978-1-4614-5711-4_16
Published:
Publisher Name: Humana Press, New York, NY
Print ISBN: 978-1-4614-5710-7
Online ISBN: 978-1-4614-5711-4
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)