
Abstract
It is well established that many different protein modifications such as phosphorylation, oxidation, and degradation can affect meat quality and it is therefore important to implement and develop proteomics methods in meat science to understand the mechanism behind the influence of these protein modifications on the quality of meat and meat products. We can expect that high-throughput modification-specific proteomics will be employed to analyze systematically the qualitative and quantitative differences of these protein modifications in meat concerning the development, genetic background, processing, and storage that will contribute greatly to our understanding of the mechanisms underlying meat quality difference.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Introduction
During the post-mortem (PM) conversion of muscle into meat, a significant series of events occurs in response to the stoppage of the respiratory system and blood circulation; the metabolism in the muscle is known to be changed substantially (Scheffler and Gerrard 2007). The rate and extent of post-mortem metabolic processes could greatly influence many important meat quality properties such as tenderness, water-holding capacity, and color. Underlying these processes, muscle proteins are the fundamental targets and factors contributing to the above-mentioned changes and properties. In PM muscle, proteins are subjected to a series of protein modifications, such as reversible phosphorylation, oxidation, degradation, and denaturation, and all these modifications are critical for the formation of different meat quality traits. Protein modifications could fundamentally affect the biological and chemical properties of proteins, and thereby they can be involved in all aspects of meat quality development.
In recent years, protein modifications have been studied by several research groups that have looked at the potential effects on muscle foods. They revealed that muscle protein modifications could significantly affect meat quality development. Oxidation of proteins in processed meat products leads to poor meat quality and lesser nutritional value (Lund et al. 2007; Morzel et al. 2006; Rowe et al. 2004). Protein phosphorylation in PM meat was supposed to regulate the activity of enzymes and rigor mortis development, consequently affecting pH decline rate and meat quality development (Huang et al. 2011; Lametsch et al. 2011; Muroya et al. 2007). It is generally accepted that degradation and denaturation of proteins in PM meat are responsible for the tenderness development of meat (Koohmaraie 1996). Oxidation, phosphorylation, and degradation are the three protein modifications mostly studied in muscle foods, and proteomics are the key tool for analysis of these muscle protein modifications.
Proteomics are the systematically unbiased study of entire protein complements expressed by the genome of an organism (proteome) for the high-throughput discovery of protein alterations. Mass spectrometry (MS) -based proteomics are dedicated to the global analysis of protein composition, modifications (PTMs), and the dynamic range of expression levels (Aebersold and Mann 2003; Yates et al. 2009). In the meat science area, proteomics were confirmed to be a powerful tool for studies with regard to different qualities or processing parameters, as described in several reviews (Bendixen 2005; Bendixen et al. 2011; Hollung et al. 2007). Proteomics can provide valuable information for a better understanding of the mechanisms influencing the different quality traits. This information can be used to optimize meat production and improve meat quality. Advanced MS-based proteomics are also a central tool for detection, site mapping, and quantification of modifications on proteins (Witze et al. 2007). Both gel-based and gel-free proteomic approaches were developed for the analysis of different protein modifications in biological samples, and these approaches were also employed for studying the protein modifications in muscle food, mainly focusing on protein oxidation, phosphorylation, and degradation.
This chapter outlines current achievements in the study of protein modifications in muscle food using proteomic approaches. First we describe the general knowledge of protein modifications and then the development of gel-based and gel-free proteomic approaches for the characterization of such modifications. Finally, we elucidate the effects of protein modifications on muscle foods and devote our main attention to the application of proteomic approaches for the analysis of these modifications.
2 Protein Modifications
Most proteins are subjected to some form of protein modifications that can occur on protein both in vivo as post-translational modifications and in response to different environmental factors. Protein modifications increase the diversity of proteins because many types of modifications are usually covalently present at different amino acid residues of the protein. The heterogeneous modifications at distinct amino acid residues lead to further complexity at the protein level (Jensen 2006). Protein modifications can be divided into two categories according to their modification forms: covalent modification of a nucleophilic amino acid side chain by an electrophilic fragment of a cosubstrate, and cleavage of a protein backbone at a specific peptide bond (Walsh et al. 2005).
The most widely studied protein modifications in biological science include protein phosphorylation, acylation, glycosylation, ubiquitination, acetylation, oxidation, and so on. One protein is often subjected to several modifications at a time. However, PTMs are usually present at substoichiometric levels, because a protein modification at a given site is often present in a small fraction of the protein. PTMs can affect the activity, structure, location, and lifetime of proteins, thereby playing essential roles in most cellular processes such as the maintenance of protein structure and integrity, regulation of metabolism and defense processes, cell signaling pathways, and protein spatial–temporal distribution (Jensen 2006).
Protein modifications can affect many food properties such as shelf-life, nutritional value, digestibility, health benefits, and consumer appeal (Kerwin and Remmele 2007). For instance, thermal treatment is widely used in processing and manufacturing steps for many foods and ingredients, including dairy, meat, and cereal products. However, thermal treatment can lead to protein oxidation, the Maillard reaction, protein aggregates, and cross-linking modifications that have been implicated in quality deterioration, nutritional damage, and adverse health effects (Promeyrat et al. 2010; Silvestre et al. 2006).
3 Proteomic Approaches for Detection of Protein Modifications
Compared to the total proteins, the proteins with site-specific modifications are only present at substoichiometric levels, and it is therefore a great analytical challenge in proteomics to analyze the specific modifications. In the past two decades, many proteomic strategies were developed for the analysis of protein modification in complex samples ranging from the traditional gel-based Western-blot to the high-throughput advanced MS-based proteomic strategies. Typically, the identification and characterization of protein modification are achieved by combining protein-extraction methods, affinity enrichment, and chromatographic and/or electrophoretic separation with peptide-mass determination and amino acid sequencing by high-performance MS (Jensen 2006; Witze et al. 2007).
3.1 Gel-Based Proteomic Approaches
Western-blot analysis is a widespread and traditional method for analyzing protein modifications. Western-blot can be specific and relatively quantitative, and specific antibodies can be utilized to study the modified forms of proteins, such as protein cleavage and post-translational modifications. However, Western-blot analysis relies heavily on prior knowledge of the type and position of specific modifications in proteins and is limited by the availability and specificity of antibodies.
One- and two-dimensional gel electrophoresis (1DE and 2DE) -based proteomic strategies are widely used for the detection of variation in protein modification, because such modifications can result in the shift of the isoelectric point or molecular weight of protein. Protein phosphorylation could affect the isoelectric point, as neutral hydroxyl groups on serines, threonines, or tyrosines are replaced with negatively charged phosphates, and cause the protein to shift towards the acid end of the 2-D gel creating the “beads-on-a-string” look for multiple phosphates. If the proteins are modified by degradation and/or glycolyzation, the molecular weight of the resulting fragments will change and migrate differently in the second dimension. Therefore, the proteins with modifications would show a different mobility pattern on a 2-D gel, and different isoforms of the same protein can be visible as different spots on the 2DE gel.
Different types of nonradioactive fluorescence staining dyes have been developed for the in-gel detection of proteins with specific modifications. The fluorescent dyes have a good dynamic range of quantification and good compatibility with subsequent protein characterization and identification (Patton 2002). Pro-Q Diamond phosphoprotein stain (Molecular Probes) was developed for the detection of phosphoserine-, phosphothreonine-, and phosphotyrosine-containing proteins on 1D and 2-D gels, electroblots, and protein microarrays (Steinberg et al. 2003). Recently, it was shown that a combination of one- and two-dimensional gel electrophoresis stained with Pro-Q Diamond could be used to detect changes in protein phosphorylation in PM porcine meat samples (Huang et al. 2011, 2012). This strategy can be applied for semi-quantitative analysis of protein phosphorylation changes in multiple samples (Fig. 7.1). Protein oxidation can be analyzed by detection of carbonyl derivatives which involves a pre-derivatization of the carbonyl groups with dinitrophenylhydrazine (DNPH) prior to electrophoresis, followed by immunoblotting with an anti-DNP antibody (Nakamura and Goto 1996). Many protein oxidation assay kits are commercially available, such as the OxyBlot Protein Oxidation Detection Kit. After modification-specific staining and imaging, the same gel will be subjected to total-protein stains, such as SYPRO Ruby Fluorescent dye; the images from both modification staining and total protein staining can be obtained with gel scanners with multiple laser excitation sources and analyzed with proteomic image software. Changes at the protein modification level can be semi-quantified by comparing the intensity of the protein in the modification staining image and its intensity in the total protein image.

The scheme for gel-based phosphoproteomic analysis of postmortem porcine muscle
After gel separation, the proteins will usually be cut off from the gel and subjected to ingel digestion and MS identification. From the MS data it is possible to identify the protein modification sites and type (Jensen 2004). It is also possible to predict the cleavage site using tandem MS if the protein is degraded (Lametsch et al. 2002; Larsen et al. 2001).
3.2 Gel-Free Proteomic Approaches
Compared to gel-based approaches, gel-free liquid chromatography coupled with tandem mass spectrometry (LC-MS/MS) approaches seems to be much more powerful for high-throughput modification-specific proteomics, inasmuch as many specific techniques for modified proteins or peptides can be incorporated prior to LC-MS/MS analysis, and it is also possible for quantitative study by introducing different isotopic labelings into the protein or peptides. For noncomplicated samples, it is possible to detect the protein modification with shotgun sequencing. The protein samples are treated with multiple proteases to generate complementary and redundant sets of overlapping peptides, and then subjected to shotgun sequencing by LC-MS/MS; finally extensive data analysis is needed. This strategy is suitable for subproteomes (Wu et al. 2003), simple protein mixtures, and protein complexes. However, for most of the complicated biological samples, it is difficult to detect and characterize protein modification using common global proteomics strategies. A key to protein modification-specific proteomics approaches is to enrich and purify protein or peptide species with specific modification prior to characterization by MS/MS.
Different enrichment methods for specific modification in combination with LC/MS/MS approaches are the most popular strategy for high-throughput protein modification-specific proteomics. The enrichment of modified proteins or peptides is often achieved by using affinity chromatographic techniques. Immunoprecipitation with modification-specific antibodies is highly helpful for enriching the corresponding modified proteins. Commercially available phosphotyrosine (pTyr)-specific antibodies are widely used for cell-signaling study (Rush et al. 2005). Phosphopeptides can also be enriched by immobilized metal affinity chromatography (IMAC) (Ficarro et al. 2002; Posewitz and Tempst 1999), or by titanium dioxide (TiO2) columns (Larsen et al. 2005; Thingholm et al. 2006) prior to MS/MS analysis. Strong cation exchange and anion exchange chromatography can be used to reduce peptide complexity (Beausoleil et al. 2004; Nuhse et al. 2003). Glycoproteins can be enriched by using sugar-specific antibody lectins (Gabius et al. 2002; Yang and Hancock 2004) and TiO2 columns (Larsen et al. 2007). Glycosidase D/H treatment and MS/MS facilitated protein identification and assignment of glycosylation sites (Hagglund et al. 2004). Acetylated peptides can be enriched by using resin-coupled antibodies to acetyllysine (Kim et al. 2006). Chemistry-based methods for modification-specific covalent capture of proteins and/or peptides are another choice for characterization of protein modification. These methods are often based on β-elimination/Michael addition chemistry, phosphoramidate chemistry, or other PTM-specific chemistries. The targeted PTMs can be converted to stable and manageable status and then identified by MS/MS sequencing (Oda et al. 2001; Wells et al. 2002).
Most modifications can result in a mass increment or a mass deficit relative to the nascent unmodified protein, as summarized by Jensen (Jensen 2006). For example, the phosphorylation of a Tyr residue can increase its mass from 163 Da to 243 Da by the addition of an HPO3 group (80 Da). After enrichment and pre-fractionation, the modification site can be assigned through the observation of a discrete mass change of the peptide in MS or of the residue in MS/MS (Larsen et al. 2006). The data acquisition software can then be programmed to monitor and sequence all of the pre-defined candidate modified peptides. Huge amounts of data are normally generated from LC-MS/MS; how to extract the useful information and interpret the data is also a great challenge. The integration of computational tools into modification-specific proteomics studies is a prerequisite for the interpretation of large-scale datasets into meaningful biological information; the explosive accumulation of large-scale data and development of a PTM database will strongly accelerate this step (Jensen 2006; Witze et al. 2007).
4 Impacts of Protein Modifications on Muscle Food Quality
During the conversion of muscle into meat, the occurrence of distinct protein modifications is also involved in the whole process of meat quality development. Protein modifications in meat are influenced by conditions prior to and after animal slaughter. Prior to slaughter, physiological reactions to stress may influence the rate and extent of PM pH decline and alter the protein modification pattern, thereby affecting meat quality traits (Ferguson and Warner 2008). Shortly after slaughter, drastic metabolic changes can result in the activation of different protein modification mechanisms (mainly PTMs) to regulate the activity of enzymes and structural proteins in response to the shortage of ATP and the development of rigor mortis. For example, the phosphorylation levels of many muscle proteins were known to change significantly during PM 1–24 h, and the reversible phosphorylation can indirectly affect the pH decline rate and the development of rigor mortis (Huang et al. 2011, 2012; Lametsch et al. 2011). Protein oxidation occurs during meat processing and has numerous negative effects on meat quality and it is therefore important to control this oxidation (Lund et al. 2011). The Maillard reaction occurring during the cooking of meat is important for the development of meat flavor and color.
5 Proteomic Analysis of Protein Modifications in Muscle Food
In muscle, intensive studies were performed using proteomic approaches for studying protein modifications of muscle protein, such as oxidation (Feng et al. 2008), phosphorylation (Hojlund et al. 2009), nitration (Kanski et al. 2005), and glycosylation (Martin-Rendon and Blake 2003). These modifications were found to play essential roles in metabolism regulation and muscle contraction. Whereas until recently protein modifications were largely unexplored in muscle food systems, more attention was drawn to characterizing protein modifications in meat during intensive exploration of the mechanisms of regulating meat quality development and storage. Specifically, researchers focus much more on oxidation and phosphorylation. Meanwhile, protein degradation has also been intensively studied in the meat science area, as it plays a key role in regulating the development of meat tenderness.
5.1 Protein Phosphorylation
Reversible protein phosphorylation is one of the most widespread regulatory mechanisms in nature. Phosphorylation and dephosphorylation of proteins regulate critical biological processes including metabolism, signaling transduction, proliferation, and differentiation (Graves and Krebs 1999; Hunter 2000). In recent years, with the development of phosphoproteomic methods, protein phosphorylation has been comprehensively studied in various muscle samples (Gannon et al. 2008; Hojlund et al. 2009; Hou et al. 2010). Many sarcoplasmic and myofibrillar proteins were identified to be phosphorylated, and the phosphorylation could affect the metabolism and contraction of muscle. In PM muscle, enzymes catalyzing the glycolysis reactions affect the rate and extent of pH decline (Scheffler and Gerrard 2007). Most of the glycolytic enzymes are phosphoproteins; several studies that focused on individual or few glycolytic enzymes indicated that protein phosphorylation plays critical roles in several key steps of glycolysis in PM muscle. Phosphorylase kinase can phosphorylate glycogen phosphorylase b on serine 14, change the structure, and transform it into the active form (Johnson 1992; Sprang et al. 1988). Phosphorylation of pyruvate kinase could result in an additional, more acid stable enzyme isoform and maintain high activity in PSE meat (Schwagele et al. 1996). The phosphorylation status of AMP-activated protein kinase (AMPK) could indirectly influence the glycolysis and pH decline in PM muscle (Shen and Du 2005). It has also been reported that myosin regulatory light chain 2 (MyLC2) became doubly phosphorylated during rigor formation in bovine longissimus (Muroya et al. 2007). Studying the dynamic changes of protein phosphorylation in PM meat can lead to the identification of candidate regulatory proteins from a new perspective.
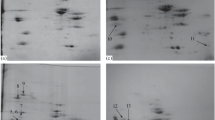
A combination of one- and two-dimensional gel electrophoresis (1DE and 2DE) coupled with Pro-Q Diamond-SYPRO Ruby staining and tandem MS strategy was employed to semi-quantitatively analyze the protein phosphorylation changes of sarcoplasmic proteins (Fig. 7.1) (Huang et al. 2011, 2012). The staining method showed high specificity for both phosphoprotein and total protein in 1DE and 2DE (Fig. 7.2). The results revealed that the fast pH decline group had the highest global phosphorylation level at PM 1 h, but lowest at 24 h, whereas the slow pH decline group showed the reverse case. The phosphorylation levels of many proteins were significantly affected by the synergy effects of pH and time (p < 0.05). Most of the phosphoproteins were identified as glycometabolism-related enzymes. The phosphorylation of pyruvate kinase and triosephosphate isomerase-1 were shown to be related to the PM muscle pH decline rate. It was suggested that the phosphorylation level might indicate the activity of corresponding proteins in PM muscle. A similar study was also performed for the myofibrillar proteins, MyLC2, troponin T, and tropomyosin, and several other structural proteins were identified as highly phosphorylated and changed with PM time. Interestingly, unlike the sarcoplasmic proteins, the phosphorylation pattern of myofibrillar proteins in PM muscle is mainly changed with PM time, but only to a minor extent influenced by the rate of pH decline, suggesting the phosphorylation of myofibrillar proteins may be related to the meat rigor mortis and quality development (Huang et al. 2012).

1DE (up) and 2 DE (down) images of sarcoplasmic proteins. (a) ProQ Diamond staining for phosphoprotein. (b) SYPRO Ruby staining for total protein (Adapted from Huang et al. 2011)
PM changes in porcine muscle protein phosphorylation in relation to the RN− and normal genotypes have also been investigated (Lametsch et al. 2011). Glycogen phosphorylase, phosphofructokinase, and pyruvate kinase were found in protein bands affected by the RN- genotype, the protein phosphorylation level of the muscle proteins could be interpreted as a global metabolic fingerprint containing information about the activity status of the enzymes in the PM metabolism. Another related study used this approach to analyze the response of sarcoplasmic proteins in PM bovine longissimus muscle to electrical stimulation and its effects on meat tenderization; the proteomic analysis showed that ES resulted in lower (p < 0.05) phosphorylation levels of creatine kinase M chain, fructose bisphosphate aldolase C-A, β-enolase, and pyruvate kinase at PM 3 h (Li et al. 2011).
5.2 Protein Oxidation
Protein oxidation in muscle foods results in the loss of meat product quality, such as reduced water-holding capacity (WHC), tenderness, and juiciness (Lund et al. 2007; Rowe et al. 2004). Oxidation of proteins may affect the susceptibility of protein substrates to proteolytic enzymes and result in low digestibility and poor nutritional value (Morzel et al. 2006). Oxidation of numerous amino acids leads to the formation of carbonyl groups and other derivatives, and causes a depletion of essential amino acids in muscle foods.
Direct oxidation of the side chains from lysine, threonine, arginine, and proline has been highlighted as the main route for protein carbonylation and the main mechanism that has been proved to yield carbonyls from meat proteins (Estevez 2011). The 2,4- dinitrophenylhydrazine (DNPH)-method (Oliver et al. 1987) combined with proteomic approaches has been widely used for evaluating protein carbonylation in multiple muscle foods. A combined immunologic and 1D and 2-D proteomic approach was employed to address protein oxidation in chicken muscles (Stagsted et al. 2004). Specific proteins containing carbonyls and/or 3-nitrotyrosine (3-NT) were detected by the DNPH method and antibody against 3-NT. It was found that enolase was the predominant carbonyl-reactive species among the water-soluble muscle proteins, and several other proteins (actin, heat shock protein 70, and creatine kinase) also contained carbonyls and/or 3-nitrotyrosine. Additionally, the effect of feed on protein oxidation was checked as well. A similar study used DNPH and anti-DNP antibody to detect oxidized myofibrillar proteins from PM porcine muscle on 2-D gel and identified about 70 oxidatively modified proteins (Bernevic et al. 2011). Another study employed 2DE combined with DNPH and fluorescent thiosemicarbazide (FTSC) staining and MS/MS to detect the protein carbonyls from bonito muscle during storage; the oxidation status of enolase, aldolase, and L-lactate dehydrogenase A chain (LDH-A) were identified as changing during storage (Kinoshita et al. 2007). Significant correlations (p < 0.05) were observed between the level of carbonyl groups and the intensities of 52 proteins on the 2-D electrophoresis of PM porcine sarcoplasmic proteins (Promeyrat et al. 2011).
In addition to the gel-based approaches, some recent studies started to perform LC-MS/MS-based proteomics to analyze oxidation in meat. Researchers used LC-MS/MS to characterize the interesting protein spots cut from 2-D gel, which led to the identification of several unique oxidation sites on creatine kinase, actin, and triosephosphate isomerise (Bernevic et al. 2011). Myoglobin (Mb) redox status affects meat color and is destabilized by lipid oxidation products. Researchers utilized LC-ESI-MS/MS and other proteomic methods to investigate oxymyoglobin (OxyMb) oxidation in Mb from pork and beef; they identified several adducted histidine (HIS) residues (more in beef), and revealed that HNE-induced HIS residues adduction occurred in a species-specific preferential manner, Preferential HNE adduction at HIS 93 was exclusively observed in bovine OxyMb (Suman et al. 2007).
5.3 Protein Degradation
Tenderness has been considered as the most important quality attribute of meat; it is well established that PM proteolysis is mainly responsible for meat tenderization (Koohmaraie and Geesink 2006). Proteomics has been widely used to investigate PM protein degradation and has provided global insight into the process of proteolysis.
Using the classic 2-D method, many proteins were found to be fragmented as a consequence of protein degradation in PM pork; identified proteins included structural proteins (such as actin, myosin heavy chain, troponin T, etc.) and sarcoplasmic proteins (such as creatine kinase, carbonate dehydratase, triosephosphate isomerase, pyruvate kinase, etc.) (Hwang et al. 2005; Lametsch et al. 2002). Moreover, the actin and the myosin heavy chain fragments have been found to correlate significantly with meat tenderness (Hwang et al. 2005; Lametsch et al. 2003), indicating that the PM degradation of structural proteins contributes to meat tenderization. It is speculated that myosin heavy chain degradation leads to disruption of the myosin–actin interaction, and has an effect on the integrity of the thin flament, which results in meat tenderization. Recently, the combination of 2DE and FTICR-MS was utilized for the identification of muscle protein degradation products in pork (Bernevic et al. 2011), and several truncation forms of creatine kinase and troponin T were identified and confirmed the former results. PM proteolysis in muscle has also been analyzed using proteomics in beef. In M. longissimus dorsi (LD) and M. semitendinosis (ST) muscle from 0 to 24 h, the intact form of cofilin, lactoylglutathione lyase, and substrate protein of mitochondrial ATP-dependent proteinase SP-22, HSP27, and HSP20 were detected as decreasing (Jia et al. 2006). It was also found that the abundance of fragments of actin, creatine kinase, HSP27, and crystallin increased, whereas the amount of intact molecules decreased over 14 days of cold storage (Morzel et al. 2008). The fragmentation of creatine kinase and other proteins was also observed in a proteomic study of fish (carp) during cold acclimation (McLean et al. 2007).
6 Conclusion
Even though it is still a very challenging task to characterize the protein modifications in complex food and biological systems, over the past 10 years development in the areas of fluorescent detection, affinity-enrichment techniques, and quantitative protein and advanced MS greatly accelerate the application of modification-specific proteomics (mainly PTMs) to reveal the molecular features and functions of proteins in life science. The global and systematic characterization and interpretation of protein modifications can be performed through the integration of computational tools into large-scale high-throughput modification-specific proteomics studies, and will offer meaningful information of biological events. Compared to the life science area, protein modifications in food science are rarely explored. The application of proteomic approaches to analyze food protein modification is predicted to become increasingly important in the area of general food science, quality assurance, and product differentiation. The knowledge and approaches of modification-specific proteomics developed in the life sciences area can be referenced and will contribute to the knowledge development of protein modifications in food sciences.
The proteins in PM muscle are subjected to both post-translational modifications and extracellular modifications. Importantly, these protein modifications mainly determine the biochemical and physical properties of meat proteins and thereby affect final meat quality. Until now, protein modification studies in meat science are mainly limited to phosphorylation, oxidation, and degradation, and to some extent, knowledge of these modifications in meat is still at the preliminary level. We can expect that high-throughput modification-specific proteomics will be employed to systematically analyze the qualitative and quantitative differences of these protein modifications in meat concerning the development, genetic background, processing, and storage, which will greatly contribute to our understanding of the mechanisms underlying meat quality difference. In addition, it is also of great interest to explore the potential roles of other important protein modifications in meat systems using modification-specific proteomics.
References
Aebersold R, Mann M (2003) Mass spectrometry-based proteomics. Nature 422(6928):198–207
Beausoleil SA, Jedrychowski M, Schwartz D, Elias JE, Villen J, Li JX, Cohn MA, Cantley LC, Gygi SP (2004) Large-scale characterization of HeLa cell nuclear phosphoproteins. Proc Natl Acad Sci USA 101(33):12130–12135
Bendixen E (2005) The use of proteomics in meat science. Meat Sci 71(1):138–149
Bendixen E, Danielsen M, Hollung K, Gianazza E, Miller I (2011) Farm animal proteomics – a review. J Proteomics 74(3):282–293
Bernevic B, Petre BA, Galetskiy D, Werner C, Wicke M, Schellander K, Przybylski M (2011) Degradation and oxidation postmortem of myofibrillar proteins in porcine skeleton muscle revealed by high resolution mass spectrometric proteome analysis. Int J Mass Spectrom 305(2–3):217–227
Estevez M (2011) Protein carbonyls in meat systems: a review. Meat Sci 89(3):259–279
Feng J, Xie HW, Meany DL, Thompson LV, Arriaga EA, Griffin TJ (2008) Quantitative proteomic profiling of muscle type-dependent and age-dependent protein carbonylation in rat skeletal muscle mitochondria. J Gerontol Ser A Biol 63(11):1137–1152
Ferguson DM, Warner RD (2008) Have we underestimated the impact of pre-slaughter stress on meat quality in ruminants? Meat Sci 80(1):12–19
Ficarro SB, McCleland ML, Stukenberg PT, Burke DJ, Ross MM, Shabanowitz J, Hunt DF, White FM (2002) Phosphoproteome analysis by mass spectrometry and its application to Saccharomyces cerevisiae. Nat Biotechnol 20(3):301–305
Gabius HJ, Andre S, Kaltner H, Siebert HC (2002) The sugar code: functional lectinomics. BBA Gen Subjects 1572(2–3):165–177
Gannon J, Staunton L, O’Connell K, Doran P, Ohlendieck K (2008) Phosphoproteomic analysis of aged skeletal muscle. Int J Mol Med 22(1):33–42
Graves JD, Krebs EG (1999) Protein phosphorylation and signal transduction. Pharmacol Ther 82(2–3):111–121
Hagglund P, Bunkenborg J, Elortza F, Jensen ON, Roepstorff P (2004) A new strategy for identification of N-glycosylated proteins and unambiguous assignment of their glycosylation sites using HILIC enrichment and partial deglycosylation. J Proteome Res 3(3):556–566
Hojlund K, Bowen BP, Hwang H, Flynn CR, Madireddy L, Geetha T, Langlais P, Meyer C, Mandarino LJ, Yi ZP (2009) In vivo phosphoproteome of human skeletal muscle revealed by phosphopeptide enrichment and HPLC-ESI-MS/MS. J Proteome Res 8(11):4954–4965
Hollung K, Veiseth E, Jia XH, Faergestad EM, Hildrum KI (2007) Application of proteomics to understand the molecular mechanisms behind meat quality. Meat Sci 77(1):97–104
Hou JJ, Cui ZY, Xie ZS, Xue P, Wu P, Chen XL, Li J, Cai TX, Yang FQ (2010) Phosphoproteome analysis of rat L6 myotubes using reversed-phase C18 prefractionation and titanium dioxide enrichment. J Proteome Res 9(2):777–788
Huang HG, Larsen MR, Karlsson AH, Pomponio L, Costa LN, Lametsch R (2011) Gel-based phosphoproteomics analysis of sarcoplasmic proteins in postmortem porcine muscle with pH decline rate and time differences. Proteomics 11(20):4063–4076
Huang HG, Larsen MR, Lametsch R (2012) Changes in phosphorylation of myofibrillar proteins during postmortem development of porcine muscle. Food Chem. doi: http://dx.doi.org/10.1016/j.foodchem.2012.03.132
Hunter T (2000) Signaling – 2000 and beyond. Cell 100(1):113–127
Hwang IH, Park BY, Kim JH, Cho SH, Lee JM (2005) Assessment of postmortem proteolysis by gel-based proteome analysis and its relationship to meat quality traits in pig longissimus. Meat Sci 69(1):79–91
Jensen ON (2004) Modification-specific proteomics: characterization of post-translational modifications by mass spectrometry. Curr Opin Chem Biol 8(1):33–41
Jensen ON (2006) Interpreting the protein language using proteomics. Nat Rev Mol Cell Biol 7(6):391–403
Jia XH, Hollung K, Therkildsen M, Hildrum KI, Bendixen E (2006) Proteome analysis of early post-mortem changes in two bovine muscle types: M-longissimus dorsi and M-semitendinosis. Proteomics 6(3):936–944
Johnson LN (1992) Glycogen-phosphorylase – control by phosphorylation and allosteric effectors. FASEB J 6(6):2274–2282
Kanski J, Hong SJ, Schoneich C (2005) Proteomic analysis of protein nitration in aging skeletal muscle and identification of nitrotyrosine-containing sequences in vivo by nanoelectrospray ionization tandem mass spectrometry. J Biol Chem 280(25):24261–24266
Kerwin BA, Remmele RL (2007) Protect from light: photodegradation and protein biologics. J Pharm Sci 96(6):1468–1479
Kim SC, Sprung R, Chen Y, Xu YD, Ball H, Pei JM, Cheng TL, Kho Y, Xiao H, Xiao L, Grishin NV, White M, Yang XJ, Zhao YM (2006) Substrate and functional diversity of lysine acetylation revealed by a proteomics survey. Mol Cell 23(4):607–618
Kinoshita Y, Sato T, Naitou H, Ohashi N, Kumazawa S (2007) Proteomic studies on protein oxidation in bonito (Katsuwonus pelamis) muscle. Food Sci Technol Res 13(2):133–138
Koohmaraie M (1996) Biochemical factors regulating the toughening and tenderization processes of meat. Meat Sci 43:S193–S201
Koohmaraie M, Geesink GH (2006) Contribution of postmortem muscle biochemistry to the delivery of consistent meat quality with particular focus on the calpain system. Meat Sci 74(1):34–43
Lametsch R, Roepstorff P, Bendixen E (2002) Identification of protein degradation during post-mortem storage of pig meat. J Agric Food Chem 50(20):5508–5512
Lametsch R, Karlsson A, Rosenvold K, Andersen HJ, Roepstorff P, Bendixen E (2003) Postmortem proteome changes of porcine muscle related to tenderness. J Agric Food Chem 51(24):6992–6997
Lametsch R, Larsen MR, Essen-Gustavsson B, Jensen-Waern M, Lundstrom K, Lindahl G (2011) Postmortem changes in pork muscle protein phosphorylation in relation to the RN genotype. J Agric Food Chem 59(21):11608–11615
Larsen MR, Larsen PM, Fey SJ, Roepstorff P (2001) Characterization of differently processed forms of enolase 2 from Saccharomyces cerevisiae by two-dimensional gel electrophoresis and mass spectrometry. Electrophoresis 22(3):566–575
Larsen MR, Thingholm TE, Jensen ON, Roepstorff P, Jorgensen TJD (2005) Highly selective enrichment of phosphorylated peptides from peptide mixtures using titanium dioxide microcolumns. Mol Cell Proteomics 4(7):873–886
Larsen MR, Trelle MB, Thingholm TE, Jensen ON (2006) Analysis of posttranslational modifications of proteins by tandem mass spectrometry. Biotechniques 40(6):790–798
Larsen MR, Jensen SS, Jakobsen LA, Heegaard NHH (2007) Exploring the sialiome using titanium dioxide chromatography and mass spectrometry. Mol Cell Proteomics 6(10):1778–1787
Li CB, Li J, Zhou GH, Lametsch R, Ertbjerg P, Bruggemann DA, Huang HG, Karlsson AH, Hviid M, Lundstrom K (2011) Electrical stimulation affects metabolic enzyme phosphorylation, protease activation and meat tenderization in beef. J Anim Sci 90(5):1638–1649
Lund MN, Lametsch R, Hviid MS, Jensen ON, Skibsted LH (2007) High-oxygen packaging atmosphere influences protein oxidation and tenderness of porcine longissimus dorsi during chill storage. Meat Sci 77(3):295–303
Lund MN, Heinonen M, Baron CP, Estevez M (2011) Protein oxidation in muscle foods: a review. Mol Nutr Food Res 55(1):83–95
Martin-Rendon E, Blake DJ (2003) Protein glycosylation in disease: new insights into the congenital muscular dystrophies. Trends Pharmacol Sci 24(4):178–183
McLean L, Young IS, Doherty MK, Robertson DHL, Cossins AR, Gracey AY, Beynon RJ, Whitfield PD (2007) Global cooling: cold acclimation and the expression of soluble proteins in carp skeletal muscle. Proteomics 7(15):2667–2681
Morzel M, Gatellier P, Sayd T, Renerre M, Laville E (2006) Chemical oxidation decreases proteolytic susceptibility of skeletal muscle myofibrillar proteins. Meat Sci 73(3):536–543
Morzel M, Terlouw C, Chambon C, Micol D, Picard B (2008) Muscle proteome and meat eating qualities of Longissimus thoracis of “Blonde d’Aquitaine” young bulls: a central role of HSP27 isoforms. Meat Sci 78(3):297–304
Muroya S, Ohnishi-Kameyama M, Oe M, Nakajima I, Shibata M, Chikuni K (2007) Double phosphorylation of the myosin regulatory light chain during rigor mortis of bovine longissimus muscle. J Agric Food Chem 55(10):3998–4004
Nakamura A, Goto S (1996) Analysis of protein carbonyls with 2,4-dinitrophenyl hydrazine and its antibodies by immunoblot in two-dimensional gel electrophoresis. J Biochem 119(4):768–774
Nuhse TS, Stensballe A, Jensen ON, Peck SC (2003) Large-scale analysis of in vivo phosphorylated membrane proteins by immobilized metal ion affinity chromatography and mass spectrometry. Mol Cell Proteomics 2(11):1234–1243
Oda Y, Nagasu T, Chait BT (2001) Enrichment analysis of phosphorylated proteins as a tool for probing the phosphoproteome. Nat Biotechnol 19(4):379–382
Oliver CN, Ahn BW, Moerman EJ, Goldstein S, Stadtman ER (1987) Age-related-changes in oxidized proteins. J Biol Chem 262(12):5488–5491
Patton WF (2002) Detection technologies in proteome analysis. J Chromatogr B Analyt Technol Biomed Life Sci 771(1–2):3–31
Posewitz MC, Tempst P (1999) Immobilized gallium(III) affinity chromatography of phosphopeptides. Anal Chem 71(14):2883–2892
Promeyrat A, Gatellier P, Lebret B, Kajak-Siemaszko K, Aubry L, Sante-Lhoutellier V (2010) Evaluation of protein aggregation in cooked meat. Food Chem 121(2):412–417
Promeyrat A, Sayd T, Laville E, Chambon C, Lebret B, Gatellier P (2011) Early post-mortem sarcoplasmic proteome of porcine muscle related to protein oxidation. Food Chem 127(3):1097–1104
Rowe LJ, Maddock KR, Lonergan SM, Huff-Lonergan E (2004) Influence of early postmortem protein oxidation on beef quality. J Anim Sci 82(3):785–793
Rush J, Moritz A, Lee KA, Guo A, Goss VL, Spek EJ, Zhang H, Zha XM, Polakiewicz RD, Comb MJ (2005) Immunoaffinity profiling of tyrosine phosphorylation in cancer cells. Nat Biotechnol 23(1):94–101
Scheffler TL, Gerrard DE (2007) Mechanisms controlling pork quality development: the biochemistry controlling postmortem energy metabolism. Meat Sci 77(1):7–16
Schwagele F, Haschke C, Honikel KO, Krauss G (1996) Enzymological investigations on the causes for the PSE-syndrome, 1. Comparative studies on pyruvate kinase from PSE- and normal pig muscles. Meat Sci 44(1–2):27–40
Shen QW, Du M (2005) Role of AMP-activated protein kinase in the glycolysis of postmortem muscle. J Sci Food Agric 85(14):2401–2406
Silvestre D, Ferrer E, Gaya J, Jareno E, Miranda M, Muriach M, Romero FJ (2006) Available lysine content in human milk: stability during manipulation prior to ingestion. Biofactors 26(1):71–79
Sprang SR, Acharya KR, Goldsmith EJ, Stuart DI, Varvill K, Fletterick RJ, Madsen NB, Johnson LN (1988) Structural-changes in glycogen-phosphorylase induced by phosphorylation. Nature 336(6196):215–221
Stagsted J, Bendixen E, Andersen HJ (2004) Identification of specific oxidatively modified proteins in chicken muscles using a combined immunologic and proteomic approach. J Agric Food Chem 52(12):3967–3974
Steinberg TH, Agnew BJ, Gee KR, Leung WY, Goodman T, Schulenberg B, Hendrickson J, Beechem JM, Haugland RP, Patton WF (2003) Global quantitative phosphoprotein analysis using multiplexed proteomics technology. Proteomics 3(7):1128–1144
Suman SP, Faustman C, Stamer SL, Liebler DC (2007) Proteomics of lipid oxidation-induced oxidation of porcine and bovine oxymyoglobins. Proteomics 7(4):628–640
Thingholm TE, Jorgensen TJD, Jensen ON, Larsen MR (2006) Highly selective enrichment of phosphorylated peptides using titanium dioxide. Nat Protoc 1(4):1929–1935
Walsh CT, Garneau-Tsodikova S, Gatto GJ (2005) Protein posttranslational modifications: the chemistry of proteome diversifications. Angew Chem Int Edit 44(45):7342–7372
Wells L, Vosseller K, Cole RN, Cronshaw JM, Matunis MJ, Hart GW (2002) Mapping sites of O-GlcNAc modification using affinity tags for serine and threonine post-translational modifications. Mol Cell Proteomics 1(10):791–804
Witze ES, Old WM, Resing KA, Ahn NG (2007) Mapping protein post-translational modifications with mass spectrometry. Nat Methods 4(10):798–806
Wu CC, MacCoss MJ, Howell KE, Yates JR (2003) A method for the comprehensive proteomic analysis of membrane proteins. Nat Biotechnol 21(5):532–538
Yang ZP, Hancock WS (2004) Approach to the comprehensive analysis of glycoproteins isolated from human serum using a multi-lectin affinity column. J Chromatogr A 1053(1–2):79–88
Yates JR, Ruse CI, Nakorchevsky A (2009) Proteomics by mass spectrometry: approaches, advances, and applications. Annu Rev Biomed Eng 11:49–79
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer Science+Business Media New York
About this chapter
Cite this chapter
Huang, H., Lametsch, R. (2013). Application of Proteomics for Analysis of Protein Modifications in Postmortem Meat. In: Toldrá, F., Nollet, L. (eds) Proteomics in Foods. Food Microbiology and Food Safety, vol 2. Springer, Boston, MA. https://doi.org/10.1007/978-1-4614-5626-1_7
Download citation
DOI: https://doi.org/10.1007/978-1-4614-5626-1_7
Published:
Publisher Name: Springer, Boston, MA
Print ISBN: 978-1-4614-5625-4
Online ISBN: 978-1-4614-5626-1
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)