
Abstract
This text provides a brief overview of specific issues in the meat industry which may be resolved using the current proteomics-based methodology. Proteins as key to understanding biological processes as well as to change the characteristics of meat products offer enormous potential in the context of meat science. The quantification, analysis and curation of proteomics data, and particularly, the industrial applications of these new findings will become a more essential tool in the coming years.
The methodology described in this chapter presents the basis for quantification, analysis, and curation of proteomics data, representing the pillars of main proteomic methods. The main steps include 1. Extraction and quantification of proteins; 2. Separation of proteins by isoelectric point; 3. Separation of proteins by molecular weight; 4. Staining of proteins; 5. Tryptic digestion with enzymes; and 6. identification of proteins by mass spectrometry.
More recent proteomics approaches are beyond the scope of this chapter. Therefore, all steps are described in detail so that the conditions proposed by us can be reproduced by other researcher groups. Although protocols and methodology are specifically obtained from our lab, these can be easily adapted for every laboratory.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Key words
1 Introduction
The protein content of meat products has a critical influence on nutritional quality, sensory and textural properties. Important quality traits of fresh meat, like tenderness, color, and water holding capacity are closely linked to their proteins and posttranslational modifications. Indeed, during meat aging, the rate of pH fall is influencing the protein denaturation, myofibrillar structures, and cellular shrinkage causing variations in meat quality [1, 2]. Moreover, protein changes such as protein oxidation has an important role in sensory attributes and other quality traits of the meat products [3], as well as proteolysis caused by enzymes during the ripening of dry-cured meat products [4]. Other processes like protein carbonylation, breakdown, and aggregation also imply solubility and functionality changes that lead to a reduction of digestibility, gelation, emulsification, and water holding capacity [5]. On the other hand, the quality of processed meats is particularly linked to the nutritional value of proteins, composed of essential amino acids and bioactive peptides. Indeed, it has been reported that peptides released by digestion gastric or enzymatic hydrolysis from animal proteins may achieve a high biological activity and bioavailability [6]. Within the meat industry, quality control should be performed using protein biomarkers determining phenotypes and predicting meat quality features. The aim is to identify proteins whose expression or abundance is linked to a phenotypic trait [7], influencing the final meat product quality. Briefly, it has been described an overview of specific issues in the meat industry which may be resolved using the current proteomics-based methodology. For the above reasons, the knowledge of meat proteins and their modifications need to be investigated further for the improvement of meat quality in all these issues.
The growing use of omics technologies including proteomics brings significant benefits to food sciences. In this regard, proteomics as the large-scale analysis of proteins has mainly been developed for gel-based techniques and mass spectrometry in these years. The identification of protein patterns using two-dimensional electrophoresis (2-DE) for protein separation implies a first separation based on their isoelectric point (pI) and a second step separated based on molecular weight (Mr). These two parameters are independent resulting in an effective separation method. The analysis of 2-DE gel is performed by software comparing the images to identify different spots in quantitatively and qualitatively terms. Afterwards, the preferred method to identify the proteins in gels is Peptide Mass Fingerprint (PMF) using matrix-assisted laser/desorption ionization-time of flight (MALDI-TOF) mass spectrometry. For this purpose, it is necessary to carry out enzymatic digestion that breaks down protein with great specificity. This specific fingerprint is used to achieve the identification of the protein searching in databases.
The in-depth knowledge of protocols for each food matrix is a key step towards reliable data of proteins. 2-DE is the most common technique used in food proteomics as it can separate different isoforms or even detecting post-translation modifications of proteins by using commercial dyes. However, variability associated with the technique should be minimized through a rigorous procedure in the laboratory. The main steps (Fig. 1) are: (1) Extraction of proteins and initial quantification by commercial kit; (2) First dimension or separation of proteins by pI; (3) Second dimension or separation of proteins by Mr.; (4) Staining of gels and comparison of gel images using a specific software package; (5) Tryptic digestion with commercial enzymes; and (6) Identification of proteins by mass spectrometry (e.g., MALDI-TOF ).

Main steps to carry out separation and identification of proteins
Particularly in muscle sample tissues, before the proteomic analysis, a protein extraction protocol for 2-DE analysis should be carried out by TCA-acetone precipitation avoiding contaminants (mainly salts and detergents) and increasing concentration of the proteins in solution [8]. The proteins should be solubilized in a denaturing buffer with neutral chaotropic, zwitterionic or neutral detergent and reducing agents. Currently, the first dimension (known as isoelectric focusing) is often performed on an immobilized pH gradient (IPG) submitted to a high voltage where the proteins can move towards the pH produces a zero-net charge on protein. The second-dimension gel is based on the movement of the unfolded protein which contains SDS through a gel pore resulting in a separation by protein size. Inside the gel, the proteins are stained using Coomassie blue, SYPRO, or silver stain producing gel imaging. Thus, the protein spots could be detected by gel image analysis and the quantitative changes measured reflecting the expression of the proteins. After all, mass spectrometry as MALDI-TOF MS is being used to identify the proteins after tryptic digestion.
To sum up, it has been presented an overview of proteomic methodology to identify and quantify proteins in meat products. In the next sections, detailed information about the main steps will be provided. The methodology described in this chapter presents the basis for quantification, analysis, and curation of proteomics data, representing the pillars of main proteomic methods. Thus, protocols and methodology were specifically obtained from our lab, these can be easily adapted for every laboratory.
2 Material and Methods
In the next sections, we will provide a detailed description of the main steps indicated in Fig. 1, developed in our lab.
2.1 Extraction and Measurement of Protein Content in the Solution
After the collection of meat samples, they must be excised in small pieces, lyophilized and maintained at −80 °C to ensure the conservation before protein extraction. Afterwards, meat proteins will be precipitated using the TCA-acetone protocol. The protein content in the solution will be carried out to know exactly the amount of protein necessary to carry out the proteomic experiment. In our lab, we use the commercial CB-X protein assay kit (GBiosciences, USA) according to the instructions of the manufacturer using a microplate reader, but other protein quantification kits would also be valid.
Solutions and buffers: Prepare all solutions using ultrapure water and analytical grade reagents.
-
Acetone (90%).
-
Extraction buffer (10% TCA and 0.07% dithiothreitol (DTT) in acetone): Dissolve 6 g of TCA in 60 mL of acetone and add 4.2 mg of DTT.
-
Lysis buffer (7 M urea, 2 M thiourea, 4% CHAPS, 10 mM DTT and 2% Pharmalytes): Dissolve 10.5 g of urea, 3.8 g of thiourea, 1 g of CHAPS, 38 mg of DTT, and 0.5 mL of Pharmalyte™ pH 3–10 (GE Healthcare, Uppsala, Sweden) in 25 mL and make up to 25 mL with water. Stir for hours to dissolve completely at room temperature.
-
Wash buffer (0.07% DTT in acetone): Dissolve 14 mg of DTT in acetone.
2.1.1 Protocol
-
1.
An amount of 50 mg of lyophilized meat tissue is suspended in 10 mL of extraction buffer (10% TCA and 0.07% DTT in acetone).
-
2.
A sonication step should be considered for cell disruption and homogenization of the solution in refrigerated conditions. The sonication should be in short pulses at 0 °C.
-
3.
The solution is maintained at −20 °C for at least 1 h.
-
4.
The solution is centrifuged at 4500 × g at 4 °C for 30 min and the pellet is recovered.
-
5.
The pellet is rinsed two times with 1 mL of wash buffer (0.07% DTT in acetone) followed by centrifugation as described in step 4.
-
6.
The last washing is using acetone (90%) and centrifugation (step 4).
-
7.
The final pellet is eluted in 1 mL of lysis buffer (7 M urea, 2 M thiourea, 4% CHAPS, 10 mM DTT, and 2% Pharmalytes) which should be stirred to dissolve completely in an Eppendorf Thermomixer.
-
8.
The concentration of protein in the final solution should be determined using the commercial CB-X protein assay kit (GBiosciences, USA) according to the instructions of the manufacturer in a microplate reader. A microplate reader is necessary to look up the absorbance at 595 nm. This method is compatible with detergents and other substances like reducing sugars, thiols, and chelating agents (protein standard is specifically provided to prepare a calibration plot).
2.2 First Dimension or Separation of Proteins by pI
Isoelectric focusing with IEF by using Protean IEF Cell (Bio-Rad Laboratories, Inc., USA) and immobilized pH gradients is employed for the first-dimension separation (Fig. 2). The protein can be loaded on 11-cm, 18-cm and 24-cm Ready Strip IPGs (Bio-Rad Laboratories). In this sense, IPG strips with different linear pH linear or non-linear gradients (3–10 and 4–7) can be chosen according to the experiment.

Protean IEF Cell (Bio-Rad Laboratories)
Solutions and buffers: Prepare all solutions using ultrapure water and analytical grade reagents.
-
Tris solution (50 mM Tris pH 8.8): Dissolve 181.5 g of Tris in 750 mL of ultrapure water, adjust the pH to 8.8, and make up to 1000 mL with water.
-
Equilibration solution (50 mM Tris pH 8.8, 6 M urea, 30% glycerol, and 2% SDS): 6.7 mL of Tris solution, 72.1 g of urea, and 60 mL glycerol are vigorously stirred for 30 min. Finally, 4 g of SDS is added and made up to 200 mL with ultrapure water. In the case of equilibration solution with 1% of DTT, dissolve 60 mg of DDT in 6 mL of equilibration solution for each strip, and in case of 2.5% of iodoacetamide, dissolve 150 mg of iodoacetamide in each 6 mL of equilibration solution for each strip.
2.2.1 Protocol
-
1.
According to the previous protein quantification, a volume containing 250 μg of total protein is dissolved in lysis buffer up to the total volume recommended by the manufacturer for each strip length (Table 1).
-
2.
The rehydration solution is completed by the addition of 0.6% DTT and 1% IPG buffer (Bio-Rad Laboratories). A complete dissolution takes place after a vigorous agitation.
-
3.
This sample should be distributed along a lane of the focusing tray from one electrode to another. Carefully place the IPG strips contacting the gel side with the sample. Finally, cover with mineral oil along the lane to avoid evaporation during the first electrophoresis according to Table 1. Place the plastic lid onto the focusing tray and introduce it into the Protean IEF Cell (Bio-Rad Laboratories, Inc., USA).
-
4.
The focusing conditions and ramping protocols have been optimized for this quantity of meat samples. A previous step of active rehydration with a voltage of 50 V during 12 h should be performed and the settings for IEF are shown in Table 2. The final voltage depends on the length and pH of each IPG strip, therefore, necessary to consult the manual.
-
5.
The equilibration of the strips is necessary to run the second dimension ensuring that the cysteines are reduced and alkylated. The strips should be placed in the equilibration tray for better treatment. The focused strips should be incubated firstly in equilibration solution (50 mM Tris pH 8.8, 6 M urea, 30% glycerol, and 2% SDS) with 1% DTT for 15 min and secondly in equilibration solution (50 mM Tris pH 8.8, 6 M urea, 30% glycerol, and 2% SDS) with 2.5% iodoacetamide for 15 min.
2.3 Second Dimension or Separation of Proteins by Mr.
For SDS-polyacrylamide gel electrophoresis (SDS-PAGE ), the protein unfolded and charged by anionic detergent SDS is carried out in an Ettan Dalt six gel system (GE Healthcare, Uppsala, Sweden) (Fig. 3). The gel pore size could be differently adjusted by percentages of polyacrylamide and cross-linker. In this regard, the more common percentage ranged from 10% to 15% w/v polyacrylamide gels.

Ettan Dalt six gel system (GE Healthcare, Uppsala, Sweden)
Solutions and buffers: Prepare all solutions using ultrapure water and analytical grade reagents.
-
Acrylamide solution: Dissolve 7.8 g of bis-acrylamide and 292.2 g of acrylamide in ultrapure water to reach a total volume of 1 L in dark conditions.
-
Polymerization solution: The polyacrylamide gels consist of the polymerization from acrylamide and bis-acrylamide initiated by TEMED and PSA to free radicals. Thus, the mixture for different percentages of acrylamide should contain the amounts shown in Table 3. This solution should be prepared with constant agitation and without interruptions. Finally, the solution is poured into the Gel Caster and covered with water-saturated isobutanol. The polymerization occurs for approximately 6 h at room temperature.
-
10× Electrophoresis buffer (50 mM Tris base, 384 mM glycine and 0.2% SDS): Dissolve 30.2 g of Tris, 144.2 g of glycine, and 10 g of SDS in distilled water reaching a total volume of 1 L. Thus, 1× electrophoresis buffer is obtained by adding 1 part of this solution to 9 parts of the distilled water and 2× electrophoresis buffer by adding 2 parts of this to 8 parts of the distilled water.
-
Agarose solution: Heat to boiling 0.5 g of agarose in 100 mL of 1× electrophoresis buffer for its dissolution and add traces of bromophenol blue.
2.3.1 Protocol
-
1.
The IPG strips should be transferred to gels polymerized according to the previous section. It should be embedded the strip on the top of the second dimension and sealing the place, with molten agarose solution and bromophenol blue to ensure good contact between the gel and the IPG strip and to track the electrophoresis with the dye.
-
2.
The electrophoresis unit (Ettan Dalt six gel system, GE Healthcare, Uppsala, Sweden) is assembled according to the manual.
-
3.
The upper chamber is filled with 1 L of 2× electrophoresis buffer meanwhile the lower chamber needs 4.5 L of 1× electrophoresis buffer.
-
4.
Electrophoresis runs applying 5 mA/gel, 2 h; 10 mA/gel for 3 h and finally 16 mA/gel until the color blue reaches the gel bottom indicating the end of the electrophoresis.
-
5.
After the electrophoresis, the gels are placed in plastic boxes to proceed to the staining.
2.4 Staining of Gels and Comparison of Gel Images Using a Specific Software Package
Gel staining protocols should be chosen depending on the specific needs. Protein staining should be mainly carried out using Coomassie blue, silver staining or Sypro Ruby® stain (Bio-Rad Laboratories). In the case of silver staining, it is a very excellent sensitive method as well as very simple and cheap. The method is based on the selective reduction of silver in the proximity of protein molecules [9]. However, it is less used due to its incompatibility with the mass spectrometry necessary for protein identification. In this sense, coomassie blue has been widely employed to stain the proteins in-gel for many years. The main benefit of this stain is economical, and the equipment used is a simple densitometer, but the protein detection is poor about 30 ng. Hence, this methodology is being implemented to improve these aspects [10]. On the other hand, fluorescent staining is often preferred by its high sensitivity and dynamic range. Sypro Ruby® stain (Bio-Rad Laboratories) is the most widely used achieving a protein detection of 2 ng, but it is necessary the use of GelDoc XR System (Bio-Rad Laboratories) (Fig. 4) or similar at least.

GelDoc XR System (Bio-Rad Laboratories)
2.4.1 SYPRO® Ruby Protein Stain According to the Instruction Manual
Gels are stained with SYPRO Ruby® stain (Bio-Rad Laboratories), following the manufacturer indications, and digitalized using the GelDoc XR System (Bio-Rad Laboratories). The volume of solutions will be determined by box-sized considering that the gel must be immersed in the solution. Furthermore, a rocker is necessary for gentle mixing of the gel in the solutions.
2.4.1.1 Protocol
-
1.
Gel is rinsed in a fixed solution (ethanol 40% and acid acetic 10%) for 3 h.
-
2.
Gel is washed with distilled water for 30 min three times.
-
3.
In dark conditions, the gel is immersed in fluorescent stain overnight.
-
4.
Finally, the gel is rinsed twice with distilled water in darkness for 30 min.
-
5.
The gel is digitalized in GelDoc XR System (Bio-Rad Laboratories).
Afterwards, the 2-DE images can be analyzed using PDQuest 2-D Analysis Software (Bio-Rad Laboratories).
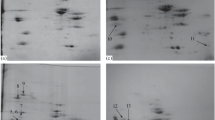
In Fig. 5 two examples of polyacrylamide gels (12%), employing strips of 24 cm in the pH range 4–7 stained with SYPRO Ruby and obtained from our lab are displayed. The pI and Mr. of spots are determined from their position on the IEF-strips and standard molecular mass markers ranging from 15 to 200 kDa (Fermentas, Ontario) could be added, respectively.

Representative 2-DE gel protein profiles of fresh meat from Longissimus thoracis of Bos taurus (a) and dry-cured ham of Sus scrofa (b)
2.5 Tryptic Digestion of Gel Spots
In a comparative proteomic analysis, the protein spots are chosen to be identified by mass spectrometry, thus, the protein spots are manually excised from the gel using a pipette tip or similar.
Solution:
-
Ambic buffer: 50 mM ammonium bicarbonate in 50% methanol.
2.5.1 Protocol
-
1.
The excised spots are cut into pieces and washed with Milli-Q water.
-
2.
The pieces are twice rinsed with Ambic buffer for 20 min after centrifugation (600 rpm), the supernatant is discarded.
-
3.
The gel pieces are dehydrated with 100 μL acetonitrile for 10 min and subsequently dried in a vacuum centrifuge.
-
4.
Following a reduction step with 10 mM DTT in Ambic buffer for 30 min at 56 °C after centrifugation (600 rpm), the supernatant is discarded.
-
5.
Repeat step 2.
-
6.
The alkylation step is produced by 55 mM iodoacetamide in Ambic buffer freshly prepared for 20 min in darkness.
-
7.
Repeat step 2.
-
8.
Modified porcine trypsin (Promega) is added to the dry gel pieces at a final concentration of 20 ng/μL in Ambic buffer incubating it at 37 °C for 16 h. Afterwards, centrifugation (600 rpm) is used to discard the supernatant.
-
9.
Peptides are extracted thrice by 20 min incubation in 40 μL of 60% acetonitrile in 0.5% HCOOH under constant agitation. The supernatant is dried and stored at −20 °C for further analysis.
In the next and final main step, a MALDI-TOF MS is used to identify the proteins.
2.6 Identification of Proteins by Mass Spectrometry
Matrix solution is prepared using 0.5 μL of peptides and 3 mg CHCA dissolved in 1 mL of 50% acetonitrile in 0.1% TFA. The final solution is placed on the Opti-TOF MALDI plate (Applied Biosystems). The mass spectrometry was carried out using a 4800 MALDI-TOF /TOF analyzer (Applied Biosystems) by reflectron positive-ion mode with a Nd:YAG, 355 nm wavelength laser, averaging 1000 laser shots and at least three trypsin autolysis peaks used as internal calibration. For tandem mass spectrometry (MS /MS ), the data are performed by resolution of 300 (FWHM) and metastable suppression. GPS Explorer Software v3.6 using Mascot software v2.1. (Matrix Science) identify the proteins by peptide mass fingerprinting (PMF) and peptide fragmentation spectra data in a protein database. The parameters most used are 30 ppm precursor tolerance, 0.35 Da MSMS fragment tolerance, CAM (carbamidomethyl cysteine) as fixed modification, oxidized methionine as variable modification and allowing 1 missed cleavage. Finally, only the protein identifications are considered in the case of the score are greater than 56 and when protein score CI (Confidence Interval) was above 98%. For MS /MS , the total ion score CI should achieve above 95%.
Abbreviations
- 2-DE :
-
Two-dimensional electrophoresis
- CHAPS detergent:
-
(3-((3-cholamidopropyl) dimethylammonium)-1-propanesulfonate)
- DTT:
-
Dithiothreitol
- IPG:
-
Immobilized pH gradient
- MALDI-TOF :
-
Matrix-assisted laser/desorption ionization-time of flight
- Mr.:
-
Molecular weight
- pI:
-
Isoelectric point
- PMF:
-
Peptide Mass Fingerprint
- PSA:
-
Persulfate ammonium
- SDS:
-
Sodium dodecyl sulfate
- SDS-PAGE :
-
Sodium dodecyl sulfate polyacrylamide gel electrophoresis
- TCA:
-
Trichloroacetic
- TEMED:
-
Tetramethylethylenediamine
- Tris:
-
Tris(hydroxymethyl)aminomethane
References
Li X, Zhang D, Ren C et al (2021) Effects of protein posttranslational modifications on meat quality: a review. Compr Rev Food Sci Food Saf 20:289–331
Hughes JM, Oiseth SK, Purslow PP et al (2014) A structural approach to understanding the interactions between colour, water-holding capacity and tenderness. Meat Sci 98:520–532
Estévez M (2011) Protein carbonyls in meat systems: a review. Meat Sci 89:259–279
López-Pedrouso M, Pérez-Santaescolástica C, Franco D et al (2019) Molecular insight into taste and aroma of sliced dry-cured ham induced by protein degradation undergone high-pressure conditions. Food Res Int 122:635–642
Berardo A, Claeys E, Vossen E et al (2015) Protein oxidation affects proteolysis in a meat model system. Meat Sci 106:78–84
Xing L, Liu R, Cao S et al (2019) Meat protein based bioactive peptides and their potential functional activity: a review. Int J Food Sci Technol 54:1956–1966
Picard B, Lefèvre F, Lebret B (2012) Meat and fish flesh quality improvement with proteomic applications. Anim Front 2:18–25
Franco D, Mato A, Salgado FJ et al (2015) Tackling proteome changes in the longissimus thoracis bovine muscle in response to pre-slaughter stress. J Proteome 122:73–85
Kumar G (2012) Principle and method of silver staining of proteins separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis. In: Kurien BT, Scofield RH (eds) Protein gel detection and imaging. Humana Press, pp 451–464
Gauci VJ, Padula MP, Coorssen JR (2013) Coomassie blue staining for high sensitivity gel-based proteomics. J Proteome 90:96–106
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2022 The Author(s), under exclusive license to Springer Science+Business Media, LLC, part of Springer Nature
About this chapter

Cite this chapter
López-Pedrouso, M., Lorenzo, J.M., Franco, D. (2022). Proteomics. In: Lorenzo, J.M., Domínguez, R., Pateiro, M., Munekata, P.E. (eds) Methods to Assess the Quality of Meat Products. Methods and Protocols in Food Science . Humana, New York, NY. https://doi.org/10.1007/978-1-0716-2002-1_13
Download citation
DOI: https://doi.org/10.1007/978-1-0716-2002-1_13
Published:
Publisher Name: Humana, New York, NY
Print ISBN: 978-1-0716-2001-4
Online ISBN: 978-1-0716-2002-1
eBook Packages: Springer Protocols