
Abstract
Parathyroid hormone (PTH) causes bone loss when produced in a continuous fashion. However, PTH induces a potent bone anabolic effect when injected intermittently. The mechanism of action of PTH remains largely unknown. This article reviews the evidence in favor of the hypothesis that T cells play an unexpected critical role in the mechanism of action of PTH in bone.
Access provided by Autonomous University of Puebla. Download conference paper PDF
Similar content being viewed by others


Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Introduction
Parathyroid hormone (PTH) causes bone loss when produced in a continuous fashion. However, PTH induces a potent bone anabolic effect when injected intermittently. The mechanism of action of PTH remains largely unknown. This article reviews the evidence in favor of the hypothesis that T cells play an unexpected critical role in the mechanism of action of PTH in bone.
2 Effects of PTH on Bone
Primary hyperparathyroidism is a common bone disease caused by continuous overproduction of PTH. This disorder causes cortical bone loss [1] and leads to a loss or gain of trabecular bone, depending on its severity, duration, and age of the patient [1–3]. Primary hyperparathyroidism is modeled by continuous PTH (cPTH) infusion, which, like hyperparathyroidism, stimulates bone resorption and causes cortical bone loss [4–6]. cPTH treatment may lead to modest gain or loss of cancellous bone depending on the age of the mouse and the dose and duration of the cPTH treatment [4–6]. By contrast, intermittent PTH (iPTH) treatment markedly increases bone volume and strength in the cortical and trabecular compartments.
Both cPTH and iPTH increase bone turnover [6–9]. The stimulation of bone formation induced by iPTH far exceeds bone resorption, leading to a net bone anabolic effect in the cortical and trabecular compartments [10]. By contrast, the stimulation of bone formation induced by cPTH is not sufficient to offset the increase in resorption, leading to a net cortical bone loss, which, in some conditions, is associated to trabecular bone loss [10]. PTH stimulates bone resorption by enhancing the production of RANKL, and M-CSF, and decreasing the production of OPG by stromal cells (SCs) and osteoblasts (OBs) [11, 12]. Recently, we have shown that another critical mechanism is the capacity of PTH to increase the production of TNF by T cells [5]. Enhanced bone resorption is accompanied by a stimulation of bone formation driven by an increase in the number of OBs [13–15] achieved through activation of quiescent lining cells [16], increased OB proliferation [17 18] and differentiation [17, 19, 20], attenuation of OB apoptosis [21–24], and signaling in osteocytes [25]. However, the specific contribution of each of these effects of PTH remains controversial. The expansion of the osteoblastic pool induced by PTH is initiated by the release from the matrix undergoing resorption of TGFβ, IGF-1 and other growth factors that recruit SCs to remodeling areas [26–29]. Subsequent events are driven primarily by the activation of Wnt signaling in osteoblastic cells [30]. Activation of Wnt signaling induces OB proliferation [31] and differentiation [30, 32], prevents OB apoptosis [23, 24, 33], and augments OB production of OPG [34]. Wnt proteins initiate a canonical signaling cascade by binding to receptors of the Frizzled family together with the coreceptors LRP4-5-6, which results in the stabilization of cytosolic β-catenin. A nuclear complex of beta-catenin and the T cell factor/lymphoid enhancer factor (TCF/LEF) family of transcription factors then interacts with DNA to regulate the transcription of Wnt target genes [35]. Wnt proteins also signal through noncanonical pathways which involve the Src/ERK and Pi3K/Akt cascades [23].
PTH is a canonical Wnt signaling agonist which increases β catenin levels in osteoblastic cells [36, 37], an effect which occurs through modulation of both the protein kinase A and protein kinase C pathways [36]. PTH, once bound to PPR, is also capable of forming a complex with LRP6 which results in LRP6 signaling and β catenin activation [38]. Thus, PTH activates Wnt signaling in osteoblastic cells through both Wnt ligands-dependent and Wnt ligands-independent mechanisms. Moreover, PTH down regulates the production of sclerostin, an osteocyte-derived Wnt antagonist which blocks Wnt signaling by binding to LRP5 and LRP6 [39, 40]. Recently, convincing evidence has emerged that PTH receptor signaling in osteocytes and the resulting direct regulation of sclerostin production play a particularly relevant role in the anabolic activity of PTH (O’Brien, 2008 #11387). PTH also regulates Dickkopf-1, a soluble LRP5 and LRP6 signaling inhibitor [37], and Sfrp-4, a factor which binds Wnt proteins thus antagonizing both canonical and noncanonical Wnt signaling [41]. Uncertainty remains with regard to the identity and the source of Wnt ligands which activate Wnt signaling in response to PTH treatment are not completely understood.
3 Role of T Cells in the Anabolic Activity of Intermittent PTH Treatment
T cells express functional the PTH receptor PPR [42, 43] and respond to PTH. This prompted us to investigate whether T cells contribute to the anabolic response to iPTH. Studies were conducted in four strains of T cell-deficient mice (TCRβ−/−, RAG2−/−, class I and II MHC double KO mice, and nude mice). Analysis by DEXA and μCT revealed that in mice lacking T cells, iPTH induced a ∼ 50 % smaller increase in bone density and bone volume as compared to T cell-replete controls [43]. Furthermore, adoptive transfer of T cells into T cell-deficient mice restored a normal response to iPTH. T cells were found to augment the capacity of iPTH to improve architecture in trabecular but not in cortical bone. Although the reason of this selectivity is unknown, a lack of access of T cells to cortical surfaces is not a likely explanation, as T cells reach endosteal and periosteal bone surface through blood vessels and recirculate in and out of the BM. In addition, direct measurements of bone strength by 4-point bending revealed that the capacity of iPTH to improve bone strength was abolished in T cell-deficient mice. Although the reason for this discrepancy is unknown, it is possible that T cells might be required to improve the material property of bone.
With regard of the mechanism by which T cells potentiate the bone anabolic activity of iPTH, studies have disclosed that in the absence of T cells iPTH is unable to increase the commitment of SCs to the osteoblastic lineage, induce OB proliferation and differentiation, and mitigate OB apoptosis. All of these actions of PTH were found to hinge on the capacity of T cells to activate Wnt signaling in osteoblastic cells [43]. Although it is well established that Wnt activation is a key mechanism by which iPTH expands the osteoblastic pool, little information is available on the nature and the source of the Wnt ligand required to activate Wnt signaling in OBs. We have found that PTH stimulates BM CD8+ T cells to produce large amounts of Wnt10b [43], a Wnt protein which activates Wnt signaling in SCs and OBs, thus increasing OB proliferation, differentiation, and life span Treatment with iPTH also caused a small increase in the production of Wnt10b by BM CD4+ cells which was associated with a slightly diminished anabolic response in class II MHC−/− mice, suggesting that production of Wnt10b by CD4+ cells contributes, in small part, to the anabolic activity of iPTH. The relevance of CD8+ cells was demonstrated by the inability of iPTH to promote bone anabolism in class I MHC−/− mice, a strain that lacks CD8+ cells [43]. Additional studies revealed that iPTH does not improve bone architecture in T cell-deficient mice reconstituted with CD4+ cells, while it does so in mice adoptively transferred with CD8+ cells [43]. The pivotal role of T cell-produced Wnt10b was revealed by the hampered effect of iPTH on bone volume in TCRβ−/− mice reconstituted with T cells from Wnt10b−/− mice. It is likely that iPTH directly targets CD8+ T cells and stimulates their production of Wnt10b as in vitro PTH treatment potently stimulates Wnt10b production by T cells.
While in vitro PTH treatment increased Wnt10b production by all T cells, iPTH upregulated Wnt10b production only by BM T cells. This diversity might be explained by the different dose and time of exposure to PTH. However, since adoptive transfer of spleen T cells into TCRβ−/− mice was followed by a restoration of a full responsiveness to iPTH, the data suggest that the capacity of T cells to upregulate their production of Wnt10b in response to iPTH is not an intrinsic feature of T cells, but rather is induced by environmental cues.
Together the data indicate that CD8+ T cells potentiate the anabolic activity of PTH by providing Wnt10b, which is a critical Wnt ligand required for activating Wnt signaling in osteoblastic cells Therefore in the absence of CD8+ cells, stimulation of osteoblastic cells by PTH is not sufficient to elicit maximal Wnt activation due to the lack of a critical Wnt ligand (Fig. 1). The residual bone anabolic activity of PTH observed in T cell-deficient mice is presumably due to ligand-independent activation of LRP6 [38], and suppressed production of sclerostin [39, 40, 44].

Schematic representation of the role of T cells in the mechanism by which intermittent PTH treatment stimulates bone formation. PTH stimulates T cells to secrete Wnt10b, a Wnt ligand required to activate Wnt signaling in SCs and OBs. In the presence of T cell-produced Wnt10b, stimulation of osteoblastic cells by PTH result in the activation of the Wnt signaling pathway. This event leads to increased commitment of mesenchimal stem cells to the osteoblastic lineage, increased osteoblast proliferation and differentiation, and decreased osteoblast apoptosis
The anabolic activity of iPTH is not identical in all strains of T cell-deficient mice. In fact, while some strains had no increase in bone volume in response to PTH, other exhibited a blunted but not a completely absent response. Osteoblastic cells produce several bone anabolic Wnt ligands including Wnt10b, Wnt7a and Wnt3b [45, 46]. These factors are likely to contribute to the T cell-independent anabolic activity of iPTH, and quantitative differences in their production may account, in part, to the strain-dependent variability in the response to iPTH observed herein. Furthermore, the magnitude of the anabolic response to iPTH in T cell null mice may be related to a strain- and age-dependent capacity of iPTH to inhibit the bone cells production of Wnt inhibitors such as sclerostin, [39, 40], Dickkopf-1 [37] and Sfrp-4 [41]. These factors have been shown to contribute to the anabolic activity of iPTH through T cell-independent mechanisms. Since B cells are regulated by PTH [47], the response of RAG2−/− mice to iPTH might also have been determined by the lack of B cells which is a feature of RAG2−/− mice.
The enhancement of bone formation induced by iPTH is accompanied by a stimulation of bone resorption which is driven by increased production of RANKL and decreased release of OPG in the bone microenvironment [48]. The direct effects of PTH on RANKL/OPG production are mitigated, in part, by the iPTH-induced activation of β catenin in OBs, as this transcriptional regulator stimulates their production of OPG [49] and represses that of RANKL [50]. The latter is one of the mechanisms that prevent bone resorption from offsetting the anabolic activity of iPTH.
Osteoblastic cells from WT mice treated with iPTH in vivo exhibited increased commitment to the osteoblastic lineage, proliferation, differentiation, and life span in vitro, as compared to the corresponding cells from T cell-deficient mice. Thus, T cells, like PTH, affect all aspects of OB life cycle. Remarkably, these differences were demonstrated in OBs purified from BM cultured for 7 days without the addition of PTH, suggesting that in vivo the hormone regulates early commitment steps of SCs and their osteoblastic progeny through T cell-produced Wnt10b, and that these steps are not reversed by the absence of PTH and T cells in vitro. This model is consistent with the capacity of Wnt signaling to guide cell fate determination [51]. A similar paradigm has been described in ovariectomized mice, a model where estrogen withdrawal in vivo leads to the formation of SCs which exhibit an increased osteoclastogenic activity which persists in vitro for 4 weeks [52].
4 T Cells and PTH-Induced Bone Loss
Studies designed to investigate the role of T cells in the bone catabolic activity of cPTH revealed that an infusion of cPTH that mimics hyperparathyroidism fails to induce OC formation, bone resorption, and cortical bone loss in mice lacking T cells [35]. A second study conducted in older mice, a model in which cPTH causes cortical and cancellous bone loss, revealed that T cell-deficient mice are also protected against trabecular bone loss [5].
An important finding of these studies was that cPTH equally stimulated bone formation in T cell-replete and T cell-deficient mice [35] [5]. It should be noted that while the stimulation of bone formation induced by cPTH was completely T cell independent, the lack of T cells blocked the increase in bone formation induced by iPTH. The reason for this critical difference remains to be determined.
These studies further revealed the existence of a cross-talk between T cells and SCs mediated by the CD40L/CD40 signaling system. T cells provide proliferative and survival cues to SCs and sensitize SCs to PTH through CD40L, a surface molecule of activated T cells that induces CD40 signaling in SCs. An important element of this regulatory loop is the capacity of PTH to upregulate the expression of CD40 in SC from T cell-replete mice but not from T cell-deficient mice [35]. Thus T cells contribute to the CD40L/CD40-mediated exchange of information between T cells and SCs in two ways: first, by providing CD40L and secondly, by upregulating the expression of CD40 on SCs. As a result, mice lacking T cells or T cell-expressed CD40L have lower number of SCs. Furthermore, these SCs produce lower amount of RANKL and have an even smaller suppression of OPG secretion in response to PTH. Therefore, SCs from T cell-deficient mice have a lower capacity to support OC formation in vivo and in vitro. The alteration in SC function is the ultimate reason why deletion of T cells or T cell-expressed CD40L blunts the bone catabolic activity of PTH [35].
Studies have also shown that cPTH increases the T cell production of TNF. This cytokine not only increases OC formation directly, but also TNF upregulates the expression of CD40 in SCs, thus increasing their response to T cell expressed CD40L. Attesting to the relevance TNF, cPTH fails to induce bone loss and stimulate bone resorption in mice lacking T cell TNF production [5].
To determine whether PTH targets T cells directly, we have conditionally silenced the PTH receptor PPR in T cells. We found that removal of PPR signaling in T cells blunts the stimulation of bone resorption induced by cPTH without affecting bone formation. As a result, silencing of PPR signaling in T cells prevents the loss of cortical bone induced by cPTH. Strikingly, the disruption of PPR signaling in T cells converts the effects of cPTH in trabecular bone from catabolic to anabolic [5].
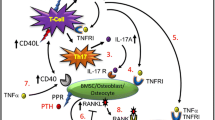
Collectively, our data reveal that the effects of cPTH on bone are the result of a mechanism that involves PPR activation and TNF production in T cells (Fig. 2). T cell-produced TNF stimulates bone resorption directly by potentiating the sensitivity of maturing OCs to RANKL. In addition, TNF enhances CD40L/CD40 signaling from T cells to SCs by upregulating CD40 expression, an effect resulting in the increased capacity of SCs to support OC formation. Thus, a complex cross-talk between T cells and the osteoclastogenic machinery of the BM is central for the bone catabolic activity of cPTH.

Schematic representation of the role of T cells in the mechanism by which continuous PTH stimulates OC formation. Continuous PPR signaling in T cells induced by continuous PTH (cPTH) treatment stimulates the production of TNF. This cytokine increases CD40 expression by SCs. Binding to CD40 of T cell expressed CD40L increases SC sensitivity to PTH resulting in enhanced SC production of RANKL and diminished secretion of OPG in response to PTH. T cell-produced TNF further stimulates OC formation through its direct effects on maturing OC precursors. The red arrows represent the main modifications induced by activation of PPR signaling in T cells
References
Kong YY, Feige U, Sarosi I, Bolon B, Tafuri A, Morony S, Capparelli C, Li J, Elliott R, McCabe S et al (1999) Activated T cells regulate bone loss and joint destruction in adjuvant arthritis through osteoprotegerin ligand. Nature 402:304–309
Di Rosa F, Santoni A (2002) Bone marrow CD8 T cells are in a different activation state than those in lymphoid periphery. Eur J Immunol 32:1873–1880
Westermann J, Pabst R (1992) Distribution of lymphocyte subsets and natural killer cells in the human body. Clin Investig 70:539–544
Kotake S, Udagawa N, Takahashi N, Matsuzaki K, Itoh K, Ishiyama S, Saito S, Inoue K, Kamatani N, Gillespie MT et al (1999) IL-17 in synovial fluids from patients with rheumatoid arthritis is a potent stimulator of osteoclastogenesis. J Clin Invest 103:1345–1352
Sipkins DA, Wei X, Wu JW, Runnels JM, Cote D, Means TK, Luster AD, Scadden DT, Lin CP (2005) In vivo imaging of specialized bone marrow endothelial microdomains for tumour engraftment. Nature 435:969–973
Becker TC, Coley SM, Wherry EJ, Ahmed R (2005) Bone marrow is a preferred site for homeostatic proliferation of memory CD8 T cells. J Immunol 174:1269–1273
Wilson A, Trumpp A (2006) Bone-marrow haematopoietic-stem-cell niches. Nat Rev Immunol 6:93–106
Tokoyoda K, Zehentmeier S, Chang HD, Radbruch A (2009) Organization and maintenance of immunological memory by stroma niches. Eur J Immunol 39:2095–2099
Uccelli A, Moretta L, Pistoia V (2008) Mesenchymal stem cells in health and disease. Nat Rev Immunol 8:726–736
Mak TW, Ferrick DA (1998) The gammadelta T-cell bridge: linking innate and acquired immunity. Nat Med 4:764–765
Nauta AJ, Fibbe WE (2007) Immunomodulatory properties of mesenchymal stromal cells. Blood 110:3499–3506
Ren G, Zhang L, Zhao X, Xu G, Zhang Y, Roberts AI, Zhao RC, Shi Y (2008) Mesenchymal stem cell-mediated immunosuppression occurs via concerted action of chemokines and nitric oxide. Cell Stem Cell 2:141–150
Le Blanc K, Frassoni F, Ball L, Locatelli F, Roelofs H, Lewis I, Lanino E, Sundberg B, Bernardo ME, Remberger M et al (2008) Mesenchymal stem cells for treatment of steroid-resistant, severe, acute graft-versus-host disease: a phase II study. Lancet 371:1579–1586
Huang W, La Russa V, Alzoubi A, Schwarzenberger P (2006) Interleukin-17A: a T-cell-derived growth factor for murine and human mesenchymal stem cells. Stem Cells 24:1512–1518
Yamaza T, Miura Y, Bi Y, Liu Y, Akiyama K, Sonoyama W, Patel V, Gutkind S, Young M, Gronthos S et al (2008) Pharmacologic stem cell based intervention as a new approach to osteoporosis treatment in rodents. PLoS One 3:e2615
Grewal IS, Flavell RA (1998) CD40 and CD154 in cell-mediated immunity. Annu Rev Immunol 16:111–135
Quezada SA, Jarvinen LZ, Lind EF, Noelle RJ (2004) CD40/CD154 interactions at the interface of tolerance and immunity. Annu Rev Immunol 22:307–328
Andre P, Prasad KS, Denis CV, He M, Papalia JM, Hynes RO, Phillips DR, Wagner DD (2002) CD40L stabilizes arterial thrombi by a beta3 integrin–dependent mechanism. Nat Med 8:247–252
Prasad KS, Andre P, He M, Bao M, Manganello J, Phillips DR (2003) Soluble CD40 ligand induces beta3 integrin tyrosine phosphorylation and triggers platelet activation by outside-in signaling. Proc Natl Acad Sci USA 100:12367–12371
Zirlik A, Maier C, Gerdes N, MacFarlane L, Soosairajah J, Bavendiek U, Ahrens I, Ernst S, Bassler N, Missiou A et al (2007) CD40 ligand mediates inflammation independently of CD40 by interaction with Mac-1. Circulation 115:1571–1580
Leveille C, Bouillon M, Guo W, Bolduc J, Sharif-Askari E, El-Fakhry Y, Reyes-Moreno C, Lapointe R, Merhi Y, Wilkins JA et al (2007) CD40 ligand binds to alpha5beta1 integrin and triggers cell signaling. J Biol Chem 282:5143–5151
Grammer AC, Lipsky PE (2000) CD40-mediated regulation of immune responses by TRAF-dependent and TRAF-independent signaling mechanisms. Adv Immunol 76:61–178
Saeland S, Duvert V, Caux C, Pandrau D, Favre C, Valle A, Durand I, Charbord P, de Vries J, Banchereau J (1992) Distribution of surface-membrane molecules on bone marrow and cord blood CD34+ hematopoietic cells. Exp Hematol 20:24–33
Ahuja SS, Zhao S, Bellido T, Plotkin LI, Jimenez F, Bonewald LF (2003) CD40 ligand blocks apoptosis induced by tumor necrosis factor alpha, glucocorticoids, and etoposide in osteoblasts and the osteocyte-like cell line murine long bone osteocyte-Y4. Endocrinology 144:1761–1769
Sato K, Suematsu A, Okamoto K, Yamaguchi A, Morishita Y, Kadono Y, Tanaka S, Kodama T, Akira S, Iwakura Y et al (2006) Th17 functions as an osteoclastogenic helper T cell subset that links T cell activation and bone destruction. J Exp Med 203:2673–2682
Grewal IS, Flavell RA (1996) The role of CD40 ligand in costimulation and T-cell activation. Immunol Rev 153:85–106
Solanilla A, Dechanet J, El Andaloussi A, Dupouy M, Godard F, Chabrol J, Charbord P, Reiffers J, Nurden AT, Weksler B et al (2000) CD40-ligand stimulates myelopoiesis by regulating flt3-ligand and thrombopoietin production in bone marrow stromal cells. Blood 95:3758–3764
Funakoshi S, Taub DD, Anver MR, Raziuddin A, Asai O, Reddy V, Rager H, Fanslow WC, Longo DL, Murphy WJ (1997) Immunologic and hematopoietic effects of CD40 stimulation after syngeneic bone marrow transplantation in mice. J Clin Invest 99:484–491
Teitelbaum SL (2000) Osteoclasts, integrins, and osteoporosis. J Bone Miner Metab 18:344–349
Haas W, Pereira P, Tonegawa S (1993) Gamma/delta cells. Annu Rev Immunol 11:637–685
Yokota T, Oritani K, Mitsui H, Aoyama K, Ishikawa J, Sugahara H, Matsumura I, Tsai S, Tomiyama Y, Kanakura Y et al (1998) Growth-supporting activities of fibronectin on hematopoietic stem/progenitor cells in vitro and in vivo: structural requirement for fibronectin activities of CS1 and cell-binding domains. Blood 91:3263–3272
Cantor JM, Ginsberg MH, Rose DM (2008) Integrin-associated proteins as potential therapeutic targets. Immunol Rev 223:236–251
Li Y, Toraldo G, Li A, Yang X, Zhang H, Qian WP, Weitzmann MN (2007) B cells and T cells are critical for the preservation of bone homeostasis and attainment of peak bone mass in vivo. Blood 109:3839–3848
Lopez-Granados E, Temmerman ST, Wu L, Reynolds JC, Follmann D, Liu S, Nelson DL, Rauch F, Jain A (2007) Osteopenia in X-linked hyper-IgM syndrome reveals a regulatory role for CD40 ligand in osteoclastogenesis. Proc Natl Acad Sci USA 104:5056–5061
Gao Y, Wu X, Terauchi M, Li JY, Grassi F, Galley S, Yang X, Weitzmann MN, Pacifici R (2008) T cells potentiate PTH-induced cortical bone loss through CD40L signaling. Cell Metab 8:132–145
Tobimatsu T, Kaji H, Sowa H, Naito J, Canaff L, Hendy GN, Sugimoto T, Chihara K (2006) Parathyroid hormone increases beta-catenin levels through Smad3 in mouse osteoblastic cells. Endocrinology 147:2583–2590
Kulkarni NH, Halladay DL, Miles RR, Gilbert LM, Frolik CA, Galvin RJ, Martin TJ, Gillespie MT, Onyia JE (2005) Effects of parathyroid hormone on Wnt signaling pathway in bone. J Cell Biochem 95:1178–1190
Wan M, Yang C, Li J, Wu X, Yuan H, Ma H, He X, Nie S, Chang C, Cao X (2008) Parathyroid hormone signaling through low-density lipoprotein-related protein 6. Genes Dev 22:2968–2979
Bellido T, Ali AA, Gubrij I, Plotkin LI, Fu Q, O’Brien CA, Manolagas SC, Jilka RL (2005) Chronic elevation of parathyroid hormone in mice reduces expression of sclerostin by osteocytes: a novel mechanism for hormonal control of osteoblastogenesis. Endocrinology 146:4577–4583
Keller H, Kneissel M (2005) SOST is a target gene for PTH in bone. Bone 37:148–158
Qin L, Qiu P, Wang L, Li X, Swarthout JT, Soteropoulos P, Tolias P, Partridge NC (2003) Gene expression profiles and transcription factors involved in parathyroid hormone signaling in osteoblasts revealed by microarray and bioinformatics. J Biol Chem 278:19723–19731
Young N, Mikhalkevich N, Yan Y, Chen D, Zheng WP (2005) Differential regulation of osteoblast activity by Th cell subsets mediated by parathyroid hormone and IFN-gamma. J Immunol 175:8287–8295
Terauchi M, Li JY, Bedi B, Baek KH, Tawfeek H, Galley S, Gilbert L, Nanes MS, Zayzafoon M, Guldberg R et al (2009) T lymphocytes amplify the anabolic activity of parathyroid hormone through Wnt10b signaling. Cell Metab 10:229–240
O’Brien CA, Plotkin LI, Galli C, Goellner JJ, Gortazar AR, Allen MR, Robling AG, Bouxsein M, Schipani E, Turner CH et al (2008) Control of bone mass and remodeling by PTH receptor signaling in osteocytes. PLoS One 3:e2942
Rawadi G, Vayssiere B, Dunn F, Baron R, Roman-Roman S (2003) BMP-2 controls alkaline phosphatase expression and osteoblast mineralization by a Wnt autocrine loop. J Bone Miner Res 18:1842–1853
Luo Q, Kang Q, Si W, Jiang W, Park JK, Peng Y, Li X, Luu HH, Luo J, Montag AG et al (2004) Connective tissue growth factor (CTGF) is regulated by Wnt and bone morphogenetic proteins signaling in osteoblast differentiation of mesenchymal stem cells. J Biol Chem 279:55958–55968
Alexiewicz JM, Klinger M, Pitts TO, Gaciong Z, Linker-Israeli M, Massry SG (1990) Parathyroid hormone inhibits B cell proliferation: implications in chronic renal failure. J Am Soc Nephrol 1:236–244
Ma YL, Cain RL, Halladay DL, Yang X, Zeng Q, Miles RR, Chandrasekhar S, Martin TJ, Onyia JE (2001) Catabolic effects of continuous human PTH (1–38) in vivo is associated with sustained stimulation of RANKL and inhibition of osteoprotegerin and gene-associated bone formation. Endocrinology 142:4047–4054
Glass DA 2nd, Bialek P, Ahn JD, Starbuck M, Patel MS, Clevers H, Taketo MM, Long F, McMahon AP, Lang RA et al (2005) Canonical Wnt signaling in differentiated osteoblasts controls osteoclast differentiation. Dev Cell 8:751–764
Spencer GJ, Utting JC, Etheridge SL, Arnett TR, Genever PG (2006) Wnt signalling in osteoblasts regulates expression of the receptor activator of NFkappaB ligand and inhibits osteoclastogenesis in vitro. J Cell Sci 119:1283–1296
Moon RT, Bowerman B, Boutros M, Perrimon N (2002) The promise and perils of Wnt signaling through beta-catenin. Science 296:1644–1646
Kimble RB, Srivastava S, Ross FP, Matayoshi A, Pacifici R (1996) Estrogen deficiency increases the ability of stromal cells to support murine osteoclastogenesis via an interleukin-1and tumor necrosis factor-mediated stimulation of macrophage colony-stimulating factor production. J Biol Chem 271:28890–28897
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer New York
About this paper
Cite this paper
Pacifici, R. (2013). T Cells Mediate the Effects of PTH in Bone. In: Choi, Y. (eds) Osteoimmunology. Springer, New York, NY. https://doi.org/10.1007/978-1-4614-5366-6_9
Download citation
DOI: https://doi.org/10.1007/978-1-4614-5366-6_9
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4614-5365-9
Online ISBN: 978-1-4614-5366-6
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)