
Abstract
Salt-retaining states are common medical problems, particularly as clinical manifestations of chronic heart failure (CHF) and chronic kidney disease (CKD). Nephrotic syndrome (NS) in adults is a relatively less common manifestation of kidney disease, with an estimated annual incidence of three new cases per 100,000 [1]. In childhood it is most commonly due to minimal change disease. Most cases of NS in adults are due to primary glomerular diseases, but some are due to secondary glomerular diseases such as diabetes mellitus, systemic lupus erythematosus and amyloidosis [1]. Cardinal features are proteinuria, hypoalbuminaemia and oedema.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Nephrotic Syndrome
- Atrial Natriuretic Peptide
- Acute Decompensated Heart Failure
- Minimal Change Disease
- Tubular Lumen
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
How Comparable Are the Different Salt-Retaining States?
Salt-retaining states are common medical problems, particularly as clinical manifestations of chronic heart failure (CHF) and chronic kidney disease (CKD). Nephrotic syndrome (NS) in adults is a relatively less common manifestation of kidney disease, with an estimated annual incidence of three new cases per 100,000 [1]. In childhood it is most commonly due to minimal change disease. Most cases of NS in adults are due to primary glomerular diseases, but some are due to secondary glomerular diseases such as diabetes mellitus, systemic lupus erythematosus and amyloidosis [1]. Cardinal features are proteinuria, hypoalbuminaemia and oedema.
Expansion of extracellular fluid (ECF) volume can be very disabling, with peripheral oedema, pulmonary oedema, gut oedema and ascites. If disease-specific therapies do not rapidly induce a remission, it is often difficult to bring these symptoms under control with generic therapies. Patients with NS may then seem to be ‘diuretic resistant’, in much the same way as some patients with CHF or CKD are considered to so be [2]. However, it may be a mistake to view all oedematous patients who are ‘slow’ to respond to initial therapies as being ‘the same’ just because the general principles of therapy are similar. It is important to recognise that the pathophysiological basis of oedema differs between different underlying conditions and this may have implications for choices in therapy. Furthermore, differences in drug pharmacokinetics and pharmacodynamics influence response to treatment [2–5].
An example of the increasing interest in more precise characterisation and treatment of oedematous states has been that focused on acute decompensated heart failure (ADHF) and associated cardio-renal syndromes (CRS) [6]. This acknowledges pathophysiological interactions between the heart and the kidney as well as the effects of neurohumeral activation [7]. Salt restriction, use of diuretics and use of other agents are demonstrated to exhibit a complex interplay in such cases. Some of these factors will overlap with the pathophysiology/treatment of ECF expansion in NS, some will not. Important trials on how to use diuretics in ADHF have been published [8]. The findings of these studies may not translate directly to strategies for treating oedematous NS patients.
Why Do Nephrotic Patients Become Oedematous?
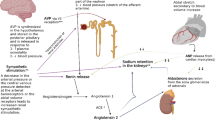
The traditional explanation has been that proteinuria leads to hypoalbuminaemia, which causes a decrease in plasma oncotic pressure. The consequent imbalance in Starling forces across the capillary wall causes fluid to ‘leak’ into the interstitium. Effective hypovolaemia follows and this triggers activation of the renin-angiotensin-aldosterone system, sympathetic nervous system and arginine vasopressin system, with inhibition of the release of atrial natriuretic peptide. All of this leads to secondary renal salt and water retention. However, observations in clinical cases and in experimental models have called this ‘underfill’ hypothesis into question, and whilst it may play some role, contemporary opinion favours a greater role for a specific renal salt retention process coupled with an alteration in capillary permeability independent of changes in oncotic gradients [9, 10].
No compelling and consistent explanation for the dysregulation in sodium balance in NS has yet been universally accepted, but it is notable that many studies indicate an up-regulation of epithelial sodium channel (ENaC) expression in the distal nephron, independent of aldosterone and other systemic hormones [10]. Recent studies postulate a role for plasminogen and plasmin, both of which appear in nephrotic urine. It is postulated that, in NS, plasminogen enters the urine through more permeable glomerular capillaries; that it is then activated to plasmin by urokinase; and that plasmin activates distal nephron ENaC channels [11]. Thus, a prominent aspect of NS is a particular salt avidity in the distal nephron, mediated through ENaC and occurring in concert with other mechanisms (possibly reflecting the ‘underfill’ hypothesis) enhancing salt retention at other sites.
What Are the Clinical Goals of Specific Management of NS?
These are dealt with elsewhere in this book and relate to inducing a remission of proteinuria and preserving glomerular filtration rate. There are extensive reviews and guidelines on potential therapies to induce remission/reduce proteinuria [12]. All make reference to the necessity for salt restriction and use of diuretics, but there is heterogeneity in clinical practice strategies, with a need to individualise such treatments. Consensus on how to achieve this is not prominent in the literature; in one very extensive guideline [12], the word ‘diuretic’ appears only 13 times in a 15-page document!
What Are the Clinical Goals of Generic Management of NS?
The objective of generic therapy is to initiate and sustain an increased natriuresis until the patient has returned to clinical euvolaemia. It should then be easier to maintain homeostasis at the new desired steady state, particularly if dietary salt restriction is appropriately introduced.
Total body salt and water will not decrease unless excretion exceeds intake. Dietary salt intake frequently exceeds 100 mmol/day (~6 g salt/day). Thus, for example, a patient will need to excrete 300 mmol of sodium over and above that needed to balance daily intake if he/she is to lose 2 kg of excess ECF volume.
Whilst salt avidity persists, this requires the use of (various) natriuretic agents – usually referred to as ‘diuretics’ – with specific pharmacokinetic and pharmacodynamic properties, in different doses and combinations.
Dietary salt intake also needs to be restricted, ideally to <80 mmol/day, but this may be difficult to achieve. Most salt intake is not ‘elective’, but occurs because of addition in food processing. Furthermore, many patients find diets containing less than 80 mmol/day to be bland and unpalatable.
Unexpected failure to lose weight/ECF volume in a patient on a seemingly appropriate diuretic dose should prompt enquiry into salt intake. Measurement of 24-h sodium excretion may help. A patient passing 150 mmol/day or more but not losing weight is likely to have an excess intake.
What Are the Different Classes of Diuretics and How Do They Work?
Diuretics are different classes of drugs that inhibit sodium reabsorption at different sites along the nephron and by increasing natriuresis achieve clinical benefit [2–5]. However, clinical goals may not always be easily achieved.
Site of action classifies the commonly used agents (Table 15.1). Although most (60–70 %) filtered sodium is reabsorbed in the proximal tubule, agents acting at this site (e.g. acetazolamide) are of relatively little clinical use in oedematous states because the increased sodium loss is offset by increased reabsorption further down the nephron in the thick ascending loop of Henle. The same principle applies to the proximal tubular effect of some thiazide diuretics.
Loop diuretics (e.g. furosemide, bumetanide, torasemide) are organic anions, secreted into the tubular lumen by the organic anion transporter (OAT1) in the proximal tubule (Table 15.2). They act on the luminal aspect of the thick ascending loop of Henle where they exhibit high affinity for the chloride-binding site of the sodium-potassium-2 chloride (NKCC2) transporter – a member of the solute carrier family 12 group of proteins [2, 4]. This directly inhibits sodium and chloride reabsorption and indirectly leads to decreased reabsorption of calcium and magnesium. Up to 20 % of filtered sodium can be excreted using these agents.
Thiazides and related compounds (e.g. bendroflumethiazide, hydrochlorothiazide, chlorthalidone, indapamide, metolazone) are organic anions also secreted by OAT1 in the proximal tubule. They act on the distal tubule and connecting segment where they bind to a number of transporters, principally the sodium-chloride cotransporter (NCC) – another member of the solute carrier family 12 protein group – directly inhibiting sodium reabsorption (Table 15.2). This indirectly increases calcium reabsorption. The maximum natriuresis is less than that achieved with loop diuretics, but a combination of these classes can be especially potent [13].
The potassium-sparing diuretics include amiloride, triamterene and spironolactone [2–5]. These have slightly different modes of action. Amiloride and triamterene are organic cations secreted into the lumen of the proximal tubule, but acting on the luminal aspect of the epithelial sodium channel (ENaC) in the cortical collecting duct. Spironolactone and eplerenone, by contrast, enter the principal cells of the cortical collecting duct from the plasma and interfere with the activation of the intracellular aldosterone receptor. This leads to a reduction in the activity of the baso-lateral sodium-potassium ATPase and a reduction in luminal expression of ENaC.
What Are the Pharmacokinetic Barriers to Achieving Therapeutic Objectives?
The primary driver of natriuresis is the rate of excretion of diuretic in the tubular fluid. This relationship exhibits a threshold phenomenon, following which the rate of sodium excretion reflects diuretic excretion in a linear dose-dependent pattern [3, 4]. Failure to deliver a sufficient dose of diuretic to exceed the natriuretic threshold may be described as ‘diuretic resistance’, but more usually reflects a failure to appreciate pharmacokinetic principles (Table 15.3).
Most prescribing choices to address pharmacokinetic issues involve administering larger doses of diuretic or enhancing bioavailability.
The first step is to ensure that an adequate dose of diuretic enters the bloodstream and is delivered to the kidney for excretion into the tubular lumen. Diuretics differ in their oral bioavailability. The oral bioavailability of furosemide ranges from 20 to 70 %, decreasing with increased gut oedema. On the other hand, bumetanide has an oral bioavailability approaching 80 %. When faced with a very oedematous patient, administering a higher dose of oral furosemide and switching to bumetanide or administering the agent intravenously are all rational therapeutic choices.
Loop and thiazide diuretics are transported bound to albumin and other plasma proteins. In NS, levels of albumin and other plasma proteins are often extremely low, and the consequent increased volume of distribution decreases the amount delivered to the kidney [14]. Increasing the dose administered is the appropriate response to this; coadministration of albumin with diuretic has not been consistently demonstrated as being of additional benefit [15, 16].
Diuretics compete with other anions for excretion by OAT1 into the proximal tubule [3–5]. Such anions accumulate particularly in the presence of renal failure and hepatic failure. In such circumstances the expected dose–response to loop and thiazide diuretics may be less than anticipated. Certain drugs (such as cimetidine) also compete for excretion. This problem does not occur with spironolactone, which does not require to be excreted into the tubular lumen, and its diuretic effect is less affected by liver failure.
A fall in GFR, particularly when combined with a low cardiac output, will decrease diurectic delivery and also initial filtered sodium load making a substantial naturiesis even more difficult to achieve. It was previously postulated that urinary protein bound to diuretic in the tubular lumen decreased its effectiveness. This view has not been substantiated by experimental studies [17].
Therefore, there are many factors active in NS that act as additional pharmacokinetic ‘hurdles’ to achieving a degree of diuretic excretion sufficient to initiate a natriuresis. In most circumstances, increasing the prescribed dose or otherwise enhancing the bioavailability of that dose is the appropriate strategy. An illogical, but common, error is to repeat the same ineffective dose more frequently. If there is doubt as to whether or not a natriuresis has been initiated, measurement of 24-h sodium excretion (or even 6-h excretion following the dose) is a rational choice.
What Are the Pharmacodynamic Barriers to Achieving Therapeutic Objectives?
Once a natriuresis is initiated, it needs to be sustained until the patient has been restored to the desired steady state. Once diuretics are administered and natriuresis achieved, there is a rapid functional and structural response in the nephron that acts to reduce the degree of enhanced natriuresis [2–5]. This can be viewed as ‘diuretic blunting’ and reflects pharmacodynamic principles.
Most of the adaptation occurs downstream from the site of action of the initially deployed diuretic. Changes in the expression and activity of transporters in the distal tubule and the cortical collecting duct occur within days [3, 4]. It is now apparent that allelic variations, particularly in the genes encoding for the SLC12A3 protein (NCC) and the β-subunit of the SCNN1 protein (ENaC) (Table 15.2) may explain the variance in response between patients [18].
In addition, there is evidence that chronic exposure to both loop and thiazide/thiazide-like diuretics increases the expression of their respective target transporters as well as of OAT1 [19].
The clinician needs to anticipate these changes. In the first instance, once a natriuresis has been initiated, one can prescribe the effective dose more frequently. Although the response to consecutive doses will progressively decline, more net natriuresis will be achieved with twice daily, thrice daily or a continuous infusion of diuretic. It is unclear if a continuous infusion achieves a greater daily natriuresis than the same total dose given as boluses [8, 20].
However, the most effective strategy to adopt is the early initiation of sequential nephron blockade, using a combination of diuretic agents to target multiple sites down the nephron [2–5]. This blocks the adaptation in the distal tubule and cortical collecting duct to the natriuretic effect of increased inhibition of the NKCC2. Combination of loop diuretics with thiazide/thiazide-like diuretics is effective, even in the presence of advanced renal dysfunction and in advanced heart failure [13, 21]. In addition, the early prescription of potassium-sparing diuretics will minimise the kaliuresis/hypokalaemia that will occur with successful blockade of the NKCC2/NCC systems [2–5].
There is now also some interest in supplementing the use of standard natriuretic agents with human atrial natriuretic peptide analogues such as carperitide [22]. These are not yet part of the mainstay of therapy.
Summary
In patients with NS in whom an early remission with specific therapy (such as in steroid-responsive minimal change disease) is unlikely, and in whom drugs inhibiting the RAAS have been already deployed, the following steps should achieve the generic goals of natriuretic therapy:
-
1.
On clinical examination determine the probable extent of ECF volume expansion; express this in kg; set a target weight at which one can anticipate that the patient will be restored to euvolaemia.
-
2.
Initiate dietary salt restriction to a target of 80 mmol/day or less (a trained dietician is very helpful for this).
-
3.
Administer a loop diuretic, estimating dose/agent/mode of administration based on degree of oedema, level of hypoproteinaemia, level of renal and cardiac function and presence of liver disease.
-
4.
Progressively increase the dose until a natriuresis is initiated (either on clinical evidence or with a measurement of urinary sodium excretion).
-
5.
Once natriuresis is established, administer the same dose more frequently, or as a continuous infusion.
-
6.
Rapidly (within 2–3 days, or immediately if the patient has already been on loop diuretics for some time) initiate sequential nephron blockade with thiazide/thiazide-like agents and potassium-sparing diuretics.
References
Hull RP, Goldsmith DJA. Nephrotic syndrome in adults. BMJ. 2008;336:1185–9.
Plant L. Clinical use of diuretics. In: Barratt J, Harris K, Topham P, editors. Oxford desk reference nephrology, chapt 18.3. Oxford: Oxford University Press; 2009. p. 708–12.
Brater DC. Pharmacology of diuretics. Am J Med Sci. 2000;319:38–50.
Brater DC. Update in diuretic therapy: clinical pharmacology. Semin Nephrol. 2011;31:483–94.
Sica DA. Diuretic use in renal disease. Nat Rev Nephrol. 2012;8:100–9.
Acute Dialysis Quality Initiative (ADQI) consensus group. Cardio-renal syndromes: an executive summary from the consensus conference of the Acute Dialysis Quality Initiative (ADQI) Consensus Conference. Contrib Nephrol. 2010;165:54–7.
Ronco C, Cicoira M, McCullough PA. Cardiorenal syndrome type 1: pathophysiological crosstalk leading to combined heart and kidney dysfunction in the setting of acutely decompensated heart failure. J Am Coll Cardiol. 2012;60:1031–42.
Felker GM, Lee KL, Bull DA, Redfield MM, Stevenson LW, Goldsmith SR, LeWinter MM, Deswal A, Rouleau JL, Ofili EO, Anstrom KA, Hernandez AF, McNulty SE, Velazquez EJ, Kfoury AG, Chen HH, Givertz MM, Semigran MJ, Bart BA, Mascette AM, Braunwald E, O’Connor CM, for the NHLBI Heart Failure Clinical Research Network. Diuretic strategies in patients with acute decompensated heart failure. N Engl J Med. 2011;364:797–805.
Rondon-Berrios H. New insights into the pathophysiology of oedema in nephrotic syndrome. Nefrologia. 2011;31:148–514.
Doucet A, Favre G, Deschenes G. Molecular mechanism of edema formation in nephrotic syndrome: therapeutic implications. Pediatr Nephrol. 2007;22:1983–90.
Svenningsen P, Bistrup C, Friis UG, Bertog M, Harteis S, Krueger N, Stubbe J, Nørregrad Jensen O, Thiesson HC, Uhrenholt TR, Jespersen B, Jensen BL, Korbmacher C, Skøtt O. Plasmin in nephrotic urine activates the epithelial sodium channel. J Am Soc Nephrol. 2009;20:299–310.
Kidney Disease: Improving Global Outcomes (KDIGO) Glomerulonephritis Work Group. KDIGO clinical practice guideline for glomerulonephritis. Kidney Int. 2012;2(Suppl):139–74.
Fliser D, Schröter M, Neubeck M, Ritz E. Coadministration of thiazides increases the efficacy of loop diuretics even in patients with advanced renal failure. Kidney Int. 1994;46:482–8.
Pichette V, Geadah D, du Souich P. Role of plasma protein binding on renal metabolism and dynamics of furosemide in the rabbit. Drug Metab Dispos. 1999;27:81–5.
Akcicek F, Yalniz T, Basci A, Ok E, Mees EJ. Diuretic effect of furosemide in patients with nephrotic syndrome: is it potentiated by intravenous albumin? BMJ. 1995;310:162–3.
Fliser D, Zurbrüggen I, Mutschler E, Bischoff I, Nussburger J, Franek E, Ritz E. Coadministration of albumin and furosemide in the nephrotic syndrome. Kidney Int. 1999;55:629–34.
Agarwal R, Gorski JC, Sundblad K, Brater DC. Urinary protein binding does not affect response to furosemide in patients with nephrotic syndrome. J Am Soc Nephrol. 2000;11:1100–5.
Vormfelde SV, Sehrt D, Toliat MR, Schirmer M, Meineke I, Tzvetlov M, Nürnberg P, Brockmöller J. Genetic variation in the renal sodium transporters NKCC2, NCC, and ENaC in relation to the effects of loop diuretic drugs. Clin Pharmacol Ther. 2007;82:300–9.
Jim JH. Long-term adaptation of renal ion transporters to chronic diuretic treatment. Am J Nephrol. 2004;24:595–605.
Salvador DR, Rey NR, Ramos GC, Punzalan FE. Continuous infusion versus bolus injection of loop diuretics in congestive heart failure. Cochrane Database Syst Rev. 2005;(3):CD003178.
Channer KS, McLean KA, Lawson-Matthew P, Richardson M. Combination diuretic treatment in severe heart failure: a randomized controlled trial. Br Heart J. 1994;71:146–50.
Kanzaki M, Wada J, Kikumoto Y, Akagi S, Nakao K, Sugiyama H, Makino H. The therapeutic potential of synthetic human atrial natriuretic peptide in nephrotic syndrome: a randomized controlled trial. In J Nephrol Renovasc Dis. 2012;5:91–6.
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer-Verlag London
About this chapter
Cite this chapter
Plant, L. (2014). Management of the Nephrotic Patient: Treatment of ECF Volume Expansion Due to Nephrotic Syndrome in Adults. In: Harber, M. (eds) Practical Nephrology. Springer, London. https://doi.org/10.1007/978-1-4471-5547-8_15
Download citation
DOI: https://doi.org/10.1007/978-1-4471-5547-8_15
Published:
Publisher Name: Springer, London
Print ISBN: 978-1-4471-5546-1
Online ISBN: 978-1-4471-5547-8
eBook Packages: MedicineMedicine (R0)