
Abstract
Maintaining a steady balance between energy production and consumption is a cornerstone of all living cells. Failure to maintain this balance affects most, if not all, cellular activities as these processes are normally tightly coupled to the energy status of the cell. It is beginning to emerge that human diseases such as obesity, Type 2 diabetes, and even certain types of cancer may be linked to underlying defects in the regulation of energy balance. Since the evolution of eukaryotes, the AMP-activated protein kinase (AMPK) system has played a pivotal role in maintaining energy homeostasis by regulating the enzymes that control flux through virtually every branch of metabolism. AMPK functions primarily as a fuel gauge monitoring the ratio of AMP to ATP, which can be regarded as a molecular read-out of cellular energy status. It becomes activated when energy utilisation overtakes energy production, and serves to rectify this imbalance by upregulating ATP-producing pathways while switching off ATP-consuming pathways such as lipid, carbohydrate and protein biosynthesis. In addition to regulating energy balance at the cellular level, AMPK is also a central regulator of whole-body energy homeostasis, integrating a variety of hormonal and nutritional signals in the central nervous system and periphery to control feeding behaviour and body weight. Increasing our understanding of the regulation and physiological roles of AMPK promises to open new avenues for the treatment of a whole range of debilitating human diseases.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Fatty Acid Oxidation
- Mitochondrial Biogenesis
- Glycogen Phosphorylase
- Hormone Sensitive Lipase
- Upstream Kinase
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
9.1 Introduction
Living cells require a constant input of energy to perform useful work such as synthesis of macromolecules (protein, lipids, carbohydrate), active transport of metabolites and ions, and cell movement. Most of these processes use adenosine-5′-triphosphate (ATP) as their energy source, which is the universal currency of energy in virtually all biological systems. Energy is liberated from ATP by hydrolysis of one or more of its phosphoester bonds (yielding ADP or AMP), which is then harnessed to power biochemical reactions that are thermodynamically unfavourable. Cells normally maintain a high ratio of ATP to ADP (or AMP) in order that these cellular processes are kept far from equilibrium; therefore complex mechanisms have evolved to ensure that ATP-production (catabolism) is balanced with ATP-consumption (anabolism). In eukaryotes, the AMP-activated protein kinase (AMPK) is a critical sensor and regulator of cellular energy charge that co-ordinates metabolic processes to balance energy supply with demand. AMPK monitors the cellular AMP to ATP ratio and is activated by metabolic stresses that either inhibit ATP production (hypoxia, fuel deprivation) or stimulate increased ATP consumption (exercise) (Scott et al. 2009). The reason that AMPK senses the AMP/ATP ratio rather than the ADP/ATP ratio is not obvious at first glance, however, the former is a much more sensitive measure of cellular energy charge despite the fact that ADP is the major product of ATP hydrolysis. The explanation for this is due to the action of adenylate kinase, a highly active enzyme found in all eukaryotes that catalyses the reaction 2ADP « AMP + ATP (Hardie and Hawley 2001). Adenylate kinase maintains this reaction close to equilibrium therefore the AMP/ATP ratio tends to vary as the square of the ADP/ATP ratio (for example, a 5-fold rise in the ADP/ATP ratio equates to a 25-fold rise in the AMP/ATP ratio). In other words, the concentration of AMP in the cell rises more dramatically than ADP when ATP levels fall, therefore it stands to reason why natural selection favoured an energy monitoring system that senses the AMP/ATP ratio.
The primary function of AMPK when activated is to conserve cellular ATP by upregulating ATP-producing pathways such as fatty acid oxidation and glycolysis, while inhibiting ATP-consuming processes including fatty acid, sterol, carbohydrate and protein synthesis (Fig. 9.1) (Scott et al. 2009). It achieves this in the main by direct phosphorylation of enzymes that catalyse the rate-limiting steps of these major metabolic pathways. AMPK also plays an adaptive role to chronic energy challenges, similar to those that occur during exercise training, by reprogramming expression of metabolic genes via phosphorylation of transcription factors and coactivators/regulators (McGee and Hargreaves 2008). It is therefore likely that AMPK is at least partly responsible for some of the beneficial metabolic effects associated with regular physical activity. In addition to physiological and pathological stresses that deplete ATP, AMPK is also activated by drugs used to treat Type 2 diabetes including the biguanides and thiazolidinediones (Zhou et al. 2009). Furthermore, a number of natural plant and herbal compounds such as resveratrol and berberine (present in grapes), and triterpenoids (present in bitter melon), all of which have health-giving properties that include prevention of obesity and insulin resistance, also activate AMPK (Baur et al. 2006; Lee et al. 2006b; Tan et al. 2008). These discoveries have propelled AMPK to the forefront as a potential drug target to treat a range of energy balance disorders and provide a compelling argument for the development of novel, specific activators of AMPK.

Regulation of energy metabolism by AMPK. Activation of AMPK by metabolic stresses, hormones and various drugs leads to activation and inhibition of catabolic and anabolic pathways, respectively
AMPK was defined historically as a sensor and regulator of energy balance at the cellular level, and the fact that AMPK orthologues are found in single celled eukaryotes is compatible with this being the function for which it evolved. However, AMPK is now also recognised as a key regulator of energy homeostasis at the whole-body level, responding to appetite controlling hormones such as leptin and ghrelin, as well as other secretory factors such as adiponectin (Kahn et al. 2005). AMPK processes signals from these hormones in both the central nervous system and peripheral tissues in order to control appetite and regulate whole-body energy expenditure. Understanding the role of AMPK in the control of energy homeostasis at both the cellular and whole body levels is crucial to developing new strategies to treat energy balance diseases.
9.2 AMPK Structure and Function
AMPK exists as a αβγ heterotrimer composed of an α catalytic subunit and regulatory β and γ subunits. In mammals, each subunit is encoded by multiple genes that can also be subject to alternative splicing, therefore at least 12 heterotrimeric combinations are possible giving rise to differences in subcellular localisation, tissue distribution, regulation and function. Orthologues of each subunit occur in all eukaryotes including plants, fungi and even the primitive protist Giardia lamblia, which highlights the importance and antiquity of the AMPK system and suggests it emerged very early during eukaryotic evolution.
The mammalian α subunits (α1 and α2) both have conventional Ser/Thr kinase domains at their N-terminus, followed by an autoinhibitory sequence and a C-terminal region that is involved in heterotrimeric formation (Fig. 9.2) (Crute et al. 1998). The kinase domain (PDB ID: 2H6D) adopts a canonical bilobal structure that is typical of the eukaryotic protein kinase superfamily, with the active site located in a cleft between the two lobes (reviewed in Scott et al. 2009). The small N-terminal lobe is predominantly responsible for ATP-binding whereas the large C-terminal lobe contains determinants and structural features that dictate binding of protein substrates. The catalytic loop in the large lobe contains an invariant aspartate residue that functions as the base for catalysis and is absolutely required for catalytic activity. AMPK is also catalytically inactive unless phosphorylated on Thr172 located on the activation loop (or T-loop) of the kinase domain by upstream kinases (Hawley et al. 1996). Protein kinases regulated by phosphorylation of the activation loop, without exception, have an arginine residue immediately preceding the catalytic aspartate, which is required to be neutralised by the presence of a phosphate on the activation loop in order to promote the correct folding and orientation of the ATP-binding site and active site residues (Johnson et al. 1996).

Domain structure of AMPK subunits. AIS autoinhibitory sequence, β-SID β subunit interacting domain, Myr myristoyl moiety, CBM carbohydrate-binding module, αγ-SBS αγ-subunit binding sequence, CBS cystathionine-β-synthase motif
In terms of protein substrate recognition, the kinase domain of AMPK belongs to the basophilic class of protein kinases, requiring the presence of positively charged residues in the sequence N-terminal to the site of phosphorylation on target proteins. The consensus motif for AMPK substrate recognition is H(X,B)XXS/TXXXH where H is a hydrophobic residue, B is a basic residue and S/T serine or threonine residues (Scott et al. 2002). Although the basic residue is critical for recognition, it can be positioned at either the P-3 or P-4 position (the P-/P+ nomenclature denotes the residue position N- or C-terminal to the phosphorylated residue, respectively), whereas there is an absolute requirement for a hydrophobic residue at the P-5 position. The P-3/P-4 basic residue forms an ionic interaction with a conserved acidic patch located at the interface between the small and large lobes of the kinase domain, while the P-5 residue binds in a hydrophobic pocket situated on the large C-terminal lobe. There are a number of other determinants that lie out with the core consensus motif that, although not essential, significantly enhance substrate binding to the kinase domain such as a basic residue at the P-6 and P+3 positions (Scott et al. 2002). Many protein kinases autoregulate their activity through their own structural elements that either directly or indirectly block the catalytic site. Both AMPK α subunit isoforms contain an autoinhibitory sequence immediately C-terminal to the kinase domain, however the autoinhibitory sequence does not block the active site like those found in other ligand regulated kinases such as the Ca2+/calmodulin dependent protein kinase (CaMK) family, but rather binds across the back of the catalytic domain holding the enzyme in an open inactive conformation (Pang et al. 2007). The extreme C-terminus of the α subunit is involved in heterotrimer assembly and binds to the C-terminus of the β subunit (Xiao et al. 2007).
The β subunits (β1 and β2) play a key role in mediating heterotrimeric complex formation, and are also important in mediating subcellular localisation of AMPK to lipid membranes and glycogen (Fig. 9.2). It has also become evident that the β subunits can allosterically regulate AMPK activity in a manner distinct from AMP (McBride et al. 2009; Sanders et al. 2007a; Scott et al. 2008). The β subunits have divergent N-terminal regions of poorly defined function; however the β1 isoform is myristoylated on a glycine residue at the extreme N-terminus (Mitchelhill et al. 1997). The myristoyl group has an autoinhibitory effect on AMPK as removal of the modification by point mutation dramatically increases catalytic activity (Warden et al. 2001). Moreover, removal of the myristoyl group also causes a marked redistribution of β1-containing AMPK complexes from lipid membranes to the cytoplasm. The non-myristoylated AMPK mutants are still activated by AMP, suggesting that the molecular mechanisms by which AMP and myristoylation regulate AMPK activity are distinct. These findings indicate that myristoylation of the β1 subunit confers a secondary level of autoinhibition upon the AMPK heterotrimer, which is removed when the myristoyl group becomes embedded within a lipid bilayer. This mechanism would ensure that β1-containing AMPK complexes only become fully activated when localised to particular membranes or subcellular compartments.
The β subunits also have internal carbohydrate binding modules (CBM) that are related to domains found in enzymes involved in glycogen and starch metabolism. These domains are non-catalytic and are involved in targeting the enzymes in which they are found to polysaccharide structures and in the case of the β subunit CBM, mediates the association of AMPK with glycogen particles (Hudson et al. 2003; Polekhina et al. 2003). The crystal structure of the β1 subunit CBM has been solved in complex with the cyclic sugar β-cyclodextrin, which revealed that binding of sugars to the CBM is largely dependent on hydrophobic interactions mediated by two conserved tryptophan residues (Trp100 and Trp133) that flank the sugar-binding pocket (Polekhina et al. 2005). The β1-CBM preferentially binds oligosaccharides containing between five and seven glucose units connected through an α(1→4) linkage with a single glucose sugar in an α(1→6) branch (Koay et al. 2007). This suggests that AMPK recruitment may be enhanced during periods of glycogen depletion, where α(1→6) branched sugars become exposed after glycogen phosphorylase mediated degradation of α(1→4) linked sugars. In fact, it has been shown that AMPK does not bind to liver α-glycogen particles that are replete with α(1→4) linkages on their surface (Parker et al. 2007). It has become evident that the CBM, rather than just localising AMPK to glycogen, is also a regulatory domain that allosterically inhibits catalytic activity when glycogen is bound (McBride et al. 2009). The extent of branching is important as AMPK is more potently inhibited by highly branched glycogen, which is consistent with the finding that the CBM binds more tightly to branched α(1→6) linked sugars.
Glycogen binding also inhibits phosphorylation and activation of AMPK by upstream kinases but not dephosphorylation by protein phosphatases. This contrasts with the effect of AMP, which inhibits dephosphorylation by protein phosphatases without affecting phosphorylation by the upstream kinases (Suter et al. 2006). The C-terminal domain of the β subunits functions as a scaffold to anchor the α and γ subunits, and is termed the αγ-subunit binding sequence (αγ-SBS) (Iseli et al. 2005). The αγ-SBS forms a network of hydrophobic contacts with the C-terminus of the α subunit whereas the βγ-interface is formed mainly by a hydrogen bonding network between the αγ-SBS and the N-terminus of the γ subunit (Iseli et al. 2008; Xiao et al. 2007). Minimal contacts exist between the α and γ subunits at the heterotrimeric core interface, suggesting that a stable interaction between the α and γ subunits is unlikely in the absence of the β subunit.
The γ subunits (γ1, γ2 and γ3) contain variable N-terminal sequences of unknown function, followed by four tandem repeats of a CBS motif. These motifs invariably occur in tandem pairs and are found in a number of unrelated proteins including IMP dehydrogenase, the CLC chloride channel family and cystathionione-β-synthase (from which the acronym CBS is derived) (Bateman 1997). Pairs of CBS motifs form a discrete structural unit called Bateman domains, which function as adenine nucleotide binding modules (Scott et al. 2004). The γ subunit Bateman domains serve as the allosteric binding sites for AMP and ATP, and are responsible for the energy-sensing properties of AMPK (Fig. 9.2).
The structure of the entire mammalian γ subunit in complex with either AMP or ATP has been solved, which has provided a detailed insight into the molecular basis of nucleotide binding (Xiao et al. 2007). Although there are four potential binding sites for AMP, only three of the sites are able to bind nucleotides as the unoccupied site lacks a conserved aspartate residue that interacts with the 2′ and 3′-hydroxyls of the ribose sugar (Fig. 9.3). Surprisingly, the site equivalent to the unoccupied site in the γ subunit orthologue from Schizosaccharomyces pombe can bind ADP by virtue of an interaction between the 2′ and 3′-ribose hydroxyls of ADP and an asparate residue from the β subunit (Jin et al. 2007). Interestingly, there is an aspartate in the corresponding position that is conserved in the sequence of the mammalian β subunit isoforms, raising the possibility that this site in mammalian AMPK may become occupied by a similar mechanism. Two of the three sites bound with AMP can readily exchange with ATP and are therefore the energy-sensing sites, whereas the third molecule of AMP is tightly bound and is non-exchangeable with ATP; however, the function of the non-exchangeable site is unknown (Xiao et al. 2007). The negatively charged phosphate moieties of AMP and ATP are held in place by an array of positively charged residues, most of which are located at the mouth of the binding pocket. ATP binding to the exchangeable sites results in only a minor rearrangement of these basic residues despite the fact that ATP has two additional phosphate groups compared with AMP, therefore it is unclear how the occupancy state of the exchangeable binding sites effects allosteric activation of the α catalytic domain.

The mammalian αβγ core complex structure. The C-terminal α subunit structure is shown in blue, the C-terminal β subunit structure in red, and the entire γ subunit in green. The three AMP molecules evident in the structure are shown in magenta
An interesting feature of the γ subunit nucleotide binding mechanism is that the second CBS motif (CBS2) contributes the majority of the basic residues that interact with the nucleotide phosphate groups. One interpretation of this feature is that CBS2 may be particularly important in the interactions between the α and γ subunits that regulate catalytic activity. Indeed, this possibility is supported by the identification of a pseudosubstrate sequence that occurs in the CBS2 motif of all mammalian γ subunits (Scott et al. 2007). Pseudosubstrate sequences act as competitive inhibitors, blocking access of protein substrates to the active site in the absence of activating ligands. These motifs are usually found adjacent to or overlapping with regulatory ligand-binding sites. There are a number of protein kinases that are regulated by internal pseudosubstrates including cAMP and cGMP-dependent protein kinases (PKA, PKG), the Ca2+/calmodulin dependent protein kinases (CaMK), and the protein kinase C (PKC) family (Kemp et al. 1994). Positively charged residues within the pseudosubstrate of CBS2 that are predicted to be involved in interacting with the substrate-binding site on the kinase domain, are also critical in binding the phosphate group of AMP. Since these two interactions would be unlikely to occur simultaneously, this suggests a simple mechanism whereby in the absence of AMP, the pseudosubstrate would occupy the substrate-binding site on the kinase domain and inhibit catalytic activity. Despite kinetic evidence in favour of this mode of activation, extended crystal structures of the AMPK complex that include the kinase domain will be required to validate this mechanism.
9.3 Regulation of AMPK
AMPK is principally activated by phosphorylation of Thr172 in the activation loop of the α subunit catalytic domain by at least three upstream protein kinases (Fig. 9.4), highlighting the potential for multiple signals to feed into the AMPK system. Conversely, AMPK is inactivated by dephosphorylation of Thr172 by several protein phosphatases including PP2A, PP2C and glycogen bound PP1 in skeletal muscle (Davies et al. 1995; McBride et al. 2009; Paterson et al. 2008). An important question in recent years has been how AMP promotes phosphorylation and activation of AMPK. Initial studies suggested that AMP stimulated phosphorylation of AMPK by two distinct mechanisms: (1) making AMPK a better substrate for activating upstream kinases; (2) making AMPK a worse substrate for inactivating protein phosphatases (Davies et al. 1995; Hawley et al. 2005). However, more recent studies using recombinant AMPK suggest that the former effect is an artefact caused by contamination by protein phosphatases, and that AMP simply protects against dephosphorylation (Suter et al. 2006). In addition to stimulating phosphorylation of Thr172, AMP also directly activates AMPK by an allosteric mechanism, however this effect is relatively modest compared with phosphorylation. Nevertheless, the combination of these two effects of AMP results in a greater than 1,000-fold increase in AMPK activity (Suter et al. 2006). Importantly, both effects of AMP are antagonised by high concentrations of ATP, therefore AMPK acts as a sensor of the AMP/ATP ratio rather than AMP in itself (Corton et al. 1995; Davies et al. 1995).

Regulation of AMPK by AMP and upstream kinases. AMPK regulated by reversible phosphorylation of Thr172 by various upstream kinases and protein phosphatases. AMP allosterically activates phosphorylated AMPK as well as inhibiting dephosphorylation of Thr172
The LKB1 complex is the major upstream AMPK kinase in most mammalian tissues, which exists a heterotrimer consisting of the protein kinase LKB1 and two regulatory subunits STRAD and MO25 (Hawley et al. 2003). STRAD and MO25 are essential for LKB1 to be functional and are also involved in targeting the complex to the cytoplasm (Boudeau et al. 2003). STRAD is termed a pseudokinase as it shares a high degree of sequence homology to the eukaryotic protein kinase superfamily but lacks key residues required for catalytic activity, whereas MO25 is structurally related to Armadillo repeat proteins. LKB1 was first described as a tumour suppressor that is mutated in Peutz-Jeghers syndrome, a rare dominantly inherited disease in humans that is characterised by benign intestinal polyps (or hamartomas) and pigmentation of the skin and mucous membranes (Hemminki et al. 1998). Peutz-Jeghers patients also have significantly increased risk of developing malignant tumours. LKB1 phosphorylates and activates 12 other protein kinases in addition to AMPK, all of which are members of the AMPK-related protein kinase subfamily (Lizcano et al. 2004). These other protein kinases share sequence similarity with the catalytic domain of the AMPK α subunit but are functionally distinct. LKB1 is constitutively active despite the fact that it is phosphorylated by cAMP-dependent protein kinase and p90 ribosomal S6 kinase at multiple sites; however none of these phosphorylation events alters LKB1 activity directly or affects its ability to phosphorylate AMPK (Fogarty and Hardie 2009; Sapkota et al. 2001). Therefore, the current model follows that AMPK is continuously phosphorylated and dephosphorylated by LKB1 and protein phosphatases, respectively, in a futile cycle that is shifted in favour of phosphorylation only when AMP levels rise (Sanders et al. 2007b). Although it would seem counterintuitive for an energy conserving system to wastefully consume ATP in a futile cycle, it does have the advantage of allowing rapid changes in the activity of AMPK to occur in response to perturbations in cellular energy charge.
In addition to changes in cellular energy, increases in intracellular Ca2+ also stimulate phosphorylation and activation of AMPK via the Ca2+-calmodulin-dependent protein kinase kinases (CaMKK), regardless of the intracellular AMP concentration (Hawley et al. 2005; Hurley et al. 2005; Woods et al. 2005). There are two CaMKK isoforms (α and β), both of which can phosphorylate AMPK in vitro, however a number of studies suggest that CaMKKβ is the physiological activator of AMPK (Anderson et al. 2008; Stahmann et al. 2006). CaMKKβ differs from CaMKKα mainly in the N-terminal region preceding the catalytic domain, in which there is a regulatory segment in CaMKKβ that renders the enzyme virtually Ca2+-calmodulin independent; although whether this N-terminal region is important for the regulation of AMPK remains to be determined (Tokumitsu et al. 2001). CaMKKβ is predominantly expressed in neuronal tissue but is also found in a small number of other tissues including endothelial and T cells, therefore Ca2+-CaMKK mediated regulation of AMPK is more restricted than LKB1, which is expressed ubiquitously.
Increased cytosolic Ca2+ is often accompanied by a surge in demand for ATP, for instance, activation of motor proteins involved in muscle contraction or extrusion of Ca2+ from the cytosol, which is predominantly driven by ATP-dependent Ca2+ pumps. Under these circumstances, activation of AMPK could be considered as playing a protective role, ensuring that energy metabolism is co-ordinated to meet the increase in demand for ATP that typically follows Ca2+ release. In support of this idea, it has been shown that AMPK is activated in response to T-cell receptor (TCR) stimulation in T cells in a Ca2+-CaMKKβ dependent manner (Tamas et al. 2006). TCR activation results in a large influx of Ca2+ into the cytosol, which acts as a second messenger to stimulate T cell proliferation and effector function, both of which are energy intensive processes (Fox et al. 2005). Activated T cells rapidly increase their ATP production by promoting glycolysis and oxidative phosphorylation, and it is likely that the CaMKK-AMPK signalling pathway is at least partly responsible for these metabolic effects.
In addition to LKB1 and CaMKKβ, a screen for protein kinases that activate AMPK using a mammalian expression library in yeast identified transforming growth factor-β-activated protein kinase-1 (TAK1) as a potential upstream kinase (Momcilovic et al. 2006). TAK1 is an important component of the signalling pathways that regulate the activities of the nuclear factor-kappa B (NFκB) and activator protein-1 (AP-1) transcription factors in response to cytokines and microbial pathogens (Scheidereit 2006). It acts upstream of several other protein kinases including the IκB kinase (IKK) complex and members of the stress-activated protein kinase family. Although AMPK is activated in response to TGFβ, interleukin-1 (IL-1) and tumour necrosis factor-α (TNFα) (Momcilovic et al. 2006; Suzuki et al. 2005), all of which are known activators of TAK1 signalling, the question of whether TAK1 regulates AMPK in a physiological setting remains to be defined.
9.4 Regulation of Lipid Metabolism by AMPK
As mentioned earlier, the principle function of AMPK in the cell is to co-ordinate metabolic processes to ensure that ATP production remains balanced with ATP consumption. In general, activation of AMPK stimulates ATP production and inhibits ATP consumption primarily by direct phosphorylation of metabolic enzymes that control anabolic (ATP-consuming) and catabolic (ATP-producing) pathways, which allows metabolic flux through these pathways to be rapidly altered. One of the most energy-intensive processes in the cell is the biosynthesis of fatty acids and lipids, and it is now clear that AMPK regulates lipid metabolism at multiple levels not only in an acute manner, but also in a chronic fashion via alterations in the expression pattern of lipogenic genes.
Perhaps the best characterised substrate for AMPK is acetyl-CoA carboxylase (ACC), the rate-limiting enzyme for fatty acid synthesis and an important regulator of fatty acid oxidation (Carling et al. 1989). ACC catalyses the synthesis of malonyl-CoA in an ATP-consuming reaction, and is the first committed step of fatty acid synthesis. There are two ACC isoforms (1 and 2), both of which play different roles and vary in their tissue expression (Abu-Elheiga et al. 1995). ACC1 is predominantly expressed in liver and adipose tissue and is primarily responsible for determining the rate of fatty acid synthesis (Iverson et al. 1990). On the other hand, ACC2 is abundantly expressed in skeletal muscle and, unlike ACC1, is localised to the outer membrane of mitochondria by virtue of an N-terminal targeting sequence (Abu-Elheiga et al. 2000). ACC2 is an important regulator of fatty acid oxidation, and plays a pivotal role in mediating the activity of carnitine palmitoyl transferase-1 (CPT-1), the transporter responsible for shuttling fatty acids into the mitochondria. Allosteric inhibition of CPT-1 by ACC2 derived malonyl-CoA is considered the most important regulatory step in determining the rate of fatty acid oxidation. In fact, genetic deletion of ACC2 results in high levels of fatty acid oxidation and decreased accumulation of triglycerides (Abu-Elheiga et al. 2001). AMPK phosphorylates and inactivates both ACC isoforms, resulting in decreased fatty acid synthesis and increased fatty acid oxidation. Activation of AMPK also promotes fatty acid uptake by stimulating relocalisation of fatty acid translocase (FAT/CD36) from intracellular vesicles to the plasma membrane (Luiken et al. 2003). As well as these acute effects, AMPK also decreases the expression of key lipogenic genes in response to long term energetic challenges. AMPK activation reduces the expression of SREBP1c, which functions as a transcriptional regulator of a number of lipogenic genes including fatty acid synthase (FAS), stearoyl-CoA desaturase-1 (SCD1), and Spot 14 (Foretz et al. 1998; Zhou et al. 2001). This occurs via an indirect mechanism as AMPK does not phosphorylate SREBP1c directly, however the signalling components that link AMPK to SREBP1c are unknown.
AMPK regulates other aspects of lipid metabolism such as triglyceride esterification and hydrolysis particularly in liver and adipose tissue, respectively. The control of these pathways has become an area of intense interest as lipid intermediates in the form of diacylglycerols, ceramides and long-chain fatty acyl-CoA molecules have been reported to activate protein kinases and phosphatases that negatively regulate the insulin signalling pathway (Kraegen and Cooney 2008). This raises the prospect that accumulation of these reactive lipid intermediates in obesity may be a causal factor underlying the development of insulin resistance. Triglyceride esterification is regulated by glycerol-3-phosphate-acyl-transferase (GPAT), which performs the first committed step in triglyceride synthesis and catalyses the formation of lysophosphatidic acid. Although activation of AMPK in the liver reduces GPAT activity and triglyceride esterification, it has yet to be demonstrated whether GPAT is a direct substrate of AMPK (Muoio et al. 1999). The hydrolysis of triglycerides to diglycerides is mediated by hormone sensitive lipase (HSL), which is activated in response to adrenergic factors that stimulate PKA activity (Anthonsen et al. 1998). AMPK phosphorylates and inhibits HSL in adipocytes thereby preventing lipolysis, which contradicts the paradigm that AMPK stimulates catabolic pathways and inhibits anabolic processes (Garton et al. 1989; Garton and Yeaman 1990). However, if fatty acids released by lipolysis are not exported from the adipocyte, they are recycled back into triglyceride in an ATP-consuming manner. Therefore, inhibition of HSL by AMPK appears to be a mechanism to restrict recycling and ensure the rate at which fatty acids are released by lipolysis does not surpass the rate at which they are disposed of by export or mitochondrial oxidation.
3-Hydroxy-3-methylglutaryl-CoA reductase (HMGR) is the rate-limiting enzyme of the cholesterol synthesis (mevalonate) pathway and a substrate for AMPK. Cholesterol is the precursor of steroid hormones such as estradiol, testosterone and the glucocorticoids, whereas geranylgeranyl and farnesyl groups derived from the mevalonate pathway play an important role in providing lipid anchors for many signalling proteins including several members of the small G-protein family (McTaggart 2006). The expression of HMGR is highly regulated at both the transcriptional and translational levels by a negative-feedback control mechanism mediated by sterols and non-sterol metabolites derived from mevalonate (Clarke et al. 1983). In addition, AMPK phosphorylates and inactivates HMGR activity, which results in decreased cholesterol synthesis (Beg et al. 1978; Clarke and Hardie 1990). Interestingly, activation of AMPK by adiponectin in a rodent model of atherosclerosis reduced cholesterol synthesis but also reduced the incidence of atherosclerotic plaques, suggesting that hormonal regulation of HMGR via AMPK may be critical for modulating whole-body cholesterol metabolism and also provide new opportunities for treating cardiovascular disease (Ouchi et al. 2001).
9.5 Regulation of Carbohydrate Metabolism by AMPK
Carbohydrates are an important fuel for virtually all organisms as they provide a rapid source of energy, especially as they are relatively simple to metabolise in comparison with other fuels such as fatty acids and proteins. Glucose is the most important carbohydrate in mammals, and complex cellular and hormonal signalling networks have evolved to ensure that its levels are maintained within narrow limits. The first hint that AMPK was likely to be involved carbohydrate metabolism came from the discovery that its a functional orthologue of the SNF1 protein kinase complex from Saccharomyces cerevisiae, which is activated in response to glucose limitation and orchestrates the switch in metabolism that permits growth on alternate carbon sources such as sucrose (Mitchelhill et al. 1994; Woods et al. 1994). Indeed, in mammals, AMPK regulates virtually all aspects of glucose metabolism including glucose uptake, gluconeogenesis and glycogen storage. AMPK also stimulates glycolysis in certain tissues, especially in the heart (see Sect. 9.8), but also in activated monocytes of the immune system (Marsin et al. 2000, 2002).
The uptake of glucose across the plasma membrane is largely mediated by the glucose transporter (GLUT) family of transmembrane proteins, with translocation of GLUT4 from intracellular vesicles to the plasma membrane considered the major mechanism by which glucose transport is stimulated in response to insulin (Sakamoto and Holman 2008). Pharmacological activation of AMPK with AICAR (which is taken into cells and converted to ZMP, an AMP-mimetic) stimulates GLUT4 translocation and glucose uptake independent of the insulin-signalling pathway, a finding that has generated significant interest in developing activators of AMPK as therapeutics to circumvent insulin resistance (Merrill et al. 1997). This concept is reinforced by the fact that genetic deletion of the AMPK α2 subunit (the predominant isoform in skeletal muscle) but not the α1 subunit abolishes AICAR-stimulated glucose uptake in muscle (Jorgensen et al. 2004b). The signalling pathway by which AMPK stimulates GLUT4 translocation is beginning to be unravelled and appears to involve the Rab GTPase-activating proteins (GAP) TBC1D1 and TBC1D4 (AS160), both of which are phosphorylated in response to AMPK activation in skeletal muscle (Kramer et al. 2006; Treebak et al. 2006). Phosphorylation of TBC1D1 and TBC1D4 results in the recruitment of 14-3-3 proteins, which in turn controls the rate of GLUT4 vesicle recycling (Chavez et al. 2008; Chen et al. 2008; Geraghty et al. 2007). Interestingly, TBC1D1 has been identified as a potential candidate gene for severe obesity, suggesting that defects in TBC1D1 signalling may be a contributory factor in obesity-induced insulin resistance (Lee et al. 2006a).
The major site of gluconeogenesis in mammals is the liver, and regulation of hepatic glucose production is crucial for maintaining whole-body glucose homeostasis and is critical for survival. Such is the importance of this process, it is regulated by number of diverse and redundant regulatory cues, which allow for the storage of glucose as glycogen and lipids following a meal and conversely to increase glucose production during a fast or intense exercise. Activation of AMPK in the liver suppresses hepatic glucose production by lowering the expression of gluconeogenic enzymes rather than by directly phosphorylating the enzymes themselves (Bergeron et al. 2001a). AMPK negatively regulates the transcription of the gluconeogenic genes L-type pyruvate kinase (PK), phosphoenolpyruvate carboxykinase (PEPCK), and glucose-6-phosphatase (G6P) by phosphorylation of various transcription factors and coactivators (da Silva Xavier et al. 2000; Leclerc et al. 1998; Lochhead et al. 2000; Woods et al. 2000).
The CREB-regulated transcription coactivator-2 (CRTC2) has emerged as an important regulator of gluconeogenic gene transcription and has been identified as a downstream target of AMPK (Koo et al. 2005). Glucagon stimulates the transcription of gluconeogenic genes when glucose is low via the cAMP-responsive factor CREB (CRE-binding protein) and subsequent recruitment of the coactivator CBP and CRTC2 to the nucleus. This nuclear translocation results in the expression of PGC-1α, which in turn promotes the transcription of PEPCK and G6P. Phosphorylation of CRTC2 by AMPK promotes binding to 14-3-3 proteins in the cytoplasm and prevents relocalisation of CRTC2 to the nucleus, thereby lowering CREB-dependent expression of PEPCK and G6P (Koo et al. 2005). The expression of PK is regulated by the transcription factor hepatic nuclear factor 4α (HNF-4α), and recent evidence suggests that HNF-4α protein is a direct substrate of AMPK, as phosphorylation of HNF-4α by AMPK in vitro reduces its ability to form homodimers that are required for DNA binding (Hong et al. 2003; Leclerc et al. 2001). The importance of AMPK in the regulation of hepatic glucose production is most convincingly demonstrated by findings from rodent models that lack the α2 subunit, which exhibit fasting hyperglycaemia, glucose intolerance and increased hepatic glucose output (Andreelli et al. 2006; Viollet et al. 2003).
Glucose is primarily stored in tissues as glycogen and is the most readily mobilised large-scale source of energy in the cell. The levels of glycogen in the cell are determined by both the rate of production (glycogenesis) and degradation (glycogenolysis), which are mediated by the enzymes glycogen synthase and glycogen phosphorylase, respectively. Glycogen synthesis and degradation are tightly co-ordinated so that glycogen synthase is almost completely inactive when glycogen phosphorylase is maximally active, and vice-versa. It has been known for several years that AMPK phosphorylates and inactivates glycogen synthase in vitro (Carling and Hardie 1989), however the role of AMPK in regulating glycogen synthase activity in vivo is more complex. Paradoxically, activation of AMPK in skeletal muscle actually increases glycogen content and glycogen synthase activity, however this effect is due to stimulation of glucose uptake and the consequent accumulation of glucose-6-phosphate, which allosterically activates glycogen synthase independent of phosphorylation (Aschenbach et al. 2002). However, in support of the initial observation, glycogen synthase has increased activity due to decreased phosphorylation in skeletal muscle lacking the α2 subunit of AMPK (Jorgensen et al. 2004a). Therefore, while phosphorylation of glycogen synthase by AMPK does inhibit its activity under basal conditions, stimuli that increase the intracellular levels of glucose-6-phosphate are capable of overriding the inhibitory effects of phosphorylation by AMPK.
9.6 Regulation of AMPK by Hormones and Cytokines
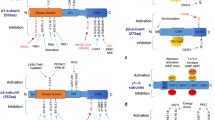
The importance of hormonal factors in the regulation of energy metabolism has been recognised for several decades, however only in recent years has the interplay between these hormones and AMPK come to light. Although AMPK was originally viewed merely as a sensor of cellular energy, it is now regarded as an integral player in processing the signals from these hormones not only in peripheral tissues such as liver, adipose and skeletal muscle, but also in the central nervous system and especially the hypothalamus (Fig. 9.5).

Hormonal control of AMPK. In the hypothalamus, AMPK is bidirectionally regulated by hormones to control appetite. Hormonal control of AMPK in peripheral tissues regulates fatty acid oxidation, glucose uptake and gluconeogenesis
One of the key factors in controlling feeding behaviour and whole-body energy expenditure in mammals is leptin, a protein hormone secreted from adipose tissue that suppresses food intake and prevents lipid accumulation in non-adipose tissue (Kahn et al. 2005). Defects in leptin production or the leptin receptor cause morbid obesity that typically results in the development of Type 2 diabetes (Chen et al. 1996). It is now clear that AMPK mediates many of the systemic metabolic effects of leptin. Classical leptin signalling via the leptin receptor involves recruitment of the JAK2 tyrosine kinase, resulting in phosphorylation of the cytoplasmic tail of the receptor and subsequent binding and activation of STAT proteins (Ghilardi et al. 1996). The STAT proteins are transcriptional activators that translocate to the nucleus when activated and primarily mediate the long term effects of leptin, however the precise signalling pathway that couples the leptin receptor to AMPK has yet to be elucidated. Nevertheless, in skeletal muscle leptin activates AMPK in a biphasic manner involving a rapid transient activation, followed by a more sustained activation that lasts a few hours (Minokoshi et al. 2002). The former effect occurs by leptin acting directly at the level of the muscle leptin receptor, whereas the latter effect is mediated through the hypothalamic-sympathetic nervous system culminating in stimulation of muscle α adrenergic receptors. Activation of AMPK in skeletal muscle by leptin results in suppression of ACC2 activity, therefore increasing the rate of fatty acid oxidation. In the longer term, leptin also promotes expression of the AMPK α2 and β2 subunits, both of which are particularly abundant in skeletal muscle, resulting in a long term decrease in intramuscular lipid storage (Steinberg et al. 2003). As well as skeletal muscle, leptin also activates AMPK in adipocytes and, in addition to stimulating fatty acid oxidation, also promotes mitochondrial biogenesis thus increasing the oxidative capacity of this tissue (Wang et al. 2005). Rather surprisingly, leptin has no effect on AMPK activity in the liver despite the fact that the leptin receptor is expressed in this tissue (Brabant et al. 2005; Lee et al. 2002).
Perhaps the most exciting effect of leptin on AMPK occurs in the hypothalamus, where the leptin-AMPK signalling axis plays an important role in controlling appetite. The control of food intake is regulated by distinct neuronal populations in the hypothalamus that process signals from various appetite-regulating hormones and nutrients such as glucose. In contrast to peripheral tissues, leptin inhibits AMPK activity in the periventricular and arcuate nucleus of the hypothalamus, and consequently suppresses food intake by reducing expression of the appetite-stimulating neuropeptides, NPY and agouti-related protein (AgRP) (Minokoshi et al. 2004). Inhibition of AMPK in the hypothalamus by leptin is mediated through melanocortin-4 (MC4) receptor signalling, as agonists for the MC4 receptor mimic the effects of leptin, whereas genetic deletion of the receptor prevents inhibition of AMPK by leptin. Further downstream, the activation state of the AMPK substrate ACC is also important for leptin signalling, as blockade of hypothalamic ACC with pharmacological inhibitors prevents the anorexigenic effects of leptin (Gao et al. 2007). The mechanism by which leptin inhibits AMPK in the hypothalamus while activating AMPK in other tissues is unclear, but it perhaps reflects differences in expression of AMPK subunit isoforms, upstream kinases and phosphatases between tissues. Although leptin has robust effects on AMPK activity and lipid metabolism in skeletal muscle and adipocytes, leptin resistance is common in obesity therefore drugs that activate AMPK, which is downstream of leptin, may be effective in circumventing leptin resistance.
Another adipocyte derived hormone that is an important regulator of whole-body energy expenditure is adiponectin, a protein factor that exists as multimers of various sizes ranging from low molecular mass trimers to high molecular mass dodecamers (Richards et al. 2006). Activation of AMPK by adiponectin is dependent on the AdipoR1 receptor, however the mechanism by which it signals to AMPK is poorly understood although it appears to involve increases in intracellular AMP (Yamauchi et al. 2002, 2007). Adiponectin activates AMPK in skeletal muscle and promotes fatty acid oxidation and glucose uptake, in addition to enhancing insulin sensitivity and promoting mitochondrial biogenesis (Civitarese et al. 2006; Tomas et al. 2002; Wang et al. 2007; Yamauchi et al. 2002). It also activates AMPK in liver and adipocytes resulting in decreased gluconeogenesis and increased glucose uptake, respectively (Wu et al. 2003; Yamauchi et al. 2002). In contrast with leptin, adiponectin activates hypothalamic AMPK to stimulate food intake and this effect is mediated by the low molecular mass trimers and hexamer forms of adiponectin, which can cross the blood-brain barrier unlike the high molecular mass species (Kubota et al. 2007). Crucially, adiponectin is able to reverse the inhibitory effect of leptin on AMPK activity, demonstrating the reciprocal role of these hormones in the control of appetite and whole-body energy expenditure.
Ghrelin is a gut derived hormone that is secreted by the cells lining the fundus of the stomach, which serves to stimulate appetite in response to fasting (Kola et al. 2006). It is a short peptide hormone that binds to the G-protein coupled growth hormone secretagogue (GSH) receptor. While ghrelin has no effect on AMPK in skeletal muscle, it decreases AMPK activity in both liver and adipose tissue (Kola et al. 2005). Similar to adiponectin and in contrast with leptin, ghrelin stimulates AMPK activity in the hypothalamus resulting in increased food intake (Andersson et al. 2004). Cannabinoids also activate hypothalamic AMPK via the GSH receptor to stimulate appetite, which likely explains the phenomenon popularly known as the “munchies” that is associated with cannabis use.
A growing number of other hormones and inflammatory cytokines including tumour necrosis factor α (TNFα) and ciliary neurotrophic factor (CNTF) have all been shown to modulate AMPK activity in a variety of tissues (Steinberg et al. 2009). The effect of inflammatory cytokines on AMPK activity is particularly relevant, as the expression and release into the circulation of a number of these factors is increased with obesity and Type 2 diabetes. Increased TNFα levels are associated with insulin resistance and genetic deletion of TNFα has been shown to enhance insulin sensitivity (Gonzalez-Gay et al. 2006; Saghizadeh et al. 1996). TNFα decreases AMPK activity chronically in skeletal muscle by upregulating the expression of the deactivating protein phosphatase PP2C, resulting in reduced fatty acid oxidation, which may be connected to decreased insulin sensitivity (Steinberg et al. 2006). CNTF is a cytokine secreted by glial cells that principally acts as a survival factor for motor and sensory neurones (Matthews and Febbraio 2008). The CNTF receptor is expressed in a number of peripheral tissues including skeletal muscle and adipose tissue, highlighting that CNTF can also regulate signalling pathways outwith the central nervous system. CNTF has central and peripheral effects on AMPK activity similar to leptin, however it is still able to modulate AMPK under conditions of leptin resistance, which is significant as bypassing leptin resistance is a major obstacle for treating obesity (Watt et al. 2006).
9.7 AMPK and Exercise
Exercise is arguably the most powerful physiological activator of AMPK, particularly as it stimulates a huge surge in demand for ATP and rapidly increases the fuel requirements of the working muscle. Skeletal muscle is an important tissue with respect to the regulation of whole-body energy metabolism as it makes up a relatively large proportion of body mass, and accounts for a sizeable portion of fuel consumption both during exercise and at rest. It is now well documented that physical inactivity has profound negative effects on whole-body energy metabolism and is a major risk factor for obesity and Type 2 diabetes. Activation of AMPK has been shown to improve the metabolic abnormalities associated with these diseases, therefore it has been postulated that some of the therapeutic metabolic effects of regular physical activity may be attributable to stimulation of AMPK.
AMPK is activated during exercise in an intensity-dependent manner, typically at exercise intensities above 60% of maximal aerobic activity, although it can also be activated at lower intensities if exercise is prolonged (Chen et al. 2003; Fujii et al. 2000; Wojtaszewski et al. 2000, 2002). As mentioned earlier, pharmacological activation of AMPK in skeletal muscle with AICAR stimulates glucose uptake independent of the insulin-signalling pathway (Merrill et al. 1997). Therefore, the role of AMPK in regulating exercise-induced glucose uptake has been an area of intense interest due to the therapeutic potential of stimulating glucose uptake in insulin-resistant muscle. Despite the strong correlation between muscle contraction, AMPK activity, and glucose uptake, the evidence so far paints a complex picture. For instance, contraction-stimulated glucose uptake is normal in skeletal muscle lacking either the α2 or γ3 subunits of AMPK whereas it is impaired in muscle expressing a dominant-negative variant of α2 (Barnes et al. 2004; Jorgensen et al. 2004b; Mu et al. 2001). In spite of these contradictory findings, AMPK is critical for the stimulation of glucose uptake in response to AICAR as this effect is abolished in both α2 and γ3 null skeletal muscle. The effect of AMPK on fatty acid oxidation during exercise also appears to be more complex than anticipated and, similar to the glucose uptake effect, looks likely to involve a number of redundant signalling pathways (Dzamko et al. 2008).
An exciting discovery relating to AMPK and exercise is the observation that chronic activation with AICAR induces a number of metabolic adaptations in skeletal muscle that resemble those that occur with exercise training. These adaptations include increased expression of proteins involved in glucose metabolism, such as GLUT4 and hexokinase, as well as increased mitochondrial biogenesis, which increases the capacity of the muscle to generate ATP by oxidative metabolism (Holmes et al. 1999; Winder et al. 2000). The ability to manipulate mitochondrial biogenesis is important as a reduction in mitochondrial density is thought to be a critical factor contributing to the accumulation of intramuscular lipids in obesity and to the development of insulin resistance. Mitochondrial biogenesis is regulated by various transcription factors and coactivators including nuclear respiratory factors-1 and 2 (NRF-1, NRF-2) and PGC-1α (Mootha et al. 2003; Virbasius and Scarpulla 1994). The expression of NRF-1 and NRF-2 is increased by chronic activation of AMPK, and both proteins are key transcriptional regulators of all five electron transport chain complexes (Bergeron et al. 2001b). On the other hand, PGC-1α is a direct substrate of AMPK and is required for expression of GLUT4 and the mitochondrial proteins cytochrome c and uncoupling protein-1 (UCP-1) (Jager et al. 2007).
Although these metabolic effects are observed in skeletal muscle in response to exercise, the question of whether AMPK is necessary is controversial as these responses are unaffected in skeletal muscle lacking either LKB1 or the α2 subunit of AMPK (Jorgensen et al. 2007; Thomson et al. 2007). This is not entirely surprising as activation of calcineurin, protein kinase D (PKD), and p38 MAP kinase have all been shown to be sufficient to increase mitochondrial biogenesis, which highlights the number of redundant signalling pathways involved in regulating oxidative capacity (Akimoto et al. 2005; Kim et al. 2008; Ryder et al. 2003). In spite of this, activation of AMPK is sufficient to induce exercise-type adaptations as evidenced by the fact that skeletal muscle expressing AMPK-activating γ1 or γ3 mutants have increased mitochondrial expression (Garcia-Roves et al. 2008; Nilsson et al. 2006; Rockl et al. 2007). Interestingly, lack of AMPK activity in skeletal muscle is associated with poor exercise performance however whether this is due to defects in fuel delivery or reduced mitochondrial capacity is unknown (Fujii et al. 2007).
9.8 AMPK in the Heart
The heart has perpetually high demands for energy in order to sustain critical processes such as contractile function and ion homeostasis. Control of energy metabolism in the heart is vital as it has a limited capacity to store fuel, therefore it is obligatory that cardiac energy homeostasis is tightly regulated to ensure energy production matches cardiac workload.
In the healthy heart, the large amounts of ATP required to maintain contractile function is derived mainly from oxidative metabolism of lipids in the mitochondria, and to a lesser extent from glucose metabolism (Wisneski et al. 1987). AMPK promotes fatty acid uptake and oxidation in the heart in response to contraction by stimulating translocation of the fatty acid transporter FAT/CD36 from intracellular vesicles to the plasma membrane, as well as enhancing transport of fatty acids into the mitochondria for oxidation via inhibition of ACC2 (Habets et al. 2007; Luiken et al. 2003). However, mitochondrial oxidative metabolism becomes severely attenuated during myocardial ischaemia, therefore under these conditions the heart must quickly switch to non-oxidative sources of ATP (Kloner and Jennings 2001). The steep decline in ATP levels that accompanies cardiac ischaemia rapidly activates AMPK, which orchestrates a shift from oxidative metabolism to anaerobic glucose metabolism by stimulating glucose uptake, as well as increasing flux through glycolysis (Dyck et al. 1999). It enhances the rate of glycolysis in the ischaemic heart by phosphorylating and stimulating phosphofructokinase-2 (PFK2) to produce fructose-2,6-bisphosphate, which in turn allosterically activates phosphofructokinase-1 (PFK1), the rate-limiting enzyme in glycolysis (Marsin et al. 2000). This effect of AMPK also provides an elegant explanation for the so-called Pasteur effect, which is an increased rate of glycolysis in response to hypoxia, a phenomenon first noted by Louis Pasteur.
Although glycolysis is a minor source of ATP in the heart under normal conditions, the small of amounts of ATP that are generated by glycolysis during ischaemia are critical to maintain essential processes such as ion homeostasis via ATP-dependent ion pumps (Xu et al. 1995). Enhancing anaerobic glucose metabolism during myocardial ischaemia is therefore a crucial metabolic response, so much so that inhibition of glucose transport or glycolysis drastically decreases the ability of the heart to withstand ischaemic insults. Although activation of AMPK in response to ischaemia is undoubtedly beneficial for the short term survival of the heart, there is evidence to suggest that it may be harmful to the recovery of the heart during reperfusion (Lopaschuk et al. 1993). This stems from the fact that the AMPK mediated increase in fatty acid oxidation that occurs during reperfusion de-couples glycolysis and glucose oxidation through inhibition of the pyruvate dehydrogenase complex, a phenomenon known as the Randle cycle, thereby causing pyruvate to be converted to the dead end product lactate resulting in lactic acidosis. Sustained increases in lactate can seriously impair cardiac function and efficiency, as it causes a diversion of ATP away from myocardial contraction towards restoring ion balance, which is necessary to counteract the effects of the acidosis and prevent Ca2+ overload. In this respect, decreasing fatty acid oxidation through inhibition of malonyl-CoA decarboxylase improves the recovery of contractile function after ischaemia (Dyck et al. 2004). However, future studies are required to definitively answer whether AMPK activation during ischaemia-reperfusion is beneficial or detrimental.
Naturally occurring mutations in the human γ2 subunit of AMPK cause a hypertrophic cardiomyopathy that is characterised by excessive glycogen storage in the cardiomyocyte, which results in the development of a ventricular pre-excitation disorder known as Wolff-Parkinson-White (WPW) syndrome (Akman et al. 2007; Arad et al. 2002; Burwinkel et al. 2005). Excessive glycogen in cardiac myocytes affects the development of the electrically insulating layer between the atria and ventricles, leading to accessory conductance pathways that compromise contractile function. The glycogen storage cardiomyopathy caused by the γ2 subunit mutations is similar to that observed in Pompe’s diseases, which also causes WPW syndrome due to impaired glycogenolysis (Bulkley and Hutchins 1978). All of the γ2 subunit mutations occur in the allosteric AMP and ATP binding sites and, with a few exceptions, affect the residues directly involved in nucleotide binding, particularly the positively charged residues that co-ordinate binding of the nucleotide phosphate groups (Xiao et al. 2007). The effect of the γ2 mutations on AMPK activity are complex because, as well as interfering with (and in some cases abolishing) allosteric activation by AMP (a loss of function effect), they also disrupt allosteric inhibition by ATP (a gain of function effect) (Adams et al. 2004; Scott et al. 2004). Therefore, although the mutations prevent activation by AMP, they also increase the basal activity of AMPK in the absence of any stimuli, which explains why these mutations are dominant in nature (Arad et al. 2002; Burwinkel et al. 2005). Constitutive activation of AMPK also provides a simple explanation for the observed glycogen storage phenotype. The increase in AMPK basal activity caused by the mutations stimulates glucose uptake that, in the absence of increased energy demand, causes a buildup of glucose-6-phosphate levels, which is preferentially diverted towards glycogen synthesis (Luptak et al. 2007). The accumulation of glucose-6-phosphate is further exacerbated by the fact that increased AMPK activity also stimulates fatty acid oxidation, which in turn inhibits glucose oxidation as a consequence of the Randle cycle. Although activation of AMPK results in phosphorylation and inhibition of glycogen synthase, this inhibitory effect is overcome by the high levels of glucose-6-phosphate, which is an allosteric activator of glycogen synthase (Jorgensen et al. 2004a). Some of the γ2 mutations also cause elevated glycogen in skeletal muscle where the γ2 subunit is also expressed, however, this does not appear to cause any obvious defects in muscle function. No mutations in γ1 have been identified so far, possibly because they are likely to be more detrimental than mutations in γ2 or γ3 given that the γ1 subunit is the predominant isoform in most tissue types.
9.9 Role of AMPK in Cancer
Several landmark studies in recent years suggest that AMPK signalling is important for the prevention and treatment of a range of human diseases, especially disorders of energy balance such as obesity and Type 2 diabetes. However, evidence is also beginning to emerge that other diseases such as cancer may have underlying defects in energy metabolism, and that activation of AMPK may also be beneficial in treating certain types of cancer. An important characteristic of cancer cells is that they acquire the ability to evade the normal regulatory mechanisms that control cell growth and proliferation, polarity and migration, and programmed cell death (apoptosis) (Hanahan and Weinberg 2000). This transformation to an autonomous state occurs as a result of multiple changes in the genome, typically as a consequence of mutations in oncogenic and tumour suppressor genes. Cancer cells also have fundamental differences in energy metabolism compared with normal cells, for example, cancer cells have a high rate of glucose consumption and produce ATP mainly by metabolising glucose to lactate rather than through oxidative phosphorylation, a phenomenon known as the ‘Warburg effect’ (Warburg 1956). As AMPK is downstream of the tumour suppressor LKB1, it is now regarded as a potential drug target for the treatment of cancer. As proof of concept, it was recently discovered that Type 2 diabetic patients receiving metformin treatment have a reduced rate of cancer incidence compared with patients on other drug programmes (Evans et al. 2005). Metformin is a widely used anti-diabetic drug that activates AMPK and has been shown to inhibit breast cancer cell growth in an AMPK-dependent manner, primarily via inhibition of the mammalian target of rapamycin (mTOR) pathway (Dowling et al. 2007; Zakikhani et al. 2006).
The mTOR pathway plays an important role in regulating cell growth and is overactivated in a variety of cancers (Schmelzle and Hall 2000). The central component of this pathway is mTOR, which exists within two distinct multiprotein complexes called mTORC1 and mTORC2 (Wullschleger et al. 2006). mTORC1 is composed of mTOR, raptor, PRAS40, mLST8 and is dependent on cellular nutrient availability as withdrawal of glucose, amino acids or oxygen rapidly suppresses mTORC1 activity. On the other hand, the mTORC2 complex consists of mSIN1, PRR5/Protor, mLST8 and rictor and is not responsive to nutrient levels. Several tumour suppressors such as PTEN, TSC1 (hamartin) and TSC2 (tuberin), and LKB1 have all been identified as negative regulators of mTOR signalling (Wullschleger et al. 2006). TSC1 and TSC2 form a heterodimer that is mutated in tuberous sclerosis complex, an inherited genetic disorder characterised by benign tumours in multiple organs that is strikingly similar, but distinct, from Peutz-Jeghers syndrome caused by mutations in LKB1 (van Slegtenhorst et al. 1998). mTORC1 is activated by an upstream pathway involving the TSC1-TSC2 heterodimer and Rheb, a member of the small G-protein family. AMPK inhibits mTORC1 signalling by a dual mechanism involving phosphorylation of TSC2 and raptor (Fig. 9.6). The TSC2 protein has a GTPase-activating protein (GAP) domain that inhibits the ability of Rheb to activate mTORC1, and this inhibitory effect is augmented by AMPK phosphorylation (Corradetti et al. 2004; Inoki et al. 2003). Meanwhile, phosphorylation of raptor by AMPK induces binding to 14-3-3 protein, which suppresses mTORC1 activity by preventing its interaction with downstream substrates 4EBP1 and ribosomal S6 kinase, two key regulators of protein translation (Gwinn et al. 2008). mTORC1 dependent protein translation is known to switch on the expression of a number of important cell growth regulators, including cyclin D1, the hypoxia inducible factor-1α (HIF-1α) transcription factor and c-myc, all of which are highly expressed in many types of cancer (Guertin and Sabatini 2007). In contrast to AMPK, protein kinase B (PKB) phosphorylates the TSC1-TSC2 complex on alternate sites in response to activation of the insulin/insulin-like growth factor-1 (IGF-1) signalling pathway, and inhibits its ability to switch off Rheb (Inoki et al. 2002). Therefore insulin and IGF-1, which indicate the availability of nutrients, and AMPK, which signals a shortage of energy or nutrients, have opposing effects on cell growth.

AMPK regulation of cell cycle and growth. AMPK inhibits cell growth via the mTORC1 complex by dual mechanism: direct inhibition and via the TSC1-TSC2 complex. Phosphorylation of p53 by AMPK induces cell cycle arrest
One of the most commonly mutated tumour suppressors in human cancers is p53, which is frequently referred to as the ‘guardian of the genome’ (Levine 1997). Expression of p53 is normally very low in the cell however conditions such as DNA damage caused by ionising radiation induce p53 transcription, which leads to the expression of multiple target genes that promote cell cycle arrest or apoptosis. AMPK phosphorylates p53 under conditions of low glucose but not ionising radiation, leading to cell cycle arrest in the G1/S phase and protection from apoptosis, whereas under prolonged glucose starvation AMPK promotes p53-dependent cell senescence (Imamura et al. 2001; Jones et al. 2005). AMPK is also activated by sestrin-1 and sestrin-2, both of which are transcriptionally upregulated by p53 in response to DNA damage and oxidative stress, however the mechanism by which the sestrins promote AMPK phosphorylation and activation is unknown (Budanov and Karin 2008). It has been suggested that the AMPK-p53 signalling axis may function as a metabolic checkpoint for cell growth that ensures energy supplies are adequate for cell cycle progression.
An unexpected role for AMPK that has recently emerged is in the regulation of cell polarity. Establishing and maintaining cell polarity is essential for organism development and tissue function, and loss of cell polarity is frequently associated with tumour invasion, particularly epithelial cell-derived malignant cancers (Williams and Brenman 2008). Clues to the involvement of AMPK in the regulation of cell polarity came from the discovery that LKB1 is required for polarisation of oocytes that determine the anterior-posterior axis of embryos, which likely explains why genetic deletion of the LKB1 gene is embryonic lethal (Martin and St Johnston 2003). LKB1 is also essential for establishing apical-basal polarity in epithelial cells (Baas et al. 2004). Although LKB1 is the upstream kinase for several other protein kinases, some of which have known roles in the establishment of cell polarity (particularly the microtubule-affinity regulating kinases), genetic deletion of AMPK in epithelial cells disrupts polarity as well as increasing cell proliferation, providing direct evidence that AMPK is also involved in these processes (Lee et al. 2007). Maintaining cell polarity is an energy intensive process, therefore it may seem incompatible that AMPK, which is activated by negative energy balance, should promote cell polarity especially during energy shortages. Nevertheless, the maintenance of cell polarity is an essential aspect of normal cell function and, therefore, this may be an instance where AMPK diverts what limited energy is available to a critical survival function.
9.10 AMPK Activators as Therapeutic Drugs
Pharmacological activation of AMPK is now considered a major therapeutic strategy to treat diseases that have underlying defects in the regulation of energy balance. Interest in AMPK as a drug target gained rapid momentum since the discovery that it is activated by the anti-diabetic drug metformin and is largely responsible for mediating its therapeutic effects. Until recently, the number of known activators of AMPK was limited, however a plethora of new activating agents have since been discovered. Unfortunately, the vast majority of these agents activate AMPK indirectly giving rise to multiple off-target effects, however this is expected to be less of a problem with the development of more direct activators. Regardless of their mechanism of action most, if not all, of the activators identified to date elicit positive metabolic effects consistent with activation of AMPK.
The most widely used drug to study the downstream effects of pharmacological activation of AMPK is the riboside AICAR (also known as acadesine) (Fig. 9.7). As mentioned previously, AICAR is a pro-drug that is phosphorylated by adenosine kinase to generate the AMP-mimetic, ZMP (Corton et al. 1995). Although ZMP mimics all the effects of AMP on the AMPK system, it also affects other AMP-sensitive enzymes including fructose-1,6-bisphosphatase and glycogen phosphorylase, both of which are important regulators of glucose metabolism (Vincent et al. 1991; Young et al. 1996). These selectivity problems and poor physicochemical properties have made AICAR and related nucleotide analogues unsuitable leads for further drug development (Dixon et al. 1991).

Activators of AMPK. AICAR is a pro-drug that is converted to the AMP-mimetic, ZMP. Metformin indirectly activates AMPK by inhibiting mitochondrial respiration. A769662, like AMP, directly activates AMPK but independently of the AMP-binding sites
AMPK is also activated by the glucose lowering drug metformin, which is the most commonly used pharmaceutical to treat Type 2 diabetes (Zhou et al. 2001). The therapeutic effects of metformin are primarily due to repression of gluconeogenesis in the liver and this effect is dependent on the presence of LKB1, which phosphorylates and activates AMPK (Shaw et al. 2005). There is also growing evidence that metformin is a potential anti-cancer drug, which may be related to the fact that LKB1 is a known tumour suppressor (Evans et al. 2005). Despite being able to activate AMPK in intact cells, metformin does not activate or affect the phosphorylation or dephosphorylation of AMPK by upstream kinases or phosphatases in cell-free assays (Hawley et al. 2002). Instead, metformin has been shown to be an inhibitor of complex I of the respiratory chain, which suggests that it activates AMPK by decreasing intracellular ATP and increasing AMP (Owen et al. 2000). Indeed, significant decreases in intracellular ATP have been detected in hepatocytes cultured with metformin, and in other cell types cultured with its more potent analogue phenformin, which likely explains some of their undesirable side-effects such as lactic acidosis (Guigas et al. 2006; Hawley et al. 2005). Inhibition of mitochondrial respiration is likely to be a common mechanism of action for the majority of indirect AMPK activators. In fact, the thiazolidinediones (another class of glucose lowering drug) and a number of natural plant compounds, such as berberine and resveratrol, have all been shown to activate AMPK by inhibiting mitochondrial respiration in much the same manner as metformin (Baur et al. 2006; Brunmair et al. 2004; Turner et al. 2008).
The first direct activator of AMPK to be discovered was the non-nucleoside thienopyridone, A769662 (Fig. 9.7) (Cool et al. 2006). AMPK activation by A769662 is similar to AMP, encompassing not only direct allosteric activation but also protection against dephosphorylation of Thr172 by protein phosphatases (Goransson et al. 2007; Sanders et al. 2007a). Intriguingly, its mechanism of action appears to be independent of the AMP-binding sites on the γ subunit since it can activate an AMP-insensitive mutant. Furthermore, A769662 selectively activates AMPK complexes containing the β1 subunit isoform but is without effect on β2 complexes (Scott et al. 2008). At present, the molecular basis of this selectivity is unknown, although activation is dependent on the β1 subunit carbohydrate-binding module. These findings are important as it highlights that isoform-specific activation of AMPK is achievable. In terms of metabolic effects, A769662 enhances whole-body fatty acid oxidation, lowers glucose and promotes weight loss predominantly via activation of AMPK in the liver (Cool et al. 2006). Notwithstanding these positive effects, A769662 has poor pharmacokinetic properties therefore it is unlikely to proceed as a potential therapy; however it does provide a useful lead for further drug development.
9.11 Concluding Remarks
Since its discovery over three decades ago, AMPK has ascended from being regarded simply as a sensor of cellular energy, to being recognised as a key player in the regulation of whole-body energy balance, earning the monicker of ‘metabolic master switch’. Although it is likely that AMPK emerged as a nutrient sensor in ancient unicellular eukaryotes, it appears to have become adapted during the evolution of multicellular organisms to become not only sensitive to cellular energy status, but also responsive to hormones that regulate whole-body energy balance. The broad spectrum of positive effects that AMPK exerts on both cellular and whole-body energy metabolism has made it an attractive target for new drugs to treat a range of metabolic disorders, particularly obesity and Type 2 diabetes, but perhaps also other diseases such as cancer. Significant strides have been made in the search for novel and direct activators of AMPK, however given that there are 12 possible combinations of AMPK heterotrimer with varying tissue expression and physiological effects, the challenge in the future will be to develop isoform-specific activators that target AMPK in particular tissues. Although this is an ambitious task, the discovery that the thienopyridone A769662 selectively activates AMPK complexes containing the β1 subunit provides proof of concept that the development of isoform specific activators is feasible. Future research in the AMPK field will undoubtedly continue to unravel the intricate mechanisms that regulate energy balance at both the cellular and whole-body levels, and provide critical information to understand and tackle the growing problem of metabolic diseases.
Abbreviations
- AMPK:
-
AMP-activated protein kinase
- ACC:
-
Acetyl CoA carboxylase
- AMP:
-
Adenosine 5’-monophosphate
- ATP:
-
Adenosine 5’-triphosphate
- CaMKK:
-
Ca2+/calmodulin dependent protein kinase kinase
- CBS:
-
Cystathionine-β-synthase domain
- CBM:
-
Carbohydrate-binding module
References
Abu-Elheiga, L., Jayakumar, A., Baldini, A., et al. (1995). Human acetyl-CoA carboxylase: characterization, molecular cloning, and evidence for two isoforms. Proc. Natl. Acad. Sci. USA 92:4011–4015.
Abu-Elheiga, L., Brinkley, W.R., Zhong, L., et al. (2000). The subcellular localization of acetyl-CoA carboxylase 2. Proc. Natl. Acad. Sci. USA 97:1444–1449.
Abu-Elheiga, L., Matzuk, M.M., Abo-Hashema, K.A., et al. (2001). Continuous fatty acid oxidation and reduced fat storage in mice lacking acetyl-CoA carboxylase 2. Science 291:2613–2616.
Adams, J., Chen, Z.P., Van Denderen, et al. (2004). Intrasteric control of AMPK via the gamma1 subunit AMP allosteric regulatory site. Protein Sci. 13:155–165.
Akimoto, T., Pohnert, S.C., Li, P., Zhang, M., et al. (2005). Exercise stimulates Pgc-1alpha transcription in skeletal muscle through activation of the p38 MAPK pathway. J. Biol. Chem. 280:19587–19593.
Akman, H.O., Sampayo, J.N., Ross, F.A., et al. (2007). Fatal infantile cardiac glycogenosis with phosphorylase kinase deficiency and a mutation in the gamma2-subunit of AMP-activated protein kinase. Pediatr. Res. 62:499–504.
Anderson, K.A., Ribar, T.J., Lin, F., et al. (2008). Hypothalamic CaMKK2 contributes to the regulation of energy balance. Cell Metab. 7:377–388.
Andersson, U., Filipsson, K., Abbott, C.R., et al. (2004). AMP-activated protein kinase plays a role in the control of food intake. J. Biol. Chem. 279:12005–12008.
Andreelli, F., Foretz, M., Knauf, C., et al. (2006). Liver adenosine monophosphate-activated kinase-alpha2 catalytic subunit is a key target for the control of hepatic glucose production by adiponectin and leptin but not insulin. Endocrinology 147:2432–2441.
Anthonsen, M.W., Ronnstrand, L., Wernstedt, C., et al. (1998). Identification of novel phosphorylation sites in hormone-sensitive lipase that are phosphorylated in response to isoproterenol and govern activation properties in vitro. J. Biol. Chem. 273:215–221.
Arad, M., Benson, D.W., Perez-Atayde, et al. (2002). Constitutively active AMP kinase mutations cause glycogen storage disease mimicking hypertrophic cardiomyopathy. J. Clin. Invest. 109:357–362.
Aschenbach, W.G., Hirshman, M.F., Fujii, N., et al. (2002). Effect of AICAR treatment on glycogen metabolism in skeletal muscle. Diabetes 51:567–573.
Baas, A.F., Kuipers, J., van der Wel, N.N., et al. (2004). Complete polarization of single intestinal epithelial cells upon activation of LKB1 by STRAD. Cell 116:457–466.
Barnes, B.R., Marklund, S., Steiler, T.L., et al. (2004). The 5’-AMP-activated protein kinase gamma3 isoform has a key role in carbohydrate and lipid metabolism in glycolytic skeletal muscle. J. Biol. Chem. 279:38441–38447.
Bateman, A. (1997). The structure of a domain common to archaebacteria and the homocystinuria disease protein. Trends Biochem. Sci. 22:12–13.
Baur, J.A., Pearson, K.J., Price, N.L., et al. (2006). Resveratrol improves health and survival of mice on a high-calorie diet. Nature 444:337–342.
Beg, Z.H., Stonik, J.A., Brewer, H.B. Jr. (1978). 3-Hydroxy-3-methylglutaryl coenzyme A reductase: regulation of enzymatic activity by phosphorylation and dephosphorylation. Proc. Natl. Acad. Sci. USA 75:3678–3682.
Bergeron, R., Previs, S.F., Cline, G.W., et al. (2001a). Effect of 5-aminoimidazole-4-carboxamide-1-beta-D-ribofuranoside infusion on in vivo glucose and lipid metabolism in lean and obese Zucker rats. Diabetes 50:1076–1082.
Bergeron, R., Ren, J.M., Cadman, K.S., et al. (2001b). Chronic activation of AMP kinase results in NRF-1 activation and mitochondrial biogenesis. Am. J. Physiol. Endocrinol. Metab. 281:E1340–1346.
Boudeau, J., Baas, A.F., Deak, M., et al. (2003). MO25alpha/beta interact with STRADalpha/beta enhancing their ability to bind, activate and localize LKB1 in the cytoplasm. EMBO J. 22:5102–5114.
Brabant, G., Muller, G., Horn, R., et al. (2005). Hepatic leptin signaling in obesity. FASEB J. 19:1048–1050.
Brunmair, B., Staniek, K., Gras, F., et al. (2004). Thiazolidinediones, like metformin, inhibit respiratory complex I: a common mechanism contributing to their antidiabetic actions? Diabetes 53:1052–1059.
Budanov, A.V. and Karin, M. (2008). p53 target genes sestrin1 and sestrin2 connect genotoxic stress and mTOR signaling. Cell 134:451–460.
Bulkley, B.H., and Hutchins, G.M. (1978). Pompe’s disease presenting as hypertrophic myocardiopathy with Wolff-Parkinson-White syndrome. Am. Heart. J. 96:246–252.
Burwinkel, B., Scott, J.W., Buhrer, C., et al. (2005). Fatal congenital heart glycogenosis caused by a recurrent activating R531Q mutation in the gamma 2-subunit of AMP-activated protein kinase (PRKAG2), not by phosphorylase kinase deficiency. Am. J. Hum. Genet. 76:1034–1049.
Carling, D. and Hardie, D.G. (1989). The substrate and sequence specificity of the AMP-activated protein kinase. Phosphorylation of glycogen synthase and phosphorylase kinase. Biochim. Biophys. Acta 1012:81–86.
Carling, D., Clarke, P.R., Zammit, V.A., et al. (1989). Purification and characterization of the AMP-activated protein kinase. Copurification of acetyl-CoA carboxylase kinase and 3-hydroxy-3-methylglutaryl-CoA reductase kinase activities. Eur. J. Biochem. 186:129–136.
Chavez, J.A., Roach, W.G., Keller, S.R., et al. (2008). Inhibition of GLUT4 translocation by Tbc1d1, a Rab GTPase-activating protein abundant in skeletal muscle, is partially relieved by AMP-activated protein kinase activation. J. Biol. Chem. 283:9187–9195.
Chen, H., Charlat, O., Tartaglia, L.A., et al. (1996). Evidence that the diabetes gene encodes the leptin receptor: identification of a mutation in the leptin receptor gene in db/db mice. Cell 84:491–495.
Chen, Z.P., Stephens, T.J., Murthy, S., et al. (2003). Effect of exercise intensity on skeletal muscle AMPK signaling in humans. Diabetes 52:2205–2212.
Chen, S., Murphy, J., Toth, R., et al. (2008). Complementary regulation of TBC1D1 and AS160 by growth factors, insulin and AMPK activators. Biochem. J. 409:449–459.
Civitarese, A.E., Ukropcova, B., Carling, S., et al. (2006). Role of adiponectin in human skeletal muscle bioenergetics. Cell Metab. 4:75–87.
Clarke, P.R. and Hardie, D.G. (1990). Regulation of HMG-CoA reductase: identification of the site phosphorylated by the AMP-activated protein kinase in vitro and in intact rat liver. EMBO J. 9:2439–2446.
Clarke, C.F., Edwards, P.A., Lan, S.F., et al. (1983). Regulation of 3-hydroxy-3-methylglutaryl-coenzyme A reductase mRNA levels in rat liver. Proc. Natl. Acad. Sci. USA 80:3305–3308.
Cool, B., Zinker, B., Chiou, W., et al. (2006). Identification and characterization of a small molecule AMPK activator that treats key components of type 2 diabetes and the metabolic syndrome. Cell Metab. 3:403–416.
Corradetti, M.N., Inoki, K., Bardeesy, N., et al. (2004). Regulation of the TSC pathway by LKB1: evidence of a molecular link between tuberous sclerosis complex and Peutz-Jeghers syndrome. Genes Dev. 18:1533–1538.
Corton, J.M., Gillespie, J.G., Hawley, S.A., et al. (1995). 5-aminoimidazole-4-carboxamide ribonucleoside. A specific method for activating AMP-activated protein kinase in intact cells? Eur. J. Biochem. 229:558–565.
Crute, B.E., Seefeld, K., Gamble, J., et al. (1998). Functional domains of the alpha1 catalytic subunit of the AMP-activated protein kinase. J. Biol. Chem. 273:35347–35354.
da Silva Xavier, G., Leclerc, I., Salt, I.P., et al. (2000). Role of AMP-activated protein kinase in the regulation by glucose of islet beta cell gene expression. Proc. Natl. Acad. Sci. USA 97:4023–4028.
Davies, S.P., Helps, N.R., Cohen, P.T., et al. (1995). 5’-AMP inhibits dephosphorylation, as well as promoting phosphorylation, of the AMP-activated protein kinase. Studies using bacterially expressed human protein phosphatase-2C alpha and native bovine protein phosphatase-2AC. FEBS Lett. 377:421–425.
Dixon, R., Gourzis, J., McDermott, D., et al. (1991). AICA-riboside: safety, tolerance, and pharmacokinetics of a novel adenosine-regulating agent. J. Clin. Pharmacol. 31:342–347.
Dowling, R.J., Zakikhani, M., Fantus, I.G., et al. (2007). Metformin inhibits mammalian target of rapamycin-dependent translation initiation in breast cancer cells. Cancer Res. 67:10804–10812.
Dyck, J.R., Kudo, N., Barr, A.J., et al. (1999). Phosphorylation control of cardiac acetyl-CoA carboxylase by cAMP-dependent protein kinase and 5’-AMP activated protein kinase. Eur. J. Biochem. 262:184–190.
Dyck, J.R., Cheng, J.F., Stanley, W.C., et al. (2004). Malonyl coenzyme a decarboxylase inhibition protects the ischemic heart by inhibiting fatty acid oxidation and stimulating glucose oxidation. Circ. Res. 94:e78–84.
Dzamko, N., Schertzer, J.D., Ryall, J.G., et al. (2008). AMPK-independent pathways regulate skeletal muscle fatty acid oxidation. J. Physiol. 586:5819–5831.
Evans, J.M., Donnelly, L.A., Emslie-Smith, A.M., et al. (2005). Metformin and reduced risk of cancer in diabetic patients. BMJ 330:1304–1305.
Fogarty, S. and Hardie, D.G. (2009). C-terminal phosphorylation of LKB1 is not required for regulation of AMP-activated protein kinase, BRSK1, BRSK2, or cell cycle arrest. J. Biol. Chem. 284:77–84.
Foretz, M., Carling, D., Guichard, C., et al. (1998). AMP-activated protein kinase inhibits the glucose-activated expression of fatty acid synthase gene in rat hepatocytes. J. Biol. Chem. 273:14767–14771.
Fox, C.J., Hammerman, P.S., Thompson, C.B. (2005). Fuel feeds function: energy metabolism and the T-cell response. Nat. Rev. Immunol. 5:844–852.
Fujii, N., Hayashi, T., Hirshman, M.F., et al. (2000). Exercise induces isoform-specific increase in 5’AMP-activated protein kinase activity in human skeletal muscle. Biochem. Biophys. Res. Commun. 273:1150–1155.
Fujii, N., Seifert, M.M., Kane, E.M., et al. (2007). Role of AMP-activated protein kinase in exercise capacity, whole body glucose homeostasis, and glucose transport in skeletal muscle -insight from analysis of a transgenic mouse model. Diabetes Res. Clin. Pract. 77 Suppl 1:S92–S98.
Gao, S., Kinzig, K.P., Aja, S., et al. (2007). Leptin activates hypothalamic acetyl-CoA carboxylase to inhibit food intake. Proc. Natl. Acad. Sci. USA 104:17358–17363.
Garcia-Roves, P.M., Osler, M.E., Holmstrom, M.H., et al. (2008). Gain-of-function R225Q mutation in AMP-activated protein kinase gamma3 subunit increases mitochondrial biogenesis in glycolytic skeletal muscle. J. Biol. Chem. 283:35724–35734.
Garton, A.J. and Yeaman, S.J. (1990). Identification and role of the basal phosphorylation site on hormone-sensitive lipase. Eur. J. Biochem. 191:245–250.
Garton, A.J., Campbell, D.G., Carling, D., et al. (1989). Phosphorylation of bovine hormone-sensitive lipase by the AMP-activated protein kinase. A possible antilipolytic mechanism. Eur. J. Biochem. 179:249–254.
Geraghty, K.M., Chen, S., Harthill, J.E., et al. (2007). Regulation of multisite phosphorylation and 14-3-3 binding of AS160 in response to IGF-1, EGF, PMA and AICAR. Biochem. J. 407:231–241.
Ghilardi, N., Ziegler, S., Wiestner, A., et al. (1996). Defective STAT signaling by the leptin receptor in diabetic mice. Proc. Natl. Acad. Sci. USA 93:6231–6235.
Gonzalez-Gay, M.A., De Matias, J.M., Gonzalez-Juanatey, C., et al. (2006). Anti-tumor necrosis factor-alpha blockade improves insulin resistance in patients with rheumatoid arthritis. Clin. Exp. Rheumatol. 24:83–86.
Goransson, O., McBride, A., Hawley, S.A., et al. (2007). Mechanism of action of A-769662, a valuable tool for activation of AMP-activated protein kinase. J. Biol. Chem. 282:32549–32560.
Guertin, D.A. and Sabatini, D.M. (2007). Defining the role of mTOR in cancer. Cancer Cell 12:9–22.
Guigas, B., Bertrand, L., Taleux, N., et al. (2006). 5-Aminoimidazole-4-carboxamide-1-beta-D-ribofuranoside and metformin inhibit hepatic glucose phosphorylation by an AMP-activated protein kinase-independent effect on glucokinase translocation. Diabetes 55:865–874.
Gwinn, D.M., Shackelford, D.B., Egan, D.F., et al. (2008). AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell 30:214–226.
Habets, D.D., Coumans, W.A., Voshol, P.J., et al. (2007). AMPK-mediated increase in myocardial long-chain fatty acid uptake critically depends on sarcolemmal CD36. Biochem. Biophys. Res. Commun. 355:204–210.
Hanahan, D. and Weinberg, R.A. (2000). The hallmarks of cancer. Cell 100:57–70.
Hardie, D.G. and Hawley, S.A. (2001). AMP-activated protein kinase: the energy charge hypothesis revisited. Bioessays 23:1112–1119.
Hawley, S.A., Davison, M., Woods, A., et al. (1996). Characterization of the AMP-activated protein kinase kinase from rat liver and identification of threonine 172 as the major site at which it phosphorylates AMP-activated protein kinase. J. Biol. Chem. 271:27879–27887.
Hawley, S.A., Gadalla, A.E., Olsen, G.S., et al. (2002). The antidiabetic drug metformin activates the AMP-activated protein kinase cascade via an adenine nucleotide-independent mechanism. Diabetes 51:2420–2425.
Hawley, S.A., Boudeau, J., Reid, J.L., et al. (2003). Complexes between the LKB1 tumor suppressor, STRAD alpha/beta and MO25 alpha/beta are upstream kinases in the AMP-activated protein kinase cascade. J. Biol. 2:28.
Hawley, S.A., Pan, D.A., Mustard, K.J., et al. (2005). Calmodulin-dependent protein kinase kinase-beta is an alternative upstream kinase for AMP-activated protein kinase. Cell Metab. 2:9–19.
Hemminki, A., Markie, D., Tomlinson, I., et al. (1998). A serine/threonine kinase gene defective in Peutz-Jeghers syndrome. Nature 391:184–187.
Holmes, B.F., Kurth-Kraczek, E.J., and Winder, W.W. (1999). Chronic activation of 5’-AMP-activated protein kinase increases GLUT-4, hexokinase, and glycogen in muscle. J. Appl. Physiol. 87:1990–1995.
Hong, Y.H., Varanasi, U.S., Yang, W. et al. (2003). AMP-activated protein kinase regulates HNF4alpha transcriptional activity by inhibiting dimer formation and decreasing protein stability. J. Biol. Chem. 278:27495–27501.
Hudson, E.R., Pan, D.A., James, J., et al. (2003). A novel domain in AMP-activated protein kinase causes glycogen storage bodies similar to those seen in hereditary cardiac arrhythmias. Curr. Biol. 13:861–866.
Hurley, R.L., Anderson, K.A., Franzone, J.M., et al. (2005). The Ca2+/calmodulin-dependent protein kinase kinases are AMP-activated protein kinase kinases. J. Biol. Chem. 280:29060–29066.
Imamura, K., Ogura, T., Kishimoto, A., et al. (2001). Cell cycle regulation via p53 phosphorylation by a 5’-AMP activated protein kinase activator, 5-aminoimidazole-4-carboxamide-1-beta-D-ribofuranoside, in a human hepatocellular carcinoma cell line. Biochem. Biophys. Res. Commun. 287:562–567.
Inoki, K., Li, Y., Zhu, T., et al. (2002). TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat. Cell Biol. 4:648–657.
Inoki, K., Zhu, T., and Guan, K.L. (2003). TSC2 mediates cellular energy response to control cell growth and survival. Cell 115:577–590.
Iseli, T.J., Walter, M., van Denderen, B.J., et al. (2005). AMP-activated protein kinase beta subunit tethers alpha and gamma subunits via its C-terminal sequence (186-270). J. Biol. Chem. 280:13395–13400.
Iseli, T.J., Oakhill, J.S., Bailey, M.F., et al. (2008). AMP-activated protein kinase subunit interactions: beta1:gamma1 association requires beta1 Thr-263 and Tyr-267. J. Biol. Chem. 283:4799–4807.
Iverson, A.J., Bianchi, A., Nordlund, A.C., et al. (1990). Immunological analysis of acetyl-CoA carboxylase mass, tissue distribution and subunit composition. Biochem. J. 269:365–371.
Jager, S., Handschin, C., St-Pierre, J., et al. (2007). AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc. Natl. Acad. Sci. USA 104:12017–12022.
Jin, X., Townley, R., Shapiro, L. (2007). Structural insight into AMPK regulation: ADP comes into play. Structure 15:1285–1295.
Johnson, L.N., Noble, M.E., Owen, D.J. (1996). Active and inactive protein kinases: structural basis for regulation. Cell 85:149–158.
Jones, R.G., Plas, D.R., Kubek, S., et al. (2005). AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol. Cell 18:283–293.
Jorgensen, S.B., Nielsen, J.N., Birk, J.B., et al. (2004a). The alpha2-5’AMP-activated protein kinase is a site 2 glycogen synthase kinase in skeletal muscle and is responsive to glucose loading. Diabetes 53:3074–3081.
Jorgensen, S.B., Viollet, B., Andreelli, F., et al. (2004b). Knockout of the alpha2 but not alpha1 5’-AMP-activated protein kinase isoform abolishes 5-aminoimidazole-4-carboxamide-1-beta-4-ribofuranoside but not contraction-induced glucose uptake in skeletal muscle. J. Biol. Chem. 279:1070–1079.
Jorgensen, S.B., Treebak, J.T., Viollet, B., et al. (2007). Role of AMPKalpha2 in basal, training-, and AICAR-induced GLUT4, hexokinase II, and mitochondrial protein expression in mouse muscle. Am. J. Physiol. Endocrinol. Metab. 292:E331–E339.
Kahn, B.B., Alquier, T., Carling, D. et al. (2005). AMP-activated protein kinase: ancient energy gauge provides clues to modern understanding of metabolism. Cell Metab. 1:15–25.
Kemp, B.E., Parker, M.W., Hu, S., et al. (1994). Substrate and pseudosubstrate interactions with protein kinases: determinants of specificity. Trends Biochem. Sci. 19:440–444.
Kim, M.S., Fielitz, J., McAnally, J., et al. (2008). Protein kinase D1 stimulates MEF2 activity in skeletal muscle and enhances muscle performance. Mol. Cell. Biol. 28:3600–3609.
Kloner, R.A. and Jennings, R.B. (2001). Consequences of brief ischemia: stunning, preconditioning, and their clinical implications: part 1. Circulation 104:2981–2989.
Koay, A., Rimmer, K.A., Mertens, H.D., et al. (2007). Oligosaccharide recognition and binding to the carbohydrate binding module of AMP-activated protein kinase. FEBS Lett. 581:5055–5059.
Kola, B., Hubina, E., Tucci, S.A., et al. (2005). Cannabinoids and ghrelin have both central and peripheral metabolic and cardiac effects via AMP-activated protein kinase. J. Biol. Chem. 280:25196–25201.
Kola, B., Boscaro, M., Rutter, G.A., et al. (2006). Expanding role of AMPK in endocrinology. Trends Endocrinol. Metab. 17:205–215.
Koo, S.H., Flechner, L., Qi, L., et al. (2005). The CREB coactivator TORC2 is a key regulator of fasting glucose metabolism. Nature 437:1109–1111.
Kraegen, E.W. and Cooney, G.J. (2008). Free fatty acids and skeletal muscle insulin resistance. Curr. Opin. Lipidol. 19:235–241.
Kramer, H.F., Witczak, C.A., Fujii, N., et al. (2006). Distinct signals regulate AS160 phosphorylation in response to insulin, AICAR, and contraction in mouse skeletal muscle. Diabetes 55:2067–2076.
Kubota, N., Yano, W., Kubota, T., et al. (2007). Adiponectin stimulates AMP-activated protein kinase in the hypothalamus and increases food intake. Cell Metab. 6:55–68.
Leclerc, I., Kahn, A., and Doiron, B. (1998). The 5’-AMP-activated protein kinase inhibits the transcriptional stimulation by glucose in liver cells, acting through the glucose response complex. FEBS Lett. 431:180–184.
Leclerc, I., Lenzner, C., Gourdon, L., et al. (2001). Hepatocyte nuclear factor-4alpha involved in type 1 maturity-onset diabetes of the young is a novel target of AMP-activated protein kinase. Diabetes 50:1515–1521.
Lee, Y., Yu, X., Gonzales, F., et al. (2002). PPAR alpha is necessary for the lipopenic action of hyperleptinemia on white adipose and liver tissue. Proc. Natl. Acad. Sci. USA 99:11848–11853.
Lee, Y., Naseem, R.H., Park, B.H., et al. (2006a). Alpha-lipoic acid prevents lipotoxic cardiomyopathy in acyl CoA-synthase transgenic mice. Biochem. Biophys. Res. Commun. 344:446–452.
Lee, Y.S., Kim, W.S., Kim, K.H., et al. (2006b). Berberine, a natural plant product, activates AMP-activated protein kinase with beneficial metabolic effects in diabetic and insulin-resistant states. Diabetes 55:2256–2264.
Lee, J.H., Koh, H., Kim, M., et al. (2007). Energy-dependent regulation of cell structure by AMP-activated protein kinase. Nature 447:1017–1020.
Levine, A.J. (1997). p53, the cellular gatekeeper for growth and division. Cell 88:323–331.
Lizcano, J.M., Goransson, O., Toth, R., et al. (2004). LKB1 is a master kinase that activates 13 kinases of the AMPK subfamily, including MARK/PAR-1. EMBO J. 23:833–843.
Lochhead, P.A., Salt, I.P., Walker, K.S., et al. (2000). 5-aminoimidazole-4-carboxamide riboside mimics the effects of insulin on the expression of the 2 key gluconeogenic genes PEPCK and glucose-6-phosphatase. Diabetes 49:896–903.
Lopaschuk, G.D., Wambolt, R.B., and Barr, R.L. (1993). An imbalance between glycolysis and glucose oxidation is a possible explanation for the detrimental effects of high levels of fatty acids during aerobic reperfusion of ischemic hearts. J. Pharmacol. Exp. Ther. 264:135–144.
Luiken, J.J., Coort, S.L., Willems, J., et al. (2003). Contraction-induced fatty acid translocase/CD36 translocation in rat cardiac myocytes is mediated through AMP-activated protein kinase signaling. Diabetes 52:1627–1634.
Luptak, I., Shen, M., He, H., et al. (2007). Aberrant activation of AMP-activated protein kinase remodels metabolic network in favor of cardiac glycogen storage. J. Clin. Invest. 117:1432–1439.
Marsin, A.S., Bertrand, L., Rider, M.H., et al. (2000). Phosphorylation and activation of heart PFK-2 by AMPK has a role in the stimulation of glycolysis during ischaemia. Curr. Biol. 10:1247–1255.
Marsin, A.S., Bouzin, C., Bertrand, L. et al. (2002). The stimulation of glycolysis by hypoxia in activated monocytes is mediated by AMP-activated protein kinase and inducible 6-phosphofructo-2-kinase. J. Biol. Chem. 277:30778–30783.
Martin, S.G. and St Johnston, D. (2003). A role for Drosophila LKB1 in anterior-posterior axis formation and epithelial polarity. Nature 421:379–384.
Matthews, V.B. and Febbraio, M.A. (2008). CNTF: a target therapeutic for obesity-related metabolic disease? J. Mol. Med. 86:353–361.
McBride, A., Ghilagaber, S., Nikolaev, A., et al. (2009). The glycogen-binding domain on the AMPK beta subunit allows the kinase to act as a glycogen sensor. Cell Metab. 9:23–34.
McGee, S.L. and Hargreaves, M. (2008). AMPK and transcriptional regulation. Front. Biosci. 13:3022–3033.
McTaggart, S.J. (2006). Isoprenylated proteins. Cell. Mol. Life Sci. 63:255–267.
Merrill, G.F., Kurth, E.J., Hardie, D.G., et al. (1997). AICA riboside increases AMP-activated protein kinase, fatty acid oxidation, and glucose uptake in rat muscle. Am. J. Physiol. 273:E1107–E1112.
Minokoshi, Y., Kim, Y.B., Peroni, O.D., et al. (2002). Leptin stimulates fatty-acid oxidation by activating AMP-activated protein kinase. Nature 415:339–343.
Minokoshi, Y., Alquier, T., Furukawa, N., et al. (2004). AMP-kinase regulates food intake by responding to hormonal and nutrient signals in the hypothalamus. Nature 428:569–574.
Mitchelhill, K.I., Stapleton, D., Gao, G., et al. (1994). Mammalian AMP-activated protein kinase shares structural and functional homology with the catalytic domain of yeast Snf1 protein kinase. J. Biol. Chem. 269:2361–2364.
Mitchelhill, K.I., Michell, B.J., House, C.M., et al. (1997). Posttranslational modifications of the 5’-AMP-activated protein kinase beta1 subunit. J. Biol. Chem. 272:24475–24479.
Momcilovic, M., Hong, S.P., Carlson, M. (2006). Mammalian TAK1 activates Snf1 protein kinase in yeast and phosphorylates AMP-activated protein kinase in vitro. J. Biol. Chem. 281:25336–25343.
Mootha, V.K., Lindgren, C.M., Eriksson, K.F., et al. (2003). PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 34:267–273.
Mu, J., Brozinick, J.T., Jr., Valladares, O., et al. (2001). A role for AMP-activated protein kinase in contraction- and hypoxia-regulated glucose transport in skeletal muscle. Mol. Cell 7:1085–1094.
Muoio, D.M., Seefeld, K., Witters, L.A., et al. (1999). AMP-activated kinase reciprocally regulates triacylglycerol synthesis and fatty acid oxidation in liver and muscle: evidence that sn-glycerol-3-phosphate acyltransferase is a novel target. Biochem. J. 338:783–791.
Nilsson, E.C., Long, Y.C., Martinsson, S., et al. (2006). Opposite transcriptional regulation in skeletal muscle of AMP-activated protein kinase gamma3 R225Q transgenic versus knock-out mice. J. Biol. Chem. 281:7244–7252.
Ouchi, N., Kihara, S., Arita, Y., et al. (2001). Adipocyte-derived plasma protein, adiponectin, suppresses lipid accumulation and class A scavenger receptor expression in human monocyte-derived macrophages. Circulation 103:1057–1063.
Owen, M.R., Doran, E., and Halestrap, A.P. (2000). Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem J. 348:607–614.
Pang, T., Xiong, B., Li, J.Y., et al. (2007). Conserved alpha-helix acts as autoinhibitory sequence in AMP-activated protein kinase alpha subunits. J. Biol. Chem. 282:495–506.
Parker, G.J., Koay, A., Gilbert-Wilson, R., et al. (2007). AMP-activated protein kinase does not associate with glycogen alpha-particles from rat liver. Biochem. Biophys. Res. Commun. 362:811–815.
Paterson, J., Kelsall, I.R., Cohen, P.T. (2008). Disruption of the striated muscle glycogen-targeting subunit of protein phosphatase 1: influence of the genetic background. J. Mol. Endocrinol. 40:47–59.
Polekhina, G., Gupta, A., Michell, B.J., et al. (2003). AMPK beta subunit targets metabolic stress sensing to glycogen. Curr. Biol. 13:867–871.
Polekhina, G., Gupta, A., van Denderen, B.J., et al. (2005). Structural basis for glycogen recognition by AMP-activated protein kinase. Structure 13:1453–1462.
Richards, A.A., Stephens, T., Charlton, H.K., et al. (2006). Adiponectin multimerization is dependent on conserved lysines in the collagenous domain: evidence for regulation of multimerization by alterations in posttranslational modifications. Mol. Endocrinol. 20:1673–1687.
Rockl, K.S., Hirshman, M.F., Brandauer, J., Fujii, N., Witters, L.A., and Goodyear, L.J. (2007). Skeletal muscle adaptation to exercise training: AMP-activated protein kinase mediates muscle fiber type shift. Diabetes 56:2062–2069.
Ryder, J.W., Bassel-Duby, R., Olson, E.N., et al. (2003). Skeletal muscle reprogramming by activation of calcineurin improves insulin action on metabolic pathways. J. Biol. Chem. 278:44298–44304.
Saghizadeh, M., Ong, J.M., Garvey, W.T., et al. (1996). The expression of TNF alpha by human muscle. Relationship to insulin resistance. J. Clin. Invest. 97:1111–1116.
Sakamoto, K. and Holman, G.D. (2008). Emerging role for AS160/TBC1D4 and TBC1D1 in the regulation of GLUT4 traffic. Am. J. Physiol. Endocrinol. Metab. 295:E29–E37.
Sanders, M.J., Ali, Z.S., Hegarty, B.D., et al. (2007a). Defining the mechanism of activation of AMP-activated protein kinase by the small molecule A-769662, a member of the thienopyridone family. J. Biol. Chem. 282:32539–32548.
Sanders, M.J., Grondin, P.O., Hegarty, B.D., et al. (2007b). Investigating the mechanism for AMP activation of the AMP-activated protein kinase cascade. Biochem. J. 403:139–148.
Sapkota, G.P., Kieloch, A., Lizcano, J.M., et al. (2001). Phosphorylation of the protein kinase mutated in Peutz-Jeghers cancer syndrome, LKB1/STK11, at Ser431 by p90(RSK) and cAMP-dependent protein kinase, but not its farnesylation at Cys(433), is essential for LKB1 to suppress cell growth. J. Biol. Chem. 276:19469–19482.
Scheidereit, C. (2006). IkappaB kinase complexes: gateways to NF-kappaB activation and transcription. Oncogene 25:6685–6705.
Schmelzle, T. and Hall, M.N. (2000). TOR, a central controller of cell growth. Cell 103:253–262.
Scott, J.W., Norman, D.G., Hawley, S.A., et al. (2002). Protein kinase substrate recognition studied using the recombinant catalytic domain of AMP-activated protein kinase and a model substrate. J. Mol. Biol. 317:309–323.
Scott, J.W., Hawley, S.A., Green, K.A., et al. (2004). CBS domains form energy-sensing modules whose binding of adenosine ligands is disrupted by disease mutations. J. Clin. Invest. 113:274–284.
Scott, J.W., Ross, F.A., Liu, J.K. et al. (2007). Regulation of AMP-activated protein kinase by a pseudosubstrate sequence on the gamma subunit. EMBO J. 26:806–815.
Scott, J.W., van Denderen, B.J., Jorgensen, S.B., et al. (2008). Thienopyridone drugs are selective activators of AMP-activated protein kinase beta1-containing complexes. Chem. Biol. 15:1220–1230.
Scott, J.W., Oakhill, J.S., and van Denderen, B.J. (2009). AMPK/SNF1 structure: a menage a trois of energy-sensing. Front. Biosci. 14:596–610.
Shaw, R.J., Lamia, K.A., Vasquez, D., et al. (2005). The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science 310:1642–1646.
Stahmann, N., Woods, A., Carling, D. et al. (2006). Thrombin activates AMP-activated protein kinase in endothelial cells via a pathway involving Ca2+/calmodulin-dependent protein kinase kinase beta. Mol. Cell Biol. 26:5933–5945.
Steinberg, G.R., Rush, J.W., and Dyck, D.J. (2003). AMPK expression and phosphorylation are increased in rodent muscle after chronic leptin treatment. Am. J. Physiol. Endocrinol. Metab. 284:E648–E654.
Steinberg, G.R., Michell, B.J., van Denderen, B.J., et al. (2006). Tumor necrosis factor alpha-induced skeletal muscle insulin resistance involves suppression of AMP-kinase signaling. Cell Metab. 4:465–474.
Steinberg, G.R., Watt, M.J., and Febbraio, M.A. (2009). Cytokine regulation of AMPK signalling. Front. Biosci. 14:1902–1916.
Suter, M., Riek, U., Tuerk, R., et al. (2006). Dissecting the role of 5’-AMP for allosteric stimulation, activation, and deactivation of AMP-activated protein kinase. J. Biol. Chem. 281:32207–32216.
Suzuki, A., Kusakai, G., Shimojo, Y., et al. (2005). Involvement of transforming growth factor-beta 1 signaling in hypoxia-induced tolerance to glucose starvation. J. Biol. Chem. 280:31557–31563.
Tamas, P., Hawley, S.A., Clarke, R.G., et al. (2006). Regulation of the energy sensor AMP-activated protein kinase by antigen receptor and Ca2+ in T lymphocytes. J. Exp. Med. 203:1665–1670.
Tan, M.J., Ye, J.M., Turner, N., et al. (2008). Antidiabetic activities of triterpenoids isolated from bitter melon associated with activation of the AMPK pathway. Chem. Biol. 15:263–273.
Thomson, D.M., Porter, B.B., Tall, J.H., et al. (2007). Skeletal muscle and heart LKB1 deficiency causes decreased voluntary running and reduced muscle mitochondrial marker enzyme expression in mice. Am. J. Physiol. Endocrinol. Metab. 292:E196–E202.
Tokumitsu, H., Iwabu, M., Ishikawa, Y., et al. (2001). Differential regulatory mechanism of Ca2+/calmodulin-dependent protein kinase kinase isoforms. Biochemistry 40:13925–13932.
Tomas, E., Tsao, T.S., Saha, A.K., et al. (2002). Enhanced muscle fat oxidation and glucose transport by ACRP30 globular domain: acetyl-CoA carboxylase inhibition and AMP-activated protein kinase activation. Proc. Natl. Acad. Sci. USA 99:16309–16313.
Treebak, J.T., Glund, S., Deshmukh, A., et al. (2006). AMPK-mediated AS160 phosphorylation in skeletal muscle is dependent on AMPK catalytic and regulatory subunits. Diabetes 55:2051–2058.
Turner, N., Li, J.Y., Gosby, A., et al. (2008). Berberine and its more biologically available derivative, dihydroberberine, inhibit mitochondrial respiratory complex I: a mechanism for the action of berberine to activate AMP-activated protein kinase and improve insulin action. Diabetes 57:1414–1418.
van Slegtenhorst, M., Nellist, M., Nagelkerken, B., et al. (1998). Interaction between hamartin and tuberin, the TSC1 and TSC2 gene products. Hum. Mol. Genet. 7:1053–1057.
Vincent, M.F., Marangos, P.J., Gruber, H.E., et al. (1991). Inhibition by AICA riboside of gluconeogenesis in isolated rat hepatocytes. Diabetes 40:1259–1266.
Viollet, B., Andreelli, F., Jorgensen, S.B., et al. (2003). The AMP-activated protein kinase alpha2 catalytic subunit controls whole-body insulin sensitivity. J. Clin. Invest. 111:91–98.
Virbasius, J.V. and Scarpulla, R.C. (1994). Activation of the human mitochondrial transcription factor A gene by nuclear respiratory factors: a potential regulatory link between nuclear and mitochondrial gene expression in organelle biogenesis. Proc. Natl. Acad. Sci. USA 91:1309–1313.
Wang, M.Y., Orci, L., Ravazzola, M., et al. (2005). Fat storage in adipocytes requires inactivation of leptin’s paracrine activity: implications for treatment of human obesity. Proc. Natl. Acad. Sci. USA 102:18011–18016.
Wang, C., Mao, X., Wang, L., et al. (2007). Adiponectin sensitizes insulin signaling by reducing p70 S6 kinase-mediated serine phosphorylation of IRS-1. J. Biol. Chem. 282:7991–7996.
Warburg, O. (1956). On the origin of cancer cells. Science 123:309–314.
Warden, S.M., Richardson, C., O’Donnell, J. Jr., et al. (2001). Post-translational modifications of the beta-1 subunit of AMP-activated protein kinase affect enzyme activity and cellular localization. Biochem J. 354:275–283.
Watt, M.J., Dzamko, N., Thomas, W.G., et al. (2006). CNTF reverses obesity-induced insulin resistance by activating skeletal muscle AMPK. Nat. Med. 12:541–548.
Williams, T. and Brenman, J.E. (2008). LKB1 and AMPK in cell polarity and division. Trends Cell Biol. 18:193–198.
Winder, W.W., Holmes, B.F., Rubink, D.S., et al. (2000). Activation of AMP-activated protein kinase increases mitochondrial enzymes in skeletal muscle. J. Appl. Physiol. 88:2219–2226.
Wisneski, J.A., Gertz, E.W., Neese, R.A., et al. (1987). Myocardial metabolism of free fatty acids. Studies with 14C-labeled substrates in humans. J. Clin. Invest. 79:359–366.
Wojtaszewski, J.F., Nielsen, P., Hansen, B.F., et al. (2000). Isoform-specific and exercise intensity-dependent activation of 5’-AMP-activated protein kinase in human skeletal muscle. J. Physiol. 528:221–226.
Wojtaszewski, J.F., Mourtzakis, M., Hillig, T., et al. (2002). Dissociation of AMPK activity and ACCbeta phosphorylation in human muscle during prolonged exercise. Biochem. Biophys. Res. Commun. 298:309–316.
Woods, A., Munday, M.R., Scott, J., et al. (1994). Yeast SNF1 is functionally related to mammalian AMP-activated protein kinase and regulates acetyl-CoA carboxylase in vivo. J. Biol. Chem. 269:19509–19515.
Woods, A., Azzout-Marniche, D., Foretz, M., et al. (2000). Characterization of the role of AMP-activated protein kinase in the regulation of glucose-activated gene expression using constitutively active and dominant negative forms of the kinase. Mol. Cell. Biol. 20:6704–6711.
Woods, A., Dickerson, K., Heath, R., et al. (2005). Ca2+/calmodulin-dependent protein kinase kinase-beta acts upstream of AMP-activated protein kinase in mammalian cells. Cell Metab. 2:21–33.
Wu, X., Motoshima, H., Mahadev, K., et al. (2003). Involvement of AMP-activated protein kinase in glucose uptake stimulated by the globular domain of adiponectin in primary rat adipocytes. Diabetes 52:1355–1363.
Wullschleger, S., Loewith, R., and Hall, M.N. (2006). TOR signaling in growth and metabolism. Cell, 124:471–484.
Xiao, B., Heath, R., Saiu, P., et al. (2007). Structural basis for AMP binding to mammalian AMP-activated protein kinase. Nature 449:496–500.
Xu, K.Y., Zweier, J.L., and Becker, L.C. (1995). Functional coupling between glycolysis and sarcoplasmic reticulum Ca2+ transport. Circ. Res. 77:88–97.
Yamauchi, T., Kamon, J., Minokoshi, Y., et al. (2002). Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nat. Med. 8:1288–1295.
Yamauchi, T., Nio, Y., Maki, T., et al. (2007). Targeted disruption of AdipoR1 and AdipoR2 causes abrogation of adiponectin binding and metabolic actions. Nat. Med. 13:332–339.
Young, M.E., Radda, G.K., and Leighton, B. (1996). Activation of glycogen phosphorylase and glycogenolysis in rat skeletal muscle by AICAR- an activator of AMP-activated protein kinase. FEBS Lett. 382:43–47.
Zakikhani, M., Dowling, R., Fantus, I.G., et al. (2006). Metformin is an AMP kinase-dependent growth inhibitor for breast cancer cells. Cancer Res. 66:10269–10273.
Zhou, G., Myers, R., Li, Y., et al. (2001). Role of AMP-activated protein kinase in mechanism of metformin action. J. Clin. Invest. 108:1167–1174.
Zhou, G., Sebhat, I.K., and Zhang, B.B. (2009). AMPK activators. Potential therapeutics for metabolic and other diseases. Acta Physiol. (Oxf) 196:175–190.
Acknowledgements
I would like to thank Prof Bruce Kemp for mentoring and helpful discussions. This work was funded by the Australian Research Council and the National Health and Medical Research Council.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2011 Springer Science+Business Media, LLC
About this chapter
Cite this chapter
Scott, J.W. (2011). Maintaining Energy Balance in Health and Disease: Role of the AMP-Activated Protein Kinase. In: Vidal, C. (eds) Post-Translational Modifications in Health and Disease. Protein Reviews, vol 13. Springer, New York, NY. https://doi.org/10.1007/978-1-4419-6382-6_9
Download citation
DOI: https://doi.org/10.1007/978-1-4419-6382-6_9
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4419-6381-9
Online ISBN: 978-1-4419-6382-6
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)