
Abstract
Primary hyperparathyroidism and secondary disease from chronic kidney disease are highlighted in this chapter. Up-to-date descriptions of new pathological mechanisms focus on fibroblastic growth factor 23 and its emergent importance in overall pathology. Historical, clinical, and examination findings are highlighted and compliment data on laboratory techniques, diagnostic imaging, and skeletal histology clinical workup of the patients. Treatment options at the dialysis and pharmacological levels are blended with some of the nutritional elements in phosphorus control. The integration of all elements is summarized at the end of the section to emphasize its influence on bone metabolism.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Primary hyperparathyroidism
- Insulinoma
- Gastrinoma
- Pheochromocytoma
- Hypercalcemia
- RET
- HRPT2
- Renal function
- 1,25 (OH)2 vitamin D
- Glomerular filtration
- Homeostasis
- J-shaped curve
- Chronic kidney disease
- FGF 23
- Vitamin D receptor
- RANK ligand
- Osteoclastogenesis
- Renal osteodystrophy
- Calciphylaxis
- Fetuin
- GLA protein
- Epitopes
- Serum calculation
- Ostase (bone alkaline phosphatase)
- TRAP-5b
- Osteitits fibrosa
- Parathyroidectomy
- Calcimimetics
- Ca × P product
- Randomized controlled trial
- Vascular calcification
- Venous sampling
- Parathyroid scanning
- Oxyphilic cells
- Neoplasia
- Osteoprotegerin
- Sodium thiosulfate
- Sestamibi
- Technetium 99m
- Osteoclasts
- Osteoblasts
- Trabecular
Pathophysiology and Epidemiology
Parathyroid glands are located in close proximity to the thyroid gland on their posterior surface. In normal health, the parathyroids are small (2–5 mm). Normally the four (sometimes more) glands are derived from the neural crest mesenchyme and the brachial pouch endoderm. The glands are composed of two cell types, of which the “chief cells” are the ones that generate parathyroid hormone (PTH). The larger oxyphilic cells are lighter in staining, fewer in number, and have no known physiologic role. A histological picture of an enlarged, proliferated gland is given in Fig. 5.1.

Low magnification of a hyperplastic intrathyreoidal located adenomatous parathyroid gland. Kindly contributed by Andreas Plötner, Pathological Institute, Hospital St. Georg, Leipzig
Excessive secretion of PTH is mostly a consequence of adenomatous proliferation of parathyroid glands in which case it is referred to as primary (p) hyperparathyroidism (HPT). The causative circumstance of such benign status is not known. However, in about 1% of pHPT, parathyroid carcinoma is the underlying cause [1]. Some genetic risk loci have been identified among which mutations in HRPT2 and RET [2] are the most important ones. Patients inheriting these genes develop parathyroid carcinoma and phaechromocytoma. In some occasions, pHPT may be associated syndromatically with other endocrinological disorders (multiple endocrinological neoplasia, MEN). In these rare conditions, endocrinological symptoms are often combined depending on the type of disease. In MEN type 1, pHPT coincidences with gastrointestinal tumors like insulinoma or gastrinoma. In MEN 2A, pheochromocytoma is the most frequent combination.
In pHPT, PTH oversecretion is frequently followed by impaired renal function due to hypercalcemia, which is a harm to healthy kidneys. Hypercalcemia in pHPT is stimulated by increased dietary calcium absorption and calcium mobilization from the bone hydroxyapatite depot. Therefore, in sustained and nontreated pHPT, renal failure due to hypercalcemia is typical sequelae. This condition causes secondary HPT and, therefore, mixes up with the primary problem of pHPT. Following this situation, a pHPT might not be easy to distinguish from secondary forms, if renal injury is already present. The frequency of newly diagnosed pHPT is higher than one might expect: 1 out of 500 women and 1 out 2,000 men were found to be affected among 60-year-old individuals in a population-based registration in Rochester, MN, in 1989 [3].
Functional hyperplasia and hypertrophy of the parathyroid glands due to chronic kidney disease (CKD) are characterized as “secondary” hyperparathyroidism (sHPT). However, following the subsequent pathophysiological considerations, the phrase “functional” hyperparathyroidism is a more appropriate wording. The morphological and imaging aspects of this disease are characterized in a particular chapter. From a functional point of view, sHPT is sequelae of altered mineral metabolism in CKD.
Healthy kidneys coregulate calcium homeostasis by different mechanisms. The major downstream pathway of kidney metabolism in terms of bone and mineral metabolism has been focused to the parathyroids many years ago. However, in such classical reasoning, the renal–parathyroid axis has been thought to be mediated through downregulation of vitamin D activation only. In fact, it is long known, that hydroxylation and activation of inactive vitamin D into the active form, Di-hydroxyl vitamin D (1.25 OH2D, calcitriol), take place at the tubular-interstitial compartment of kidneys. Already in early forms of CKD, that activation and subsequently the synthesis of 1.25 OH2D are abolished. The levels of 1.25 OH2D in sera of patients with CKD are inversely and linearly correlated with excretory renal function, measured by glomerular filtration rate (GFR) [4].
As this, the classical concept of sHPT reflects the adaptations to decreased 1.25 OH2D. Since calcium absorption from the bowel wall is mediated by 1.25 OH2D, a decrease of that hormone results in calcium deficit, particularly present in a reduced level of free serum calcium. In the serum, calcium is partly bound to albumin resulting in a difference between whole and acting/free calcium. Free calcium content of sera is sensed very precisely and tightly by the calcium-sensing receptors (CaR) of the parathyroids [5]. A decrease of free calcium leads to increase of PTH synthesis [6]. PTH increases serum calcium by its action on bowel vitamin D uptake and Ca mobilization from the bone hydroxyapatite depot. By that means, the decrease of 1.25 OH2D is counter-regulated and the serum-free calcium level, which has much downstream impacts, can be partly maintained through a wide range of renal insufficiency. However, the gross calcium balance including the bone depot is negative and the adaptation maintains serum homeostasis by means of decalcification of bones. During last years, these mechanisms have been discussed more and more as adaptive ones, i.e., in certain sense as a physiological response. A lot of discussion is ongoing concerning the question, if PTH elevation is a pathological process per se, or if the onset of disease-mediating mechanisms has to be determined at a certain PTH threshold. While this is controversial, from a functional viewpoint, an intact calcium–PTH regulation axis could be seen as one aspect of a truly adaptive process [7, 8]. Such classical experiments establishing a J-shaped Ca–PTH response curve [6] have recently been performed in the end-stage renal failure setting using Ca dialysis bath concentration variation and measuring PTH response [9]. The degree of PTH response, measured as relationship between set point and slope of the curve (Fig. 5.2), can be seen as a measure of appropriate regulation.

Original (panel a, left) and schematic (panel b, right) drawing of the relationship between calcium concentration and PTH response in healthy (a) and renal population (b) with still functioning response in secondary (s) and autonomous (a) HPT. Y-axis PTH concentration, X-axis Ca concentration. Response is characterized by slope, midpoint, maximum and minimum which might resemble the functioning status of the Ca–PTH regulation. Adapted from Brown EM, Gardner DG, Brennan MF et al. Calcium-regulated parathyroid hormone release in primary hyperparathyroidism: studies in vitro with dispersed parathyroid cells. Am J Med. 1979;66(6):923–31
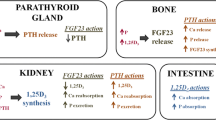
By note, serum phosphorus elevation comes into play only in later stages of CKD and acts as further stimulus of PTH elevation. Besides a remarkable degree of excretory renal failure in CKD stage 3 and 4, phosphorus (P) levels usually will be found not elevated in these stages [10]. The reason of that difference in time course (P ↑ late, Ca ↓ and PTH ↑ early) has been clarified at least in part during the last few years. Phosphaturia has been demonstrated to be promoted not only by PTH but also by fibroblast growth factor 23 (FGF-23), which is secreted mainly from bone-forming osteoblasts [11]. FGF-23 exerts a very strong phosphaturic effect [12–14]. However, it does not only stimulate phosphaturia but also diminish activation of vitamin D by a strong inhibitory effect on 1-α-hydroxylase resulting in 1.25 OH2D downregulation [14] and does not inhibit HPT [15]. Therefore, FGF-23 is likely to maintain normal phosphate homeostasis in early stages of CKD on the cost of decreased 1.25 OH2D levels comparable to PTH elevation which is the pay-off to stabilize calcium in early CKD (Fig. 5.3).

Simplified drawing of time-dependent alterations of mineral metabolism in chronic kidney disease
In such a concept, looking at PTH and FGF-23, the resulting 1.25 OH2D deficiency is the key-playing negative regulator in mineral metabolism changes due to CKD. 1.25 OH2D binds to its receptor (vitamin D receptor, VDR) which is located not only throughout the body, including parathyroids, bowel wall but also in endothelium and immune cells. Like other members of the steroid receptor family, the VDR acts as a ligand-activated transcription factor. Classical down-stream effectors of the 1.25 OH2D–VDR complex include enhancement of small-bowel calcium absorption and osteoclastogenesis and liberation of calcium from bones. The latter effect is exerted via the regulation of receptor activator of NF-B ligand (RANKL)–receptor activator of NF-B (RANK) interactions and osteoprotegerin interplay through the 1.25 OH2D–VDR complex. Yet this interaction is complex and altered by PTH. Newer findings suggests that bone forming in the presence of altered 1.25 OH2D–VDR complex only transforms to bone resorption when PTH increase is paralleled by active vitamin D deficiency [16]. Inactivation of the 25-hydroxyvitamin D 1-hydroxylase and vitamin D receptor demonstrates independent and interdependent effects of calcium and vitamin D on skeletal and mineral homeostasis. This means that if the Ca/1.25 OH2D → PTH response is not functional, bone turnover is abolished resulting in bone loss. This condition is characterized as “dead bone disease” but incorporates not only skeletal but also systemic pathological features.
Pathophysiology of Bone Disorder in CKD-MBD
“Dead bone disease” or low turnover renal osteodystrophy (ROD) is one of three particular pathologies related to mineral–parathyroid regulation in frame of CKD. While low-turnover bone disease is mostly characterized by “arrested” bone metabolism with both inactive osteoblasts and osteoclasts and low PTH, high-turnover bone disease is identified by stimulated bone metabolisms and both cell types together with increased PTH.
During recent years, the focus has moved from “bone only” to “bone and vasculature.” The hypothesis behind that reasoning is a pathological translocation of calcium salts from bones to vasculature mediated by mechanisms, among which sHPT is one of the best investigated ones. In fact, the presence of ectopic vascular calcification (VC) and the transition of smooth muscle cells (SMC) to osteoblasts represents a cornerstone of vascular risk and an independent risk factor of mortality [17]. Adjusted for age, the risk of death, is elevated up to 100-fold in patients with CKD 5 compared with healthy controls [18]. However, the molecular mechanisms leading to VC and the possible link between bone disease (somewhat mechanistic seen as “calcification minus”) and vessel disease (“calcification plus”) have not been established until very recent years. During last years, markers of mineral bone metabolism and parathyroid dysregulation have been clearly associated with patient survival [19]. This is true for the serum concentrations of calcium and phosphorus and, in smaller extent with a more J-shaped mortality association curve, for PTH [19]. The role of FGF-23 in terms of survival has to be investigated in future studies. The pathophysiological impacts, which bone disease might execute on vasculature and cardiovascular risk in renal failure, led to the newly inaugurated entity “chronic kidney disease-mineral bone disease” (CKD-MBD). This scenario comprises risks of osteodystrophy like pathological fractures and sequelae of bone decalcification as calcium salt deposition. That deposition may either occur in an elementary form, e.g., in grafted kidneys resulting in earlier transplant loss [20] or in more complex forms together with phosphorus deposition in “brown tumors” of skin and joints, which are already longer known.
Calciphylaxis
In particular, changes of the skin comprising painful ulceration and inflammation and occurring along with calcification of middle and small arteries intima-media-layers (Fig. 5.4) have been defined as calciphylaxis (Fig. 5.5), a condition inheriting 50–80% mortality. At current, there are no evidence-based treatment schemes available for such deleterious complication. However, chelation and excretion of calcium salts with sodium thiosulfate [21–28], lowering calcium concentration by intensified dialysis and pharmaceutical regimes and parathyroidectomy (PTX) yielded beneficial results in single cases and small series [29]. Calcification does not occur only in patients who are subjected to high serum levels of calcium, phosphorus, and PTH. In particular, calciphylaxis and coronary artery calcification have been observed even in patients with normal and low-normal serum calcium. Therefore, the incidence of extraosseous calcification has been thought to be mediated by other factors as calcium, too. Since precipitation of calcium salts and not the presence of dissolved free calcium is the key pathological issue, research on precipitating cofactors has been promoted recently. Among those, fetuin and matrix gla protein have been identified as precipitation inhibitors [30] and a deficiency of these agents has been hypothesized as cofactors of precipitation. In vitro precipitation experiments with fetuin supported that reasoning [30–32]. Since fetuin and matrix gla protein are related to inflammatory activation, a very attractive link has been identified connecting the frequent occurrence of calcification in patients with inflammation. Another important cofactor for precipitation of calcium salts is the inhibition of vitamin K synthesis by warfarin [33], which consequently has to be stopped in calciphylaxis.

Calcification of intima-media layer of small arteries (→ arrow) in calciphylaxis (von Kossa staining, ×100 magnification). Kindly contributed by Dr. Jens Plöthner, Pathological Institute, Hospital St. Georg, Leipzig, Germany

Ulcerated skin surface of the lower leg of a patient with calciphylaxis, concomitant with intima-media arterial calcification seen in Fig. 5.3
Inflammation, particularly subclinical microinflammation, is overrepresented in cohorts with renal disease in general. Without dissecting details, extracorporeal (dialysis) circuits, autoimmune kidney diseases, subclinical transplant rejection, and uremic inflammation are the most important headings pointing to microinflammation in these patients. Taking all recent keys together, to describe a nowadays comprehensive picture of “diseases of parathyroids” in renal patients, hyperplasia and hypertrophy of parathyroid glands with subsequent PTH oversecretion is only one aspect of a more complex disease described as CKD-MBD [34]. This complex focuses more on “end-organ-damage” of PTH in terms of calcification of vasculature, specific calcification disease of the skin (calciphylaxis), and renal bone osteodystrophy. That end-organ-damage is mediated by cofactors apart from the calcium–parathyroid axis like precipitation inhibitors and inflammation.
See Chapter 6 for detailed discussion.
Clinical Course, History, and Physical Findings
In primary HPT, which is sometimes a nondiscovered silent disease, hypocalcemia causes the most important symptoms. Central nervous effects of hypocalcemia, if not abrogated by chronic course, are the leading symptoms. Somnolence, cramps, or in lower grade fatigue together with increased serum calcium (in pHPT) must lead to PTH assessment and enable the diagnosis.
An obstacle to withstand a correct and early diagnosis is the later renal effects of hypocalcemia. If pHPT is not diagnosed early, renal calcinosis is a frequent late squeal. In such a situation, as in any CKD, sHPT due to vitamin D deficiency decreases serum calcium and mixes up with the pHPT being the underlying disease in such cases. In general, in more chronic cases of any HPT, clinical symptoms are not very characteristic but must be evaluated by biochemical studies. A summary of symptoms with most observed frequencies is listed in Table 5.1 [35, 36].
Diagnostic Techniques
Imaging
The role of preoperative imaging in HPT is controversial due to varying success rates reported in studies [37]. In the more recent years, ultrasound (US) and technetium 99m-methoxyisobutylisonitrile (sestamibi) scintigraphy scan has been used most frequently. Mihai et al. [38] found in an evidence-based analysis, that sestamibi is a recommended primary test, but ultrasound in experienced hands turned out to be a valuable alternative (see example in Fig. 5.6). The authors concluded, that if both investigations are concordant, a minimal invasive surgical approach is indicated, if only one of the two investigations is positive, a unilateral exploration with intraoperative PTH assay should be performed. In case of two negative investigations, a bilateral exploration should be planned [38]. The technological development of US devices (resolution down to 1 mm) currently contributes to use of such methodology. If glands are located atypically, CT scans (Fig. 5.7) might be helpful.

(a) Ultrasonography of right adenomatous parathyroid gland (+ picture marks of enlarged gland). (b) Ultrasonography of left adenomatous parathyroid gland, note central inhomogenous structure (+ picture marks of enlarged gland, ACI = internal carotid artery, VJi = internal jugular vene, Li Lappen = left thyroid lobe)

CT scan of atypical localization of parathyroidal glands (between trachea and vena cava)
Laboratory Techniques
Most optimal, patients suffering from sHPT and or CKD-MBD should be diagnosed by screening measures during the course of CKD. One of the most discussed issues was establishing thresholds, from which sHPT should be regarded or even treated. Recent guidelines of the Kidney disease global outcome initiative KDIGO [39] discarded further CKD stage-dependent PTH limits and use threshold of up to ninefold “normal” PTH, where normal refers to local reference. This is meaningful, since PTH assays are yet not standardized between different systems and laboratories.
PTH secretion underlies a both short frequent and circadian rhythm. Therefore, for diagnostic studies, standardized assessment time points must be used. Laboratories do use different assay techniques, which target different epitopes of the 84-amino acid protein. The level of PTH compared to baseline can be used to establish surgical success (Table 5.2).
Serum calcium can be measured as free serum calcium or as whole calcium including its albumin-bound proportion. In the latter case, for correct assessment, it must be corrected for plasma albumin concentration
Assessment of Bone Disease
Although no clinical routine method, evaluation of bone status is important to distinguish between conditions of either arrested or stimulated turnover of cellular bone-forming mechanisms. That cellular turnover is denoted by a steady state of activity and number of osteoclasts and osteoblasts at the front of trabecular mineralization. It is meaningful, since therapeutic measures are different in such states. Measuring bone density alone cannot help to distinguish between these entities since it gives only information about content of mineral salts. The histomorphometric approach is based on standardized parameters, which were described in 1987 [40]. Bone histology must be processed following certain procedures both in harvesting, transfer and preparation of the samples. Prior to biopsy, patients should be given tetracycline 250 mg bid at day minus 20 and minus 5 before biopsy. That substance accumulates at the endosteal mineralization front and allows quantification by spontaneous fluorescence. Bone sample biopsy is usually done using a mechanical-driven drill device. Since such approach includes relevant patients discomfort and pain, we use a 4-mm diameter bone marrow biopsy trochar (Pajunk™), which allows a safe and uncomplicated biopsy with satisfying biopsy samples. As biopsy site, we use the posterior upper iliac spine. Samples must be transferred to acetone solution (using mineral glassware) and can be shipped without cooling to bone histology. For embedding, methyl-methacrylate will be used. Pathologists use different staining procedures, with Masson-Goldner and Giemsa being the most important ones. Quantitative histomorphometry is usually done by manual methods of Merz and Schenk [41] or semiautomatically following Malluche [42]. A pathological bone status in CKD is considered from a histopathological point of view as ROD (Table 5.3, Figs. 5.8– 5.11). However in recent years, with advanced understanding of systemic implications of pathological bone state, the more comprehensive term CKD-MBD has been used for clinical perception.

Type 1 ROD. High-turnover variant of osteitis fibrosa with massive osteoclastogenesis (→), high resorptive activity, Masson-Goldner. Kindly contributed by Dr. Gabriele Lehmann, University Hospital Jena, Germany

Type 3 ROD mixed osteitis fibrosa, peritrabecular fibrosis (→) and osteoclastic activity (→), Masson-Goldner. Kindly contributed by Dr. Gabriele Lehmann, University Hospital Jena, Germany

Osteomalacia, intratrabecular disturbed mineralization (→), Masson-Goldner. Kindly contributed by Dr. Gabriele Lehmann, University Hospital Jena, Germany

Adynamic bone disease, only small osteoid volume (→) detectable; decreased cellular activity, Masson-Goldner. Kindly contributed by Dr. Gabriele Lehmann, University Hospital Jena, Germany
Biochemical markers of bone status have been studied widely. Although none of these markers have been shown to fit perfectly with the gold standard bone histomorphometry, measuring the bone alkaline phosphatase (ostase) is the most used surrogate. Combination with PTH yielded about 60% fit with histology [43]. Other bone markers include urine cross-links, i.e., fragmentation products of bone collagen. The diagnostic value of these markers is not completely accepted by all. Following trends maybe more helpful than a single-point measurement in time. Therefore, they are promising but must be handled with caution.
Alternative Diagnostic Tools
Biochemical Fine Needle Aspiration
A biochemical fine needle aspiration with PTH measurement allows differentiation of parathyroid tissue from lymph nodes and thyroid tissue with a specificity of 100%. The sample is drawn through a 25-gauge needle, the aspirate is diluted with 1 ml saline solution; after centrifugation for 10 min, the supernatant is used for the PTH measurement [44]. This technique allows prompt tissue identification without frozen section, thereby limiting the preparation of nonparathyroid tissue. It has been proposed for preoperative procedures although with a weak level of evidence but recommendations at grade B [38].
Differential Internal Jugular Venous Sampling
When the preoperative localization techniques are equivocal, a venous sampling may guide the surgeon to the side harboring the hyperfunctioning gland. It has been proposed to analyze simultaneously samples from both jugular veins and from a peripheral vein. In most cases, indirect regionalization, however, should be reserved for preoperative procedures particularly after an extended previous exploration.
Treatment
Therapeutic intervention in dysbalanced mineral-metabolic pathways should consider multiple pathways as outlined in the pathophysiology chapter. In brief and principle, intervention can be done at subsequent levels. Vitamin D replacement in “hormonal doses” acknowledges the central role of that hormone in CKD-MBD. Abolishment of the activity of the parathyroid can be performed by either excision of the glands totally or in parts, by pharmacological intervention in PTH secretion by means of calcimimetics or by “pharmacological vitamin D dose intervention” with the active, double hydroxylated form of vitamin D (calcitriol or analogs/derivatives). Not very acknowledged, but helpful, is treating CKD-MBD by modified dialysis conditions, which will be considered first.
Impact of Dialysis Techniques
Both hemo (HD) and peritoneal dialysis (PD) fluids do use external solute conditions which have been based on a positive net calcium balance to suppress PTH secretion of patients in earlier era. In hemodialysis, calcium bath has been set to 1.5 or 1.75 mEq/l, in PD to 1.2 mEq/l. Nowadays, with the focus on avoidance of PTH oversuppression along with low turnover ROD, calcium baths will be fixed at 1.0–1.5 mEq/l in HD or at 0.9–1.2 mEq/l in PD targeting a neutral calcium balance. That goal, however, is influenced by further dialysis conditions and might be difficult to accomplish. Since patients exhibit different bone status, individualization with regard to clinical condition is a necessary tool to consider [45].
Vitamin D Analogues
Prospective studies have shown that pharmacological doses of Calcitriol and other active vitamin D derivatives suppress parathyroid function and lower PTH levels in CKD and reduced even mortality in retrospective data analysis [46]. Osseous effects include a reduction in bone turnover, improvement of osteitits fibrosa, and stimulation of bone mineralization. Side effects comprise hypercalcemia and hyperphosphatemia. Oral and intravenous forms are on the market, which can be adapted to the kind of treatment, to which patients are subjected (Table 5.4).
The correction of vitamin D deficiency (as assessed by low 25 vitamin D 1-25 OH2D ) in CKD patients has not yet been proven to result in better clinical outcomes. However in routine clinical practice, a vitamin D 1-25 OH2D level of 45 ng/ml is currently considered optimal and can be reached by daily or weekly application of 1,000 or 10,000 international units, respectively. Hypocalcemia, which is on the other hand a frequent side effect of active vitamin D (i.e., calcitriol), is not common with low inactive vitamin D 1-25 OH2D (i.e., low 25 vitamin D) and the therapy is not costly. Therefore, many nephrologists treat their patients routinely with vitamin D2 or D3 during European and American winter seasons with low light conditions.
Parathyroidectomy
In primary HPT due to adenoma, surgery is the primary therapy of choice to avoid secondary end-organ damage with renal calcinosis being the first line (indications in Table 5.5). Indications were defined by a 2002 National Institute’s of Health summary statement [47]. A clear indication is given along with high-turnover ROD (types 1 and 3) refractory to active vitamin D and calcimimetics. In non- or oligosymptomatic patients, particularly with psychiatric symptoms, decision-making has to incorporate individual aspects of the patients (Table 5.6). These “nonspecific” symptoms, however, can be sufficiently treated by PTX. Furthermore, reduction of blood pressure, left ventricular hypertrophy after PTX have been reported [48–53]. In young patients with established but not sufficient effective conservative therapy, indication for surgery should be established more liberally compared to older and multimorbid patients.
Despite the introduction of new therapeutics like calcimimetics or vitamin D derivatives, these classical indications are still valid in the presence of drug resistance [54]. A recent study has shown the increased mortality due to cardiovascular events in chronic renal failure patients [55].
HPT can resolve after kidney transplantation, but persistent disease may occur in 10–50% of the patients [56]. Recently, the consequences of abnormal mineral metabolism after kidney transplantation have been evaluated in a retrospective study. 11–25% of patients showed abnormal serum calcium or an increased Ca × P product within the first year after kidney transplantation. A calcium >10.5 mg/dl at 3 months was identified as an independent risk factor for recipient death (OR 3.0; 95% CI: 1.2–7.4); a Ca >10.5 mg/dl at 12 months was an independent risk factor for death censored graft loss (OR 4.0; 95% CI: 1.2–14) [57]. Other retrospective studies showed reduced graft survival after PTX following kidney transplantation [58], and was augmented by another German [59] but contradicted by Dutch study [49, 60]
Operative Technique
In case of a conventional surgical procedure, a Kocher’s incision is the standard approach to the thyroid and the parathyroids. The strap muscles are divided in the midline, the parathyroids can then be approached on either side. A precise anatomic knowledge is the key for a rapid and safe exploration (Fig. 5.12).

Operative figure of an adenomatous parathyroid gland within the thyreothymic ligament
In case of a positive preoperative localization in one or two investigations, a unilateral preparation is justified. After the removal of one enlarged gland, an intraoperative PTH allows to decide whether or not a further neck exploration is required. In contrast to a systematic bilateral exploration, an adequate drop of the PTH in the PTH assay after extirpation of one enlarged gland allows to finish the operation. However the interpretation of the results remains controversial in some details, the success rates of parathyroid surgery using this technique reach 96–98%.
In case of secondary HPT, a bilateral exploration is mandatory in every case. There have been some controversial debates whether to perform a total or a subtotal PTX. Lorenz et al. evaluated the results of total versus subtotal PTX in renal hyperparathyroidism. They showed that measurable PTH following total PTX is the consequence of isolated cell nests that may be responsible for recurrences with time. The authors favored in their conclusions the advantage of total PTX and cryopreservation with regard to the prevention of recurrence [61].
Follow-up of surgical removal must include close monitoring of serum calcium. Serum calcium level decreases due to recalcification of bones (“hungry bone syndrome”) in a typical course within first days and weeks after surgery with the frequent sequela of symptomatic hypocalcemia (Fig. 5.13). Typical symptoms include diathesis and muscular cramps, i.e., the clinical picture of tetanus. Therefore, calcium must be supported orally or even intravenously, necessitating a central venous line. Intravenous calcium infusion raises the danger of cardiac arrhythmia and must not be avoided by using infusion rates less than 5 me/h. Oral calcium frequently causes nausea and vomiting. To overcome these shortcomings, active vitamin D beginning with a (high) daily dose of 2 micg is the therapy of best choice and should be titrated to low-to-normal serum calcium concentration. Since “hungry bone” resolves after weeks to months, that active D therapy has to be adapted following laboratory controls every 1–4 weeks.

Course of serum calcium after parathyroidectomy in renal transplant patients
Cryopreservation of Parathyroid Tissue
In 1975, S.A. Wells reported his first experiences in autotransplantation of parathyroid tissue [62]. In 29 patients, Wells et al. demonstrated the functional status of the glands 1 year after transplantation. The technique of immediate transplantation of parathyroid tissue during total thyroidectomy of cancer has been proposed in 1977 [63]. The risk of recurrent hyperparathyroidism following transplantation of parathyroid tissue from a parathyroid adenoma has been shown first by Brennan et al. [64]. One case report has shown the occurrence of HPT after transplantation of a normal parathyroid [65].
Questions about the use of cryopreserved tissue and the viability after long-term storage are still matter of debate [66]. Brennan et al. first described viable grafted parathyroids after as long as 18 months of cryopreservation [67].
Independent of the surgical approach, a cryopreservation of parathyroid tissue should be planned in every case of secondary and sporadic or hereditary multiglandular hyperparathyroidism. A frozen section of the parathyroids to be cryopreserved is recommended. The tissue is placed in ice-cold physiologic saline solution, thereafter it is placed into a Petri dish with RPMI 1640 culture medium (RPMI: Roswell Park Memorial Institute). With a sharp scalpel, fragments of 1–2 mm are prepared, 5–8 pieces are stored in one tube, several tubes may be used [68].
Calcimimetics
Calcimimetics interact with the CaR and increase its sensitivity to calcium to lower PTH secretion without increasing calcium serum concentration, which was significantly shown in a controlled, randomized clinical trial [69]. They, moreover, enhance the expression of CaR [70] and VDR [71] and inhibit parathyroid gland proliferation [72]. This differentiates calcimimetics from the vitamin D analogs, which enhance calcium and phosphate resorption from the intestinal tract and increase serum calcium and serum phosphate. The EVOLVE study, a randomized placebo-controlled outcome trial of 3,883 patients to evaluate the influence of cinacalcet on mortality is ongoing and results are expected at the end of 2011 [73]. In a meta-analysis of already conducted studies, the frequency of bone fractures and cardiovascular morbidity (hospital admissions) was reported to be reduced [74]. The KDIGO guidelines [39] give recommendations for using calcimimetics to lower PTH levels.
Calcimimetics after Renal Transplantation
After renal transplantation with a sufficient kidney graft, in the vast majority of cases, sHPT resolves together with reductions of serum calcium and phosphorus. In a few cases that process does not occur appropriately, since parathyroid glands have been proliferated due to long lasting stimulation. That process is comparable to primary HPT with low phosphorus and high calcium both in opposite to sHPT. That condition is called transplant-persistent HPT and should be treated thoroughly since grafts are in danger due to interstitial calcification. Therapy must consider specific pathophysiological features of such state, what usually makes active vitamin D inappropriate due to related hypercalcemia. Cinacalcet has been applied successfully [75–79] but is yet not approved in most European countries and the USA. PTX, in particular, in patients with already injured renal function boosts the risk of graft loss [58] but must be considered anyway in some cases. To overcome the pitfalls, it must be recommended to work-up sHPT in patients awaiting renal transplantation. Patients in need of intensive medical therapy (active vitamin D and cinacalcet >30 mg/day) should be treated with PTX and cryoconservation of glands for later autologous retransplantation.
Phosphorus Control in CKD
Hyperphosphatemia is a key issue in chronic renal disease as outlined in the introductory pathophysiology chapter. Since 50% uptake from the intestine is passive and uncontrolled, renal excretion is of crucial impact for phosphate homeostasis. However, while normophosphatemia is maintained through a wide range of renal failure presumably by the early exert of FGF-23, the association of increased phosphate levels with mortality is out of any doubt. This was shown in dialysis-dependent patients [80], in renal transplantation [57], and even in healthy subjects like the Framingham population [81, 82]. On the other hand, no randomized controlled trial (RCT) has ever been conducted comparing phosphate lowering versus no measure in terms of hard endpoint (mortality). There were, however, numerous efforts to decrease phosphate levels by means of both nonpharmaceutical and pharmaceutical measures.
As for nonpharmaceutical action taking, it is important to recognize that modification of diet with avoidance of phosphate intake is crucial to help patients maintain their phosphate homeostasis. Physicians should convey detailed information to their patients displaying the phosphorus content of daily nutrients. Of note, convenience and preconditioned food is stabilized with phosphate-containing ingredients. Gross amounts of phosphate are carried with Coke (soft drink) of any manufacturer, tinned food and softened cheese, to mention (not comprehensively) the most important ones. Phosphorus can hardly be avoided completely since it comes along with protein-rich food in general. A list of phosphate content of nutrients can be retrieved at a list of the NIH [83]. Patients in stage 5 CKD necessitating dialysis do benefit from formalized nutrition programs which declare phosphorus content and aim to reduce nutritional intake [84]. Phosphate control is of particular importance in patients undergoing peritoneal dialysis compared to hemodialysis. The overall phosphorus excretion is higher in HD compared to PD [85] and can be increased further by choosing convective procedure modalities like hemodiafiltration and usage of larger dialysis filters. However, the best measure to increase phosphorus elimination is prolongation of dialysis time, e.g., use of nocturnal dialysis three times per week for 8 h [86].
Following the need of protein-rich nutrition in hemodialysis, in a majority of patients, nutritional intake must exceed excretion capability and therefore must be coped by measures to decrease intestinal absorption. Historically (until the mid-1980s), aluminum was the cornerstone of phosphate binding. However, oral administration was complicated by systemic toxicity, which was mainly related to accumulation in brain, bone, and joints. Therefore, aluminum is nowadays mostly excluded from clinical use and clinical research, too. If such neglect is fully justified can only be speculated [87] since the poisonous properties were largely interlinked with covariates like nonsterile dialysis water or bone turnover oversuppression, which have changed significantly in recent times. The first substitute of aluminum, introduced in the late 1980s years, was calcium-based binders [88], namely calcium carbonate and calcium acetate. These drugs have medium phosphate-binding capacity compared to aluminum and were never tested for outcome in RCT. However, they are the most applied binders until recent times. Along with the discussion of extraosseous calcification in frame of CKD-MBD, calcium-containing phosphate binders have been incriminated to promote calcification-related morbidity and mortality [89, 90]. In a small RCT (n = 27) comparing calcium carbonate and a calcium-free alternative (sevelamer), attenuation of vascular calcification has been reported [91]. Sevelamer was introduced in 1997 and acts through its anion-exchange capabilities within the bowel. Their phosphate-binding capacity is smaller compared to calcium salts and aluminum, and moderate gastrointestinal side effects are common. However, sevelamer showed in RCTs and retrospective studies slowing of calcification and amelioration of bone disease [92–96]. One RCT exploring mortality could not show a significant effect of sevelamer versus calcium salts [97].
A further calcium and aluminum-free compound is lanthanum carbonate. As aluminum, lanthanum is a triple charged metal and has been therefore discussed widely envisioning cerebral and bone accumulation and toxicity. In fact it deposits in bones, but not at the mineralization front. In animal toxicological experiments, with multiplies of human dosage, it could be detected in rat brains [98, 99] while no human central nervous system toxicity was reported so far [99, 100]. Phosphate-binding capacity is high, so the substance is in principle highly needed nonaluminum, noncalcium choice. RCTs resulted in substantial phosphate lowering capacity and showed no severe side effects [101] but a high incidence of substance discontinuation in verum groups. No RCT was targeted and powered to demonstrate mortality benefit.
Taking these issues together, there is currently no “ideal” phosphate binder available. Our current practice is summarized in Table 5.7 and can be appreciated further in a current comprehensive review from Tonelli et al. [102].
Research on new substances does focus on iron-based binders, niacin, and chitosan chewing gums while no such substances are being currently available on the market. In a phase 1 study, an iron-containing compound (SBR 759) showed phosphate reduction into the KDIGO target using a dose between 3.5 and 15 g [103], which translates into a pill burden of up to 20 tablets per day. Niacin, in opposite to direct intestinal binding of all other substances, inhibits the phosphate uptake into the bowel wall by inhibiting the small bowel phosphate cotransporter [104]. Chewing gums containing chitosan [105, 106] have experimentally shown to absorb phosphate already with salivation in the mouth.
Influencing Bone Metabolism
Serum parameters of mineral metabolism do not depict the whole complexity of CKD-MBD, in particular, the bone features end the vascular injury permitted by such disease. To target these aspects, influencing bone turnover has come into scope during recent years again after two decades of neglect along with diminished use of bone biopsy. While having information’s about ROD type, influence on bone turnover, i.e., activity of osteoclasts or osteoblasts might be a sufficient choice to treat patients with CKD-MBD.
In low-turnover ROD (type 4), osteo-arresting therapies like bisphosphonates, convey enhancement of bone disease. They are, however, currently widely applied in postmature osteoporosis even in CKD without information on bone status. Bisphosphonates might be helpful in ROD type 1 or 3, which should be established initially by bone histomorphometry. In two studies, alendronat has been proved to influence bone diseases effectively in patients with primary HPT [107, 108]. In a clinical analog, pHPT could be translated in sHPT without low-turnover aspects and with high HPT and therefore be treated with bisphosphonates, although no RCT for such reasoning is available
In low-turnover ROD type 4, arrest of osteoblasts must be resolved. In patients with CKD 4 or 5, most frequently, oversuppression of PTH by active vitamin D and/or positive calcium intake by phosphate binders and/or inadequately high calcium dialysis bath concentration (down to 1.0 mEq/l) must be stopped or adopted, respectively. Teriparatide, i.e., synthetic PTH, resembles a pharmaceutical alternative for enhancing bone turnover. The substance has been used for fracture-prone osteoporosis [109] and could be considered in CKD-MBD associated with abolished PTH secretion as, for instance, after PTX. In a small open-label study in seven patients undergoing chronic hemodialysis with low-turnover ROD, bone mineral density but not coronary calcification scores were improved [110].
Integrative CKD-MBD Therapy
In summary, treatment of secondary hyperparathyroidism must not target PTH levels above certain numerical thresholds alone but has to envision the concept of parathyroid function as a variable of complex regulation of the endocrine–vasculature–bone axis in CKD. A schematic proposal of integrative therapy is given in Fig. 5.14. To conduct such time-dependent and multiple-level approach, continuous monitoring of parameters of CKD-MBD is mandatory. According to recent KDIG guidelines, monthly laboratory monitoring of serum calcium and phosphorus levels in patients dependent from renal replacement therapy is recommended. PTH should be regarded at least every 3 months. Although not directly included in the guidelines, we would recommend additional monitoring of “bone markers” like Ostase every 3 months. Bone biopsy and histomorphometry should be done, when differentiation between high and low-turnover bone disease cannot be done clinically and sufficient therapy choice can be considered. PTX is the therapy of choice in irreversible, autonomous parathyroid hypersecretion along with high-turnover bone disease with a particular regard to patients awaiting renal transplantation.

Schematic drawing of integrative therapeutic approach in secondary HPT
Overall, the field evolves very rapidly and further implication, e.g., of adipose and muscle tissue can be awaited during the near future. With regard to the excess mortality in CKD, physicians must make any effort, to overcome or at least lighten such disease burden in their patients.
References
Marcocci C, Cetani F, Rubin MR, Silverberg SJ, Pinchera A, Bilezikian JP. Parathyroid carcinoma. J Bone Miner Res. 2008;23(12):1869–80.
Kouvaraki MA, Shapiro SE, Perrier ND, et al. RET proto-oncogene: a review and update of genotype-phenotype correlations in hereditary medullary thyroid cancer and associated endocrine tumors. Thyroid. 2005;15(6):531–44.
Heath III H, Hodgson SF, Kennedy MA. Primary hyperparathyroidism. Incidence, morbidity, and potential economic impact in a community. N Engl J Med. 1980;302(4):189–93.
Mak RH, Wong JH. The vitamin D/parathyroid hormone axis in the pathogenesis of hypertension and insulin resistance in uremia. Miner Electrolyte Metab. 1992;18(2–5):156–9.
Brown EM. PTH secretion in vivo and in vitro. Regulation by calcium and other secretagogues. Miner Electrolyte Metab. 1982;8(3–4):130–50.
Brown EM, Wilson RE, Eastman RC, Pallotta J, Marynick SP. Abnormal regulation of parathyroid hormone release by calcium in secondary hyperparathyroidism due to chronic renal failure. J Clin Endocrinol Metab. 1982;54(1):172–9.
Rodriguez M, Canalejo A, Garfia B, Aguilera E, Almaden Y. Pathogenesis of refractory secondary hyperparathyroidism. Kidney Int Suppl. 2002;80: 155–60.
Felsenfeld AJ, Rodriguez M, guilera-Tejero E. Dynamics of parathyroid hormone secretion in health and secondary hyperparathyroidism. Clin J Am Soc Nephrol. 2007;2(6):1283–305.
Holgado R, Haire H, Ross D, et al. Effect of a low calcium dialysate on parathyroid hormone secretion in diabetic patients on maintenance hemodialysis. J Bone Miner Res. 2000;15(5):927–35.
Gutierrez O, Isakova T, Rhee E, et al. Fibroblast growth factor-23 mitigates hyperphosphatemia but accentuates calcitriol deficiency in chronic kidney disease. J Am Soc Nephrol. 2005;16(7): 2205–15.
Benet-Pages A, Lorenz-Depiereux B, Zischka H, White KE, Econs MJ, Strom TM. FGF23 is processed by proprotein convertases but not by PHEX. Bone. 2004;35(2):455–62.
Rodriguez M, Felsenfeld AJ. PTH, FGF-23 and early CKD. Nephrol Dial Transplant. 2008;23(11): 3391–3.
Quarles LD. FGF23, PHEX, and MEPE regulation of phosphate homeostasis and skeletal mineralization. Am J Physiol Endocrinol Metab. 2003;285(1): E1–9.
Yu X, White KE. FGF23 and disorders of phosphate homeostasis. Cytokine Growth Factor Rev. 2005;16(2):221–32.
Canalejo R, Canalejo A, Martinez-Moreno JM, et al. FGF23 fails to inhibit uremic parathyroid glands. J Am Soc Nephrol. 2010;21(7):1125–35.
Panda DK, Miao D, Bolivar I, et al. Inactivation of the 25-hydroxyvitamin D 1alpha-hydroxylase and vitamin D receptor demonstrates independent and interdependent effects of calcium and vitamin D on skeletal and mineral homeostasis. J Biol Chem. 2004;279(16):16754–66.
Wang AY, Ho SS, Wang M, et al. Cardiac valvular calcification as a marker of atherosclerosis and arterial calcification in end-stage renal disease. Arch Intern Med. 2005;165(3):327–32.
Foley RN, Parfrey PS. Cardiovascular disease and mortality in ESRD. J Nephrol. 1998;11(5):239–45.
Block GA, Hulbert-Shearon TE, Levin NW, Port FK. Association of serum phosphorus and calcium x phosphate product with mortality risk in chronic hemodialysis patients: a national study. Am J Kidney Dis. 1998;31(4):607–17.
Schwarz A, Mengel M, Gwinner W, et al. Risk factors for chronic allograft nephropathy after renal transplantation: a protocol biopsy study. Kidney Int. 2005;67(1):341–8.
Cicone JS, Petronis JB, Embert CD, Spector DA. Successful treatment of calciphylaxis with intravenous sodium thiosulfate. Am J Kidney Dis. 2004;43(6):1104–8.
Araya CE, Fennell RS, Neiberger RE, Dharnidharka VR. Sodium thiosulfate treatment for calcific uremic arteriolopathy in children and young adults. Clin J Am Soc Nephrol. 2006;1(6):1161–6.
Tokashiki K, Ishida A, Kouchi M, et al. Successful management of critical limb ischemia with intravenous sodium thiosulfate in a chronic hemodialysis patient. Clin Nephrol. 2006;66(2):140–3.
Meissner M, Bauer R, Beier C, et al. Sodium thiosulphate as a promising therapeutic option to treat calciphylaxis. Dermatology. 2006;212(4):373–6.
Ackermann F, Levy A, Daugas E, et al. Sodium thiosulfate as first-line treatment for calciphylaxis. Arch Dermatol. 2007;143(10):1336–7.
Pasch A, Schaffner T, Huynh-Do U, Frey BM, Frey FJ, Farese S. Sodium thiosulfate prevents vascular calcifications in uremic rats. Kidney Int. 2008;74(11): 1444–53.
Musso CG, Enz P, Vidal F, et al. Oral sodium thiosulfate solution as a secondary preventive treatment for calciphylaxis in dialysis patients. Saudi J Kidney Dis Transpl. 2008;19(5):820–1.
Hayden MR, Goldsmith DJ. Sodium thiosulfate: new hope for the treatment of calciphylaxis. Semin Dial. 2010;23(3):258–62.
Velasco N, MacGregor MS, Innes A, MacKay IG. Successful treatment of calciphylaxis with cinacalcet-an alternative to parathyroidectomy? Nephrol Dial Transplant. 2006;21(7):1999–2004.
Hermans MM, Brandenburg V, Ketteler M, et al. Association of serum fetuin-A levels with mortality in dialysis patients. Kidney Int. 2007;72(2):202–7.
Kuzniar J, Porazko T, Klinger M. Relationship between fetuin-A concentration, elevated levels of inflammatory markers, and arterial wall stiffness in end-stage kidney disease. J Ren Nutr. 2008;18(1): 83–6.
Schafer C, Heiss A, Schwarz A, et al. The serum protein alpha 2-Heremans-Schmid glycoprotein/fetuin-A is a systemically acting inhibitor of ectopic calcification. J Clin Invest. 2003;112(3): 357–66.
Koos R, Krueger T, Westenfeld R, et al. Relation of circulating Matrix Gla-Protein and anticoagulation status in patients with aortic valve calcification. Thromb Haemost. 2009;101(4):706–13.
Moe SM, Chen NX. Pathophysiology of vascular calcification in chronic kidney disease. Circ Res. 2004;95(6):560–7.
Bilezikian JP, Rubin M, Silverberg SJ. Asymptomatic primary hyperparathyroidism. Arq Bras Endocrinol Metabol. 2006;50(4):647–56.
Bilezikian JP, Silverberg SJ. Clinical spectrum of primary hyperparathyroidism. Rev Endocr Metab Disord. 2000;1(4):237–45.
Gayed IW, Kim EE, Broussard WF, et al. The value of 99mTc-sestamibi SPECT/CT over conventional SPECT in the evaluation of parathyroid adenomas or hyperplasia. J Nucl Med. 2005;46(2):248–52.
Mihai R, Simon D, Hellman P. Imaging for primary hyperparathyroidism—an evidence-based analysis. Langenbecks Arch Surg. 2009;394(5):765–84.
Kidney Disease: Improving Global Outcomes (KDIGO) CKD-MBD Work Group. KDIGO clinical practice guideline for the diagnosis, evaluation, prevention, and treatment of Chronic Kidney Disease-Mineral and Bone Disorder (CKD-MBD). Kidney Int Suppl. 2009;113:S1–130.
Klein KB, Orwoll ES, Lieberman DA, Meier DE, McClung MR, Parfitt AM. Metabolic bone disease in asymptomatic men after partial gastrectomy with Billroth II anastomosis. Gastroenterology. 1987;92: 608–16.
Merz WA, Schenk RK. Quantitative structural analysis of human cancellous bone. Acta Anat (Basel). 1970;75(1):54–66.
Malluche HH, Sherman D, Meyer W, Massry SG. A new semiautomatic method for quantitative static and dynamic bone histology. Calcif Tissue Int. 1982;34(5):439–48.
Lehmann G, Ott U, Kaemmerer D, Schuetze J, Wolf G. Bone histomorphometry and biochemical markers of bone turnover in patients with chronic kidney disease Stages 3–5. Clin Nephrol. 2008;70(4): 296–305.
Irvin III GL, Carneiro D. Parathyroid hyperplasia: parathyroidectomy. Philadelphia: Elsevier Saunders (Textbook of Endocrine Surgery); 2005. p. 472–80. Ref Type: Serial (Book,Monograph).
Gotch F, Levin NW, Kotanko P. Calcium balance in dialysis is best managed by adjusting dialysate calcium guided by kinetic modeling of the interrelationship between calcium intake, dose of vitamin D analogues and the dialysate calcium concentration. Blood Purif. 2010;29(2):163–76.
Teng M, Wolf M, Lowrie E, Ofsthun N, Lazarus JM, Thadhani R. Survival of patients undergoing hemodialysis with paricalcitol or calcitriol therapy. N Engl J Med. 2003;349(5):446–56.
Bilezikian JP, Khan AA, Potts Jr JT. Guidelines for the management of asymptomatic primary hyperparathyroidism: summary statement from the third international workshop. J Clin Endocrinol Metab. 2009;94(2):335–9.
Al-Hilali N, Hussain N, Ataia AI, Al-Azmi M, Al-Helal B, Johny KV. Hypertension and hyperparathyroidism are associated with left ventricular hypertrophy in patients on hemodialysis. Indian J Nephrol. 2009;19(4):153–7.
Evenepoel P, Claes K, Kuypers D, Maes B, Vanrenterghem Y. Impact of parathyroidectomy on renal graft function, blood pressure and serum lipids in kidney transplant recipients: a single centre study. Nephrol Dial Transplant. 2005;20(8):1714–20.
Nasri H, Baradaran A, Naderi AS. Close association between parathyroid hormone and left ventricular function and structure in end-stage renal failure patients under maintenance hemodialysis. Acta Med Austriaca. 2004;31(3):67–72.
Piovesan A, Molineri N, Casasso F, et al. Left ventricular hypertrophy in primary hyperparathyroidism. Effects of successful parathyroidectomy. Clin Endocrinol (Oxf). 1999;50(3):321–8.
Rosenthal FD, Roy S. Hypertension and hyperparathyroidism. Br Med J. 1972;4(837):396–7.
Rubin MR, Maurer MS, McMahon DJ, Bilezikian JP, Silverberg SJ. Arterial stiffness in mild primary hyperparathyroidism. J Clin Endocrinol Metab. 2005;90(6):3326–30.
Dotzenrath C. Indications for parathyroidectomy in renal hyperparathyroidism: comments on the significance of new therapeutics. Chirurg. 2010;81(10): 902–8.
Costa-Hong V, Jorgetti V, Gowdak LH, Moyses RM, Krieger EM, De Lima JJ. Parathyroidectomy reduces cardiovascular events and mortality in renal hyperparathyroidism. Surgery. 2007;142(5): 699–703.
Alfrey AC, Jenkins D, Groth CG, Schorr WS, Gecelter L, Ogden DA. Resolution of hyperparathyroidism, renal osteodystrophy and metastatic calcification after renal homotransplantation. N Engl J Med. 1968;279(25):1349–56.
Egbuna OI, Taylor JG, Bushinsky DA, Zand MS. Elevated calcium phosphate product after renal transplantation is a risk factor for graft failure. Clin Transplant. 2007;21(4):558–66.
Lee PP, Schiffmann L, Offermann G, Beige J. Effects of parathyroidectomy on renal allograft survival. Kidney Blood Press Res. 2004;27(3):191–6.
Schwarz A, Rustien G, Merkel S, Radermacher J, Haller H. Decreased renal transplant function after parathyroidectomy. Nephrol Dial Transplant. 2007;22(2):584–91.
Evenepoel P, Claes K, Kuypers DR, Debruyne F, Vanrenterghem Y. Parathyroidectomy after successful kidney transplantation: a single centre study. Nephrol Dial Transplant. 2007;22(6):1730–7.
Lorenz K, Ukkat J, Sekulla C, Gimm O, Brauckhoff M, Dralle H. Total parathyroidectomy without autotransplantation for renal hyperparathyroidism: experience with a qPTH-controlled protocol. World J Surg. 2006;30(5):743–51.
Wells Jr SA, Gunnells JC, Shelburne JD, Schneider AB, Sherwood LM. Transplantation of the parathyroid glands in man: clinical indications and results. Surgery. 1975;78(1):34–44.
Paloyan E, Lawrence AM, Paloyan D. Successful autotransplantation of the parathyroid glands during total thyroidectomy for carcinoma. Surg Gynecol Obstet. 1977;145(3):364–8.
Brennan MF, Brown EM, Marx SJ, et al. Recurrent hyperparathyroidism from an autotransplanted parathyroid adenoma. N Engl J Med. 1978;299(19):1057–9.
D’Avanzo A, Parangi S, Morita E, Duh QY, Siperstein AE, Clark OH. Hyperparathyroidism after thyroid surgery and autotransplantation of histologically normal parathyroid glands. J Am Coll Surg. 2000;190(5):546–52.
de Menezes Montenegro FL, Custodio MR, Arap SS, et al. Successful implant of long-term cryopreserved parathyroid glands after total parathyroidectomy. Head Neck. 2007;29(3):296–300.
Brennan MF, Brown EM, Spiegel AM, et al. Autotransplantation of cryopreserved parathyroid tissue in man. Ann Surg. 1979;189(2):139–42.
Al-Sobhi SCO. Parathyroid hyperplasia: parathyroidectomy. Philadelphia: Elsevier Saunders (Textbook of Endocrine Surgery); 2005. p. 481–8. Ref Type: Serial (Book, Monograph).
Block GA, Martin KJ, de Francisco AL, et al. Cinacalcet for secondary hyperparathyroidism in patients receiving hemodialysis. N Engl J Med. 2004;350(15):1516–25.
Mizobuchi M, Hatamura I, Ogata H, et al. Calcimimetic compound upregulates decreased calcium-sensing receptor expression level in parathyroid glands of rats with chronic renal insufficiency. J Am Soc Nephrol. 2004;15(10):2579–87.
Rodriguez M, Nemeth E, Martin D. The calcium-sensing receptor: a key factor in the pathogenesis of secondary hyperparathyroidism. Am J Physiol Renal Physiol. 2005;288(2):F253–64.
Colloton M, Shatzen E, Miller G, et al. Cinacalcet HCl attenuates parathyroid hyperplasia in a rat model of secondary hyperparathyroidism. Kidney Int. 2005;67(2):467–76.
Chertow GM, Pupim LB, Block GA, et al. Evaluation of Cinacalcet Therapy to Lower Cardiovascular Events (EVOLVE): rationale and design overview. Clin J Am Soc Nephrol. 2007;2(5):898–905.
Cunningham J, Danese M, Olson K, Klassen P, Chertow GM. Effects of the calcimimetic cinacalcet HCl on cardiovascular disease, fracture, and health-related quality of life in secondary hyperparathyroidism. Kidney Int. 2005;68(4):1793–800.
Kruse AE, Eisenberger U, Frey FJ, Mohaupt MG. The calcimimetic cinacalcet normalizes serum calcium in renal transplant patients with persistent hyperparathyroidism. Nephrol Dial Transplant. 2005;20(7):1311–4.
Apostolou T, Damianou L, Kotsiev V, Drakopoulos S, Hadjiconstantinou V. Treatment of severe hypocalcaemia due to refractory hyperparathyroidism in renal transplant patients with the calcimimetic agent cinacalcet. Clin Nephrol. 2006;65(5):374–7.
Borchhardt KA, Heinzl H, Mayerwoger E, Horl WH, Haas M, Sunder-Plassmann G. Cinacalcet increases calcium excretion in hypercalcemic hyperparathyroidism after kidney transplantation. Transplantation. 2008;86(7):919–24.
Lopez V, Toledo R, Sola E, et al. Treatment with cinacalcet in 29 kidney transplant patients with persistent hyperparathyroidism. Transplant Proc. 2009;41(6):2394–5.
Serra AL, Savoca R, Huber AR, et al. Effective control of persistent hyperparathyroidism with cinacalcet in renal allograft recipients. Nephrol Dial Transplant. 2007;22(2):577–83.
Block GA, Klassen PS, Lazarus JM, Ofsthun N, Lowrie EG, Chertow GM. Mineral metabolism, mortality, and morbidity in maintenance hemodialysis. J Am Soc Nephrol. 2004;15(8):2208–18.
Dhingra R, Sullivan LM, Fox CS, et al. Relations of serum phosphorus and calcium levels to the incidence of cardiovascular disease in the community. Arch Intern Med. 2007;167(9):879–85.
Dhingra R, Gona P, Benjamin EJ, et al. Relations of serum phosphorus levels to echocardiographic left ventricular mass and incidence of heart failure in the community. Eur J Heart Fail. 2010;12(8): 812–8.
USDA National Nutrient Database for Standard Reference, Release 17. http://dietarysupplementdatabase.usda.nih.gov/. Accessed at Nov 2nd 2010.
Kuhlmann MK. Practical approaches to management of hyperphosphatemia: can we improve the current situation? Blood Purif. 2007;25(1):120–4.
Granja CA, Juergensen P, Finkelstein FO. Phosphate balance in peritoneal dialysis patients: role of ultrafiltration. Contrib Nephrol. 2009;163:198–205.
Mucsi I, Hercz G, Uldall R, Ouwendyk M, Francoeur R, Pierratos A. Control of serum phosphate without any phosphate binders in patients treated with nocturnal hemodialysis. Kidney Int. 1998;53:1399–404.
Jenkins DA, Gouldesbrough D, Smith GD, Cowie JF, Winney RJ. Can low-dosage aluminium hydroxide control the plasma phosphate without bone toxicity? Nephrol Dial Transplant. 1989;4(1):51–6.
Nakamura M, Suzuki H, Ohno Y, et al. Oral calcium carbonate administration ameliorates the progression of renal failure in rats with hypertension. Am J Kidney Dis. 1995;25:910–7.
Chertow GM, Raggi P, Chasan-Taber S, Bommer J, Holzer H, Burke SK. Determinants of progressive vascular calcification in haemodialysis patients. Nephrol Dial Transplant. 2004;19(6):1489–96.
Zacharias JM, Fontaine B, Fine A. Calcium use increases risk of calciphylaxis: a case-control study. Perit Dial Int. 1999;19(3):248–52.
Asmus HG, Braun J, Krause R, et al. Two year comparison of sevelamer and calcium carbonate effects on cardiovascular calcification and bone density. Nephrol Dial Transplant. 2005;20(8):1653–61.
Ferreira A, Frazao JM, Monier-Faugere MC, et al. Effects of sevelamer hydrochloride and calcium carbonate on renal osteodystrophy in hemodialysis patients. J Am Soc Nephrol. 2008;19(2):405–12.
Izumi M, Morita S, Nishian Y, et al. Switching from calcium carbonate to sevelamer hydrochloride has suppressive effects on the progression of aortic calcification in hemodialysis patients: assessment using plain chest X-ray films. Ren Fail. 2008;30(10):952–8.
McIntyre CW, Patel V, Taylor GS, Fluck RJ. A prospective study of combination therapy for hyperphosphataemia with calcium-containing phosphate binders and sevelamer in hypercalcaemic haemodialysis patients. Nephrol Dial Transplant. 2002;17(9):1643–8.
Nagano N, Miyata S, Abe M, Wakita S, Kobayashi N, Wada M. Effects of intermittent treatment with sevelamer hydrochloride on parathyroid hyperplasia and vascular calcification in rats with chronic kidney disease. Clin Calcium. 2005;15 Suppl 1:35–9. discussion 39–40.
Takei T, Otsubo S, Uchida K, et al. Effects of sevelamer on the progression of vascular calcification in patients on chronic haemodialysis. Nephron Clin Pract. 2008;108(4):c278–83.
Suki WN, Zabaneh R, Cangiano JL, et al. Effects of sevelamer and calcium-based phosphate binders on mortality in hemodialysis patients. Kidney Int. 2007;72(9):1130–7.
Loghman-Adham M. Safety of new phosphate binders for chronic renal failure. Drug Saf. 2003;26(15):1093–115.
Feng L, Xiao H, He X, et al. Neurotoxicological consequence of long-term exposure to lanthanum. Toxicol Lett. 2006;165(2):112–20.
Finn WF. Lanthanum carbonate versus standard therapy for the treatment of hyperphosphatemia: safety and efficacy in chronic maintenance hemodialysis patients. Clin Nephrol. 2006;65(3):191–202.
Albaaj F, Hutchison AJ. Lanthanum carbonate for the treatment of hyperphosphataemia in renal failure and dialysis patients. Expert Opin Pharmacother. 2005;6(2):319–28.
Tonelli M, Pannu N, Manns B. Oral phosphate binders in patients with kidney failure. N Engl J Med. 2010;362(14):1312–24.
Block GA, Brillhart SL, Persky MS, Amer A, Slade AJ. Efficacy and safety of SBR759, a new iron-based phosphate binder. Kidney Int. 2010;77(10): 897–903.
Ahmed MH. Niacin as potential treatment for dyslipidemia and hyperphosphatemia associated with chronic renal failure: the need for clinical trials. Ren Fail. 2010;32(5):642–6.
Eknoyan G. Salivary phosphorus binding: a novel approach to control hyperphosphatemia. J Am Soc Nephrol. 2009;20(3):460–2.
Savica V, Calo LA, Monardo P, et al. Salivary phosphate-binding chewing gum reduces hyperphosphatemia in dialysis patients. J Am Soc Nephrol. 2009;20(3):639–44.
Rossini M, Gatti D, Isaia G, Sartori L, Braga V, Adami S. Effects of oral alendronate in elderly patients with osteoporosis and mild primary hyperparathyroidism. J Bone Miner Res. 2001;16(1): 113–9.
Khan AA, Bilezikian JP, Kung AW, et al. Alendronate in primary hyperparathyroidism: a double-blind, randomized, placebo-controlled trial. J Clin Endocrinol Metab. 2004;89(7):3319–25.
Gallacher SJ, Dixon T. Impact of Treatments for postmenopausal osteoporosis (bisphosphonates, parathyroid hormone, strontium ranelate, and denosumab) on bone quality: a systematic review. Calcif Tissue Int. 2010;87(6):469–84.
Cejka D, Kodras K, Bader T, Haas M. Treatment of Hemodialysis-Associated Adynamic Bone Disease with Teriparatide (PTH(1-34)): A Pilot Study. Kidney Blood Press Res. 2010;33(3):221–6.
Acknowledgments
The authors want to thank Dr. Gabriele Lehmann (Jena), for critical review of the bone section and gratification of histological pictures as well as Andreas Plötner, MD (Leipzig), for gratification of Fig. 5.1.
My wife (J.B.), Karin Kaori Beige, bolstered this work by fine grasp and patience.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2012 Springer Science+Business Media, LLC
About this chapter
Cite this chapter
Beige, J., Lamesch, P. (2012). New Concepts for Primary and Secondary Hyperparathyroidism. In: Licata, A., Lerma, E. (eds) Diseases of the Parathyroid Glands. Springer, New York, NY. https://doi.org/10.1007/978-1-4419-5550-0_5
Download citation
DOI: https://doi.org/10.1007/978-1-4419-5550-0_5
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4419-5549-4
Online ISBN: 978-1-4419-5550-0
eBook Packages: MedicineMedicine (R0)