
Abstract
Management of chronic kidney disease is often fraught with derangements in calcium and phosphorus metabolism. The development of secondary hyperparathyroidism begins in the early stage of CKD and usually progresses as the level of renal impairment advances. Treatment of SHPT is challenging and maintenance of calcium and phosphorus level is an important determinant of cardiovascular and bone health. In this chapter, we focus on the pathogenesis, etiology, and treatment of secondary hyperparathyroidism secondary to chronic kidney disease.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others
Keywords
- Secondary hyperparathyroidism
- Chronic kidney disease (CKD)
- Vitamin D receptor agonist/analogs
- Vitamin D
- Calcimimetic agents
- Cinacalcet
- Phosphate binders
- Parathyroidectomy
Introduction
Patients with chronic renal disease often times have derangements in calcium and phosphorus levels with resultant increases in PTH. These abnormalities in mineral metabolism are important determinants of bone and cardiovascular health. The current Kidney Disease: Improving Global Outcome (KDIGO) guidelines recommend individualized treatment and targets. This chapter reviews the action of PTH and calcium regulation as well as the pathogenesis, etiology, and treatment of secondary hyperparathyroidism of renal origin.
Physiology of PTH Action and Calcium Regulation
The parathyroid hormone is a single-peptide chain consisting of 84 amino acids. The amino terminal portion of this molecule is predominantly responsible for its actions. The rate of clearance of the 1–84, biologically active, amino acid peptide is faster than the inactive fragments. Earlier assays only provided an insight into the gland activity and not necessarily the function as they measured both the active and inactive fragments. The newer double antibody assays that are more widely used these days, measure only the intact fragment.
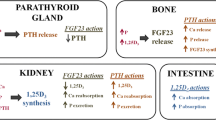
The primary function of parathyroid hormone (PTH) is the maintenance of the extracellular calcium concentration. Parathyroid hormone acts directly on the bone and kidney and indirectly on the intestine in order to maintain calcium homeostasis. As extracellular calcium concentrations decline and PTH levels rise, there is release of calcium from the bone into the blood. At the level of the kidney, PTH exerts its effect by increasing reabsorption of calcium from the glomerulus. Indirectly, PTH increases the production of 1, 25(OH) D2 which in turn increases the intestinal absorption of calcium. The action of PTH on these organs is critical in maintaining calcium homeostasis. Ionized calcium is the most important determinant of PTH secretion. In addition to hypocalcemia, mild hypomagnesemia, hyperphosphatemia, and low calcitriol levels stimulate the secretion of parathyroid hormone.
Hypocalcemia results in an increase in the level of parathyroid hormone up to five times the basal rates of secretion. This in turn results in (1) increase in the calcium release from bone into the blood, (2) increased calcium reabsorption at the level of the kidney and (3) vitamin D mediated increased absorption of calcium through the intestines. The parathyroid glands are stimulated via a calcium sensor, a G protein-linked receptor located on the plasma membrane of the parathyroid glands. Under conditions where all glands are functioning normally, excesses in calcium concentration are effectively corrected by the changes in parathyroid levels. The decline in calcium levels is counteracted by an increase in the secretion of parathyroid hormone. In situations where the organs are unable to function normally, such as in kidney disease, the elevations in PTH fail to maintain calcium and phosphate homeostasis and secondary hyperparathyroidism ensues.
In chronic kidney disease , due to a decrease in renal mass and subsequent depletion of 1 α hydroxylase, the production of 1, 25(OH) D2 (calcitriol) is decreased and phosphate levels increase due to impaired phosphate excretion. The phosphatonin fibroblast growth factor FGF23 also increases which in turn down regulates the enzyme 1 α hydroxylase and thereby exacerbates the deficiency of calcitriol. This leads to hypocalcemia and as an adaptive response, the parathyroid levels increase. Continuous stimulation of the parathyroid gland results in diffuse polyclonal hyperplasia followed by monoclonal nodular hyperplasia [1, 2]. In early kidney disease, abnormalities in mineral metabolism are not frequently observed. It is likely that FGF23 plays a role in maintaining this calcium and phosphate homeostasis [3]. FGF-23 reduces serum phosphate by directly inhibiting the phosphate absorption in the proximal tubule and indirectly by decreasing calcitriol synthesis. As kidney disease advances, the above mechanisms are unable to maintain the phosphate balance. The resultant hyperphosphatemia that ensues may cause extra osseous deposition of calcium and phosphate. In SHPT of renal origin, calcium levels are often within or below the reference range. Uremia may further affect the calcium levels by impairing the intestinal absorption of calcium.
FGF23 decreases PTH secretion and cell proliferation in normal glands but it does not have an effect on a hyperplastic gland. Parathyroid hormone levels begin to rise once creatinine clearance falls below 80 mL/min [4].
Klotho is a transmembrane protein with decreased production in CKD and is likely responsible for the changes in vasculature, bone and skin in these individuals. In the kidneys, this gene is located on the proximal tubule cells, brush border, and urinary lumen. Klotho acts as a cofactor for FGF23, and regulates phosphate metabolism. In the absence of Klotho , FGF23 signaling is impaired [5]. It is postulated that the decline in Klotho causes resistance of FGF23 actions on the kidney and the parathyroid gland. As a result, FGF23 levels continue to rise, further increasing PTH and reducing vitamin D. This results in a vicious cycle and contributes to the progression of SHPT in CKD. Elevations in calcium and phosphate levels associated with SHPT may result in vascular calcifications and an increase in morbidity as well as mortality. The high FGF23 levels seen in CKD and are associated with increased all-cause mortality in hemodialysis patients as well as poor cardiovascular outcomes [6–8].
Physiology of Vitamin D
Upon exposure to sunlight, ultraviolet radiation enters the epidermis and 7-dehydrocholesterol is converted to pre-vitamin D3. This compound is biologically inactive and within 24 h, it is converted to Vitamin D3 in the epidermis. In the presence of vitamin D binding proteins, the synthesized Vitamin D is translocated into the circulation. From there, it is transported to the liver and metabolized to 25OHD by hepatic enzymes. 25OHD is not active at physiologic levels. It is transported to the kidney for additional hydroxylation. The kidney plays a vital role in the conversion of 25OHD to an active metabolite 1, 25, (OH) D2 by the enzyme 1 alpha hydroxylase. In the presence of hypocalcemia, PTH normally stimulates the synthesis of 1,25(OH)2D. In mild to moderate renal failure EGFR > 30 ml/min, hyperphosphatemia and decreased phosphate clearance suppress 1,25(OH) D2. This occurs even despite the elevation seen in PTH.
Clinical History and Diagnosis
In SHPT, calcium levels are usually normal to slightly low and individuals may be asymptomatic. Symptoms include mild perioral numbness and tingling, cramping in hands and feet. Uremic symptoms may also be noted. Chvostek and Trousseau’s sign is positive on physical examination. Often times the cause of hypocalcemia is obtained from the history itself. In Table 7.1, we have shown the causes for secondary hypoparathyroidism and the appropriate test where applicable to help in the diagnosis.
In the case of SHPT due to renal disease, additional history of renal insufficiency, serum creatinine, phosphorus, BUN, assessment of urine output, and comorbidities is essential. The measurement of PTH is required and is typically high in the presence of a normal to mildly low serum calcium concentration. Due to the chronicity of the disease, serum albumin should be routinely measured to obtain a corrected calcium level. Elevated serum calcium excludes SHPT of renal origin.
Guidelines for the Management of SHPT
Treatment of SHPT is aimed at minimizing all-cause morbidity and mortality, abnormal mineral metabolism, and bone disease as well as preventing extra-skeletal calcium deposits including vascular calcification.
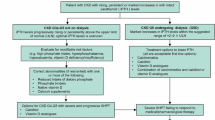
In 2009, the Kidney Disease: Improving Global Outcome (KDIGO) developed practice guidelines which provided recommendations for the evaluation and management of chronic kidney disease-mineral and bone disorder (CKD-MBD) . The KDIGO does not recommend specific targets for PTH based on the stage of renal disease. They recommend using intact PTH as an assay. There is a greater momentum towards early detection and treatment of SHPT. In CKD stage III {GFR 30–59 mL/min/1.73 m2] PTH should be monitored every 12 months along with annual calcium and phosphorus assessments. Therapy should be initiated if the PTH rises steadily or persistently remains above the upper limit of normal. In CKD stage IV [GFR 15–29 mL/min/1.73 m2], PTH measurements are recommended every 3 months. In CKD V not yet on dialysis, PTH should be assessed every 3 months while calcium and phosphorus levels are assessed monthly. The goal is to maintain calcium and phosphorus levels within the normal range.
In 2003, the Kidney Disease Outcome Quality Initiative KDOQI specified target goals for PTH, calcium, and phosphorus. For patients with CKD stage V [below 15 mL/min/1.73 m2], the KDOQI guidelines recommended a target intact PTH (first-generation immunometric assay) of between 150 and 300 pg/mL, calcium phosphorus product of less than 55, serum calcium in the lower half of the normal range [8.4–9.5 mg/dL] and serum phosphorus between 3.5 and 5.5 mg/dL. The standard treatments used in order to achieve these targets were limited by drug side effects and thus these goals remained largely unmet [9].
The newer KDIGO CKD-MBD guidelines have used more of an evidence-based approach and have individualized treatment and represent the most current clinical management guidelines. Target PTH levels should be based on the levels of renal dysfunction to avoid high bone turnover and maintain near normal levels of alkaline phosphatase. This goal can be achieved by keeping PTH near the upper limit of normal in CKD stages 3–4 and up to nine times normal in the dialysis population [10]. However, a uniform classification system in this area is lacking.
Current KDIGO guidelines do not recommend routine measurements of bone mineral density in CKD, although suggest that serum PTH or bone-specific alkaline phosphatase levels should be assessed to predict bone turnover [10].
Bone biopsy is the gold standard procedure to assess CKD-MBD but is invasive and not widely available. Vascular calcification can be assessed by lateral abdominal radiograph, echocardiogram, or computed tomography (CT) scanning.
Treatment of SHPT
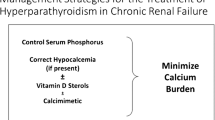
The goal of treatment is to lower PTH while maintaining normal calcium and phosphorus levels. This is often times very challenging due to the complex interrelationship of hormones, bone health, diet, and mineral balances within the body. It is to be kept in mind that the treatment options discussed below improve biochemical parameters and bone histology although the evidence on the impact on patient-related outcomes is lacking.
The use of standard treatment measures such as calcium supplementation, phosphate binders, and vitamin D analogs are limited by the development of hypercalcemia, hyperphosphatemia, increased Ca × P product with resultant increase in vascular calcifications.
Calcium Supplements
Calcium supplements are only moderately effective in controlling PTH levels in those with SHPT on dialysis or early stages of renal failure. Calcium carbonate alone versus oral or IV calcitriol did not show a significant decrease in the levels of PTH [11, 12]. High doses of calcium supplementation are limited by calcifications that occur once the Ca X P product exceeds 55. Calcium supplements are limited to 1–2 g per day due to this concern.
Restriction of Dietary Phosphorus and Phosphate Binders
The mainstay of treatment is the correction of hyperphosphatemia and prevention of a positive phosphate balance. This in turn limits the development of hyperparathyroidism and its related effects.
Phosphate control is still an unmet need in CKD. Serum phosphorus is an independent risk factor for mortality in ESRD [13, 14] In mild CKD , restriction of dietary phosphate reduces PTH levels [15]. Phosphate restriction can be achieved via eliminating food preservatives and other additives. In order to maintain a balance between protein and phosphorus levels, foods with high biological value such as eggs and meats are preferred. Phosphorus intake should be limited to 900 mg/day.
In addition to dietary interventions, phosphate binders are used to control serum phosphate levels. As the disease advances, dietary restriction alone is insufficient to reduce hyperphosphatemia and the resultant SHPT. The currently available phosphate binders bind about 250 mg/day of phosphate [16]. With use of a single phosphate binder, about 30–50 % of ESRD patients remain hyperphosphatemic. Combination therapy with two different binders, increases the phosphate binding capacity and maintains serum phosphorus levels within acceptable ranges. Both calcium and non-calcium-based phosphate binders have been used in the treatment of SHPT. Aluminum is no longer used due to its serious toxicities. The use of calcium-based binders increase the risk of hypercalcemia, calciphylaxis, and vascular calcification [17]. The calcium free phosphate binders such as, sevelamer and lanthanum carbonate, decrease serum phosphate levels without causing hypercalcemia but they are not potent in lowering PTH levels [18]. Ferric citrate is an iron-based oral phosphate binder that effectively lowers serum phosphorus levels and has been shown to have safety profile similar to sevelamer and calcium acetate in hemodialysis patient [19]. Niacin reduces phosphate absorption by blocking the active sodium-phosphate co-transporters in the small intestines and has shown promising results [20]. Calcium containing binders are less costly and readily available and can be used in patients with CKD who have hypocalcemia. Non calcium containing phosphate binders are preferred in individuals with CKD and normal or high calcium levels.
Calcium as well as non-calcium-containing phosphate binders were shown to comparably lower FGF23 levels [21]. Overall, both calcium and non-calcium phosphate binders are shown to be effective in lowering phosphorus levels but the impact of these agents on all-cause mortality and cardiovascular mortality in CKD is unclear [22]. In a recently published meta-analysis, non-calcium-containing phosphate binders were associated with lower all-cause mortality [23]. Available phosphate binders can increase calcifications in coronary artery and abdominal aorta [24]. Dietary phosphate restriction, with or without calcium carbonate treatment, resulted in progression of vascular calcifications , although this effect was not seen in those treated with sevelamer [25]. However, data supporting improved clinical outcomes by limiting the progression of vascular calcifications is lacking.
Vitamin D and VDR Agonist
Adequate levels of vitamin D are required for intestinal absorption of calcium. In the presence of low vitamin D, intestinal absorption of calcium is reduced which results in elevated PTH levels and subsequently parathyroid gland hyperplasia [26]. In CKD stages 3–4, calcium and phosphorus levels are usually in the physiologic range [27]. The majority of patients with CKD have low vitamin D [28]. The KDOQI guidelines recommend correction with vitamin D especially in stages 3 and 4 as these low levels may trigger the development of hyperparathyroidism. Supplementation of vitamin D with either ergocalciferol or cholecalciferol increases the level of 25HD and 1,25 (OH)D2 and may suppress but not necessarily normalize PTH in stages 3–4. In Stage 5, supplementation with vitamin D is generally ineffective in suppressing PTH levels.
The deficiency of endogenous calcitriol production is often treated with biologically active VDR agonist such as calcitriol, paricalcitol, alfacalcidol (not approved for use in the United States), and doxercalciferol. These active sterols increase the absorption of calcium and phosphorus from the intestines and in turn decrease the synthesis of PTH in a dose dependent manner regardless of the stage of CKD. Dialysis patients have impaired uptake and metabolism of 25HD. Calcitriol not only replete the levels of 1,25-D but also increases the uptake of 25HD [29]. However, These drugs are limited by the development of hypercalcemia and hyperphosphatemia, especially at higher doses and have a narrow therapeutic index [30]. The currently available synthetic analogs reduce PTH to a similar extent although paricalcitol achieves this reduction sooner than the other drugs [31]. In addition, paricalcitol showed significant and sustained control of PTH, with fewer episodes of hypercalcemia [32].
Intravenous calcitriol has been used since the late 1980s as an alternative therapy to either oral calcitriol or parathyroidectomy in adult dialysis patients with SHPT. Long term treatment showed reductions in PTH as well as alkaline phosphatase [33]. In addition, lowering PTH was also shown to be cardioprotective [34]. Widespread use of intravenous calcitriol has resulted in fewer parathyroidectomies [35].
Low vitamin D and 1,25 (OH)D2 levels correlate with increased cardiovascular disease and deaths, while the use of VDR agonist therapy may be cardioprotective. There are few prospective studies evaluating the effects of VDR agonist on survival. Previous meta-analysis showed Vitamin D supplementation was beneficial in lowering cardiovascular and all-cause mortality in patients with CKD [36]. Paricalcitriol has a greater survival advantage over calcitriol [36]. Contrary to the above, current evidence on Vitamin D supplementation does not support a benefit in survival or cardiovascular mortality in patients with CKD [37].
Continuous use of a VDR agonist results in lowering PTH, preserving bone mass, and lowering markers of bone remodeling such as bone specific alkaline phosphatase and osteocalcin [38, 39]. There is a lack of data on the effectiveness of the interrupted use of VDR agonist. Continuous therapy is recommended in order to maintain PTH suppression.
Therapy and goals for optimal PTH suppression need to be individualized. Occurrence of hypercalcemia due to VDR agonist may suggest the development of adynamic bone, a form of renal osteodystrophy characterized by a low bone turnover state [40]. VDR agonists can be used to achieve PTH suppression while ensuring that hypercalcemia and hyperphosphatemia do not occur.
Calcium-Sensing Receptor Agonist: Cinacalcet
The calcium-sensing receptor (CaSR) is a G protein-coupled receptor located on the parathyroid chief cell membrane and represents the pivotal mechanism regulating PTH secretion. This receptor was initially cloned from bovine parathyroid cells and described by Brown et al. [41]. Activation of this receptor by an increase in extracellular calcium, the endogenous ligand, decreases PTH secretion [42]. The lack of a drug capable of directly altering PTH secretion without affecting serum calcium levels made the CaSR a high priority molecular target [43].
Ligands that simulate or potentiate the effects of extracellular calcium at the CaSR have been termed calcimimetics. There are two mechanistically distinct types. Type I calcimimetics are inorganic and organic polycations that act as agonists. Type II calcimimetics are l-amino acids and phenylalkamines that function as allosteric activators [44]. These type II drugs interact with membrane-spanning portions of the CaSR and induce conformational change in the receptor. This conformational change lowers the threshold for CaSR activation by plasma calcium, thereby reducing PTH secretion without a change in the serum calcium level [45].
The first-generation calcimimetic drug candidate was NPS R-568, a phenylalkamine type II compound. NPS R-568 was shown to selectively activate the parathyroid CaSR and inhibit PTH secretion both in vitro and in vivo [46]. However this compound demonstrated a variable pharmacokinetic and molecular profile [47]. This prompted the development of cinacalcet HCl, or (αR)-(−)-α-methyl-N-[3-[3-[trifluoromethylphenyl]propyl]-1-napthalenemethanamine hydrochloride, a second-generation analog of NPS R-568 that possesses the same safety and efficacy with improved bioavailability.
Cinacalcet (Sensipar, Amgen, Thousand Oaks, CA) has been approved by the FDA for use in the US since 2004. Its indications include (1) treatment of SHPT in adult patients on dialysis for chronic kidney disease, (2) hypercalcemia in patients with parathyroid carcinoma, and (3) severe hypercalcemia in patients with primary HPT.
In SHPT, cinacalcet treatment results in a decrease in FGF23 levels and the effect on FGF23 is independent of the changes in PTH levels [48]. Orally administered cinacalcet has been shown to reduce PTH and this effect can be maintained long term [49]. In CKD patients not on dialysis, cinacalcet decreases the levels of PTH but can cause hypocalcemia, hyperphosphatemia, and hypercalciuria. Therefore close monitoring of these laboratory values is required [50, 51].
Long-term administration of cinacalcet is associated with reduced progression of abdominal aortic calcification and uremic arteriolopathy [52]. Appropriate calcium and phosphorus levels may be achieved and together these changes reduce the rates of cardiovascular events and mortality in patients on hemodialysis [52, 53]. Cinacalcet did not reduce the risk of death or cardiovascular morbidity in those individuals on hemodialysis with moderate to severe hyperparathyroidism [54]. Nevertheless, addition of cinacalcet to standard therapy in adults with EGFR < 15 % (Stage 5) disease who were on dialysis, helped prevent surgical parathyroidectomy .
Cinacalcet resulted in a higher incidence of hypocalcemia. The data is robust for individuals with EGFR < 15 but sparse for those with EGFR between 15 and 60 ml/min/1.73 m2 and kidney transplant recipients [55]. Cinacalcet does not reduce the rate of clinical fractures in patients on hemodialysis [56].
Combination Therapy
Low-dose cinacalcet plus calcitriol is more effective than calcitriol alone for the treatment of moderate and severe SHPT in chronic dialysis patients. Combination therapy resulted in fewer episodes of hyperphosphatemia and hypercalcemia [57]. When combined with VDR agonist, cinacalcet has been shown to reduce the parathyroid gland volume [58].
In head-to-head trial with a VDR agonist , cinacalcet was shown to be inferior to paricalcitol in suppressing bone turnover markers [59]. In adult patients on hemodialysis, intravenous paricalcitol was found to be more cost-effective than cinacalcet plus low dose vitamin D [60, 61].
Primary Hyperparathyroidism
A growing body of evidence exists to support the efficacy of cinacalcet in lowering plasma PTH and serum calcium levels in patients with PHPT [62, 63]. A phase 3 multi-center trial demonstrated that cinacalcet resulted in a significant plasma PTH reduction and serum calcium normalization compared to placebo [64]. Cinacalcet is effective and durable for multiple years for PHPT patients with varying degrees of disease severity [65]. In patients with PHPT and nephrolithiasis, the addition of cinacalcet to standard therapy and dietary measures led to a significant reduction in both the number and size of renal stones [66]. However, bone mineral density does not improve with cinacalcet therapy [67].
Cincalcet has also been shown to correct hypercalcemia and hyperparathyroidism in kidney transplant recipients [68].
Recent data have emerged to indicate that patients with mild, asymptomatic PHPT who do not meet surgical criteria for parathyroidectomy experience more effective biochemical control with cinacalcet compared to patients with surgical criteria. Patients without surgery criteria are more likely to reach normocalcemia at the end of the initiation phase, they maintain significantly lower serum calcium levels throughout treatment, and they experience stronger reductions in PTH level when compared to patients with surgical criteria [69]. This represents an active area of investigation, as neither the FDA nor the European Medical Agency currently approves cinacalcet for the treatment of PHPT with surgical criteria.
Surgical Intervention
Parathyroidectomy for SHPT is generally considered if the serum PTH levels are >1000 pg/ml with associated hypercalcemia, the volume of at least one hyperplastic gland is >500 mm3 or SHPT is refractory to treatment.
Patients undergoing parathyroidectomy achieved the target KDOQI ranges 14–43 % of the time for calcium and 65–76 % for phosphate. However, most patients had a PTH below target [70]. Several smaller studies have looked at the survival benefit of parathyroidectomy versus medical treatment and have shown better cardiovascular and all-cause mortality with surgical intervention [71, 72]. There is ongoing debate about the preferred method of surgery; subtotal parathyroidectomy versus total parathyroidectomy with auto transplantation [73]. Currently total parathyroidectomy with or without autotransplantation is considered safe in patients with uncontrolled SHPT.
Summary
In this chapter, we have reviewed the etiology and treatment of secondary hyperparathyroidism. Secondary hyperparathyroidism is characterized by elevated PTH levels for an appropriate stimulus of hypocalcemia. In chronic kidney disease, the elevations in PTH fail to maintain calcium and phosphorus homeostasis. Current treatment options aimed at maintaining the phosphate balance and prevention of hyperphosphatemia. The new KDIGO guidelines do not recommend a specific target range for PTH, calcium, or phosphorus but rather individualize targets based on renal dysfuntion.
Expert Opinion
Early diagnosis and treatment of SHPT is key. Dietary phosphate restriction and vitamin D supplementation is an effective treatment for CKD patients who are not on dialysis.
Vitamin D analogs and calcimimetics have shown to be effective in lowering PTH levels and can be used alone or in combination. Cost of these treatments can be prohibitive. Regular laboratory evaluations are needed. For SHPT that is refractory to medical treatment, parathyroidectomy remains a viable option.
Society Guidelines
As reviewed above.
Best Practices:
N/A
References
Druke TB. Cell biology of parathyroid gland hyperplasia in chronic renal failure. J Am Soc Nephrol. 2000;11:1141–52. Basic Science review, Level 1, Grade C.
Tominnaga Y, Tajagi J. Molecular genetics of hyperparathyroid disease. Curr Opin Nephrol Hypertens. 1996;5:336–41. Basic Science review, Level 1, Grade C.
Fliser D, Kollerits B, Neyer U, Ankerst DP, Lhotta K, Lingenhel A, Ritz E, Kronenberg F, Kuen E, Konig P, Kraatz G, Mann JF, Muller GA, Kohler H, Riegler P. Fibroblast growth factor 23 (FGF23) predicts progression of chronic kidney disease: the Mild to Moderate Kidney Disease (MMKD) study. J Am Soc Nephrol. 2007;18:2600–8. Clinical Investigation, Level 3, Grade A.
Martinez I, Saracho R, Montenegro J, Llach F. The importance of dietary calcium and phosphorous in the secondary hyperparathyroidism of patients with early renal failure. Am J Kidney Dis. 1997;29(4):496–502. Clinical Investigation, Level 3, Grade A.
Hu MC, Shi M, Zhang J, Pastor J, Nakatani T, et al. Klotho: a novel phsophaturic substance acting as an autocrine enzyme in the renal proximal tubule. FASEB J. 2010;24(9):3438–50. Basic Science, Level 1, Grade A.
Gutiérrez OM, Mannstadt M, Isakova T, Rauh-Hain JA, Tamez H, Shah A, Smith K, Lee H, Thadhani R, Jüppner H, Wolf M. Fibroblast growth factor 23 and mortality among patients undergoing hemodialysis. N Engl J Med. 2008;359(6):584–92. Clinical Investigation, Level 2, Grade A.
Gutiérrez OM, Januzzi JL, Isakova T, Laliberte K, Smith K, Collerone G, Sarwar A, Hoffmann U, Coglianese E, Christenson R, Wang TJ, de Filippi C, Wolf M. Fibroblast growth factor 23 and left ventricular hypertrophy in chronic kidney disease. Circulation. 2009;119(19):2545–52. Clinical Investigation, Level 2, Grade A.
Jongbloed F, Galassi A, Cozzolino M, Zietse R, Chiarelli G, Cusi D, Brancaccio D, Gallieni M. Clinical significance of FGF-23 measurement in dialysis patients. Clin Nephrol. 2011;76(3):201–9. Clinical Investigation, Level 3, Grade A.
Moe SM, Chertow GM, Coburn JW, Quarles LD, Goodman WG, Block GA, Drüeke TB, Cunningham J, Sherrard DJ, McCary LC, Olson KA, Turner SA, Martin KJ. Achieving NKF-K/DOQI bone metabolism and disease treatment goals with cinacalcet HCl. Kidney Int. 2005;67(2):760–71. Clinical Investigation, Level 1, Grade C.
Kidney Disease: Improving Global Outcomes (KDIGO) CKD-MBD Work Group. KDIGO clinical practice guideline for the diagnosis, evaluation, prevention, and treatment of Chronic Kidney Disease-Mineral and Bone Disorder (CKD-MBD). Kidney Int Suppl. 2009;(113):S1–130. doi:10.1038/ki.2009.188. Clinical Review, Level 1, Grade C (GUIDELINES).
Indridason OS, Quarles LD. Comparison of treatments for mild secondary hyperparathyroidism in hemodialysis patients. Durham Renal Osteodystrophy Study Group. Kidney Int. 2000;57(1):282–92. Clinical Investigation, Level 1, Grade A.
Teruel JL, Tenorio MT, Rodríguez JR, n Marc R, Orofino L, Rivera M, Ortuño J. Treatment of secondary hyperparathyroidism in hemodialyzed patients with high-dose calcium carbonate without vitamin D3 supplements. Am J Nephrol. 1999;19(3):428–32. Clinical Investigation, Level 3, Grade A.
Kalantar-Zadeh K, Kuwae N, Regidor DL, Kovesdy CP, Kilpatrick RD, Shinaberger CS, McAllister CJ, Budoff MJ, Salusky IB, Kopple JD. Survival predictability of time-varying indicators of bone disease in maintenance hemodialysis patients. Kidney Int. 2006;70(4):771–80. Clinical Investigation, Level 3, Grade A.
Block GA, Klassen PS, Lazarus JM, Ofsthun N, Lowrie EG, Chertow GM. Mineral metabolism, mortality, and morbidity in maintenance hemodialysis. J Am Soc Nephrol. 2004;15(8):2208–18. Clinical Investigation, Level 3, Grade B.
Llach F, Massry SG. On the mechanism of secondary hyperparathyroidism in moderate renal insufficiency. J Clin Endocrinol Metab. 1985;61(4):601–6. Clinical Investigation, Level 3, Grade A.
Huml AM, Sullivan CM, Leon JB, Sehgal AR. The adequacy of phosphorus binder prescriptions among American hemodialysis patients. Ren Fail. 2012;34(10):1258–63. Clinical Investigation, Level 3, Grade A.
Meric F, Yap P, Bia MJ. Etiology of hypercalcemia in hemodialysis patients on calcium carbonate therapy. Am J Kidney Dis. 1990;16(5):459–64. Clinical Investigation, Level 2, Grade B.
Bleyer AJ, Burke SK, Dillon M, Garrett B, Kant KS, Lynch D, Rahman SN, Schoenfeld P, Teitelbaum I, Zeig S, Slatopolsky E. A comparison of the calcium-free phosphate binder sevelamer hydrochloride with calcium acetate in the treatment of hyperphosphatemia in hemodialysis patients. Am J Kidney Dis. 1999;33(4):694–701. Clinical Investigation, Level 1, Grade A.
Van Buren PN, Lewis JB, Dwyer JP, Greene T, Middleton J, Sika M, Umanath K, Abraham JD, Arfeen SS, Bowline IG, Chernin G, Fadem SZ, Goral S, Koury M, Sinsakul MV, Weiner DE, Collaborative Study Group. The phosphate binder ferric citrate and mineral metabolism and inflammatory markers in maintenance dialysis patients: results from prespecified analyses of a randomized clinical trial. Am J Kidney Dis. 2015;66(3):479–88. Clinical Investigation, Level 1, Grade A.
Eto N, Miyata Y, Ohno H, Yamashita T. Nicotinamide prevents the development of hyperphosphataemia by suppressing intestinal sodium-dependent phosphate transporter in rats with adenine-induced renal failure. Nephrol Dial Transplant. 2005;20(7):1378–84. Basic Science, Level 3, Grade A.
Covic A, Passlick-Deetjen J, Kroczak M, Büschges-Seraphin B, Ghenu A, Ponce P, Marzell B, de Francisco AL. A comparison of calcium acetate/magnesium carbonate and sevelamer-hydrochloride effects on fibroblast growth factor-23 and bone markers: post hoc evaluation from a controlled, randomized study. Nephrol Dial Transplant. 2013;28(9):2383–92. Clinical Investigation, Level 1, Grade A.
Navaneethan SD, Palmer SC, Vecchio M, Craig JC, Elder GJ, Strippoli GF. Phosphate binders for preventing and treating bone disease in chronic kidney disease patients. Cochrane Database Syst Rev. 2011;(2):CD006023. Clinical Review, Level 1, Grade C.
Jamal SA, Vandermeer B, Raggi P, Mendelssohn DC, Chatterley T, Dorgan M, Lok CE, Fitchett D, Tsuyuki RT. Effect of calcium-based versus non-calcium-based phosphate binders on mortality in patients with chronic kidney disease: an updated systematic review and meta-analysis. Lancet. 2013;382(9900):1268–77. Clinical Review, Level 1, Grade C.
Block GA, Wheeler DC, Persky MS, Kestenbaum B, Ketteler M, Spiegel DM, Allison MA, Asplin J, Smits G, Hoofnagle AN, Kooienga L, Thadhani R, Mannstadt M, Wolf M, Chertow GM. Effects of phosphate binders in moderate CKD. J Am Soc Nephrol. 2012;23(8):1407–15. Clinical Investigation, Level 1, Grade A.
Russo D, Miranda I, Ruocco C, Battaglia Y, Buonanno E, Manzi S, Russo L, Scafarto A, Andreucci VE. The progression of coronary artery calcification in predialysis patients on calcium carbonate or sevelamer. Kidney Int. 2007;72(10):1255–61. Clinical Investigation, Level 2, Grade C.
Slatopolsky E, Brown A, Dusso A. Pathogenesis of secondary hyperparathyroidism. Kidney Int Suppl. 1999;73:S14–9. Clinical Review, Level 5.
Craver L, Marco MP, Martínez I, Rue M, Borràs M, Martín ML, Sarró F, Valdivielso JM, Fernández E. Mineral metabolism parameters throughout chronic kidney disease stages 1-5—achievement of K/DOQI target ranges. Nephrol Dial Transplant. 2007;22(4):1171–6. Clinical Investigation, Level 3, Grade B.
González EA, Sachdeva A, Oliver DA, Martin KJ. Vitamin D insufficiency and deficiency in chronic kidney disease. A single center observational study. Am J Nephrol. 2004;24(5):503–10. Clinical Investigation, Level 3, Grade B.
Gallieni M, Kamimura S, Ahmed A, Bravo E, Delmez J, Slatopolsky E, Dusso A. Kinetics of monocyte 1 alpha-hydroxylase in renal failure. Am J Physiol. 1995;268(4 Pt 2):F746–53. Basic Science, Level 3, Grade A.
Brown AJ, Dusso AS, Slatopolsky E. Vitamin D analogues for secondary hyperparathyroidism. Nephrol Dial Transplant. 2002;17 Suppl 10:10–9. Basic Science Review, Level 2, Grade C.
Sprague SM, Llach F, Amdahl M, Taccetta C, Batlle D. Paricalcitol versus calcitriol in the treatment of secondary hyperparathyroidism. Kidney Int. 2003;63(4):1483–90. Clinical Investigation, Level 1, Grade A.
Coyne D, Acharya M, Qiu P, Abboud H, Batlle D, Rosansky S, Fadem S, Levine B, Williams L, Andress DL, Sprague SM. Paricalcitol capsule for the treatment of secondary hyperparathyroidism in stages 3 and 4 CKD. Am J Kidney Dis. 2006;47(2):263–76. Clinical Investigation, Level 1, Grade C.
Andress DL, Norris KC, Coburn JW, Slatopolsky EA, Sherrard DJ. Intravenous calcitriol in the treatment of refractory osteitis fibrosa of chronic renal failure. N Engl J Med. 1989;321(5):274–9. Clinical Investigation, Level 3, Grade A.
Park CW, Oh YS, Shin YS, Kim CM, Kim YS, Kim SY, Choi EJ, Chang YS, Bang BK. Intravenous calcitriol regresses myocardial hypertrophy in hemodialysis patients with secondary hyperparathyroidism. Am J Kidney Dis. 1999;33(1):73–81. Basic Science, Level 1, Grade B.
Kestenbaum B, Seliger SL, Gillen DL, Wasse H, Young B, Sherrard DJ, Weiss NS, Stehman-Breen CO. Parathyroidectomy rates among United States dialysis patients: 1990-1999. Kidney Int. 2004;65(1):282–8. Clinical Investigation, Level 3, Grade B.
Zheng Z, Shi H, Jia J, Li D, Lin S. Vitamin D supplementation and mortality risk in chronic kidney disease: a meta-analysis of 20 observational studies. BMC Nephrol. 2013;14:199. doi:10.1186/1471-2369-14-199. Clinical Investigation, Level 1, Grade C.
Mann MC, Hobbs AJ, Hemmelgarn BR, Roberts DJ, Ahmed SB, Rabi DM. Effect of oral vitamin D analogs on mortality and cardiovascular outcomes among adults with chronic kidney disease: a meta-analysis. Clin Kidney J. 2015;8(1):41–8. Clinical Investigation, Level 1, Grade C.
Rix M, Eskildsen P, Olgaard K. Effect of 18 months of treatment with alfacalcidol on bone in patients with mild to moderate chronic renal failure. Nephrol Dial Transplant. 2004;19:870–6. Clinical Investigation, Level 1, Grade A.
Bianchi ML, Colantonio G, Campanini F, Rossi R, Valenti G, Ortolani S, Buccianti G. Calcitriol and calcium carbonate therapy in early chronic renal failure. Nephrol Dial Transplant. 1994;9(11):1595–9. Clinical Investigation, Level 2, Grade B.
Andress DL. Adynamic bone in patients with chronic kidney disease. Kidney Int. 2008;73(12):1345–54. Clinical Review, Level 5.
Brown EM, Gamba G, Riccardi D, Lombardi D, Butters RR, Kifor O, et al. Cloning and characterization of an extracellular Ca2+-sensing receptor from bovine parathyroid. Nature. 1993;366:575–80. Basic Science, Level 1, Grade B.
Brown EM, MacLeod RJ. Extracellular Ca2+ sensing and extracellular Ca2+ signaling. Physiol Rev. 2001;81:239–97. Basic Science Review, Level 1, Grade A.
Nemeth EF, Heaton WH, Miller M, Fox J, Balandrin MF, Van Wagenen BC, et al. Pharmacodyamics of the Type I calcimimetic compound cinacalcet HCl. J Pharmacol Exp Ther. 2004;308:627–35. Basic Science, Level 1, Grade B.
Nemeth EF. The search for calcium receptor antagonists (calcilytics). J Mol Endocrinol. 2002;29:15–22. Basic Science Review, Level 1, Grade A.
Hauache OM, Hu J, Ray K, Xie R, Jacobson KA, Spiegel AM. Effects of a calcimimetic compound and naturally activating mutations on the human Ca2+ receptor and on Ca2+ receptor/metabotropic glutamate chimeric receptors. Endocrinology. 2000;141:4156–63. Basic Science, Level 1, Grade B.
Fox J, Lowe SH, Conklin RL, Petty BA, Nemeth EF. NPS R-568: a type II calcimimetic compound that acts on parathyroid cell Ca2+ receptor of rats to reduce plasma levels of parathyroid hormone and Ca2+. J Pharmacol Exp Ther. 1999;290:473–9. Basic Science, Level 1, Grade B.
Frazão JM, Martins P, Coburn JW. The calcimimetic agents: perspectives for treatment. Kidney Int. 2002;60:S149–54. Clinical Review, Level 1, Grade C.
Sprague SM, Wetmore JB, Gurevich K, Da Roza G, Buerkert J, Reiner M, Goodman W, Cooper K. Effect of Cinacalcet and Vitamin D Analogs on Fibroblast Growth Factor-23 during the Treatment of Secondary Hyperparathyroidism.Clin J Am Soc Nephrol. 2015 Jun 5;10(6):1021–30.
Block GA, Martin KJ, de Francisco AL, Turner SA, Avram MM, Suranyi MG, Hercz G, Cunningham J, Abu-Alfa AK, Messa P, Coyne DW, Locatelli F, Cohen RM, Evenepoel P, Moe SM, Fournier A, Braun J, McCary LC, Zani VJ, Olson KA, Drüeke TB, Goodman WG. Cinacalcet for secondary hyperparathyroidism in patients receiving hemodialysis. N Engl J Med. 2004;350(15):1516–25. Clinical Investigation, Level 1, Grade A.
Chonchol M, Locatelli F, Abboud HE, Charytan C, de Francisco AL, Jolly S, Kaplan M, Roger SD, Sarkar S, Albizem MB, Mix TC, Kubo Y, Block GA. A randomized, double-blind, placebo-controlled study to assess the efficacy and safety of cinacalcet HCl in participants with CKD not receiving dialysis. Am J Kidney Dis. 2009;53(2):197–207. Clinical Investigation, Level 1, Grade A.
Charytan C, Coburn JW, Chonchol M, Herman J, Lien YH, Liu W, Klassen PS, McCary LC, Pichette V. Cinacalcet hydrochloride is an effective treatment for secondary hyperparathyroidism in patients with CKD not receiving dialysis. Am J Kidney Dis. 2005;46(1):58–67. Clinical Investigation, Level 1, Grade A.
Nakayama K, Nakao K, Takatori Y, Inoue J, Kojo S, Akagi S, Fukushima M, Wada J, Makino H. Long-term effect of cinacalcet hydrochloride on abdominal aortic calcification in patients on hemodialysis with secondary hyperparathyroidism. Int J Nephrol Renovasc Dis. 2013;7:25–33. Clinical Investigation, Level 1, Grade B.
Floege J, Kubo Y, Floege A, Chertow GM, Parfrey PS. The effect of cinacalcet on calcific uremic arteriolopathy events in patients receiving hemodialysis: The EVOLVE trial. Clin J Am Soc Nephrol. 2015;10(5):800–7. Clinical Investigation, Level 1, Grade A.
Chertow GM, Block GA, Correa-Rotter R, Drüeke TB, Floege J, Goodman WG, Herzog CA, Kubo Y, London GM, Mahaffey KW, Mix TC, Moe SM, Trotman ML, Wheeler DC, Parfrey PS, EVOLVE Trial Investigators. Effect of cinacalcet on cardiovascular disease in patients undergoing dialysis. N Engl J Med. 2012;367(26):2482–94. Clinical Investigation, Level 1, Grade A.
Ballinger AE, Palmer SC, Nistor I, Craig JC, Strippoli GF. Calcimimetics for secondary hyperparathyroidism in chronic kidney disease patients. Cochrane Database Syst Rev. 2014;(12):CD006254.
Moe SM, Abdalla S, Chertow GM, Parfrey PS, Block GA, Correa-Rotter R, Floege J, Herzog CA, London GM, Mahaffey KW, Wheeler DC, Dehmel B, Goodman WG, Drüeke TB, Evaluation of Cinacalcet HCl Therapy to Lower Cardiovascular Events (EVOLVE) Trial Investigators. Effects of cinacalcet on fracture events in patients receiving hemodialysis: The EVOLVE trial. J Am Soc Nephrol. 2015;26(6):1466–75. Clinical Investigation, Level 1, Grade B.
Lee YT, Ng HY, Kuo CC, Chen TC, Wu CS, Chiu TT, Lee WC, Lee CT. Comparison between calcitriol and calcitriol plus low-dose cinacalcet for the treatment of moderate to severe secondary hyperparathyroidism in chronic dialysis patients. Nutrients. 2013;5(4):1336–48. Clinical Investigation, Level 3, Grade A.
Yamada S, Tokumoto M, Taniguchi M, Toyonaga J, Suehiro T, Eriguchi R, Fujimi S, Ooboshi H, Kitazono T, Tsuruya K. Two years of cinacalcet hydrochloride treatment decreased parathyroid gland volume and serum parathyroid hormone level in hemodialysis patients with advanced secondary hyperparathyroidism. Ther Apher Dial. 2015;19(4):367–77.
Cozzolino M, Ketteler M, Martin KJ, Sharma A, Goldsmith D, Khan S. Paricalcitol- or cinacalcet-centred therapy affects markers of bone mineral disease in patients with secondary hyperparathyroidism receiving haemodialysis: results of the IMPACT-SHPT study. Nephrol Dial Transplant. 2014;29(4):899–905. Clinical Investigation, Level 1, Grade A.
Roggeri DP, Cozzolino M, Mazzaferro S, Brancaccio D, Paoletti E, Roggeri A, Costanzo AM, di Luzio PU, Festa V, Messa P. Evaluating targets and costs of treatment for secondary hyperparathyroidism in incident dialysis patients: the FARO-2 study. Int J Nephrol Renovasc Dis. 2014;8:1–6. Clinical Investigation, Level 1, Grade A.
Sharma A, Marshall TS, Khan SS, Johns B. Cost effectiveness of paricalcitol versus cinacalcet with low-dose vitamin D for management of secondary hyperparathyroidism in haemodialysis patients in the USA. Clin Drug Investig. 2014;34(2):107–15. Clinical Investigation, Level 2, Grade B.
Schwarz P, Body JJ, Cap J, Hofbauer LC, Farouk M, Gessl A, et al. The PRIMARA study: a prospective, descriptive, observational study to review cinacalcet use in patients with primary hyperparathyroidism in clinical practice. Eur J Endocrinol. 2014;171:727–35. Clinical Investigation, Level 1, Grade B.
Saponaro F, Faggiano A, Grimaldi F, Borretta G, Brandi ML, Minisola S, et al. Cinacalcet in the management of primary hyperparathyroidism: post marketing experience of an Italian multicenter group. Clin Endocrinol (Oxf). 2013;79:20–6. Clinical Investigation, Level 2, Grade A.
Khan A, Bilezikian J, Bone H, Gurevich A, Lakatos P, Misiorowski W, et al. Cinacalcet normalizes serum calcium in a double-blind randomized, placebo-controlled study in patients with primary hyperparathyroidism with contraindications to surgery. Eur J Endocrinol. 2015;172:527–35. Clinical Investigation, Level 1, Grade B.
Peacock M, Bilezikian JP, Bolognese MA, Borofsky M, Scumpia S, Sterling LR, et al. Cinacalcet HCl reduces hypercalcemia in primary hyperparathyroidism across a wide spectrum of disease severity. J Clin Endocrinol Metab. 2011;96:E9–18. Clinical Review, Level 1, Grade A.
Brardi S, Cevenini G, Verdacchi T, Romano G, Ponchietti R. Use of cinacalcet in nephrolithiasis associated with normocalcemic or hypercalcemic primary hyperparathyroidism: results of a prospective randomized pilot study. Arch Ital Urol Androl. 2015;87:66–71. Clinical Investigation, Level 1, Grade B.
Cetani F, Saponaro F, Banti C, Cianferotti L, Vignali E, Chiavistelli S, et al. Cinacalcet efficacy in patients with moderately severe hyperparathyroidism according to the European Medicine Agency prescription labeling. J Endocrinol Invest. 2012;35:655–60. Clinical Investigation, Level 1, Grade B.
Thiem U, Gessl A, Borchhardt K. Long-term clinical practice experience with cinacalcet for treatment of hypercalcemic hyperparathyroidism after kidney transplantation. Biomed Res Int. 2015;2015:292654.
Marotta V, Di Somma C, Rubino M, Sciammarella C, Del Prete M, Marciello F, et al. Potential role of cinacalcet hydrochloride in sporadic primary hyperparathyroidism without surgery indication. Endocrine. 2015;49:274–8. Clinical Investigation, Level 1, Grade B.
Mazzaferro S, Pasquali M, Farcomeni A, Vestri AR, Filippini A, Romani AM, Barresi G, Pugliese F. Parathyroidectomy as a therapeutic tool for targeting the recommended NKF-K/DOQI ranges for serum calcium, phosphate and parathyroid hormone in dialysis patients. Nephrol Dial Transplant. 2008;23(7):2319–23. Clinical Investigation, Level 3, Grade A.
Lin HC, Chen CL, Lin HS, Chou KJ, Fang HC, Liu SI, Hsu CY, Huang WC, Huang CW, Huang CK, Chang TY, Chang YT, Lee PT. Parathyroidectomy improves cardiovascular outcome in nondiabetic dialysis patients with secondary hyperparathyroidism. Clin Endocrinol (Oxf). 2014;80(4):508–15. Clinical Investigation, Level 1, Grade A.
Costa-Hong V, Jorgetti V, Gowdak LH, Moyses RM, Krieger EM, De Lima JJ. Parathyroidectomy reduces cardiovascular events and mortality in renal hyperparathyroidism. Surgery. 2007;142(5):699–703. Clinical Investigation, Level 3, Grade A.
Uhlig K, Berns JS, Kestenbaum B, Kumar R, Leonard MB, Martin KJ, Sprague SM, Goldfarb S. KDOQI US commentary on the 2009 KDIGO clinical practice guideline for the diagnosis, evaluation, and treatment of CKD-Mineral and Bone Disorder (CKD-MBD). Am J Kidney Dis. 2010;55(5):773–99. Clinical review, Level 5.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Choksi, P., Lawson, B.R. (2017). Secondary Hyperparathyroidism. In: Stack, Jr., B., Bodenner, D. (eds) Medical and Surgical Treatment of Parathyroid Diseases. Springer, Cham. https://doi.org/10.1007/978-3-319-26794-4_7
Download citation
DOI: https://doi.org/10.1007/978-3-319-26794-4_7
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-26792-0
Online ISBN: 978-3-319-26794-4
eBook Packages: MedicineMedicine (R0)