
Abstract
In this chapter we describe a continuum model for the cortex that includes both axon-to-dendrite chemical synapses and direct neuron-to-neuron gap-junction diffusive synapses. The effectiveness of chemical synapses is determined by the voltage of the receiving dendrite V relative to its Nernst reversal potential \(V^{\rm rev}{}\). Here we explore two alternative strategies for incorporating dendritic reversal potentials, and uncover surprising differences in their stability properties and model dynamics. In the “slow-soma” variant, the \((V^{\rm rev} - V)\) weighting is applied after the input flux has been integrated at the dendrite, while for “fast-soma”, the weighting is applied directly to the input flux, prior to dendritic integration. For the slow-soma case, we find that–-provided the inhibitory diffusion (via gap-junctions) is sufficiently strong–-the cortex generates stationary Turing patterns of cortical activity. In contrast, the fast-soma destabilizes in favor of standing-wave spatial structures that oscillate at low-gamma frequency (\(\sim\)30-Hz); these spatial patterns broaden and weaken as diffusive coupling increases, and disappear altogether at moderate levels of diffusion. We speculate that the slow- and fast-soma models might correspond respectively to the idling and active modes of the cortex, with slow-soma patterns providing the default background state, and emergence of gamma oscillations in the fast-soma case signaling the transition into the cognitive state.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


12.1 Introduction
Continuum models of the cortex aim to describe those interactions of neural populations that generate the electrical fluctuations and rhythms able to be detected directly, with scalp and cortical EEG (electroencephalogram) electrodes, or remotely, using their magnetic counterpart, via MEG (magnetoencephalogram) sensors. Because the numbers of neurons involved in these cooperative behaviors is so vast, the continuum, or mean-field, approach makes no attempt to model the detailed biophysics of individual neurons, nor does it attempt to track the birth and axonal propagation of individual spike events. Instead, neuronal properties are represented as spatial averages, averaged, say, over the population of neurons sampled by a small EEG electrode, with spiking activity being represented as an average firing rate for the population-average neuron.
When constructing a theoretical model for the cerebral cortex, there is an unavoidable tension between the competing requirements of biophysical accuracy (leading to increased complexity) versus mathematical tractability (arguing for simplicity). In the end, we must make a pragmatic assessment of model quality by asking: Is the model fit for purpose–-i.e., Is the model able to make predictions that can be tested against biological reality? And: Does the model provide fresh insight?
In this chapter we will argue that incorporation of two biophysical features–-namely, cell-reversal potentials, and direct diffusive coupling between inhibitory neurons–-has important implications for emergent nonlinear behavior with respect to oscillatory rhythms and pattern formation in the cortex. Specifically, we show that the manner in which reversal potentials enter the model determines whether cortical oscillations appear in the delta (\(\sim\)2-Hz) or in the gamma (\(\sim\)30-Hz) frequency range. Further, we will demonstrate that inclusion of gap-junction diffusive connections modifies the strength and spatial extent of the Turing and standing-wave firing-rate patterns that can form in the cortical sheet.
12.1.1 Continuum modeling of the cortex
While continuum models of the cortex have evolved considerably since the foundation work of Wilson and Cowan [31], Nunez [18], and Freeman [9], present mean-field models continue to share three simplifying assumptions: (i) neural properties can be represented as spatial averages, (ii) neural inter-connectedness decays with distance, (iii) neural firing rates can vary between zero and some maximum value, with a sigmoidal mapping from membrane voltage to firing rate.
Continuum models are expressed either as coupled partial differential equations (PDEs) , or as integro-differential equations (IDEs) , or as purely integral equations, with the choice of representation being determined by the type and dimensionality (1-D or 2-D) of the connectivity kernel . The PDE forms have the advantage of speed and ease of analysis, but have access to a restricted range of connectivity kernels, e.g., exponential decay in 1-D [11,31], modified Bessel (Macdonald)-function decay in 2-D [21]. The integral forms are slower to compute numerically, and require a large amount of storage, but have the advantage that the kernel can be chosen at will; Wright and Liley [35] use a Gaussian to represent the decreasing synaptic density with distance. For the integro-differential forms, “Mexican hat” connectivity kernels are frequently used [4,5,6,14,15].
In the PDE-based cortical model we present here, flux activity generated by excitatory and inhibitory neural populations is received at a dendritic synapse whose transmission efficiency is modulated by the difference between the membrane voltage and its reversal potential [16,20]. Following Robinson et al [21], axonal flux transmission is assumed to obey a 2-D wave equation with Macdonald-function connectivity. The net neuron voltage is determined not only by axono-dendritic activity at chemical synapses, but also by diffusive currents from adjacent neurons that are directly coupled to the target neuron via gap junctions.
Our parameter values for the chemical-synaptic component of the model largely match those of Rennie et al [20], but we have chosen to retain the symbols and labeling conventions used in our earlier sleep [23] and anesthesia modeling [24], which drew on work by Liley et al [16]. For the gap-junction component of the model, we adopt the values and notation we introduced in Ref. [26].
12.1.2 Reversal potentials
The size and direction of the postsynaptic potential evoked at a chemical synapse by incoming spike activity depends on the voltage state of the receiving neuron, and, in particular, on \((V^{\rm rev} - V)\), the voltage of the receiving dendrite relative to its reversal potential. If this difference is large, spike events will be more effective at transferring charge across, and eliciting a voltage response in, the post-synaptic membrane; this efficiency diminishes to zero as V approaches \(V^{\rm rev}\), the reversal potential being \(\sim\)0 mV for excitatory events (mediated by AMPA receptors) and \(\sim-\)70 mV for inhibitory events (mediated by GABA receptors) .
Although a standard feature in all Hodgkin–Huxley [13] conductance-based neuron models, surprisingly few mean-field cortical models include excitatory and inhibitory reversal potentials [16,20,25,36]. The neglect of these biophysical constraints might be justifiable if the voltage fluctuations about resting equilibrium (\(V^{\rm rest} \approx -\)60 mV) remain sufficiently small that the reversal potentials are effectively infinite. But if the fluctuations grow sufficiently large–-as can happen when the equilibrium state destabilizes in favor of a Hopf , Turing , or wave instability –-then the existence of finite reversal potentials could have a significant impact on neural behavior. In fact, we will show that a subtle change in the way in which reversal potentials are incorporated into the model leads to qualitative change in its stability properties.
12.1.3 Gap-junction diffusion
The traditional picture of neural communication requires active propagation of action potentials from the axon of the transmitting neuron to the dendrite of the receiving neuron via release of neurotransmitters at the chemical-synaptic interface.
There is accumulating evidence, however, that subthreshold voltage fluctuations can be passively communicated from neuron to neuron via electrical synapses formed from gap-junction proteins that make direct resistive connections between neighboring cells at their points of dendritic contact. This is particularly so for inhibitory neurons in the cat visual cortex where the measured density of connexin-36 (Cx36) gap-junctions is so high that Fukuda et al [10] described the result as establishing a dense and widespread network of interneurons able to be traced in a boundless chain. In addition, researchers have detected copious gap-junction couplings between interneurons and their supporting glial cells (via Cx32 connexin), and between pairs of glial cells (via Cx43) [1,17], suggesting that diffusive neuronal coupling may be augmented by glial-cell “bridges”. To date, there are no reports of dense gap-junction connectivity between pairs of excitatory neurons, suggesting that, for reasons unknown, neural tissue has evolved to strongly favor inhibitory-to-inhibitory diffusion over excitatory-to-excitatory diffusion .
In Ref. [26], we used the Fukuda measurements to estimate an upper bound for D 2, the inhibitory coupling strength, \(D_2 \approx 0.6 {\rm cm}^2\), then investigated the impact of incorporating inhibitory diffusion into a mean-field model of the cortex based on chemical synapses. We found that, provided that the D 2 inhibitory diffusion is sufficiently large, a homogeneous cortical sheet will spontaneously destabilize in favor of cm-scale stationary Turing patterns of intermixed regions of high- and low-firing activity.
In this chapter we extend this work by demonstrating that gap-junction diffusion \(D_2\nabla^2 V\) can interact with the \((V^{\rm rev} - V)\) reversal-potential terms to generate two distinct types of spatiotemporal instability: either (a) stationary Turing structures when the feedback from soma to dendrite is delayed (“slow-soma” model ); or (b) standing waves of gamma-band cortical activity when the soma-to-dendrite feedback is prompt (“fast-soma” model ). We develop the background theory for the slow- and fast-soma models in Sect. 12.2; analyze their respective linearized stability characteristics in Sect. 12.3, then verify these predictions with a series of 2-D grid simulations of the full nonlinear equations. We follow this with a comparison against an earlier model due to Rennie and colleagues [20] in Sect. 12.4, then comment on the possible biological significance of the slow- and fast-soma forms.
12.2 Theory
We present the equations of motion for a continuum model of the cortex that consists of mutually interacting populations of excitatory and inhibitory neurons, each population receiving flux inputs from spiking events arriving at excitatory and inhibitory chemical synapses. The transmission efficiency of an excitatory (inhibitory) synapse is modulated by the voltage state of the post-synaptic dendrite relative to the AMPA (GABA) reversal potential . We consider two alternative schemes for incorporating the dendritic reversal potentials , leading to the slow-soma (Sect. 12.2.1.1) and fast-soma (Sect. 12.2.1.2) variants of the model. In both cases we assume that axonal flux propagation obeys damped 2-D wave equations (Sect. 12.2.1.3), with slower local (unmyelinated gray-matter) connections and faster long-range (myelinated white-matter) connections. The cortex is stimulated by nonspecific tonic activity generated by the subcortex (Sect. 12.2.1.4). Finally, in Sect. 12.2.2 we complete the model with the addition of diffusive voltage perturbations transmitted via electrical (gap-junction ) synapses.
12.2.1 Input from chemical synapses
In deriving the equations of motion for V e and V i the soma voltages for the excitatory and inhibitory neural populations, we assume that a pre-synaptic spike event will induce a post-synaptic potential (PSP), a momentary voltage change in the receiving dendrite, whose shape can be modeled either as a biexponential (first line of Eq. (12.1)) or as an alpha-function (second line),
for \(t > 0\), where \(\alpha\) and \(\beta\) are positive constants.
If the incoming presynaptic spike rate is M [spikes/s], then the net voltage disturbance [in mV] at the dendrite will be given by \(\rho U\), where \(\rho\) is the synaptic strength [mV\(\cdot\)s]; and U is the post-synaptic response rate [s\(^{-1}\)] given by the temporal convolution-integral of the input flux M with the dendrite filter response H, scaled by a dimensionless synaptic reversal-potential factor \(\psi\),
We follow the earlier work of Wright et al [36], Liley et al [16], and Rennie et al [20] in defining the \(\psi\) scaling factor to be unity when the neuron is at rest (\(V = V^{\rm rest}\)), and zero when the membrane voltage matches the relevant synaptic reversal potential (\(V^{\rm rev}_{e} = 0\) mV for excitatory (AMPA) receptors; \(V^{\rm rev}_{i} = -70\) mV for inhibitory (GABA) receptors),
Here we have introduced subscript labels a, b, each of which stands for either e (excitatory) or i (inhibitory), indicating that there are four reversal-potential functions: \(\psi_{ee}\), \(\psi_{ei}\), \(\psi_{ie}\), \(\psi_{ii}\), where, for example, \(\psi_{ei}\) is the scaling function for excitatory flux entering an inhibitory neuron. Corresponding double-subscripts are also to be attached to the H, M, and U appearing in Eq. (12.2).
The excitatory and inhibitory voltage disturbances at the dendrite are then integrated at the soma by convolving with the exponential soma impulse-response L,
where \(\tau_b\) is the soma time-constant for neurons of type b (e or i). This second integration results in a pair of integral equations of motion for V e and V i , the soma voltages for the excitatory and inhibitory neuron populations,
The \(\rho_{e,i}\) synaptic strengths are signed quantities, with \(\rho_e > 0\) for excitatory postsynaptic potential (EPSP) events, and \(\rho_i < 0\) for inhibitory postsynaptic potentials (IPSPs) .
We wish to draw attention to the assumption, implicit in Eq. (12.2) regarding the construction of the post-synaptic rate U, by asking the question: Should the \(\psi\)-scaling by the reversal-potential weight be performed after the \(H\otimes\) dendrite integration of input flux M–-as written in Eq. (12.2)—
or, should the \(\psi\)-scaling be applied directly to the input flux, so that it is the weighted product \(\psi \cdot M\) that is integrated at the dendrite? This alternative ordering leads to a revised post-synaptic rate U,
We refer to the Eq. (12.7) form for the U post-synaptic rate as the “slow soma” case, since the somata voltage of the neuron is presumed to vary on a time-scale that is much slower than that of the synaptic input events. This limit should be valid when the synaptic inputs are sparse or weak, so that the \(\psi\) reversal-potential feedback from soma to dendrite is slow to arrive.
On the other hand, if synaptic activity is strong, or if the soma voltage changes on a time-scale similar to that of dendritic integration , then the reversal-potential feedback onto the dendrite will be prompt. In this case, the M flux input at time t should be scaled by the \(\psi\) reversal-potential weight at time t, then integrated at the dendrite; this limit gives the Eq. (12.8) “fast-soma” form for the U post-synaptic rate.
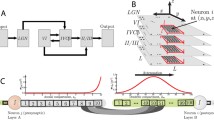
These slow- and fast-soma variants are block-diagrammed in the flow-charts of Fig. 12.1.

Dendrite-to-soma flow diagrams for (a) slow-soma and (b) fast-soma cortical models. M is the average spike-rate input arriving at the dendrite via chemical synapses; V is the resulting voltage perturbation at the soma (for simplicity, we ignore the constant \(V^{\rm rest}\) offset here); \(\rho\) is the synaptic strength; H and L are respectively the dendrite and soma impulse-response functions. The \(\psi\) reversal-potential weighting function provides immediate feedback from soma to dendrite. Symbols \(\otimes\) and \(\odot\) represent convolution and product operations respectively. (a) For slow-soma, the input flux is modulated by the reversal-weighting after integration at the dendrite filter, while for fast-soma, the reversal modulation occurs prior to dendrite integration.
We find that swapping the order of the \(\psi\cdot\) and \(H\otimes\) operations has surprising implications for cortical stability that may have biological significance. If the \(\psi\cdot\) weighting occurs after the \(H\otimes\) dendrite integration (i.e., Eq. (12.7): slow-soma), then, as reported in [26], the homogeneous 2-D cortex can destabilize in the presence of inhibitory diffusion to form Turing patterns –-stationary spatial patterns of activated and inactivated patches of cortical tissue. Footnote 1
But if the \(\psi\cdot\) weighting is applied prior to the \(H\otimes\) convolution (i.e., Eq. (12.8): fast-soma), we will see that the stationary Turing patterns are replaced by standing-wave patterns of similar spatial frequency but whose temporal frequency lies within the gamma band (\(\sim\)30–80 Hz) of EEG oscillations.
12.2.1.1 Slow-soma limit
In the limit of a slowly varying membrane potential, the soma-voltage equations (12.5) and (12.6) become,
where the \(\Phi_{eb}\), \(\Phi_{ib}\) (b = e, i) represent the four slow-soma flux convolutions of flux input M against dendrite filter H,
and where
with subcortical inputs
Equation (12.11) defines M, the total input flux of type a (e or i) entering neurons of type b (e or i). The superscript labels \(\alpha\), \(\beta\), sc indicate long-range, short-range, and subcortical chemical-synaptic inputs respectively. The \(N^{\alpha,\beta,{\rm sc}}\) are the number of synaptic connections; the \(\phi^{\alpha,\beta,{\rm sc}}\) are per-synapse flux rates. The \(\phi^{\alpha,\beta}\) fluxes obey wave equations detailed below in Eqs (12.18, 12.20). The long-range and subcortical inputs are excitatory only, so \(N_{ie}^\alpha = N_{ii}^\alpha = N_{ie}^{\rm sc} = N_{ii}^{\rm sc} = 0\), and \(\phi_{ie}^{\rm sc} = \phi_{ii}^{\rm sc} = 0\). Here s is a subcortical scaling parameter whose value can range between 0 and 1. Since \(Q_e^{\max}= 100\) s\(^{-1}\) (see Table 12.4 in the Appendix), choosing s=0.1 will ensure compatibility with the earlier modeling work by Rennie et al [20] in which the default level for per-synapse subcortical drive was set at \(\phi^{\rm sc} = 10\) s\(^{-1}\).
The slow-soma integral equations of Eq. (12.9) are equivalent to a pair of first-order differential equations of motion for soma voltage ,
Taking the biexponential form of the Eq. (12.1) postsynaptic potential, the four dendrite convolutions of Eq. (12.10) can be rewritten as four second-order ODEs in \(\Phi(t)\),
12.2.1.2 Fast-soma limit
For the fast-soma version of the cortical model, we use the revised form of the post-synaptic rate given in Eq. (12.8),
leading to two fast-soma differential equations for the V b (b=e,i) neuron voltage (cf. Eq. (12.13)),
with dendrite ODEs (cf. Eq. (12.14)),
Comparing Eqs (12.16, 12.17) with (12.13, 12.14), we see that, in the fast-soma limit, the \(\psi_{ab}\) reversal-potential weights are applied directly to the incoming M ab synaptic flux, with the product being integrated at the dendrite to give the (weighted) dendritic flux U ab ; whereas in the slow-soma model, the \(\psi_{ab}\) weights are applied after the M ab input flux has been integrated at the dendrite.
12.2.1.3 Wave equations
The axonal wave equations described here apply equally to both the slow- and fast-soma cortical models. Following Robinson et al [21], we assume that the \(\phi^\alpha\) long-range excitatory fluxes obey a pair of 2-D damped wave equations generated by excitatory sources \(Q_e({\bf r}, t)\),
where \(\Lambda^\alpha\) is the inverse-length scale for axonal connections [cm\(^{-1}\)], and \(v^\alpha\) is the axonal conduction speed [cm/s]. Q is the sigmoidal mapping from soma voltage to neuronal firing rate,
with \(C = \pi/\sqrt{3}\). Here, \(\theta_a\) is the population-average threshold for firing, \(\sigma_a\) is its standard deviation, and \(Q_a^{\max}\) is the maximum firing rate.
In previous work [23,24,25,32,33,34],
we have assumed that the short-range axonal signals propagate instantaneously, allowing local spike-rate fluxes \(\phi_{ab}^\beta\) to be replaced by their sources Q a . In this chapter we allow for finite propagation speeds by writing four wave equations for the short-range fluxes \(\phi_{ab}^\beta\) traveling on unmyelinated axons ,
12.2.1.4 Subcortical inputs
Equation (12.12) is applicable when the subcortical drive is a fixed constant. To allow noise to enter the cortex, we replace (12.12) with the stochastic form
where \(\gamma\) is a constant noise scale-factor, and the \(\xi_m\) are a pair of Gaussian-distributed, zero-mean, spatiotemporal white-noise sources that are delta-correlated in time and space,
In the grid simulations presented in Sect. 12.3, we specify s, the subcortical drive (e.g., s = 0.1), and initialize the 2-D sheet of cortical tissue at the homogeneous steady-state corresponding to this level of subcortical stimulation. Then, using Eq. (12.21), we distribute spatially-independent small-amplitude random perturbations across the cortical grid to allow the model to explore its proximal state space. If the homogeneous equilibrium is unstable, these small-scale deviations from homogeneity can organize and grow into large-scale Turing structures, Hopf oscillations, and gamma-band standing-wave patterns.
12.2.2 Input from electrical synapses
The existence of gap junctions in the mammalian brain has been known for decades, but only recently has their electrophysiological significance with respect to neuron coupling and rhythm synchronization become apparent. According to the review article by Bennett and Zukin [2], electrical transmission (via gap junctions) between neurons is “likely to be found wherever it is useful,” with its selective advantage being the communication of subthreshold potentials that facilitate synchronization.
The reported abundances of gap-junction connections in brain tissue are increasing as detection methods become more sensitive and discriminating. These connections are found between pairs of inhibitory interneurons (via connexin Cx36 channels [10]), between interneurons and their supporting glial cells (via Cx32), and between the glial cells themselves (via Cx42). The neuron-to-glia and glia-to-glia connections have been detected in all layers of the rat cerebral cortex [17]. These findings support the notion of a diffusively-intercoupled continuous scaffolding that links networks of active (neuronal) and passive (glial) cells.
Fukuda et al [10] reported that, on average, each L-type inhibitory interneuron in the cat visual cortex was coupled to \(N_{ii}^{\rm gap} = 60 \pm 12\) other L-type interneurons via Cx36 connexin channels , that the connections were randomly and uniformly distributed over a disk of radius \(\sim\)200 \(\mu\)m centered on a given neuron, and that L-type abundance was \(\sim\)400 mm\(^{-2}\), implying a connection density of \(\sim\)24 000 gap-junctions per mm\(^2\). The Fukuda measurements were specific to Cx36 connexins only, so total interneuron gap-junction interconnectivity could be considerably higher.
In [26], we used the Fukuda measurements to construct a theoretical 2-D lattice of square “Fukuda cells” , with each lattice cell representing the effective area u of diffusive influence for a single L-type interneuron; see Fig. 12.2.

Equivalent electrical circuit for nearest-neigbor gap-junction connections between neurons in a 2-D cortex. Diffusion currents \(I_{\rm N}, I_{\rm S}, I_{\rm E}, I_{\rm W}\) enter the neuron at the central node from the four neighboring nodes via gap-junction resistances R shown in bold. For clarity, chemical-synaptic currents are not shown. (Figure reproduced from [26].)
We assume that the neuron at the lattice center has resting voltage \(V^{\rm rest}\), capacitance C, membrane resistance R m , and receives diffusive current along four resistive arms, each of resistance \(R = R^{\rm gap}/\frac{1}{4}N_{ii}^{\rm gap} = R^{\rm gap}/15\), where \(R^{\rm gap}\) is the resistance of a single Cx36 gap junction .
The total current to “ground” (i.e., to the extracellular space) is the sum of membrane current \((V - V^{\rm rest})/R_m\) plus capacitive current \(C \, dV\!\!/\!dt\), and this must match the addition of chemical synaptic currents \(I^{\rm syn}\) (not shown) plus gap-junction diffusive currents \(I^{\rm gap}\),
where
The effective area u of the Fukuda cell depends on the underlying connectivity assumption. If the connectivity is uniform across the 200-\(\mu\)m radius disk, we circumscribe the circle with a square of side 0.4 mm, giving an upper bound of \(u \approx 0.16\) mm\(^2\). More realistically, we can fit a Gaussian distribution to the Fukuda data (see Appendix A of [26] for details), leading to \(u \approx 0.03\) mm\(^2\), a factor of five smaller. We note that these estimates for u will increase if subsequent determinations of gap-junction dendritic extent and distal abundance are found to be larger than those reported by Fukuda et al.
Rearranging Eq. (12.24), we obtain the differential equation for soma voltage ,
where \(\tau = R_m C\) is the membrane time-constant , \(I^{\rm syn}R_m\) is the voltage contribution at the soma arising from chemical-synaptic currents, and D ii is the diffusive coupling strength for inhibitory-to-inhibitory gap-junction currents,
In [26] we estimated \(R_m \approx 7100\) M\({\rm M}\Omega\), \(R^{\rm gap} \approx 290\) M\({\rm M}\Omega\) (corresponding to a gap-junction in its fully-open configuration). Setting \(u = 0.16\) mm\(^2\) gives \(D_{ii} \approx 0.6\) cm\(^2\), but we emphasize that all four components (\(u, N_{ii}, R_m, R^{\rm gap}\)) in the D ii expression are uncertain, and are likely to vary with time, neuromodulatory state, and stage of development: it is not implausible that the “true” value for D ii might lie within an uncertainty band that extends from \(+1\) to \(-2\) orders-of-magnitude above and below the nominal value quoted here.
12.2.2.1 Slow-soma limit with gap junctions
We incorporate the effect of gap-junction diffusion currents entering a slow-soma neuron by combining Eq. (12.26) with the slow-soma membrane Eqs (12.14) to give
where the terms in square brackets \([\ldots]\) are the contributions from chemical-synaptic flux entering the slow-soma model. Here, D bb = D ee for excitatory-to-excitatory diffusion, and D bb = D ii for inhibitory-to-inhibitory diffusion.Footnote 2
We note that direct electrical connections between pairs of same-family inhibitory interneurons are common, but apparently gap junctions between pairs of excitatory neurons are rare, so in the work below we set D ee to be a small (but non-zero) fraction of D ii , with D ee = D ii/100.
12.2.2.2 Fast-soma limit with gap junctions
Combining Eq. (12.26) with the fast-soma membrane Eqs (12.17) give the pair (\(b = e,i\)) of partial differential equations for the diffusion-enhanced fast-soma model,
12.3 Results
12.3.1 Stability predictions
The stability characteristics of the slow- and fast-soma cortical models are referenced to a homogeneous steady-state corresponding to a given value of subcortical drive s. This reference state is determined by zeroing the \(\xi_m\) noise terms in Eq. (12.21), and removing all time- and space-dependence by setting \(d/dt = \nabla^2 = 0\) in either the slow-soma differential equations (12.28, 12.14, 12.18, 12.20), or the fast-soma equations (12.29, 12.17, 12.18, 12.20). Either procedure gives a set of nonlinear simultaneous equations that we solve numerically to locate the steady-state soma voltage \((V_e^0, V_i^0)\) and firing rate \((Q_e^0, Q_i^0)\). Note that, at steady state, the distinction between the slow-soma \(U = \psi\cdot[H\otimes M]\) (Eq. (12.7)) and fast-soma \(U = H\otimes [\psi\,.\,M]\) (Eq. (12.8)) convolution forms vanishes, and therefore the equilibrium states for the slow-soma and fast-soma cortical models are identical. Yet despite their shared equilibria, we will show that the dynamical properties of the two models–-as predicted by linear eigenvalue analysis, and confirmed by nonlinear grid simulations–-are very different.
The 12 state variables for the slow-soma and fast-soma models are listed in Table 12.1; related system variables appear in Table 12.2. The 12 state variables are governed by two first-order (V e , V i ) and 10 second-order (U ab , \(\phi^\alpha_{eb}\), \(\phi^\beta_{ab}\)) differential equations, so are equivalent to 22 coupled first-order DEs, and therefore, after linearization about homogeneous steady state, own 22 eigenvalues .
The linearization proceeds by expressing each of the 22 first-order variables (12 state variables plus 10 auxiliaries) as its homogeneous equilibrium value plus a fluctuating component. For example, the excitatory soma voltage is written
where \({\bf r}\) is the 2-D position vector, and \(\delta V_e\), the fluctuation about equilibrium \(V_e^0\), has spatial Fourier transform
with \({\bf q}\) being the 2-D wave vector. This is equivalent to assuming that the voltage perturbation can be expressed as a spatiotemporal mode of the form,
where \(\Lambda\) is its (complex) eigenvalue . If \(\Lambda\) has a positive real part, the perturbation will grow, indicating that the equilibrium state is unstable.
After linearizing the 22 first-order DEs about homogeneous equilibrium, then Fourier transforming in space, we compute numerically the 22 eigenvalues of the Jacobian matrix for a range of finely-spaced wavenumbers, \(q = |{\bf q}|\). Arguing that the stability behavior of the cortical model will be dominated by the eigenvalue \(\Lambda\) whose real part is least negative (or most positive), we plot the distribution of dominant eigenvalues as a function of wavenumber, looking for regions for which Re\([\Lambda(q)] > 0\), indicating the presence of spatial modes that can destabilize the homogeneous rest state.
12.3.2 Slow-soma stability

Slow-soma dispersion curves for (a) increasing inhibitory diffusion D 2 and (b) increasing subcortical drive s
(a) Imaginary (upper thin traces) and real (lower thick traces) parts of dominant eigenvalue plotted as a function of scaled wavenumber \(q/2\pi\) for three values of diffusion strength, \(D_2 = [2.0, \, 2.5, \, 4.0]\) cm\(^2\); excitatory diffusion strength is set at 1% of inhibitory strength: \(D_1 = D_2/100\). Subcortical drive is fixed at s = 0.1 (i.e., \(\phi^{\rm sc} = 10\) s\(^{-1}\)), corresponding to homogeneous steady state \((V_e^0, V_i^0) = (-59.41, -59.41)\) mV; \((Q_e^0, Q_i^0) = (6.37, 12.74)\) s\(^{-1}\). (See Table 12.4 for parameter values.) The homogeneous state is predicted to be unstable at all spatial frequencies for which the real part of the dispersion curve is positive. Stationary Turing patterns are predicted at \(q/2\pi \approx 0.45\) cm\(^{-1}\) for \(D_2 \gtrsim 2.5\) cm\(^2\), and are enhanced by increases in D 2 coupling strength.
(b) Eigenvalue distribution for three values of subcortical drive, s = [0.1, 0.3, 0.5]; excitatory and inhibitory diffusion strength are fixed at (D 1, D 2) = (0.025, 2.5) cm\(^2\). The slow-soma Turing instability at \(q/2\pi = 0.45\) cm\(^{-1}\) is damped out by increases in subcortical tone, restoring stability to the homogenous steady-state. (Figure reproduced from Ref. [27].)
Figure 12.3 shows that, in the slow-soma limit, the homogeneous steady state can be destabilized either by increasing the inhibitory diffusion D 2 (Fig. 12.3(a)), or by decreasing the level of subcortical drive s (Fig. 12.3(b)). Instability at a given wavenumber q is predicted when its dominant eigenvalue crosses the zero-axis, changing sign from negative (decaying mode) to positive (exponentially-growing mode). For the case D 2 = 4 cm\(^{2}\), s = 0.1 (top curve of Fig. 12.3(a)), all wavenumbers in the range \(0.24 \lesssim q/2\pi \lesssim 0.7\) cm\(^{-1}\) support growing modes, with strongest growth predicted at \(q/2\pi \approx 0.4\) cm\(^{-1}\) (i.e., wavelength \(\approx 2.5\) cm), at the peak of the dispersion curve. At this wavenumber, the eigenvalue has a zero imaginary part, so the final pattern is expected to be a stationary periodic pattern in space–-a Turing structure of intermixed regions of high- and low-firing cortical activity. This spontaneous Turing emergence is similar to that reported in [26] for an earlier version of the slow-soma model that had access to three homogeneous steady states (two stable, one unstable).
12.3.3 Fast-soma stability

Fast-soma dispersion curves for (a) increasing inhibitory diffusion D 2 and (b) increasing subcortical tone s.
(a) The three pairs of eigenvalue curves correspond to three values for inhibitory diffusion, D 2 = [0.0, 0.04, 0.10] cm\(^2\), with subcortical drive kept fixed at s = 0.1. Wave instabilities of temporal frequency \(\sim\)29-Hz and spatial frequency 0.5 waves/cm are expected when \(D_2 \lesssim 0.04\) cm\(^2\).
(b) Eigenvalue distribution for three values of subcortical drive, s = [0.1, 0.3, 0.5], corresponding to subcortical flux rates of \(\phi^{\rm sc} = [10, 30, 50]\) s\(^{-1}\), giving homogeneous steady-state firing rates \(Q_e^0 = [6.37, 7.28, 8.10]\) s\(^{-1}\); excitatory and inhibitory diffusion is fixed at (D 1, D 2) = (0.0005, 0.05) cm\(^2\). For s = 0.1 (lower-thick and upper-thin solid curves), homogeneous steady-state destabilizes in favor of 29-Hz traveling waves of spatial frequency 0.49 cm\(^{-1}\). Increasing subcortical tone to 0.3 and 0.5 strengthens the wave instability , and raises its frequency slightly to 31 and 32.5 Hz respectively. For s = 0.5, the peak at \(q/2\pi = 0\) indicates that the wave pattern will be modulated by a whole-cortex Hopf instability of frequency 35 Hz. (Figure reproduced from Ref. [27].)
Figure 12.4(a) shows the set of dispersion curves obtained for the fast-soma version of the model operating at the same \(s = 0.1\) level of subcortical excitation used in Fig. 12.3(a). In marked contrast to the slow-soma case, maximum instability is obtained in the limit of zero inhibitory diffusion (D 2 = 0), with the instability being promptly damped out as the diffusion increases. We see that small increases in diffusion serve to narrow the range of spatial frequencies able to destabilize the equilibrium state. Thus, when D 2 = 0, the instability is distributed across the broad range \(0.35 < q/2\pi < 3.48\) cm\(^{-1}\) (this upper value is not shown on the Fig. 12.4 graph), shrinking to \(0.40\)–\(0.67\) cm\(^{-1}\) when D 2 = 0.04 cm\(^2\), and vanishing completely for \(D_2 \ge 0.06\) cm\(^2\).
The fact that the dominant eigenvalue has a nonzero imaginary part indicates that these fast-soma spatial instabilities will tend to oscillate in time: for spatial frequency \(q/2\pi = 0.5\) cm\(^{-1}\), the predicted temporal frequency is \(\sim\)29 Hz, at the lower end of the gamma band . Writing \(\omega = {\rm Im}[\Lambda]\), the slope of the \(\omega\)-vs-q graph is nearly flat (thin curves of Fig. 12.4(a)), implying that these wave instabilities will propagate but slowly. For example, with D 2 = 0.04 cm\(^{2}\), the group velocity at \(q/2\pi = 0.5\) cm\(^{-1}\) is \(d\omega/dq = 3.8\) cm/s, so that, over the timescale a single gamma oscillation (\(\sim\)0.034 s), the wave will travel only 1.3 mm. This suggests that the instability will manifest as a slowly-drifting standing-wave pattern of 29-Hz gamma oscillations , with wavelength \(\sim\)2 cm.
Another surprising difference in the behavior of the slow- and fast-soma models is their contrasting stability response to alterations in the level of subcortical drive s. If the subcortical tone is stepped, say, from s = 0.1 to 0.3 to 0.5, the homogeneous excitatory firing rate for both models increases slightly, from \(Q_e^0 = 6.4\) to 7.3 to 8.1 spikes/s (not shown here). In Fig. 12.4(b) we observe that this boost in subcortical tone tends to destabilize the fast-soma cortex, increasing both the strength and the frequency of the gamma wave instability –-but for the slow-soma cortex (Fig. 12.3(b)) the effect is precisely opposite, acting to damp out Turing instabilities , encouraging restoration of the homogeneous equilibrium state . We will argue in Sect. 12.4 that these interesting divergences in the slow- and fast-cortical responses are consistent with the notion that the slow-soma could describe the idling or default background state of the conscious brain, while the fast-soma could describe the genesis of gamma resonances that characterize the active, cognitive state .
12.3.4 Grid simulations
To test the linear-stability predictions of Turing and traveling-wave activity in the cortical model, we ran a series of numerical simulations of the full nonlinear slow-soma (12.28, 12.14, 12.18, 12.20) and fast-soma (12.29, 12.17, 12.18, 12.20) cortical equations. The substrate was a 240\(\times\)240 square grid, of side-length 6 cm, joined at the edges to provide toroidal boundaries. We used a forward-time, centered-space Euler algorithm custom-written in matlab 7.6, with the diffusion and wave-equation \(\nabla^2\) Laplacians implemented as wrap-around (toroidal) convolutionsFootnote 3 of the 3\(\times\)3 second-difference mask against the grid variables holding \(V_{e,i}({\bf r}, t)\), the excitatory and inhibitory membrane voltages. The grid was initialized at the homogeneous steady state corresponding to a specified value of subcortical drive s, then driven continuously by two independent sources of small-amplitude unfiltered spatiotemporal white noise representing unstructured subcortical tone \(\phi^{\rm sc}_{ee,ei}\) (see Eq. (12.21)). The timestep was set sufficiently small to ensure numerical stability, ranging from \(\Delta t = 100\) \(\mu\)s for the fast-soma (weak diffusion) runs, down to 1 \(\mu\)s for the slow-soma runs with strongest inhibitory diffusion (i.e., D 2 = 6 cm\(^2\)).
The upper bound for the slow-soma timestep was obtained by recognizing that \(D_2/\tau_i\), the ratio of diffusive strength to membrane relaxation time, defines a diffusion coefficient [units: cm/s] for inhibitory voltage change, so in time \(\Delta t\), a voltage perturbation is expected to diffuse through an rms distance \(d^{\rm rms} = \sqrt{4D_2 \Delta t/\tau_i}\). Setting \(d^{\rm rms} = \Delta x = \Delta y\), the lattice spacing, and solving for \(\Delta t\), gives
which we replaced with the more conservative
to ensure that, on average, the diffusive front would have propagated by less than one lattice spacing between consecutive timesteps.
For the fast-soma case, diffusion values are very weak, so it is the long-distance wave equations (12.18) that set the upper bound for the timestep. The Courant stability condition for the 2-D explicit-difference method requires \(v^\alpha \Delta t/\Delta x \le 1/\sqrt{2}\). Setting \(\Delta x = L_x/240 = 0.025\) cm, and \(v^\alpha = 140\) cm/s (see Table 12.4) gives \(\Delta t \le 126\) \(\mu\)s, so the fast-soma choice of \(\Delta t = 100\) \(\mu\)s is safely conservative.

[Color plate] Grid simulation for slow-soma cortical model with subcortical drive s = 0.1 (i.e., \(\phi^{\rm sc} = 10\) s\(^{-1}\)), inhibitory diffusion \(D_2 = 4.0\) cm\(^2\). Cortex is a 240\(\times\)240 square grid of side-length 6 cm, with toroidal boundaries, initialized at its homogeneous steady-state firing-rate \((Q_e^0, Q_i^0) = (6.37, 12.74)\) s\(^{-1}\), driven continuously with small-amplitude spatiotemporal white noise. Snapshots show the spatial and temporal evolution of Q e as bird’s-eye (top row) and mesh (bottom row) perspectives. Consistent with Fig. 12.3(a), cortical sheet spontaneously organizes into stationary Turing patterns of wavelength \(\sim 2.5\) cm. Turing structures grow strongly with time; see Fig. 12.6.
Grid resolution \(\Delta x = \Delta y = 0.25\) mm; timestep \(\Delta t = 1.5\) \(\mu\)s. (Reproduced from Ref. [27].)
12.3.5 Slow-soma simulations
Figure 12.5 shows a sequence of snapshots of the firing-rate patterns that evolve spontaneously in the slow-soma model when the inhibitory diffusion is sufficiently strong, here set at D 2 = 4 cm\(^2\). Starting from the homogeneous steady-state corresponding to a subcortical stimulation rate of \(\phi^{\rm sc} = 10\) s\(^{-1}\) (i.e., s = 0.1), small-amplitude white-noise perturbations (with zero mean) destabilize the uniform equilibrium in favor of a spatially-organized stationary state consisting of intermixed regions of high-firing and low-firing cortical activity. The pattern wavelength of \(\sim\)2.5 cm is consistent with the Fig. 12.3(a) prediction of maximum instability at wavenumber \(q/2\pi \approx 0.4\) cm\(^{-1}\). As is evident from Fig. 12.6, the patterns evolve promptly, with fluctuations obeying an exponential growth law \(\sim e^{\alpha t}\) with \(\alpha \approx 7.7\) s\(^{-1}\), matching the Fig. 12.3(a) prediction for the dominant eigenvalue. The Turing structures are fully formed after \(\sim\)2 s, evolving on much slower time-scales thereafter.
The Fig.-12.9 gallery of 24 snapshot images explores the sensitivity of the slow-soma cortex to changes in subcortical stimulus intensity (s increasing from left-to-right across the page), and to inhibitory diffusion (D 2 increasing from top-to-bottom). In Sect. 12.3.7 we compare and contrast these slow-soma patterns with the corresponding Fig.-12.10 gallery of images for the fast-soma case.

Time-series showing formation of Fig. 12.5 Turing patterns. (a) Q e vs time for 10 sample points distributed down the middle of the cortical sheet. Inset: Zoomed view of the first 0.7 s of evolution from the homogeneous equilibrium firing rate \(Q_e^0 = 6.3677\) s\(^{-1}\); scale bar = 0.005 s\(^{-1}\). Spatial patterns are fully developed after about 2 s. (b) Growth of fluctuations (deviations from equilibrium firing-rate) plotted on a log-scale. Dashed line shows that, for the first 1.5 s, fluctuations grow exponentially; the slope is 7.7 s\(^{-1}\), consistent with the Fig. 12.3(a) slow-soma prediction.
12.3.6 Fast-soma simulations
Figure 12.7 illustrates the response of the unstable fast-soma cortex to the imposition of continuous low-level spatiotemporal white noise . With excitatory and inhibitory diffusion strengths set at (D 1, D 2) = (0.0005, 0.05) cm\(^{2}\), and background subcortical flux at \(\phi^{\rm sc} = 30\) s\(^{-1}\), the cortical sheet organizes itself into a dynamic pattern of standing oscillations of temporal frequency \(\sim\)31 Hz and wavelength \(\sim\)2 cm, consistent with the peak in the \((s=0.3, D_2=0.05 {\rm cm}^2)\) dispersion curve of Fig. 12.4(b). The dominant spatial mode grows at the expense of higher- and lower-frequency spatial modes that either decay with time (have dominant eigenvalues whose real part is negative) or that grow more slowly than the favored mode.

[Color plate] Grid simulation for fast-soma cortical model with subcortical drive s = 0.3 (i.e., \(\phi^{\rm sc} = 30\) s\(^{-1}\)), inhibitory diffusion D 2 = 0.05 cm\(^2\). Cortex was initialized at homogeneous steady-state \((Q_e^0, Q_i^0) = (7.28, 14.55)\) s\(^{-1}\), and driven continuously with small-amplitude spatiotemporal white noise. Snapshots show the spatial and temporal evolution of Q e at 0.4-s intervals. Consistent with Fig. 12.4(b), grid evolves into a 31-Hz standing-wave pattern of wavelength \(\sim\)2.0 cm. Red = high-firing; blue = low-firing. The fluctuations grow strongly with time, with successive panels displaying amplitude excursions, in s\(^{-1}\), of (a) \(\pm\)0.005; (b) \(\pm\)0.01; (c) \(\pm\)0.05; (d) \(\pm\)0.2; (e) \(\pm\)1.3 about the 7.28-s\(^{-1}\) steady-state. Timestep \(\Delta t = 100\) \(\mu\)s; other settings as for Fig. 12.5. (Figure reproduced from Ref. [27].)

Time-series showing formation of gamma-band standing-waves of Fig. 12.7. (a) Q e vs time for three sample points in the cortical sheet. Inset: Zoomed view of the first 1 s of evolution from homogeneous equilibrium firing rate \(Q_e^0 = 7.2762\) s\(^{-1}\); scale bar = 0.01 s\(^{-1}\). Spatial patterns are fully developed after about 2.5 s. (b) Fluctuation growth plotted on a log-scale. Dashed line shows that, for the first 2 s, fluctuation amplitudes increase with an exponential growth rate of \(\sim\)3.9 s\(^{-1}\).
The semilog plot of fluctuation amplitude vs time in Fig. 12.8(b) reveals an exponential growth law \(\sim e^{\alpha t}\) with \(\alpha \approx 3.9\) s\(^{-1}\) that persists until the onset of saturation effects at \(t \approx 2.2\) s. We note that the growth rate for the standing-wave instability is about a factor of two slower than the eigenvalue prediction of Fig. 12.4(b); the reason for this anomalous slowing has not been investigated. Nevertheless, the quantitative confirmation, via nonlinear simulation , of gamma-frequency wave activity emerging at the expected spatial and temporal frequencies, is most encouraging.
12.3.7 Response to inhibitory diffusion and subcortical excitation
The bold arrows labeled “D 2 increasing” and “s increasing” in Figs 12.3 and 12.4 highlight the fact that increases in inhibitory diffusion and in subcortical driving are predicted to act in contrary directions with respect to breaking or maintaining spatial symmetry across the cortical sheet.
For the slow-soma cortex, increased ii diffusion D 2 encourages formation of Turing patterns, while increased subcortical stimulation s acts to restore stability to the uniform, unstructured state. These counteracting slow-soma tendencies–-predicted by Fig. 12.3–-are illustrated in Fig. 12.9. This figure presents a 6\(\times\)4 gallery of excitatory firing-rate images captured after 2 s of continuous white-noise stimulation. A vertical top-to-bottom traverse shows that for constant subcortical drive (e.g., s=0.01, left-most column), Turing formation is enhanced as D 2 diffusion is strengthened. In contrast, if diffusion is held constant (e.g., at \(D_2= 2.5 cm^2\), second row), a horizontal scan from left-to-right shows the Turing structures losing contrast, tending to wash out as subcortical drive is increased.
For the fast-soma cortex, Fig. 12.4 indicates that these counteracting tendencies are reversed: gamma-wave instability should be enhanced as subcortical drive is boosted, but suppressed when inhibitory diffusion is increased. These theoretical claims are verified in the Fig. 12.10 gallery of fast-soma snapshots of the gamma-band standing-wave patterns. The patterns have maximum contrast in the top-right corner where subcortical drive is strong and inhibitory diffusion is minimal. A vertical downwards traverse shows the patterns broadening and weakening as diffusion is increased, eventually disappearing altogether when diffusion is set to moderate levels. It is evident that the fast-soma gamma instabilities are vastly more sensitive to small changes in inhibitory diffusion than are the slow-soma Turing instabilities.

[Color plate] Gallery of slow-soma Turing patterns for four values of subcortical drive s (horizontal axis), six values of inhibitory diffusion D 2 (vertical axis). Cortical sheet is initialized at homogeneous equilibrium, driven continuously by low-level continuous spatiotemporal white noise, and iterated for 2 s. Red = high-firing; blue = low-firing. Settings: L x = L y = 6.0 cm; \(\Delta x = \Delta y = 0.25\) mm. Larger D 2 values required smaller timestepping: from top-to-bottom, timestep was set at \(\Delta t = [3, 2, 2, 1.5, 1, 1]\) \(\mu\)s.

[Color plate] Gallery of fast-soma standing-wave patterns for four values of subcortical drive s (horizontal axis), six values of inhibitory diffusion D 2 (vertical axis). Wave instabilities can emerge if diffusion is weak and subcortical stimulation is sufficiently strong. The wave patterns oscillate in place at \(\sim\)30 Hz, with red and blue extrema exchanging position every half-cycle. Timestep \(\Delta t = 100\) \(\mu\)s; other settings as for Fig. 12.9. (Figure reproduced from Ref. [27].)
12.4 Discussion
The crucial distinction between the slow-soma and fast-soma models is the temporal ordering of reversal-potential weighting (the scaling by \(\psi\)) relative to Dendritic integration (convolution with H): the slow-soma model assumes that the input flux M is integrated at the dendrite, then modulated by the reversal function, while the fast-soma model applies the reversal function directly to the input flux, then integrates at the dendrite. In both cases, the dendrite-filtered output flux is integrated at the soma, via convolution with the soma impulse response L, to give \(\Delta V = V - V^{\rm rest}\), the voltage perturbation from rest.
Ignoring the contribution from gap-junction diffusion, the voltage perturbation equations have the general forms,
where \(\rho\) is the (constant) synaptic strength at resting voltage. These general forms are illustrated as flow-diagrams in Fig. 12.1, with the instantaneous voltage-feedback–-from soma to the dendritic reversal-weighting function–-shown explicitly. As we have demonstrated in this chapter, swapping the order of dendrite filtering and reversal-potential weighting makes qualitative changes to the dynamical properties of the cortex.
For the slow-soma configuration (Fig. 12.1(a)), stationary Turing patterns can form if inhibitory diffusion is sufficiently strong; in addition, a low-frequency (\(\sim\)2-Hz) whole-of-cortex Hopf oscillation emerges if the inhibitory PSP decay time-constant is sufficiently prolonged (not shown here; refer to [28] for details). But no evidence of higher-frequency rhythms–-such as gamma oscillations–-has been predicted or observed in the slow-soma stability analysis or its numerical simulation.
In contrast, the fast-soma model (Fig. 12.1(b)) supports \(\sim\)30-Hz gamma rhythms as cortical standing waves distributed across the 2-D cortex, with simulation behaviors being consistent with linear eigenvalue prediction. For this configuration, gamma oscillations only arise when the inhibitory diffusion is weak or non-existent. The contrary behaviors of the slow- and fast-soma mean-field models indicate that a primary determinant of cortically-generated rhythms is the nature and timeliness of the feedback from soma to dendrite.
Earlier mean-field work by Rennie et al [20] also predicted gamma oscillations. However, although we have chosen our model constants (and corresponding steady-states) to be closely similar to theirs, the underlying convolution structures are very different. Translating the Rennie et al symbols to match those used here by way of Table 12.3, their formulation for soma voltage reads,
In the Rennie form, the incoming flux M is multiplied by a composite filter formed by convolving the reversal-potential weight \(\psi\) against the soma filter L; the soma voltage is then obtained by integrating the product at the dendrite filter H. The flow-diagram for this sequence of operations (not shown) is difficult to interpret.
What is the biological significance of these slow- and fast-soma modeling predictions? In [26] we identified the slow-soma Turing patterns with the default noncognitive background state of the cortex that manifests when there is little subcortical stimulation, and in [28] we demonstrated that these patterns can be made to oscillate in place at \(\sim\)1 Hz with a reduction in \(\gamma_{ie}\), the rate-constant for the inhibitory post-synaptic potential (equivalent to rate-constant \(\beta_{ie}\) in this chapter).
We argue that these slow patterned oscillations might relate to the even slower hemodynamic oscillations observed in the BOLD (blood-oxygen-level dependent) signals detected in fMRI (functional magnetic resonance imaging) measurements recorded from relaxed, non-engaged human subjects [7,8].
Increases in the level of subcortical activation tend to wash out the slow-soma Turing patterns. Therefore any spatial patterns of firing activity observed during times of elevated subcortical stimulation–-for example, during active cognition–-cannot be explained using a slow-soma (with its implicit slow soma-to-dendrite feedback) limit. Instead, we replace the slow feedback assumption with the prompt feedback interactions implicit in the ordering of the convolutions adopted for the fast-soma case. In this limit, increases in subcortical drive favor emergence of traveling-wave instabilities of temporal frequency \(\sim\)30 Hz, in the low-gamma band. Our grid simulations show that these gamma oscillations are coherent over distances of several centimeters, synchronized by an underlying standing-wave modulation of neuronal firing rates that provides a basis for the “instantaneous” action-at-a-distance observed in cognitive EEG experiments [22].
The contrasting sensitivity to inhibitory gap-junction diffusion predicted by the slow- and fast-soma models finds clinical support in brain-activity measurements from schizophrenic patients . The brains of schizophrenics carry excess concentration of the neuromodulator dopamine [30]. Dopamine is known to have a number of physiological impacts, one of these being a tendency to block neuronal gap junctions [12]. For the slow-soma model, the closure of gap junctions will reduce the inhibitory diffusion D 2, and therefore, for a given value of subcortical drive s, will reduce the likelihood of forming Turing pattern spatial coherences during the default noncognitive state–-e.g., consider a bottom-to-top traverse of the Fig. 12.9 slow-soma gallery. This degraded ability to form default-mode Turing structures leads to the prediction that schizophrenics should exhibit impairments in the function of their default networks, and this is confirmed in two recent clinical studies [3,19].
Applying excess dopamine to the fast-soma model, coherent gamma activity is predicted to emerge in the cognitively-active schizophrenic brain (see upper-right panel of Fig. 12.10), but these patterns will be “spindly” and less spatially generalized than those observed in a normal brain with lower dopamine levels and therefore stronger inhibitory diffusion (e.g., bottom-right panel of Fig. 12.10). This is consonant with gamma-band EEG measurements captured during cognitive tasks: compared with healthy controls, schizophrenics exhibited diminished levels of long-range phase synchrony in their gamma activity [29].
We acknowledge that, although the slow- and fast-soma models share identical steady states, the two models, as presently constructed, are not strictly comparable. This mismatch is evident in two respects. First, we find that the fast-soma model is about two orders of magnitude more sensitive to variations in inhibitory diffusion D 2 than is the slow-soma. Second, in order to bring the instability peaks in the respective dispersion graphs (Figs 12.3 and 12.4) into rough alignment at similar wavenumbers, we found it necessary to set the spatial decay-rate \(\Lambda_{eb}^\alpha\) for the slow-soma wave-equation four times larger (i.e., axonal connectivity drops off four times faster) than for the fast-soma case (see Table 12.4). Despite these somewhat arbitrary model adjustments, we consider that the qualitative findings presented in this chapter are robust, namely that:
-
delayed soma-to-dendrite feedback, via membrane reversal potentials, supports stationary or slowly fluctuating spatial firing-rate patterns
-
prompt feedback from soma to dendrite enhances spatially-coherent gamma oscillations
-
gap-junction diffusion has a strong influence on the stability and spatial extent of neural pattern coherence.
Notes
- 1.
- 2.
Later we simplify the subscripting notation for diffusion so that (D ee , D ii) \(\equiv\) (D 1, D 2).
- 3.
The 2-D circular convolution algorithm was written by David Young, Department of Informatics, University of Sussex, UK. His convolve2() matlab function can be downloaded from The MathWorks File Exchange, www.mathworks.com/matlabcentral/fileexchange.
References
Alvarez-Maubecin, V., García-Hernández, F., Williams, J.T., Van Bockstaele, E.J.: Functional coupling between neurons and glia. J. Neurosci. 20, 4091–4098 (2000)
Bennett, M.V., Zukin, R.S.: Electrical coupling and neuronal synchronization in the mammalian brain. Neuron 41, 495–511 (2004)
Bluhm, R.L., Miller, J., Lanius, R.A., Osuch, E.A., Boksman, K., Neufeld, R.W.J., Théberge, J., Schaefer, B., Williamson, P.: Spontaneous low frequency fluctuations in the BOLD signal in schizophrenic patients: Anomalies in the default network. Schizophrenia Bulletin 33(4), 1004–1012 (2007)
Bressloff, P.C.: New mechanism for neural pattern formation. Phys. Rev. Lett. 76(24), 4644–4647 (1996), doi:10.1103/PhysRevLett.76.4644
Coombes, S., Lord, G.J., Owen, M.R.: Waves and bumps in neuronal networks with axo-dendritic synaptic interactions. Physica D 178, 219–241 (2003), doi: 10.1016/S0167-2789(03)00002-2
Ermentrout, G.B., Cowan, J.D.: Temporal oscillations in neuronal nets. Journal of Mathematical Biology 7, 265–280 (1979)
Fox, M.D., Snyder, A.Z., Vincent, J.L., Corbetta, M., van Essen, D.C., Raichle, M.E.: The human brain is intrinsically organized into dynamic, anticorrelated functional networks. Proc. Natl. Acad. Sci. USA 102(27), 9673–9678 (2005), doi:10.1073/pnas.0504136102
Fransson, P.: Human spontaneous low-frequency BOLD signal fluctuations: An fMRI investigation of the resting-state default mode of brain function hypothesis. Hum. Brain Mapp. 26, 15–29 (2005), doi:10.1002/hbm.20113
Freeman, W.J.: Mass Action in the Nervous System. Academic Press, New York (1975)
Fukuda, T., Kosaka, T., Singer, W., Galuske, R.A.W.: Gap junctions among dendrites of cortical GABAergic neurons establish a dense and widespread intercolumnar network. J. Neurosci. 26, 3434–3443 (2006)
Haken, H.: Brain Dynamics: Synchronization and Activity Patterns in Pulse-Coupled Neural Nets with Delays and Noise. Springer, Berlin (2002)
Hampson, E.C.G.M., Vaney, D.I., Weile, R.: Dopaminergic modulation of gap junction permeability between amacrine cells in mammalian retina. J. Neurosci. 12, 4911–4922 (1992)
Hodgkin, A.L., Huxley, A.F.: A quantitative description of membrane current and its application to conduction and excitation in nerve. J. Physiol. (Lond.) 117, 500–544 (1952)
Hutt, A., Bestehorn, M., Wennekers, T.: Pattern formation in intracortical neuronal fields. Network: Computation in Neural Systems 14, 351–368 (2003)
Laing, C.R., Troy, W.C., Gutkins, B., Ermentrout, G.B.: Multiple bumps in a neuronal model of working memory. SIAM J. Appl. Math. 63(1), 62–97 (2002), doi:10.1137/S0036139901389495
Liley, D.T.J., Cadusch, P.J., Wright, J.J.: A continuum theory of electro-cortical activity. Neurocomputing 26–27, 795–800 (1999)
Nadarajah, B., Thomaidou, D., Evans, W.H., Parnavelas, J.G.: Gap junctions in the adult cerebral cortex; Regional differences in their distribution and cellular expression of connexins. Journal of Comparative Neurology 376, 326–342 (1996)
Nunez, P.L.: The brain wave function: A model for the EEG. Mathematical Biosciences 21, 279–297 (1974)
Ouyang, L., Deng, W., Zeng, L., Li, D., Gao, Q., Jiang, L., Zou, L., Cui, L., Ma, X., Huang, X.: Decreased spontaneous low-frequency BOLD signal fluctuation in first-episode treatment-naive schizophrenia. Int. J. Magn. Reson. Imaging 1(1), 61–64 (2007)
Rennie, C.J., Wright, J.J., Robinson, P.A.: Mechanisms for cortical electrical activity and emergence of gamma rhythm. J. Theor. Biol. 205, 17–35 (2000)
Robinson, P.A., Rennie, C.J., Wright, J.J.: Propagation and stability of waves of electrical activity in the cerebral cortex. Phys. Rev. E 56, 826–840 (1997)
Rodriguez, E., George, N., Lachaux, J.P., Martinerie, J., Renault, B., Varela, F.J.: Perception’s shadow: long-distance synchronization of human brain activity. Nature 397, 430–433 (1999)
Steyn-Ross, D.A., Steyn-Ross, M.L., Sleigh, J.W., Wilson, M.T., Gillies, I.P., Wright, J.J.: The sleep cycle modelled as a cortical phase transition. Journal of Biological Physics 31, 547–569 (2005)
Steyn-Ross, M.L., Steyn-Ross, D.A., Sleigh, J.W.: Modelling general anaesthesia as a first-order phase transition in the cortex. Progress in Biophysics and Molecular Biology 85, 369–385 (2004)
Steyn-Ross, M.L., Steyn-Ross, D.A., Sleigh, J.W., Liley, D.T.J.: Theoretical electroencephalogram stationary spectrum for a white-noise-driven cortex: Evidence for a general anesthetic-induced phase transition. Phys. Rev. E 60, 7299–7311 (1999)
Steyn-Ross, M.L., Steyn-Ross, D.A., Wilson, M.T., Sleigh, J.W.: Gap junctions mediate large-scale Turing structures in a mean-field cortex driven by subcortical noise. Phys. Rev. E 76, 011916 (2007), doi:10.1103/PhysRevE.76.011916
Steyn-Ross, M.L., Steyn-Ross, D.A., Wilson, M.T., Sleigh, J.W.: Modeling brain activation patterns for the default and cognitive states. NeuroImage 45, 298–311 (2009), doi:10.1016/j.neuroimage.2008.11.036
Steyn-Ross, M.L., Steyn-Ross, D.A., Wilson, M.T., Sleigh, J.W.: Interacting Turing and Hopf instabilities drive pattern formation in a noise-driven model cortex. In: R. Wang, F. Gu, E. Shen (eds.), Advances in Cognitive Neurodynamics ICCN 2007, chap. 40, pp. 227–232, Springer (2008)
Uhlhaas, P.J., Linden, D.E.J., Singer, W., Haenschel, C., Lindner, M., Maurer, K., Rodriguez, E.: Dysfunctional long-range coordination of neural activity during gestalt perception in schizophrenia. J. Neurosci. 26, 8168–8175 (2006)
Uhlhaas, P.J., Singer, W.: Neural synchrony in brain disorders: Relevance for cognitive dysfunctions and pathophysiology. Neuron 52(1), 155–168 (2006), doi:10.1016/j.neuron.2006.09.020
Wilson, H.R., Cowan, J.D.: A mathematical theory of the functional dynamics of cortical and thalamic nervous tissue. Kybernetik 13, 55–80 (1973)
Wilson, M.T., Sleigh, J.W., Steyn-Ross, D.A., Steyn-Ross, M.L.: General anesthetic-induced seizures can be explained by a mean-field model of cortical dynamics. Anesthesiology 104, 588–593 (2006)
Wilson, M.T., Steyn-Ross, D.A., Sleigh, J.W., Steyn-Ross, M.L., Wilcocks, L.C., Gillies, I.P.: The slow oscillation and K-complex in terms of a mean-field cortical model. Journal of Computational Neuroscience 21, 243–257 (2006)
Wilson, M.T., Steyn-Ross, M.L., Steyn-Ross, D.A., Sleigh, J.W.: Predictions and simulations of cortical dynamics during natural sleep using a continuum approach. Phys. Rev. E 72, 051910 (2005)
Wright, J.J., Liley, D.T.J.: A millimetric-scale simulation of electrocortical wave dynamics based on anatomical estimates of cortical synaptic density. Network: Comput. Neural Syst. 5, 191–202 (1994), doi:10.1088/0954-898X/5/2/005
Wright, J.J., Robinson, P.A., Rennie, C.J., Gordon, E., Bourke, P.D., Chapman, C.L., Hawthorn, N., Lees, G.J., Alexander, D.: Toward an integrated continuum model of cerebral dynamics: the cerebral rhythms, synchronous oscillation and cortical stability. BioSystems 73, 71–88 (2001)
Acknowledgment
We thank Chris Rennie for helpful discussions on convolution formulations for the cortex. This research was supported by the Royal Society of New Zealand Marsden Fund, contract 07-UOW-037.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Appendix
Appendix
Rights and permissions
Copyright information
© 2010 Springer Science+Business Media, LLC
About this chapter
Cite this chapter
Steyn-Ross, M., Steyn-Ross, D., Wilson, M., Sleigh, J. (2010). Cortical patterns and gamma genesis are modulated by reversal potentials and gap-junction diffusion. In: Steyn-Ross, D., Steyn-Ross, M. (eds) Modeling Phase Transitions in the Brain. Springer Series in Computational Neuroscience, vol 4. Springer, New York, NY. https://doi.org/10.1007/978-1-4419-0796-7_12
Download citation
DOI: https://doi.org/10.1007/978-1-4419-0796-7_12
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4419-0795-0
Online ISBN: 978-1-4419-0796-7
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)