
Abstract
Identification of very long-chain acyl-CoA dehydrogenase deficiency is possible in the expanded newborn screening (NBS) due to the increase in tetradecenoylcarnitine (C14:1) and in the C14:1/C2, C14:1/C16, C14:1/C12:1 ratios detected in dried blood spots. Nevertheless, different confirmatory tests must be performed to confirm the final diagnosis. We have revised the NBS results and the results of the confirmatory tests (plasma acylcarnitine profiles, molecular findings, and lymphocytes VLCAD activity) for 36 cases detected in three Spanish NBS centers during 4 years, correlating these with the clinical outcome and treatment. Our aim was to distinguish unambiguously true cases from disease carriers in order to obtain useful diagnostic information for clinicians that can be applied in the follow-up of neonates identified by NBS.
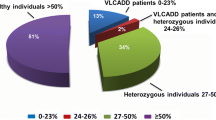
Increases in C14:1 and of the different ratios, the presence of two pathogenic mutations, and deficient enzyme activity in lymphocytes (<12% of the intra-assay control) identified 12 true-positive cases. These cases were given nutritional therapy and all of them are asymptomatic, except one. Seventeen individuals were considered disease carriers based on the mild increase in plasma C14:1, in conjunction with the presence of only one mutation and/or intermediate residual activity (18–57%). In addition, seven cases were classified as false positives, with normal biochemical parameters and no mutations in the exonic region of ACADVL. All these carriers and the false positive cases remained asymptomatic. The combined evaluation of the acylcarnitine profiles, genetic results, and residual enzyme activities have proven useful to definitively classify individuals with suspected VLCAD deficiency into true-positive cases and carriers, and to decide which cases need treatment.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

Keywords
- ACADVL mutations
- Newborn screening
- Very long-chain acyl-CoA dehydrogenase deficiency
- VLCAD enzyme activity
Introduction
Very long-chain acyl-CoA dehydrogenase (VLCAD) is a membrane-bound mitochondrial enzyme involved in the ß-oxidation of long-chain fatty acids that is encoded by ACADVL. Patients with VLCAD deficiency (VLCADD; MIM 201475) have a varied clinical phenotype: a severe early-onset form that is associated with a high incidence of cardiomyopathy and high mortality; an intermediate form with childhood onset that is usually associated with hypoketotic hypoglycemia, hepatopathy, and a more favorable outcome; and an adult-onset, myopathic form with isolated skeletal muscle involvement, rhabdomyolysis, and myoglobinuria after exercise or fasting (Andresen et al. 1999; Merinero et al. 1999). The accumulation of tetradecenoylcarnitine (C14:1) in plasma is a biomarker of this disease, and some correlations between genotype and phenotype have been established. Thus, patients with null mutations in ACADVL that provoke a loss of activity have more severe symptoms than those that retain some residual activity (Andresen et al. 1999).
The determination of acylcarnitines (AC) in dried blood spots (DBS) by tandem mass spectrometry (MS/MS) has enabled VLCADD to be identified in presymptomatic newborns through the elevation of its primary marker C14:1, and of the C14:1/C2, C14:1/C16 and C14:1/C12:1 ratios (Merritt et al. 2014). Early disease detection by newborn screening (NBS) is absolutely essential to achieve a more favorable clinical outcome and indeed, it helps to significantly reduce disease mortality and morbidity (Arnold et al. 2009; Spiekerkoetter et al. 2009). Since its introduction into NBS programs, the number of cases of VLCADD detected has increased, reaching an estimated prevalence of 1:31,500–50,000 (Boneh et al. 2006; Spiekerkoetter et al. 2010). This increase could be explained by the detection of either milder phenotypes or asymptomatic individuals, even by the identification of false-positives. In fact, the mild accumulation of long-chain AC can be observed in healthy children due to the activation of mitochondrial fatty acid oxidation during catabolism in the first days of life (Boneh et al. 2006). Thus, each newborn with a suggestive VLCADD AC profile requires further confirmatory diagnosis (ter Veld et al. 2009).
Molecular and/or enzyme assays have been recommended as second-level testing for VLCADD (Browning et al. 2005). However, genetic analysis alone may be inconclusive in predicting the risk of future metabolic decompensation as new nucleotide variants of unknown clinical significance are often identified (Coughlin and Ficicioglu 2010). Besides, as there is considerable molecular heterogeneity in VLCADD, genotyping is not always a swift and cost-effective confirmatory diagnostic technique (ter Veld et al. 2009). Despite the use of different tests, the confirmation of a VLCADD diagnosis identified in NBS still requires several rounds of discussion (Pena et al. 2016).
In this study we have revised the NBS results, the diagnostic evaluation through plasma AC profiles, the molecular findings and lymphocyte VLCAD activity, as well as the clinical outcome and treatment of 36 cases detected over 4 years in three Spanish NBS centers. The aim of the study was to unambiguously distinguish true VLCADD cases from carriers of the disease and from false-positive cases, and to provide useful diagnostic information to clinicians in the follow-up of NBS identified neonates.
Materials and Methods
Patients
A retrospective analysis was performed on the clinical evaluation and results of confirmatory tests carried out on 36 neonates with suspected VLCADD that were detected by MS/MS between April 2011 and December 2015 among 619,906 newborns in three Spanish screening centers (Madrid, Eastern Andalucía and Murcia). Each center had previously independently determined its internal NBS 99.5 percentile cut-off levels and the criteria for a positive screen of newborns at 48–72 h of life. Cut-offs for C14:1 ranged from 0.27 to 0.50 μmol/L, and for C14:1/C2 from 0.015 to 0.030, C14:1/C16 from 0.087 to 0.14 and/or of C14:1/C12:1 from 7 to 7.3.
The data for all cases were analyzed using the current Region 4 VLCADD general single post-analytical tool. The Region 4 Stork (R4S) Collaborative Project is a database developed for newborn screening quality improvement (Hall et al. 2014). This tool calculates predictive score for VLCADD based on newborn screening analytes including C14:1, C14, C14:2, C16:1, C16OH, C12, C12:1 and ratios thereof. A score of ≥100 is “very likely” to be VLCADD, >50–100 is “likely” to be VLCADD, ≥10–50 is “possibly” VLCADD, and <10 “not informative”.
The identified cases were remitted to their clinical reference units for confirmation of VLCADD between 7 and 20 days post-partum. In all cases the confirmatory tests involved measuring the plasma AC levels, the organic acids in urine and a thorough molecular analysis of ACADVL. In some selected cases, namely those with two mutations or those with only one variant of unknown clinical significance, VLCAD enzyme activity in lymphocytes was assessed by MS/MS. Parents signed a written informed consent for the mutation analysis and/or the assessment of enzyme activity.
Biochemical Analysis
Acylcarnitines were analyzed in plasma as described previously (Ferrer et al. 2007). Organic acids were determined by GC/MS as trimethylsilyl derivatives after urease treatment and ethyl acetate liquid–liquid extraction without oxymation.
VLCAD Enzyme Activity in Lymphocytes
The method used to assess VLCAD activity is based on the oxidation of palmitoyl-CoA (C16:0-CoA) in the presence of the electron acceptor, ferrocenium hexafluorophosphate (FcPF6), essentially as described for MCAD activity (Tajima et al. 2005). Lymphocytes were isolated from whole blood (3–5 mL) using Histopaque (Sigma-Aldrich, Deisenhofen, Germany) and the resulting pellet was frozen before use. The lymphocytes were resuspended in a buffer containing 125 mmol/L KH2PO4, 1 mmol/L EDTA and 9% Triton X-100, and they were then sonicated twice for 5 min, kept on ice for 30 min and finally, centrifuged at 12,000 rpm for 10 min. The reaction mixtures contained 5 mmol/L C16:0-CoA, 4 mmol/L FcFP6, in a final volume of 100 μL.
The analysis of C16:0-CoA and the 2-hexadecenoyl-CoA (C16:1-CoA) and 3-hydroxyhexadecanoyl-CoA (C16OH-CoA) reaction products was performed by LC-MS/MS (1100 Agilent Series HPLC, Santa Clara, California, USA), with the apparatus coupled to an Applied Biosystems API 2000 QTrap (Carlsbad, California, USA). A 5 μL aliquot of each sample was injected onto a Symmetry C18 column (100 mm × 2.1 mm, particle size 3.5 μm, Waters) and the mobile phase consisted of 10 mmol/L ammonium acetate in a mixture of H2O:ACN (45:55). The flow rate was 200 μL/min, and the run time was 20 min. Acyl-CoAs were analyzed in positive multiple reaction monitoring (MRM) mode, with MRM transitions of 1,006 → 499 for C16:0-CoA, 1,004 → 497 for C16:1-CoA and 1,022 → 515 for standard C16OH-CoA. Quantification was based on the peak area [sum of products areas/(sum of substrate + products areas) ratio], with the initial amount of C16:0-CoA substrate set to 20 nmol (ter Veld et al. 2009). Proteins were quantified by the Lowry method prior to adding Triton X-100.
Product formation was shown to be linear for up to 15 min and proportional to the amount of protein added in the range of 20–200 μg, with a correlation coefficient of r 2 = 0.99. The limit of C16:1-CoA or C16OH-CoA detection (signal/noise >3) was estimated to be 0.015 nmol/min/mg protein. The intra-day reproducibility of VLCAD activity was calculated in one control sample, realizing multiple replicates that revealed an imprecision coefficient of 1.8%, indicative of satisfactory reproducibility in the analysis.
Mutation Analysis
Genotypes were analyzed by exon sequencing of the ACADVL locus using conventional Sanger sequencing or massive parallel sequencing. Massive parallel sequencing was done using a targeted customized panel to capture the exome of 120 genes involved in metabolic disorders (Nextera Nature Capture, Illumina, San Diego, California, USA: the list of genes included can be sent upon request). The mean depth of coverage was 292× (99.9%) for the ACADVL gene. Mutations are named according to the HGVS nomenclature (http://www.hgvs.org/mutnomen), using the RefSeq number NM_000018.3. New single nucleotide variants (SNVs) were analyzed using Alamut Visual Software to predict their pathogenic effect and all variant changes were confirmed by Sanger sequencing in parental samples.
Clinical Parameters
A clinical questionnaire was sent to the referring physicians to collect data regarding the neonatal screening, family origin, consanguinity, pregnancy and delivery, neonatal complications, nutritional treatment (low-LCT diet, MCT, and/or carnitine supplementation), metabolic decompensation, and clinical evaluation (weight, height and head circumference parameters, routine laboratory findings, neurological and cardiological examinations).
Statistical Analysis
A non-parametric Mann–Whitney U-test (two-tailed) was used to assess the significance of the differences in AC levels between the groups, with a p value <0.05 considered significant.
Results
Newborn Screening
Fifteen cases presented a significant elevation of C14:1 (1.02–7.55 μmol/L), as well as the enhanced C14:1/C2 and C14:1/C16 ratios in DBS (P1–P10, P13–P17: Tables 1 and 2). Ratio C14:1/C12:1 was also found elevated in some of them (P1, P3, P4, P5, P7, P8, P10). The remaining individuals had elevated C14:1 ranging from 0.44 to 0.93 μmol/L and/or C12, C12:1, C14, C14:2 (data not shown), with at least one of the ratios in some cases (Tables 1 and 2). The predictive score for VLCADD was found informative in 31 out of 36 cases (condition “very likely” in P1–P10; P13, P14; P17; condition “likely” in P11; P15; P16; P18; P21; P22; and condition “possibly” in P12; P19; P20; P23–P26; P29; P31–P33; P35).
Confirmatory Testing
Classification
Cases were classified according to the results of the confirmatory tests. True-positive cases were those with abnormal plasma C14:1 levels and ratios, and with bi-allelic mutations coupled to deficient VLCAD activity in lymphocytes (<12% of intra-assay control: cases P1–P12). Individuals P11 and P12 displayed a mild increase in DBS C14:1 and they were later considered true-positive cases after completing the confirmatory testing. Disease carriers were considered P13–P29: those with only a mild increase in plasma C14:1 and of at least one ratio, the presence of only one single loss of function (LoF) mutation (P13–P20), or with one single probably missense mutation (P21–P28) and with intermediate residual activity (18–57%), and P29 (not genotyped) with just an intermediate activity of 57%. Those individuals with normal biochemical parameters and no mutations were classified as false positive cases (P30–P36).
Biochemical Confirmatory Analysis
Acylcarnitines were measured in plasma obtained between day 8 and 23 in all cases, except in cases P3 and P12 where it was obtained at 2 and 3 months of age, respectively. Sixteen cases (P1–P13, P15–P17) presented increased C14:1 levels (0.36–2.74 μmol/L; NV <0.17) with altered ratios in many of them. These parameters were normal in the remaining cases except for an isolated mild increase of C14:1 in P20 and P28 (Tables 1 and 2). Some overlapping in the plasma AC levels was observed between the true-positive cases and the disease carriers.
Significant differences in the C14:1 concentration were found between the 12 true-positive cases and the eight carriers of loss of function (LoF) mutations (P13–P20), both in DBS and plasma (p < 0.01). Similarly, the increases in the C14:1/C2, C14:1/C16, and C14:1/C12:1 ratios were also significant different in both groups (p < 0.05), with the increase in the C14:1/C16 ratio being the most significant parameter (p < 0.005) in both situations. The C14:1 concentrations were also significantly higher in both DBS and plasma (p < 0.05) among the carriers of LoF mutations (P13–P20) and the rest of heterozygotes (P21–P29), although in this case the ratios were not (Tables 1 and 2).
Secondary indicators of abnormal fatty acid oxidation, such as urine dicarboxylic and 3-hydroxydicarboxylic acids, were within normal limits in all the cases studied, except in individuals P2 and P14 who excreted medium-chain dicarboxylic acids (data not shown). The analysis of organic acids in urine proved to be uninformative.
Moreover, three asymptomatic mothers of heterozygous individuals (P13, P16, and P17) that carried the mutation found in their child, had a normal AC profile in DBS, ruling out maternal VLCADD (data not shown).
Molecular Analysis
A mutational analysis of ACADVL was performed on 35 individuals (33 unrelated families), of which 12 had bi-allelic variants (P1–P12), 16 only one pathogenic variant in the exonic region (P13–P28), and 7 carried no mutations (P30–P36, see Tables 1 and 2 for the genotypes). In total, 28 different variants were detected, including 12 that have not been previously reported: 8 potential missense changes and 4 severe LoF mutations. All the possible missense changes were predicted to be probably disease-causing mutations, affecting highly conserved residues in at least one algorithm used, and none of these were detected in control samples (Richards et al. 2015; Dopazo et al. 2016) (Table 3). All the individuals with bi-allelic changes carried at least one previously described or one LoF mutation, except P1.
Only one variant was detected in 16 individuals, 8 of whom carried a LoF mutation (Table 2), while 6 variants were new: 2 LoF (c.990_994delCAAGC; c.1102_1103delCA) and 4 probably missense mutations (c.515T>C; c.1037C>T; c.1322G>C; c.1627T>C). Genomic rearrangements were ruled out by CGH arrays in P13, P16, and P21. The most common mutations have already been described: c.848T>C (p.Val283Ala), six alleles; c.896_898delAGA (p.Lys299del), three alleles; [c.1097G>A; c.1844G>A] [p.Arg366His; p.Arg615Gln], three alleles; and c.643T>C (p.Cys215Arg), three alleles. Curiously, two mutations [p.Arg366His; p.Arg615Gln] were present in the same allele in two patients (P5, P10).
Lymphocyte Enzyme Activity
The VLCAD activity in lymphocytes from nine healthy adult subjects ranged from 0.95 to 6.85 nmol/min/mg protein, with a mean (±SD) value of 3.37 (±1.99) nmol/min/mg protein. Enzyme activity of 0.015 and 0.135 nmol/min/mg protein (0.6 and 5.8% of the intra-assay control) was determined in two confirmed adult myopathic VLCAD-deficient patients to validate the method. The enzyme activity in lymphocytes was clearly deficient in 11 individuals with abnormal NBS results and 2 bi-allelic mutations, ranging from undetectable to 11.8% of the simultaneous control (Table 1). There was residual activity in five cases with only one LoF mutation in ACADVL (P13, P15, P16, P17, and P20), ranging from 18 to 57% of the intra-assay control (Table 2). The enzyme activity was also reduced in P23 (34%), an individual carrying one novel missense variant, and in P29 (57%) that has not yet been genotyped. The enzyme activity in five parents carrying one mutation in ACADVL ranged from 0.43 to 1.31 nmol/min/mg protein (34.7–40% of the intra-assay control).
Clinical and Biochemical Outcome
Clinical questionnaires were completed for 36 cases from 33 families. The parents of most individuals were of Spanish origin (18 families), there were 4 from Morocco, 2 from Romania, 2 from China, 1 from Peru, 1 from Venezuela, and 6 mixed couples (Spain-Hispanic). Consanguinity was only recognized in the family of P1 and no incidents of interest were reported in any of the pregnancies. There were three twin pregnancies (two identical and one fraternal), and both children of two of these pregnancies (P14 and P33; P18 and P19) and only one from the third (P23) were investigated. One of these pregnancies was a preterm labor, and in the whole cohort five cesarean deliveries were documented. All cases were asymptomatic when referred to clinical units, except four cases that were admitted to hospital in the neonatal period (between 1 and 15 days) due to dehydration and weight loss (P2), paroxysmal supraventricular tachycardia (P5), jaundice (P16), or prematurity (P23, twin pregnancy), all without metabolic decompensation. Seven true-positive individuals were admitted to hospital due to different infections between 3 months and 4 years of age, usually gastroenteritis, gingival stomatitis or pneumonia, and these symptoms were accompanied by metabolic decompensation in four of these cases (P1, P3, P7 and P10). These individuals presented elevated creatine kinase (CK: 515–4,000 U/L), elevated transaminases, hypoglycemia, and/or metabolic acidosis. In addition, P3 was admitted several times for emergency treatment due to severe metabolic decompensation, usually presenting increased CK (515–1,296 U/L, N 26–308) even when stable.
The decision whether or not to treat the children, and what is the optimal treatment for each patient, is difficult as there are currently no evidence-based guidelines available. For each individual patient clinical decision making was based on the consensus recommendations of European experts (Spiekerkoetter et al. 2009). Asymptomatic cases with normal values of glucose, CK and liver enzymes were fed with a 50:50 mixture of breast milk (or infant formula) mixed with a special medium-chain triglyceride (MCT)-enriched infant formula, which was prescribed according to age in order to avoid fasting. A specific appropriate sick day regime was also provided. After confirmatory studies, dietary intervention was recommended in those cases with two mutations and low residual activity. A fat modified diet (LCT ≤10% of total caloric value and MCT 20% of total caloric value) and carnitine (15–50 mg/kg/day) were administered to 6 of the 12 children. Only MCT supplementation was provided to 3 of the 12, LCT restriction and MCT supplementation in 1, LCT restriction and carnitine in 1, and only carnitine supplementation in another (Table 1). The follow-up period of the individuals varied between 12 months and 5.5 years. All the evaluated individuals except one (P3) are in good condition, with normal growth and anthropometric parameters (weight, height and head circumference), normal psychomotor development and neurologic examination, normal cardiologic examination with normal ECG record and normal routine biochemical parameters without hepatic or muscle impairment. To date, P3 is the only clinically affected individual despite adhering to a regulated diet, with several decompensated parameters, hypertrophic cardiomyopathy, hepatopathy, myopathy, and hypotonia at 3 years of age.
The plasma AC levels were monitored periodically in ten true-positive cases and six carriers of LoF mutations. Under treatment, plasma C14:1 levels remained high (1.01–3.4 μmol/L) in cases P1, P2, P5, and P7, all with very low residual activity (<5%), who were in good clinical condition and complied well with their diet. By contrast, in six cases the plasma AC levels were only slightly higher than the controls (0.17–0.50 μmol/L): P4, P6, P8, P9, P10, and P12. Carriers that did not receive dietary interventions always had C14:1 levels in the control range (<0.17 μmol/L), except P13 who had levels slightly above those of the controls (0.21–0.50 μmol/L).
Discussion
Although the detection of VLCADD is not mandatory in Spanish expanded NBS programs, it is included in some regions because the early detection of this disease may improve the outcome if prophylactic measures are rapidly put in place. The true incidence of VLCADD in Spain is not known and in fact, between 1989 and 2010 very few cases were diagnosed based on clinical evidence: five patients in Madrid (three with severe early onset and two with adolescent myopathic presentation) (Merinero et al. 1996); and at least three cases in Barcelona (Osorio et al. 2003). In some Spanish regions, no positive cases were identified between 2000 and 2013 in more than 800,000 newborns screened in the expanded NBS programs (approximately 46% of all annual births (Rocha et al. 2014)). Hence, the high number of cases with suspected VLCADD that were detected here over a short period (between April 2011 and December 2015) was quite striking. In order to better understand how this increased incidence arose, we first set out to unambiguously distinguish the true-positive cases that need nutritional intervention and clinical monitoring from those individuals that are carriers of the disease, or even false-positive cases. To achieve this we revised the results of the NBS and of the diagnostic evaluation (plasma AC profiles, molecular findings, lymphocyte enzyme activity), as well as the clinical outcome and treatment data of 36 cases detected by NBS in the last 4 years.
Initially, the combination of plasma AC profile and mutational analysis was used to confirm the diagnosis of VLCADD. However, most of the cases presented only one exonic mutation, although other nucleotide changes in the proximal or distal promoter or in deep intronic sequences could not be ruled out because routine genetic analysis only involves the sequencing of coding exonic regions. Besides, the finding of novel mutations with uncertain clinical significance in some cases makes it more complicated to achieve a true diagnosis of VLCADD, as occurred elsewhere (Schiff et al. 2013).
The determination of enzyme activity in lymphocytes was added to the diagnostic algorithm in order to provide accurate information for clinicians. All the cases with two bi-allelic mutations had low residual activity (<12%), even the cases bearing the mild p.Val283Ala mutation (Andresen et al. 1999). By contrast, those cases with only one nucleotide change presented intermediate residual activity that ranged from 18 to 57%, suggesting that they were indeed carriers of the disease. In our hands, the determination of enzyme activity in lymphocytes proved useful as a rapid confirmatory test to definitively classify the cases into true-positive cases or carriers. There is a lack of correlation between VLCAD enzyme activity in lymphocytes and AC levels in DBS and plasma samples in our confirmed patients. Similar inconsistencies between the biochemical data, genetic results, enzyme studies, and/or presence of clinical symptoms have already been described; therefore, the need to combine different confirmatory tests. The enzyme activity also does not allow carriers to be discriminated from healthy controls, as occurs elsewhere (ter Veld et al. 2009).
The fact that with nutritional therapy 11 true-positive patients remain free of symptoms after a mean follow-up of 30 months, and only one is symptomatic, highlights the importance of early diagnosis through NBS and prompt nutritional intervention. Nevertheless, early intervention does not prevent asymptomatic cases from being at risk of presenting symptoms in the future and hence, such cases will require close clinical monitoring, especially in situations of energy demand (fasting, prolonged exercise, illness, adolescence, etc.). In this cohort the disease carriers did not present any symptoms, despite a mild increase in plasma C14:1 during the follow-up period. Thus, it would seem that they are not at risk of developing clinical complications in the future and therefore, a clinical and biochemical follow-up may not be necessary.
In our experience the R4S post-analytical tool has allowed to discriminate most true-positive cases as very likely/likely (P1–P11) except P12 and there were no false negative cases. The evaluation of five clearly false positive cases (P27, P28, P30, P34, and P36) could have been avoided. So, the R4S post-analytical tool is able to provide reliable advice on the interpretation of NBS results.
Although the increase in the number of cases with suspected VLCADD may be at least partially related to the different cut-offs used in the regional NBS centers, it seems that this disease is more common in Spain than previously thought. Based on this experience, we suggest that cases with abnormal plasma C14:1 (>1 μmol/L) and with higher ratios of C14:1/C2, C14:1/C16 and/or C14:1/C12:1 will need early dietary treatment pending the outcome of the enzymatic/molecular analysis. We recommend that enzyme analysis should be performed in all newborns with mild plasma C14:1 levels ranging from 0.36 to 1 μmol/L in order to definitively classify them as true-positive cases or disease carriers, and to decide which will require treatment, as well as clinical and biochemical monitoring. Moreover, in those children with normal plasma results in the first 6 months of life, no further biochemical studies are needed.
Conclusions
This is the first time that data has been compiled on suspected VLCADD cases identified through NBS programs in Spain. True-positive cases were defined as those with increased C14:1 and ratios, two pathogenic mutations and deficient VLCAD activity in lymphocytes. All these individuals are receiving a nutritional intervention and to date, all but one remain asymptomatic. Children with normal or only a mild increase in plasma C14:1, the presence of only one single mutation and intermediate residual activity don’t seem to be at risk of developing clinical complications in the future and a close follow-up is not necessary. Enzyme determination is essential in many cases to interpret the genetic findings and to decide which cases require treatment, as well as close clinical and biochemical monitoring. Using biochemical, enzymatic, and comprehensive gene analysis, a biochemical genetic service could rapidly evaluate potential patients.
References
Andresen BS, Olpin S, Poorthuis BJ, Scholte HR et al (1999) Clear correlation of genotype with disease phenotype in very-long-chain acyl-CoA dehydrogenase deficiency. Am J Hum Genet 64:479–494
Arnold GL, Van Hove J, Freedenberg D, Strauss A et al (2009) A Delphi clinical practice protocol for the management of very long chain acyl-CoA dehydrogenase deficiency. Mol Genet Metab 96:85–90
Boneh A, Andresen BS, Gregersen N, Ibrahim M et al (2006) VLCAD deficiency: pitfalls in newborn screening and confirmation of diagnosis by mutation analysis. Mol Genet Metab 88:166–170
Browning MF, Larson C, Strauss A, Marsden DL (2005) Normal acylcarnitine levels during confirmation of abnormal newborn screening in long-chain fatty acid oxidation defects. J Inherit Metab Dis 28:545–550
Coughlin CR 2nd, Ficicioglu C (2010) Genotype-phenotype correlations: sudden death in an infant with very-long-chain acyl-CoA dehydrogenase deficiency. J Inherit Metab Dis 33(Suppl 3):S129–S131
Dopazo J, Amadoz A, Bleda M, Garcia-Alonso L et al (2016) 267 Spanish exomes reveal population-specific differences in disease-related genetic variation. Mol Biol Evol 33:1205–1218
Ferrer I, Ruiz-Sala P, Vicente Y, Merinero B et al (2007) Separation and identification of plasma short-chain acylcarnitine isomers by HPLC/MS/MS for the differential diagnosis of fatty acid oxidation defects and organic acidemias. J Chromatogr B 860:121–126
Hall PL, Marquardt G, McHugh DM, Currier RJ et al (2014) Postanalytical tools improve performance of newborn screening by tandem mass spectrometry. Genet Med 16:889–895
Merinero B, Perez-Cerda C, Garcia MJ, Gangoiti J et al (1996) Mitochondrial very long-chain acyl-CoA dehydrogenase deficiency with a mild clinical course. J Inherit Metab Dis 19:173–176
Merinero B, Pascual Pascual SI, Perez-Cerda C, Gangoiti J et al (1999) Adolescent myopathic presentation in two sisters with very long-chain acyl-CoA dehydrogenase deficiency. J Inherit Metab Dis 22:802–810
Merritt JL 2nd, Vedal S, Abdenur JE, Au SM et al (2014) Infants suspected to have very-long chain acyl-CoA dehydrogenase deficiency from newborn screening. Mol Genet Metab 111:484–492
Osorio JH, Lluch M, Ribes A (2003) Analysis of organic acids after incubation with (16-2H3)palmitic acid in fibroblasts from patients with mitochondrial beta-oxidation defects. J Inherit Metab Dis 26:795–803
Pena LD, van Calcar SC, Hansen J, Edick MJ et al (2016) Outcomes and genotype-phenotype correlations in 52 individuals with VLCAD deficiency diagnosed by NBS and enrolled in the IBEM-IS database. Mol Genet Metab 118:272–281
Richards S, Aziz N, Bale S, Bick D et al (2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17:405–424
Rocha H, Castineiras D, Delgado C, Egea J et al (2014) Birth prevalence of fatty acid beta-oxidation disorders in Iberia. JIMD Rep 16:89–94
Schiff M, Mohsen AW, Karunanidhi A, McCracken E et al (2013) Molecular and cellular pathology of very-long-chain acyl-CoA dehydrogenase deficiency. Mol Genet Metab 109:21–27
Spiekerkoetter U, Lindner M, Santer R, Grotzke M et al (2009) Treatment recommendations in long-chain fatty acid oxidation defects: consensus from a workshop. J Inherit Metab Dis 32:498–505
Spiekerkoetter U, Haussmann U, Mueller M, ter Veld F et al (2010) Tandem mass spectrometry screening for very long-chain acyl-CoA dehydrogenase deficiency: the value of second-tier enzyme testing. J Pediatr 157:668–673
Tajima G, Sakura N, Yofune H, Nishimura Y et al (2005) Enzymatic diagnosis of medium-chain acyl-CoA dehydrogenase deficiency by detecting 2-octenoyl-CoA production using high-performance liquid chromatography: a practical confirmatory test for tandem mass spectrometry newborn screening in Japan. J Chromatogr B 823:122–130
ter Veld F, Mueller M, Kramer S, Haussmann U et al (2009) A novel tandem mass spectrometry method for rapid confirmation of medium- and very long-chain acyl-CoA dehydrogenase deficiency in newborns. PLoS One 4:e6449
Acknowledgments
The authors would like to thank the families involved in this study for giving their consent. This work was funded by a grant from the Fundación Isabel Gemio. The authors confirm independence from the sponsors; the content of the article has not been influenced by the sponsors.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Additional information
Communicated by: Piero Rinaldo, MD, PhD
Appendices
A Concise One Sentence Take-Home Message
The combined evaluation of the acylcarnitine profiles, genetic results, and residual enzyme activities have proven useful to definitively classify individuals with suspected VLCAD deficiency into true-positive cases and carriers, and to decide which cases need treatment, as well as close clinical and biochemical monitoring.
Compliance with Ethics Guidelines
Conflict of Interests
None of the authors have any conflict of interests to declare.
Informed Consent
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation and with the Helsinki Declaration of 1975, as revised in 2000 (5). This project was approved by the Universidad Autónoma Madrid Ethics Committee (reference number CEI 74-1349).
Details of the Contributions of Individuals Authors
All authors approved the final manuscript as submitted.
BM: conception and design, drafting, and coordination of the manuscript.
PA: enzyme activity determinations, statistical analysis, and writing the first draft.
EMH: treating metabolic specialist of patients, drafting of the manuscript.
AM: treating metabolic specialist of patients.
MTGS: treating metabolic specialist of patients.
PQF: treating metabolic specialist of patients.
CPG: treating metabolic specialist of patients.
ED: analysis and interpretation of newborn screening data from Madrid.
RY: analysis and interpretation of newborn screening data from Western Andalucia.
JME: analysis and interpretation of newborn screening data from Murcia.
ABQ: treating metabolic specialist of patients.
JBA: treating metabolic specialist of patients.
MLFR: analysis and interpretation of newborn screening data from Madrid.
BB: analysis of newborn screening data from Madrid.
IFL: acylcarnitines and organic acids analysis.
FL: molecular genetic analysis.
MU: critical review of the manuscript for important intellectual content.
PRS: biochemical and enzyme data analysis.
BP: molecular genetic data interpretation, drafting of the manuscript.
CPC: biochemical data interpretation, drafting of the manuscript.
Rights and permissions
Copyright information
© 2017 SSIEM and Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Merinero, B. et al. (2017). Four Years’ Experience in the Diagnosis of Very Long-Chain Acyl-CoA Dehydrogenase Deficiency in Infants Detected in Three Spanish Newborn Screening Centers. In: Morava, E., Baumgartner, M., Patterson, M., Rahman, S., Zschocke, J., Peters, V. (eds) JIMD Reports, Volume 39. JIMD Reports, vol 39. Springer, Berlin, Heidelberg. https://doi.org/10.1007/8904_2017_40
Download citation
DOI: https://doi.org/10.1007/8904_2017_40
Received:
Revised:
Accepted:
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-662-57576-5
Online ISBN: 978-3-662-57577-2
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)