
Abstract
Very long chain acyl-CoA dehydrogenase deficiency (VLCADD, OMIM #201475) has been increasingly diagnosed since the advent of expanded newborn screening (NBS). Elevated levels of tetradecenoyl-L-carnitine (C14:1) in newborn screening blood spot samples are particularly common in New Zealand, however this has not translated into increased VLCADD clinical presentations. A high proportion of screen-positive cases in NZ are of Maori or Pacific ethnicity and positive for the c.1226C > T (p.Thr409Met) ACADVL gene variant. We performed a retrospective, blinded, case–control study of 255 cases, born between 2006 and 2013, with elevated NBS C14:1 levels between 0.9 and 2.4 μmol/L, below the NZ C14:1 notification cut-off of 2.5 μmol/L. Coded healthcare records were audited for cases and age- and ethnicity- matched controls. The clinical records of those with possible VLCADD-related symptoms were reviewed. The follow-up period was 6 months to 7 years. Two of 247 cases (0.8 %) had possible VLCADD-like symptoms while four of 247 controls (2 %) had VLCADD-like symptoms (p = 0.81). Maori were overrepresented (68 % of the cohort vs 15 % of population). Targeted analysis of the c.1226 locus revealed the local increase in screening C14:1 levels is associated with the c.1226C > T variant (97/152 alleles tested), found predominantly in Maori and Pacific people. There was no increase in clinically significant childhood disease, irrespective of ethnicity. The study suggests that children with elevated C14:1, between 0.9-2.4 μmol/L, on NBS are at very low risk of clinically significant childhood disease. A minimally interventional approach to managing these patients is indicated, at least in the New Zealand population.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Very long chain acyl-CoA dehydrogenase deficiency (VLCADD, OMIM #201475) is the most common disorder of long-chain mitochondrial fatty acid beta-oxidation. This autosomal recessive disorder has a heterogeneous phenotype encompassing an early, severe presentation with hypoglycaemia-related encephalopathy, cardiomyopathy, arrhythmia and liver disease, a childhood onset form characterised by fasting hypoketotic hypoglycaemia and a late-onset form presenting with exercise-induced rhabdomyolysis and myoglobinuria (Andresen et al 1999). The deficiency in very long chain acyl-CoA dehydrogenase (VLCAD, EC:1.3.99.13) results in the accumulation of long chain (C14-C18) acylcarnitines. Tetradecenoyl-L-carnitine (C14:1), the most disease-specific marker, is included on many international expanded newborn screening (NBS) panels (Liebig et al 2006). Screening detects more cases than present clinically, increasing the incidence of VLCADD from 1:125,000 pre-NBS to 1:31,400 (Boneh et al 2006). Early diagnosis allows reduction in mortality and morbidity through dietary management. Long-chain fats are restricted, medium chain triglycerides are supplemented and prolonged fasting is avoided, however it is unclear which screen-positive patients may have remained asymptomatic throughout childhood without medical intervention (Spiekerkoetter et al 2010a, b).
Screening for VLCADD is complicated by a number of factors. Levels of C14:1 in asymptomatic individuals overlap with those of heterozygotes or healthy babies experiencing extreme catabolism (Merritt et al 2014, Spiekerkoetter et al 2010a, b). False negative newborn screening with C14:1 as low as 0.45 μmol/L can result in death from VLCADD (Spiekerkoetter et al 2012). Forced anabolism in newborns receiving intravenous glucose or supplementary feeds may normalise NBS acylcarnitine profiles (Sahai et al 2011).
There is limited consensus on treatment due to difficulties predicting phenotype severity. The mutational spectrum is wide and compound heterozygosity is common (Liebig et al 2006). Genotype-phenotype correlations are limited to the general, but not exclusive, association of a severe phenotype with two null mutations and an attenuated phenotype with two missense mutations (Andresen et al 1999). Enzymology may better predict phenotype but there remains uncertainty regarding the level of intervention required for intermediate enzyme activities (Hoffmann et al 2012, Spiekerkoetter et al 2010a, b). Cases of severe VLCADD with enzyme levels of only 6 % may present with normal NBS C14:1 (Spiekerkoetter et al 2012).
The cut-off threshold of NBS C14:1 varies internationally, with a range reported by the Region 4 Stork project of 0.34-0.69 μmol/L (http://www.clir-r4s.org). In NZ, despite a much higher NBS cut off of 2.5 μmol/L, a greater than anticipated number of newborns with elevated C14:1 have been detected, resulting in a VLCADD prevalence rate of 1:32,300 (Ryder et al 2014). The majority remain asymptomatic on a normal diet, are of Maori ethnicity and carry the c.1226C > T (p.Thr409Met) ACADVL variant (Ryder et al 2014). Here patients with a level greater than 2.5 μmol/L are immediately referred for clinical assessment, while those with levels between 1.3 and 2.5 μmol/L have had a second card requested. In the case of normal C14:1 levels on repeat testing, the family is not notified of a potential VLCADD diagnosis. It is now accepted that a repeat card, once a child is anabolic, may be falsely negative (Boneh et al 2006, Schymik et al 2006, Spiekerkoetter et al 2012). A review of the NZ protocol was thus required. Lowering the NZ threshold to within the Region 4 range would result in an additional 1000 patients to be seen over seven years, which would be overwhelming from a service resource perspective.
This paper reports the natural history of children with elevated C14:1 levels between 0.9-2.4 μmol/L in NZ and provides evidence of the frequency, ethnic association and benign nature of the c.1226C > T variant.
Study design
Subject selection
Cases and controls were obtained from NZ newborn screening records. Families were not contacted at any stage during the study. The case group comprised all babies with a screening C14:1 level between 0.9-2.4 μmol/L at 48–72 hours of age, born between 1st Dec 2006 (when NBS screening commenced in NZ) and 31st July 2013. Controls were ethnically matched babies, born at a similar time whose C14:1 levels were below 0.12 μmol/L (50th centile of the normal range of C14:1). Ethnicity data, as identified by the child’s parent, was recorded by the lead maternity carer at the time of obtaining the dried blood spot or Guthrie card. Cards from babies with a birth weight less than 2.0 kg and those in neonatal intensive care were excluded from the control group. Patients who had been clinically assessed by the metabolic service and found to have an alternative diagnosis were excluded from analysis.
Study methodology
A retrospective, case–control, observational analysis was performed with investigators blinded to case or control group through the random allocation of study numbers. The primary outcome measure was the prevalence of clinically significant VLCADD like disease or death. The secondary outcome measure was the genotype and allelic frequencies of the c.1226C > T mutation.
Clinical coding data for all hospital attendances or deaths were obtained from the Ministry of Health for both cases and controls (as coded by the International Statistical Classification of Diseases and Related Health Problems, Tenth Revision, Australian Modification (ICD-10-AM). Coding criteria considered consistent with VLCADD included hypoglycaemia, lethargy, vomiting, seizures, coma, metabolic acidosis, hepatomegaly, increased transaminases, increased CK, muscle weakness, jaundice, cardiac arrhythmias, cardiomyopathy and unexplained death. Study numbers were noted and the medical notes were reviewed for these admissions, allowing classification into one of three groups based on clinical evidence of VLCADD: no evidence, mild (known as “broad”) or strong (known as “tight”). Groups were then unblinded for statistical analysis. The sample size was selected for a power of greater than 88 %, assuming 4 % of patients were likely to be categorised as “tight” and 12 % as “broad”.
Newborn screening dried blood spot acylcarnitine profiles, the majority collected on days 2 and 3 of life for both cases and control groups (94.4 % across both groups) were measured using flow injection electrospray-tandem mass spectrometry. Cards collected since July 2011 (75 in each group) were analysed for the c.1226C > T (p.Thr409Met) variant. In July 2011, the New Zealand Guthrie card consent process changed to include anonymous research on stored samples, if approved by the NSU and a recognised ethics committee. Therefore, only cards obtained after this date underwent molecular analysis.
Genomic DNA was extracted using the prepGEM® Storage card blood method (ZyGEM, Hamilton, New Zealand). Single nucleotide polymorphism (SNP) RS identification number rs113994169 (c.1226C > T) was included in a novel Mass Array based congenital panel. The assay was designed using the Agena Bioscience, MySequenom, Assay design suite online design tool (https:///www.mysequenom.com/Tools). Genotyping was performed using the Sequenom MassARRAY iPLEX platform (Agena Bioscience, SanDiego, USA) as previously described (Gabriel et al 2009). Results were corroborated by using Sanger-based sequencing of exon 12 of the ACADVL gene (NM_000018.3; NP_000009.1) of randomly selected samples.
Region 4 post-analytical tool
The data for all cases were analysed using the current Region 4 VLCADD general single post-analytical tool. The Region 4 Stork (R4S) Collaborative Project is a database developed for newborn screening quality improvement. (http://www.clir-r4s.org/PostAlytTools/CondScoreSingle.aspx?pcID=1538)
This tool calculates a predictive score for VLCADD based on newborn screening analytes including C14:1, C14, C16, C2, C12:1 and ratios thereof. A score of > =110 is very likely to be VLCADD, 50–109 is likely to be VLCADD, 15–49 is possibly VLCADD and <15 not informative.
Statistical analysis
The controls were individually matched for age and ethnicity. The proportions of cases and controls were compared using the Fisher’s exact test.
Results
Between December 2006 and July 2013 a total of 255 infants (out of 390,000 screened) had newborn screening C14:1 levels between 0.9 and 2.5 μmol/L. Eight cases and their corresponding controls were excluded resulting in a final study group of 494 children, 247 cases and 247 matched controls.
Demographics
Cases and controls were ethnically matched with no statistically significant differences in ethnicity between groups. Maori were overrepresented at 68 % (n = 168), as opposed to the population frequencies of 15 % identifying as Maori (NZ Census data, 2013). There were 48 European (19.4 %) and nine Asian cases (3.6 %) (Table 1).
The average birth weight for cases was 3271 g (range 2150–4480 g) and for controls 3472 g (2095–4670 g); p < 0.0001. Gender was not documented.
Clinical audit
Based on the clinical coding, medical notes were reviewed for 6 % of the cohort (n = 32: 13 cases, 17 controls, two exclusions). Clinical coding which prompted notes review included hypoglycaemia, jaundice, liver disease, metabolic acidosis, developmental delay, seizures, arrhythmia, cardiomyopathy, encephalopathy and death. The number of patients in the case group categorised as “tight” and “broad” VLCADD-like presentation was one (0.4 %) of each while in the control group they were slightly higher at three (1.2 %) and one (0.4 %) respectively. The number of patients categorised as “tight” and “broad” were much lower than anticipated for our study design and consequently power was reduced. Nevertheless, with only two cases classified as either tight or broad, compared to four controls (p = 0.81, Fisher’s exact test) there was certainly no evidence of increased clinically significant early childhood VLCADD-like disease in the case compared to the control group.
Region 4 Stork (R4S) post-analytical data
A predictive score for VLCADD was calculated for all cases using the current R4S VLCADD general single post-analytical tool. Only 10 % of cases were uninformative; 14.2 % (35/247) were rated as very likely, 39.7 % (98/247) were likely and 36.4 % (90/247) were possible cases of VLCADD. None of the cases rated as “likely” or “very likely” VLCADD presented clinically. The two cases categorised as “tight” and “broad” scored just 42 and 44 respectively rating as “possible VLCADD”.
Molecular analysis
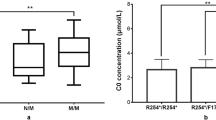
Molecular analysis was performed on 75 cases and 75 controls. This represented 30 % of the cohort. Homozygosity for the c.1226C > T variant was significantly associated with an elevated C14:1 between 0.9-2.4 μmol/L (p < 0.0001). None of the controls and 54 % of cases (41/75) were homozygous for this mutation. Twenty per cent of cases (15/75) and 11 % of controls (8/75) were heterozygous for this mutation, but only controls of Maori or Pacific ethnicity. The c.1226C > T mutation is strongly associated with Maori and to a lesser extent Pacific ethnicity; 78 % of cases of Maori ethnicity were homozygous and 18 % heterozygous for this mutation (Table 2).
Discussion
Striking the correct balance between ascertainment of true positive, potential childhood disease and unnecessary, potentially harmful, intervention in false positive or benign disease is one of the foremost challenges of NBS. Comprehensive long-term, natural history disease data is needed before management of screen positive cases can be optimised. Such data is currently lacking for most inborn errors of metabolism diagnosed by NBS. Despite this, metabolic and NBS services are reluctant to not intervene. Screen-positive cases remaining asymptomatic at follow up have therefore generally received preventative treatment such as emergency plans or dietary manipulation (Spiekerkoetter et al 2009a, b). Current screening methods do not accurately distinguish those who need treatment from those who will remain asymptomatic, and in the case of VLCADD, there is no robust molecular or biochemical enzymatic assay that will accurately predict phenotype.
This study provides important natural history data for children who had elevated levels of C14:1 at newborn screening in the NZ population. This audit of seven years of infants with NBS C14:1 levels between 0.9-2.4 μmol/L finds no increase in clinically significant early childhood disease. It therefore supports the current high screening cut off used by the NZ metabolic screening programme and questions the appropriate level of intervention in newborn screening programmes worldwide. New Zealand has a unique ethnic population, which directly impacts on the local manifestation of metabolic diseases such as VLCADD. This study provides reassurance, in the NZ population, that a less interventional approach is not just practical, but safe.
While C14:1 is generally regarded as the most informative analyte in screening for VLCADD, there can be a high incidence of false positive results. Rare false negative cases cannot be avoided. This study suggests that the R4S post-analytical tool is neither adequately specific nor sensitive for early VLCADD disease. According to the R4S tool, 54 % of cases were likely or very likely to be VLCADD, however none of these presented clinically during the study period. It is difficult to justify screening for VLCADD in NZ with a lower cut off both from a harm minimisation and resource allocation perspective.
The c.1226C > T ACADVL variant is a missense variant previously reported as pathogenic (Clinvar SVC000040367), however recent reports suggest this may be an attenuated variant. Fibroblast enzyme analysis and fatty acid oxidation flux is normal in homozygotes, although compound heterozygosity with a pathogenic mutation mildly reduces fatty acid oxidation flux (Ryder et al 2014). This study is the largest published series to date of patients with this variant and provides strong support for its categorisation as a benign variant. It is strongly associated with Maori, and to a lesser extent Pacific, ethnicity and is infrequently found in Europeans. The prevalence of this likely benign ACADVL variant accounts for Maori being overrepresented in existing NZ screen-positive VLCADD cases and largely accounts for the higher frequency of elevated NBS C14:1 levels in NZ. This study supports observations in the Hawaiian population that the c.1226C > T ACADVL variant may be a benign variant (Merritt et al 2014) and supports the view that there is some genotype-phenotype correlation (Andresen et al 1999) and that specific molecular analysis could improve second line screening specificity in some centers.
Given New Zealand’s unique ethnic demographics, these findings regarding the c.1226C > T variant may not be relevant to the international community however people of Maori and Pacific ethnicity are not uncommon in the United States, the United Kingdom and Australia, and screening units in those countries should be aware of this mutation. In addition, there were 48 European and nine Asian cases, the vast majority without the c.1226C > T mutation and with likely to have variable genetics, who also did not present with childhood disease. This strongly suggests that, no matter the ethnicity, initial newborn screening C14:1 levels between 0.9 and 2.4 μmol/L are not commonly associated with clinically significant metabolic disease.
There were a number of limitations identified in this study. Firstly, the presence of VLCADD-like disease was based heavily on clinical coding data, which may be inaccurate, therefore it is possible that cases with true disease may have been missed. Severe cases resulting in death however, would not have been missed by this audit. Secondly, there were a small number of patients within the cohort in whom key parameters such as a blood glucose level or liver enzyme tests were not recorded when they appeared to be clinically indicated. Finally, the follow up for the cohort ranges from only 6 months to 7 years; however, it is likely that the majority of severe and intermediate VLCADD phenotypes will have presented in this time. The NZ metabolic service is a national service that looks after the country and no patients have presented clinically from this cohort in the past 2 years. It is possible that some of the cases may go on to develop late onset, exercise induced muscle disease VLCADD, but the experience of the metabolic service which also covers adult disease is that adult VLCAD is rare in New Zealand and interestingly no cases with this common mutation have been diagnosed. However only continued follow up and accumulating data will reveal whether a cut-off of 2.5 μmol/L is appropriate in the New Zealand population or set too high to avoid unacceptably high mortality and morbidity in the longer term.
Conclusion
This study strongly suggests that current international screening for VLCADD is identifying and indeed treating a large number of children who are at no, or at least very low, risk of clinically significant disease. While classical VLCADD is a potentially devastating disease and symptomatic patients with relatively low C14:1 screening levels are reported, clinicians should be aware that many patients currently being treated for VLCADD may be at negligible risk of ever having clinically significant disease. This is the largest published series to date of patients with the c.1226C > T ACADVL gene mutation and provides support for its categorisation as a benign VLCADD variant.
References
Andresen BS et al (1999) Clear correlation of genotype with disease phenotype in very-long-chain acyl-CoA dehydrogenase deficiency. Am J Hum Genet 64:479–494
Boneh A et al (2006) VLCAD deficiency: pitfalls in newborn screening and confirmation of diagnosis by mutation analysis. Mol Genet and Metab 88:166–70
Gabriel S, Ziaugra L, Tabbaa D (2009) SNP genotyping using the Sequenom MassARRAY iPLEX platform. Curr Protoc Hum Genet. 2.12.1-2.12.18
Hoffmann L et al (2012) VLCAD enzyme activity determinations in newborns identified by screening: a valuable tool for risk assessment. J Inherit Metab Dis 35:269–277
Liebig M et al (2006) Neonatal screening for very long-chain acyl-CoA dehydrogenase deficiency: enzymatic and molecular evaluation of neonates with elevated C14:1-carnitine levels. Pediatrics 118:1065
Merritt JL II et al (2014) Infants suspected to have very-long chain acyl-CoA dehydrogenase deficiency from newborn screening. Mol Genet Metab 111:484–492
Ryder B et al (2014) Clinical, molecular and biochemical characteristics of patients with elevated tetradecenoylcarnitine (C14:1) detected by newborn screening in New Zealand: support for a common, likely benign mutation in the Pacific population. Poster (P035) presented at SSIEM 2014 Annual Symposium, September 2–5 Innsbruck, Austria. J Inherit Metab Dis 37(1):27–185
Sahai I et al (2011) A near-miss: very long chain acyl-CoA dehydrogenase deficiency with normal primary markers in the initial well-timed newborn screening specimen. J Ped 158(1):172–3
Schymik et al (2006) Pitfalls of neonatal screening for very-long-chain acyl-CoA dehydrogenase deficiency using tandem mass spectrometry. J Pediatr 149:128–130
Spiekerkoetter U et al (2009a) Management and outcome in 75 individuals with long-chain fatty acid oxidation defects: results from a workshop. J Inherit Metab Dis 32:488–497
Spiekerkoetter U et al (2009b) Treatment recommendations in long-chain fatty acid oxidation defects: consensus from a workshop. J Inherit Metab Dis 32:498–505
Spiekerkoetter U et al (2010a) Current issues regarding treatment of mitochondrial fatty acid oxidation disorders. J Inherit Metab Dis 33:555–561
Spiekerkoetter U et al (2010b) Tandem mass spectrometry screening for very long-chain acyl-CoA dehydrogenase deficiency: the value of second-tier enzyme testing. J Pediatr 157:668–73
Spiekerkoetter et al (2012) Lethal undiagnosed very long-chain acyl-CoA dehydrogenase deficiency with mild C14-acylcarnitine abnormalities on newborn screening. JIMD Reports 2:113–115
Acknowledgments
This study was made possible by funding from the NZ Newborn Screening Unit and an ASIEM small project grant.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2000 (5).
Conflict of interest
None.
Informed consent
The study was anonymous and informed consent was not required. No identifying information about patients is included in the article. The study received approval from the NZ Ministry of Health National Screening Unit (NSU), the National Health and Disability Ethics Committee and the Auckland District Health Board Research Committee.
Additional information
Communicated by: Verena Peters
Rights and permissions
About this article

Cite this article
Ryder, B., Knoll, D., Love, D.R. et al. The natural history of elevated tetradecenoyl-L-carnitine detected by newborn screening in New Zealand: implications for very long chain acyl-CoA dehydrogenase deficiency screening and treatment. J Inherit Metab Dis 39, 409–414 (2016). https://doi.org/10.1007/s10545-015-9911-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10545-015-9911-z