
Abstract
A 10-year-old boy with transcobalamin II (TCII) deficiency on oral cyanocobalamin therapy presented with acute right hemiparesis and sensory axonal neuropathy in the context of an intercurrent viral illness. MRI demonstrated unilateral globus pallidus stroke with normal MRA. Echocardiogram was normal. Methylmalonic acid in serum was mildly elevated at 10.29 μmol/L (normal < 0.37 μmol/L), which was an 18-fold increase from his previous baseline. The patient was switched to IM cyanocobalamin and serum methylmalonic acid levels normalized over 6 months to 0.01 μmol/L. After 4 months of IM cyanocobalamin therapy, the neuropathy had resolved. Repeat MRI 4 months after the sentinel stroke demonstrated a chronic-appearing contralateral globus pallidus stroke of uncertain timing.
Conclusions: We are describing the first case of metabolic stroke and peripheral neuropathy in TCII deficiency. The neuropathy was responsive to parenteral hydroxycobalamin. Unilateral globus pallidus stroke in the appropriate clinical context should not exclude a metabolic etiology as it may herald contralateral involvement and may provide an opportunity for early recognition and treatment. IM hydroxycobalamin should be strongly considered in all patients with TCII, particularly when they reach later childhood. This case highlights the selective vulnerability of the globus pallidus to increased levels of methylmalonic acid of various causes, which is important for both diagnosis and ultimately understanding the mechanisms of neurological injury in this group of conditions. Metabolic stroke may occur with lower levels of methylmalonic acid than previously reported in the context of an intercurrent bioenergetic stressor.
Competing interests: None declared
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Globus Pallidus
- Nerve Conduction Study
- Methylmalonic Acid
- Selective Vulnerability
- Superficial Peroneal Nerve
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
Introduction
The globus pallidus is an area of the brain that shows a selective vulnerability in a number of conditions associated with methylmalonic aciduria (MMA), including methylmalonyl-CoA mutase deficiency, cobalamin complementation group disorders, and severe pernicious anemia (Heidenreich et al. 1988; Trinh et al. 2001; Sharrief et al. 2012). The pathophysiology behind this selective vulnerability is currently unknown, although it has been postulated to result from respiratory chain dysfunction that preferentially affects the highly metabolically active globus pallidi (Heidenreich et al. 1988). Here we present the first reported case of globus pallidus stroke associated with methylmalonic aciduria in a patient with transcobalamin II (TCII) deficiency (OMIM #275350), thereby expanding the spectrum of MMA-induced pallidal stroke.
Case Report
The index patient is a 10-year-old, left-handed boy who was diagnosed with TCII deficiency at the age of 10 weeks when he presented with failure to thrive, lethargy, myoclonus, pancytopenia, and megaloblasts in his bone marrow. He had elevated methylmalonic acid and plasma homocysteine with normal serum B12 and folic acid. Enzymatic testing confirmed a defect of TCII. He was initially treated with IM hydroxycobalamin, with rapid normalization of homocysteine and methylmalonic acid levels. At 1 year of age, he was transitioned to oral cyanocobalamin (2,500 μg daily) with sustained normalization of laboratory parameters. The genetic diagnosis was later established as a homozygous sequence variant in the TCN2 gene (c.497_498de/tc). He remained systemically well following the early infantile period, with no history of metabolic decompensations. He did preferentially toe walk and on serial neurological assessments had decreased reflexes in his lower extremities; two prior nerve conduction studies were normal. Serum methylmalonic acid level performed at the age of 9 years was 0.58 μmol/L (normal < 0.37 μmol/L).
At the age of 10 years, the patient presented with a 2-day history of gradually progressive right hemiparesis (arm and leg) 2 weeks following a viral exanthem. The family had not noticed any additional weakness, encephalopathy, or seizures. Other than a moderate right hemiparesis, the examination was significant for bilateral lower extremity areflexia, with preserved reflexes in the upper extremities.
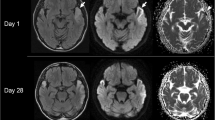
MR brain with spectroscopy (Fig. 1) demonstrated a well-defined area of restricted diffusion involving the left globus pallidus. MR angiography was normal. MRS with voxel placed in the left basal ganglia demonstrated a small lactate doublet at 1.33 ppm. Echocardiogram with bubble study was normal.

MR brain with spectroscopy at time of sentinel stroke. Diffusion-weighted imaging (a) with apparent diffusion coefficient (b) demonstrates restricted diffusion in the left globus pallidus (arrows). Proton spectroscopy (c) with voxel placed in the left globus pallidus demonstrates a small lactate doublet at 1.33 ppm (arrows)
On first presentation, serum methylmalonic acid was elevated at 8.57 μmol/L, which rose to 10.29 μmol/L over the following 4 days (normal < 0.37 μmol/L). The latter represented an 18-fold increase from his previous level at 9 years of age. Urine methylmalonic acid was also elevated at 184 mmol/mol creatinine. Quantitative acylcarnitines demonstrated only a borderline elevation of C3 at 1.11 μmol/L (normal <1.08) with normal total and free carnitine. Serum amino acids, homocysteine, hemoglobin, mean corpuscular volume (MCV), WBC count, venous blood gas, and serum immunoglobulins were normal.
Nerve conduction studies in the lower extremities were abnormal with decreased sensory nerve action potential (SNAP) amplitude of the sural nerve (with normal conduction velocity and distal latency) and absent superficial peroneal nerve SNAPs. Motor studies in the lower extremities and sensory/motor studies in the upper extremities were normal. These findings were in keeping with a length-dependent sensory axonal polyneuropathy.
The etiology of stroke in our patient at this point was not entirely clear, and both primary metabolic and vascular etiologies were entertained. The patient was switched from his oral cyanocobalamin formulation to 1 mg IM hydroxycobalamin daily. He was also started on 3 mg/kg ASA.
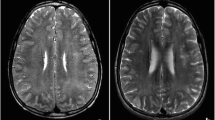
On repeat bloodwork at approximately 2, 3, and 6 months’ follow-up, the serum methylmalonic acid gradually normalized from 2.70 μmol/L to 1.99 μmol/L, and finally to 0.01 μmol/L, respectively. On clinical assessment at 4 months, the patient’s right hemiparesis was mildly improved. Reflexes were 1+ in the lower extremities. There was no evidence of dystonia or chorea. Repeat MR brain (Fig. 2) at 4 months demonstrated interval development of a new, chronic-appearing right globus pallidus stroke as well as gliotic changes of the original left globus pallidus stroke. Spectroscopy was normal. Repeat nerve conduction studies of sural and superficial peroneal nerves were normal.

MR brain at 4-month follow-up. Axial T2-weighted sequence demonstrates bilateral hyperintensities in the globus pallidi (arrows)
Discussion
Transcobalamin II (TCII) is one of the three human transporters of cobalamin (vitamin B12). It binds and transports cobalamin absorbed in the terminal ileum to tissue cells throughout the body, where it ultimately participates in cell surface adhesion and internalization of the cobalamin (Quadros 2009). TCII is also synthesized by astrocytes and is the primary enzyme involved in the neuronal uptake of cobalamin in the central nervous system (Begley et al. 1994). Cobalamin ultimately has an important role in the metabolism of both homocysteine and methylmalonic acid as a cofactor for methionine synthase and methylmalonyl-CoA mutase, respectively (Takahashi-Iñiguez et al. 2012; Banerjee and Ragsdale 2003). Methylmalonyl-CoA mutase isomerizes mitochondrial methylmalonyl-CoA to succinyl-CoA, a crucial substrate in energy provision via the citric acid cycle and complex II of the respiratory chain (Dutra et al. 1993; Okun et al. 2002). Inherited deficiency of TCII has been described in over 40 patients (Watkins and Rosenblatt 2011). TCII deficiency is an autosomal recessive condition resulting from mutations in the TCN2 gene on chromosome 22q12 (Watkins and Rosenblatt 2011; Regec et al. 1995). It typically presents in the first 2 months of life with pancytopenia, failure to thrive, and gastrointestinal symptoms (Kaikov et al. 1991; Gimpert et al. 1975). Laboratory markers typically include increased homocysteine, reduced methionine, increased methylmalonic acid, metabolic acidosis, and megaloblastic anemia (Kaikov et al. 1991). Stroke and peripheral neuropathy have not previously been reported in association with TCII deficiency.
We presume that the mechanism of stroke in our patient was MMA-induced pallidal stroke for the following reasons. Firstly, our patient presented with staggered, bilateral globus pallidus strokes, without any additional parenchymal involvement in the lateral lenticulostriate or anterior choroidal vascular territories bilaterally. The latter vessels supply the globus pallidus, as well as the putamen, caudate, and internal capsule. It would be highly implausible for a vaso-occlusive stroke to selectively affect bilateral pallidi in isolation (the identical portion of the vascular territory bilaterally), sparing the remainder of the vascular territory. This is especially the case with normal vascular imaging. The selective bilateral pallidal involvement is, therefore, more consistent with a “metabolic” stroke in a vulnerable region of the basal ganglia, as previously described in MMA of various etiologies. While globus pallidus stroke has not been specifically described in patients with TCII deficiency, it is a logical extension given the role of TCII in the metabolism of methylmalonic acid. It is admittedly unusual that our patient presented initially with unilateral globus pallidus involvement, as asymmetries have not been previously described with MMA. Repeat neuroimaging was done 4 months after the incident stroke, making it difficult to determine the precise timing of the chronic-appearing contralateral globus pallidus stroke. The asymmetry in the initial presentation may suggest subtle differences in the pallidal neuronal micromilieu in this patient. In addition, although our patient’s peak level of MMA was lower than that previously reported in association with MMA-associated metabolic stroke (lowest reported level is 89 μmol/L as per Sharrief et al. 2012), our patient’s stroke did occur in the context of an almost 20-fold elevation of MMA from his previous baseline. The threshold for metabolic stroke in our patient may have been lowered in part by the bioenergetic stress of his intercurrent viral illness.
In addition to stroke, our patient presented with a new, electrophysiologically confirmed peripheral neuropathy. A predominantly demyelinating peripheral neuropathy has been previously described in both early-onset and late-onset forms of cobalamin C deficiency and methylene tetrahydrofolate reductase deficiency (Frattini et al. 2010; Nishimura et al. 1985). In addition, sensory neuropathy has been described in patients with other forms of homocystinuria, although this has been attributed to toxicity from supplementation with pyridoxine (Ludolph et al. 1991). Dietary B12 deficiency is also associated with a reversible sensorimotor demyelinating polyneuropathy (Sakly et al. 2005). The presumed pathophysiology relates to deficient methylation of myelin basic protein, an important component of myelin that influences myelin compaction (Frattini et al. 2010). To our knowledge, this is the first report of sensory axonal peripheral neuropathy in a patient with TCII deficiency. Importantly, abnormalities on nerve conduction studies reversed with IM hydroxycobalamin.
Previous authors have suggested aggressive treatment of TCII deficiency with IM hydroxycobalamin and have cited reports of treatment failure in late childhood and adolescence in patients transitioned from IM treatment to oral cyanocobalamin. The patients with treatment failure presented with cognitive decline, visual dysfunction from maculopathy, and proprioceptive difficulties from presumed dorsal column involvement of the spinal cord. Symptoms improved when IM therapy was resumed (Schiff et al. 2010).
In conclusion, we are describing the first case of metabolic stroke and peripheral neuropathy in TCII deficiency. The neuropathy was responsive to parenteral hydroxycobalamin. This case highlights a number of important issues. Firstly, unilateral globus pallidus stroke in the appropriate clinical context should not exclude a metabolic etiology as it may herald contralateral involvement; in fact, it may provide an opportunity for early recognition and treatment. Secondly, this case suggests that IM hydroxycobalamin should be strongly considered in all patients with TCII, particularly when they reach later childhood. Finally, this case highlights the selective vulnerability of the globus pallidus to increased levels of methylmalonic acid of various causes, which is important for both diagnosis and ultimately understanding the mechanisms of neurological injury in this group of conditions. It appears that metabolic stroke may occur with lower levels of MMA than previously reported in the context of an intercurrent bioenergetic stressor.
References
Banerjee R, Ragsdale SW (2003) The many faces of vitamin B12: catalysis by cobalamin-dependent enzymes. Annu Rev Biochem 72:209–247
Begley JA, Colligan PD, Chu RC (1994) Synthesis and secretion of transcobalamin II by cultured astrocytes derived from human brain tissue. J Neurol Sci 122:57–60
Dutra JC, Dutra-Filho CS, Cardozo SE et al (1993) Inhibition of succinate dehydrogenase and beta-hydroxybutyrate dehydrogenase activities by methylmalonate in brain and liver of developing rats. J Inherit Metab Dis 16:147–153
Frattini D, Fusco C, Ucchino V, Tavazzi B, Della Giustina E (2010) Early onset methylmalonic aciduria and homocysteinuria cblC type with demyelinating neuropathy. Pediatr Neurol 43:135–138
Gimpert E, Jakob M, Hitzig WH (1975) Vitamin B12 transport in blood in congenital deficiency of transcobalamin II. Blood 45:71–82
Heidenreich R, Natowicz M, Hainline BE, Kelley RI, Hillman RE et al (1988) Acute extrapyramidal syndrome in methylmalonic academia: “metabolic stroke” involving the globus pallidus. J Pediatr 113(6):1022–1027
Kaikov Y, Wadsworth LD, Hall CA, Rogers PC (1991) Transcobalamin II deficiency: case report and review of the literature. Eur J Pediatr 150:841–843
Ludolph AC, Ullrich K, Bick U, Fahrendorf G, Przyrembel H (1991) Functional and morphological deficits in late-treated patients with homocysteinuria: a clinical, electrophysiologic, and MRI studies. Acta Neurol Scand 83:161–165
Nishimura M, Yoshino K, Tomita Y, Takashima S, Tanaka J et al (1985) Central and peripheral nervous system pathology of homocysteinuria due to 5,10 –methylene tetrahydrofolate reductase deficiency. Pediatr Neurol 1(6):375–378
Okun JG, Horster F, Farkas LM, Feyh P, Hinz A, Sauer S et al (2002) Neurodegeneration in methylmalonic aciduria involves inhibition of complex II and tricarboxylic acid cycle, and synergistically acting excitotoxicity. J Biol Chem 277:14674–14680
Quadros EV (2009) Advances in the understanding of cobalamin assimilation and metabolism. Brit J Hematol 148:195–204
Regec A, Quadros EV, Platica O, Rothenberg SP (1995) The cloning and characterization of the human transcobalamin II gene. Blood 85:2711–2719
Sakly G, Hellara O, Trabelsi A, Dogui M (2005) Reversible peripheral neuropathy induced by B12 deficiency. Neurophysiol Clin 35(5–6):149–153
Schiff M, Ogier de Baulny H, Bard G, Barlogis V, Hamil C, Moat SJ et al (2010) Should transcobalamin deficiency be treated aggressively. J Inherit Metab Dis 33:223–229
Sharrief AZ, Raffel J, Zee DS (2012) Vitamin B12 deficiency with bilateral globus pallidus abnormalities. Arch Neurol 69(6):769–772
Takahashi-Iñiguez T, Garcia-Hernandez E, Arreguin-Espinosa R, Flores ME (2012) Role of vitamin B12 on methylmalonyl-CoA mutase activity. J Zhejiang Univ-Sci B (Biomed & Biotechnol) 13(6):423–437
Trinh BC, Melhem ER, Barker PB (2001) Multi-slice proton MR spectroscopy and diffusion weighted imaging in methylmalonic academia: report of two cases and review of the literature. AJNR 22:831–833
Watkins D, Rosenblatt DS (2011) Inborn errors of cobalamin absorption and metabolism. Am J Med Genet 157:33–44
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Additional information
Communicated by: Ertan Mayatepek
Appendices
Synopsis
Transcobalamin II deficiency may present with metabolic pallidal stroke and peripheral neuropathy, reversible with IM cyanocobalamin.
Contributions
Lance H Rodan – reviewed patient, concept, manuscript preparation, literature review, discussion, manuscript writing, guarantor
Navin Mishra – reviewed patient, manuscript editing, literature review, discussion
Ivanna Yau – reviewed patient, manuscript editing, discussion
Andrea Andrade – reviewed patient, manuscript editing, discussion
Komudi Siriwardena – contributed patient and patient data and reviewed patient, manuscript editing, discussion
Ingrid Tein – reviewed patient, concept, manuscript editing, discussion, supervisor
Conflict of Interest
No authors report a conflict of interest related to the publication of this manuscript.
Rights and permissions
Copyright information
© 2013 SSIEM and Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Rodan, L.H., Mishra, N., Yau, I., Andrade, A., Siriwardena, K., Tein, I. (2013). Expanding the Spectrum of Methylmalonic Acid-Induced Pallidal Stroke: First Reported Case of Metabolic Globus Pallidus Stroke in Transcobalamin II Deficiency. In: Zschocke, J., Gibson, K., Brown, G., Morava, E., Peters, V. (eds) JIMD Reports - Volume 11. JIMD Reports, vol 11. Springer, Berlin, Heidelberg. https://doi.org/10.1007/8904_2013_215
Download citation
DOI: https://doi.org/10.1007/8904_2013_215
Received:
Revised:
Accepted:
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-642-37327-5
Online ISBN: 978-3-642-37328-2
eBook Packages: MedicineMedicine (R0)