
Abstract
Transcobalamin (transcobalamin II, TC) transports plasma vitamin B12 (cobalamin, Cbl) into cells. TC deficiency is a rare autosomal recessive disorder causing intracellular Cbl depletion, which in turn causes megaloblastic bone marrow failure, accumulation of homocysteine and methylmalonic acid, and methionine depletion. The clinical presentation reflects intracellular Cbl defects, with early-onset failure to thrive with gastrointestinal symptoms, pancytopenia, and megaloblastic anemia, sometimes followed by neurological complications. We report the clinical, biological, and molecular findings and the outcome in five TC-deficient patients. The three treated early had an initial favorable outcome, whereas the two treated inadequately had late-onset severe neuro-ophthalmological impairment. Even if the natural course of the disease over time might also result in late-onset symptoms in the aggressively treated patients, these data emphasize that TC deficiency is a severe disorder requiring early detection and probably long-term aggressive therapy. Mutation analysis revealed six unreported mutations in the TCN2 gene. In silico structural analysis showed that these mutations disrupt the Cbl-TC interaction domain and/or the putative transcobalamin–transcobalamin receptor interaction domain.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
When pancytopenia presents in early infancy, the most important diagnoses to consider are inherited disorders of cobalamin (Cbl, vitamin B12) or folate metabolism and nutritional Cbl deficiency. The latter should be especially suspected in breast-fed infants of vegan mothers. Accurate diagnosis is an emergency because early detection and treatment can be life saving. Transcobalamin (transcobalamin II, TC) is a plasma protein indispensable for absorption, transport, and cellular uptake of Cbl (Quadros 2009; Quadros et al. 2009; Rosenblatt and Fenton 2001). TC deficiency causes severe intracellular Cbl depletion leading to the hematological and metabolic abnormalities that characterize this disorder. These include early-onset pancytopenia with megaloblastic bone marrow, urinary methylmalonic acid (MMA) excretion, and most often, accumulation of homocysteine (HCy) in body fluids. About 45 TC-deficient patients have been reported (Kaikov et al. 1991; Rosenblatt and Fenton 2001). The gene coding for TC (TCN2) was identified in 1995 (Regec et al. 1995), and ten pathogenic mutations have further been reported (Haberle et al. 2009; Li et al. 1994a; Li et al. 1994b; Namour et al. 2003; Prasad et al. 2008; Qian et al. 2002; Ratschmann et al. 2009). We report herein on five TC-deficient patients who exhibited six unreported TCN2 mutations defining two important coding regions in exons 4 and 8, which code for TC-TC receptor and TC-Cbl interaction domains, respectively.
Patients and methods
Clinical, biochemical, and metabolic data were collected prospectively. All nine exons and intron/exon boundaries of the TCN2 gene were amplified by polymerase chain reaction (PCR) and sequenced on an ABI PRISM 3100 Analyzer. PCR primers are available upon request.
Results
Clinical signs, biochemical, and hematological findings at diagnosis, results of molecular analysis, and initial treatment are summarized in Table 1.
Detailed clinical and follow-up findings
Patient 1
This second child born to consanguineous parents was admitted at 4 months of age for acute gastroenteritis with extreme pallor. Her brother had died in unexplained circumstances from acute anemia at the age of 3 months. Our patient exhibited recurrent episodes of diarrhea associated with stomatitis from 1 month of age. After one red blood cell (RBC) and two platelet transfusions, she was treated with hydroxocobalamin (OHCbl) 1 mg IM daily for 1 week, with prompt normalization of hematological and metabolic parameters (Table 2). The rhythm of injections could be lengthened to 1 mg monthly with good clinical, hematological and metabolic control (Table 3). In association with OHCbl therapy, folic acid and betaine supplementations were given (Table 2). The child, at the age of 22 months, had a perfect clinical status with normalization of hematological and metabolic parameters (Tables 2 and 3).
Patient 2
This second girl, born to nonconsanguineous parents, was admitted at 3 months of age for failure to thrive, diarrhea, and vomiting. Initial bone marrow analysis led to a misdiagnosis of acute myeloid leukemia type 4. She was initially treated with OHCbl 1 mg triweekly IM with good clinical and biological outcome at 2 years of age (Table 3).
Patient 3
This child, born to nonconsanguineous parents, was admitted at 7.5 weeks of age for severe failure to thrive. After initial treatment with IM OHCbl 1 mg daily for 1 week, she was treated with 1 mg IM weekly with a good clinical outcome at 2.5 years of age and normalization of metabolic and hematological parameters (Table 3).
Patient 4
This first-born daughter of nonconsanguineous parents was admitted at the age of 7 months for acute pallor and purpura. At first, the patient was treated for 1 year with IM injections of OHCbl 1 mg every 10 days in association with oral supplementation of folic acid. Then, she received 1 mg/day of cyanocobalamin orally. Whereas initial normalization of hematological and metabolic abnormalities could be obtained (data not shown), no biological data were further available until the age of 16 years. At that age, she exhibited an acute neurological deterioration with myoclonus and posterior cordonal syndrome associated with bilateral macular dysfunction. These were attributed to metabolic imbalance (high HCy and MMA, unavailable data), and the patient was again treated with parenteral OHCbl (5 mg triweekly), with clinical and biological stabilization (data not shown).
Patient 5
This newborn girl was admitted at 1 month of age for failure to thrive, vomiting, and stomatitis. Her brother had died at 2 months of age with a pyocyanic septicemia associated with pancytopenia. She was initially treated IM with OHCbl, 2 mg/day for a few days and then 1 mg three times a week. B12 therapy was associated with oral folic acid. At 1 year of age, parenteral OHCbl was substituted with OHCbl 1 mg orally three times a week. At 7 years of age, progressive increase of MMA urinary excretion and total HCy (tHCy) accumulation in plasma (mean values MMA 250 mmol/mol and tHCy 50 µM) were recorded. At that time, OHCbl supplementation orally was increased to 1 mg daily. At 9 years of age, she exhibited severe visual impairment with macular dysfunction and mild mental retardation. Daily OHCbl was reintroduced IM (1 mg/day) and resulted in stabilization of visual and cognitive functions and normalization of metabolic parameters (Table 3).
Discussion
All our patients shared similar clinical features with patients previously reported: very early-onset gastrointestinal symptoms (diarrhea and/or vomiting) with subsequent severe failure to thrive. These symptoms associated with pallor and/or mucosal signs led to hematological investigations that disclosed moderate aregenerative anemia and/or pancytopenia with megaloblastic bone marrow. As previously reported for other TC-deficient patients (Rosenblatt and Fenton 2001), patient 2 was misdiagnosed with leukemia due to immature white blood cell precursors in marrow that was otherwise hypocellular.
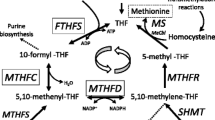
The majority of circulating Cbl is bound to haptocorrin and therefore not affected by TC deficiency. Thus, serum Cbl levels were, as previously reported (Hakami et al. 1971; Prasad et al. 2008), in the normal range, except for patient 5 who had low levels. Normal serum Cbl levels excluded nutritional Cbl deficiency, congenital intrinsic factor deficiency, or a disorder of intestinal Cbl receptors, and led to investigation of Cbl transport and intracellular metabolism. This revealed abnormal MMA urinary excretion, low-normal methioninemia, and accumulation of total or free homocysteine (patients 1, 2, 4, and 5). These metabolic abnormalities are the hallmark of Cbl intracellular depletion (Whitehead 2006). Moreover, adenosyl-Cbl is the cofactor of methylmalonyl-CoA-mutase, deficiency of which (due to intracellular adenosyl-Cbl depletion) results in methylmalonic aciduria (Rosenblatt and Fenton 2001); methyl-Cbl is the cofactor of methionine synthase, which catalyzes the remethylation of homocysteine to methionine. Intracellular methyl-Cbl depletion results in a functional deficiency of methionine synthase with defective remethylation. Subsequently, there is homocysteine accumulation as well as intracellular methionine and S-Adenosyl-methionine (SAM) depletion (Rosenblatt and Fenton 2001). Homocysteine accumulation, with subsequent prothrombotic effect, (Castro et al. 2006) and SAM depletion are the two main factors in neurological impairment (Surtees 1998). SAM plays a pivotal role in numerous methylation processes, especially in the central nervous system (CNS) (Bottiglieri 2002).
Regarding outcome, our patients fell into two categories: the first three, treated early, had a favorable initial outcome without any neurological impairment; the other two were inadequately treated (Table 3). The latter patients had both been switched from IM to orally administered OHCbl supplementation and suffered late-onset neurological deterioration associated with retinopathy due to macular dysfunction [previously reported in patient 4 (Souied et al. 2001)]. This retinal degeneration resembles the ophthalmologic complications reported in some patients with inherited intracellular Cbl type C defect (Gerth et al. 2008). An additional TC-deficient patient has been described with retinal degeneration (Dharmasena et al. 2008), which, along with neurological impairment, is possibly attributable to insufficient and/or orally administered Cbl therapy, with subsequent severe intracellular Cbl depletion and defective CNS remethylation (Hall 1992; Surtees 1998).
Long-term follow-up for our first three patients will be mandatory to confirm their favorable outcome (especially the absence of neurological and eye deterioration over time) and its possible causative link with long-term IM OHCbl therapy. Nevertheless, the eye and neurological symptoms of patients 4 and 5 could be stabilized when they were again treated with IM OHCbl. For patient 5, the same is true for the metabolic parameters that normalized (Table 3). Unfortunately, follow-up data regarding the potential benefits of long-term IM OHCbl are only available for patients 1 and 5. Patient 1 (IM OHCbl) is a 22-months old normal girl with normalized metabolic parameters (Table 2), whereas when orally treated, patient 5 had higher tHCy and MMA urinary excretion.
Treatment of TC deficiency should combine life-long parenteral pharmacological doses of OHCbl with folate supplements and probably orally administered betaine. The frequency of OHCbl injections should be monitored with hematological and metabolic parameters and should aim at normalizing not only pancytopenia but also methionine and total homocysteine (tHCy) plasma levels and MMA urinary excretion (Tables 2 and 3). Patient 1 is the only one who received betaine, which allowed normal-high methionine plasma levels and normalization of tHCy (Table 2). Even if these data are definitely not sufficient to demonstrate betaine efficacy, by analogy with CblC defect (Ogier de Baulny et al. 1998), betaine should probably be recommended in the treatment of TC deficiency. Betaine catalyzes the remethylation of homocysteine to methionine in the liver using a non-CNS alternative pathway (Delgado-Reyes et al. 2001). Thus, it theoretically reduces homocysteine accumulation while increasing systemic methionine levels and probably also methionine availability for the CNS (Strauss et al. 2007).
Finally, the molecular analysis in our patients revealed six unreported mutations in the TCN2 gene defining two important domains in exons 4 and 8 (Fig. 1). Molecular studies in the parents of each patient confirmed Mendelian inheritance. The previously reported mutations are varied, with nonsense mutations (Li et al. 1994a; Prasad et al. 2008), deletions (Haberle et al. 2009; Li et al. 1994a; Li et al. 1994b), insertions (Ratschmann et al. 2009), RNA editing (Qian et al. 2002), and splice-site mutations (Haberle et al. 2009; Namour et al. 2003), all giving rise to truncated proteins. We also observed nucleotide deletions or duplications predicted to give rise to abnormal proteins. However, in two cases (patients 3 and 5), we found a 2-bp deletion in exon 4 that theoretically induces a frameshift, giving rise to a normal-length protein. In addition, exon 4 skipping (due to a splice-site mutation) was found in patient 1. In patient 4, we found a mutation affecting exon 8 that theoretically results in a normal-length protein.

Three-dimensional view of the transcobalamin II (TC) protein showing in silico prediction of the effect of the mutations harbored by patient 4. These are located in exon 4 (TC-TC receptor interaction) and exon 8 [cobalamin (Cbl) binding site]
In silico structural analysis revealed that these mutations, as well as those previously reported (Haberle et al. 2009; Ratschmann et al. 2009), always disrupt the Cbl-TC interaction domain and/or the putative TC-TC receptor interaction domain. From these in silico studies, we confirm that exons 4 and 8 are important coding domains for TC functionality. These regions have previously been reported as structures coding for TC-TC receptor interaction and Cbl binding, respectively (Wuerges et al. 2006).
Conclusion
TC deficiency is a severe disorder with intracellular Cbl depletion. The patients who were treated early have had a favorable outcome as opposed to the inadequately treated ones, who had severe neurological and retinal impairment. Nevertheless, as the patients who had a favorable outcome are still very young (maximum 2.5 years of age), one cannot exclude that despite prompt recognition and “aggressive” treatment, the natural course of the disease over time might also result in late-onset eye and neurological symptoms in these aggressively treated patients (as described in CblC defect). In the clinical setting of early-onset pancytopenia with megaloblastic bone marrow but normal B12 serum levels, our findings emphasize the importance of prompt recognition of TC deficiency and its aggressive, probably life-long treatment, with pharmacological doses of parenteral hydroxocobalamin. Further, the molecular and in silico data in our patients confirm that TC-Cbl and TC-TCR regions are two major domains for TC function that are affected in TC-deficient patients.
References
Bottiglieri T (2002) S-Adenosyl-L-methionine (SAMe): from the bench to the bedside–molecular basis of a pleiotrophic molecule. Am J Clin Nutr 76:1151S–1157S
Castro R, Rivera I, Blom HJ, Jakobs C, Tavares de Almeida I (2006) Homocysteine metabolism, hyperhomocysteinaemia and vascular disease: an overview. J Inherit Metab Dis 29:3–20
Delgado-Reyes CV, Wallig MA, Garrow TA (2001) Immunohistochemical detection of betaine-homocysteine S-methyltransferase in human, pig, and rat liver and kidney. Arch Biochem Biophys 393:184–186
Dharmasena A, Calcagni A, Kerr AR (2008) Retinopathy in inherited transcobalamin II deficiency. Arch Ophthalmol 126:141–142
Gerth C, Morel CF, Feigenbaum A, Levin AV (2008) Ocular phenotype in patients with methylmalonic aciduria and homocystinuria, cobalamin C type. J Aapos 12:591–596
Haberle J, Pauli S, Berning C, Koch HG, Linnebank M (2009) TC II deficiency: avoidance of false-negative molecular genetics by RNA-based investigations. J Hum Genet 54:331–334
Hakami N, Neiman PE, Canellos GP, Lazerson J (1971) Neonatal megaloblastic anemia due to inherited transcobalamin II deficiency in two siblings. N Engl J Med 285:1163–1170
Hall CA (1992) The neurologic aspects of transcobalamin II deficiency. Br J Haematol 80:117–120
Kaikov Y, Wadsworth LD, Hall CA, Rogers PC (1991) Transcobalamin II deficiency: case report and review of the literature. Eur J Pediatr 150:841–843
Li N, Rosenblatt DS, Seetharam B (1994a) Nonsense mutations in human transcobalamin II deficiency. Biochem Biophys Res Commun 204:1111–1118
Li N, Rosenblatt DS, Kamen BA, Seetharam S, Seetharam B (1994b) Identification of two mutant alleles of transcobalamin II in an affected family. Hum Mol Genet 3:1835–1840
Namour F, Helfer AC, Quadros EV et al (2003) Transcobalamin deficiency due to activation of an intra exonic cryptic splice site. Br J Haematol 123:915–920
Ogier de Baulny H, Gerard M, Saudubray JM, Zittoun J (1998) Remethylation defects: guidelines for clinical diagnosis and treatment. Eur J Pediatr 157(Suppl 2):S77–S83
Prasad C, Rosenblatt DS, Corley K, Cairney AE, Rupar CA (2008) Transcobalamin (TC) deficiency—potential cause of bone marrow failure in childhood. J Inherit Metab Dis 10.1007/s10545-008-0864-3
Qian L, Quadros EV, Regec A, Zittoun J, Rothenberg SP (2002) Congenital transcobalamin II deficiency due to errors in RNA editing. Blood Cells Mol Dis 28:134–142, discussion 143–135
Quadros EV (2009) Advances in the understanding of cobalamin assimilation and metabolism. Br J Haematol 148(2):195–204
Quadros EV, Nakayama Y, Sequeira JM (2009) The protein and the gene encoding the receptor for the cellular uptake of transcobalamin-bound cobalamin. Blood 113:186–192
Ratschmann R, Minkov M, Kis A et al (2009) Transcobalamin II deficiency at birth. Mol Genet Metab 98(3):285–288
Regec A, Quadros EV, Platica O, Rothenberg SP (1995) The cloning and characterization of the human transcobalamin II gene. Blood 85:2711–2719
Rosenblatt DS, Fenton WA (2001) Inherited disorders of folate and cobalamin transport and metabolism, In: Scriver CR, Beaudet AL, Valle D, Sly WS (eds) Metabolic and molecular bases of inherited disease, 8th ed. McGraw-Hill, New-York, Vol. III, pp 3897–3933
Souied EH, Benhamou N, Sterkers M et al (2001) Retinal degeneration associated with congenital transcobalamin II deficiency. Arch Ophthalmol 119:1076–1077
Strauss KA, Morton DH, Puffenberger EG et al (2007) Prevention of brain disease from severe 5, 10-methylenetetrahydrofolate reductase deficiency. Mol Genet Metab 91:165–175
Surtees R (1998) Demyelination and inborn errors of the single carbon transfer pathway. Eur J Pediatr 157(Suppl 2):S118–S121
Whitehead VM (2006) Acquired and inherited disorders of cobalamin and folate in children. Br J Haematol 134:125–136
Wuerges J, Garau G, Geremia S, Fedosov SN, Petersen TE, Randaccio L (2006) Structural basis for mammalian vitamin B12 transport by transcobalamin. Proc Natl Acad Sci U S A 103:4386–4391
Acknowledgements
The authors are grateful to Dr. Kevin Collins for his critical review and constructive comments on this article.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by: Matthias Baumgartner
Competing interests: None declared.
Rights and permissions
About this article
Cite this article
Schiff, M., Ogier de Baulny, H., Bard, G. et al. Should transcobalamin deficiency be treated aggressively?. J Inherit Metab Dis 33, 223–229 (2010). https://doi.org/10.1007/s10545-010-9074-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10545-010-9074-x