
Abstract
Congenital disorders of glycosylation (CDG) are genetic diseases caused by abnormal protein and lipid glycosylation. In this chapter, we report the clinical, biochemical, and molecular findings in two siblings with an unidentified CDG (CDG-Ix). They are the first and the third child of healthy consanguineous Argentinean parents. Patient 1 is now a 11-year-old girl, and patient 2 died at the age of 4 months. Their clinical picture involved liver dysfunction in the neonatal period, psychomotor retardation, microcephaly, seizures, axial hypotonia, feeding difficulties, and hepatomegaly. Patient 1 also developed strabismus and cataract. They showed a type 1 pattern of serum sialotransferrin. Enzymatic analysis for phosphomannomutase and phosphomannose isomerase in leukocytes and fibroblasts excluded PMM2-CDG and MPI-CDG. Lipid-linked oligosaccharide (LLO) analysis showed a normal profile. Therefore, this result could point to a deficiency in the dolichol metabolism. In this context, ALG8-CDG, DPAGT1-CDG, and SRD5A3-CDG were analyzed and no defects were identified. In conclusion, we could not identify the genetic deficiency in these patients yet. Further studies are underway to identify the basic defect in them, taking into account the new CDG types that have been recently described.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
Introduction
Congenital disorders of glycosylation (CDG) are genetic defects in the synthesis and transfer of the glycan moiety of glycoproteins and glycolipids. About ~1% of the human genome is estimated to be involved in glycosylation processes (Jaeken 2003; Morava et al. 2008). Disorders of N-glycosylation are divided into CDG type I or II, according to the intracellular localization of the molecular defect (McKenzie et al. 2007; Morava et al. 2008; Lefeber et al. 2009; Jaeken et al. 2009). CDG-I is caused by defects in enzymes governing the synthesis and transfer of the oligosaccharide in the ER. On the other hand, defects leading to CDG-II belong to different classes: enzymes responsible for the modifications of the N-glycan chain in the Golgi apparatus, sugar transporters, and proteins with a role in intracellular trafficking, like the COG complex. A new nomenclature system indicates the specific disorder with the use of the gene name or the protein name to include all protein glycosylation disorders and to extend it to the lipid glycosylation defects (Jaeken et al. 2008). The diseases are labeled CDG-Ix as long as the basic defect remains unknown.
Since the initial description of CDG in 1980 (Jaeken et al. 1980), serum transferrin IEF with immunodetection has been widely used as a screening test. Any defect in the synthesis or processing of these glycans results in the alteration of sialotransferrin isoforms, which is detectable by IEF according to their different charges (Jaeken 2003). The analysis on HPLC allows for the separation of transferrin glycoforms based not only on the net charge of the molecule, but also on structural differences of the glycans. Both methodologies can be used for assignment of cases to either type I or type II (Sturiale et al. 2008; Quintana et al. 2009).
Application of matrix-assisted laser desorption/ionization time of flight mass spectrometry (MALDI-TOF MS) allows the high-throughput identification of proteins by the accurate mass measurement of peptides derived from total proteome digestion (Reinders et al. 2004; Guerrera and Kleiner 2005). MALDI-TOF applications in CDG aim to investigate glycosylation changes of proteins, providing structural information on the targeted protein. In addition, MS characterization of defective glycan structures is an essential step to solve defects in patients with CDG-x (Sturiale et al. 2005; Babovic-Vuksanovic and O’Brien 2007). The clinical spectrum of N-glycosylation defects is variable, ranging from severe multisystem disorders to dysfunction of specific organs. The clinical, metabolic, and molecular aspects of CDG patients have been described extensively in recent reviews (Marklová and Albahri 2007; de Lonlay et al. 2008; Barone et al. 2009). Other reports (Marklová and Albahri 2007; Morava et al. 2008) tried to find similarities in the clinical presentations of patients and to detect specific symptoms suggestive of a distinct CDG I type. Clinical features such as inverted nipples and abnormal fat distribution may be found in PMM2-CDG (CDG Ia) (OMIM 212065) patients, the most frequent type. Other clinical features such as retrognathia, low-set ears, and club foot are common findings in congenital central nervous system anomalies. In one of the studies, an overview of the unusual clinical symptoms in CDG-Ix (OMIM 212067) showed that two main subgroups could be distinguished based on the severity of the disease: one with a pure neurological presentation and the other with a neurological-multivisceral form (Morava et al. 2008). Nevertheless, other unique findings in CDG-Ix include arthrogryposis, macrocephaly, polyneuropathy, and cystic kidneys.
We want to report the clinical features, the biochemical studies, and the molecular analysis of two siblings characterized as CDG-Ix, who have been identified in the context of the first program for these pathologies in Argentina.
Case Report
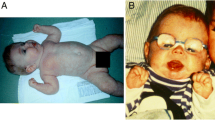
We present two sisters born as the first and the third child of healthy consanguineous Argentinean parents. One of the children is now 11 years old (patient 1) and the other (patient 2) had an early death at the age of 4 months. The clinical features of both patients, who share some common features of CDG, are illustrated in Fig. 1a, b. The clinical findings include axial hypotonia, psychomotor retardation, dysmorphic features, feeding problems, hepatomegaly, coagulopathy, and recurrent infections (Table 1). Patient 2 was born with a severe phenotype consisting of liver involvement (hepatomegaly, hypoalbuminemia, protein-losing enteropathy, recurrent vomiting, and diarrhea), seizures, progressive developmental delay, and coagulation abnormalities with recurrent infections. Unfortunately, detailed examination of the brain was not possible due to the early death (Table 1). In patient 1, ophthalmological abnormalities were observed including strabismus and nuclear-cortical cataracts (Fig. 1a). She presented liver dysfunction in the neonatal period, and has had psychomotor retardation, microcephaly, intractable seizures, feeding difficulties, and malnutrition.

Clinical features of two CDG-Ix sibling patients. Patient 1 (a and b) at 8 years old. She showed psychomotor retardation, feeding problems, hepatomegaly and ophthalmological abnormalities (strabismus and cataract). Patient 2 (c and d): she had dysmorphic features and a severe phenotype consisting of liver involvement (hepatomegaly, hypoalbuminemia, ascitis, protein-losing enteropathy, recurrent vomiting, and diarrhea), microcephaly, intractable seizures, axial hypotonia, feeding difficulties, and malnutrition. She had recurrent infections and an early death at 4 months
Materials and Methods
Blood samples from both patients were obtained after informed parental consent. EDTA blood samples were taken from patient 1 and the parents to extract genomic DNA from leukocytes with a commercially available kit (Wizard Genomic Purification Kit, Promega, Madison, WI). For cell culture, skin biopsy was obtained from patient 1, and the fibroblasts were cultivated in minimal essential medium (MEM). Approval of Human Research was obtained from the institutional review boards of CIEIS-Health Investigation Ethic Committee. Children’s Hospital, Córdoba, Argentina.
SDS-PAGE and Western Blot
Plasma sample from P1 was incubated for 30 min at room temperature with a solution of NaCl 0.9% and 10 mM ferric citrate in a ratio of 15:70:15 to saturate the transferrin with iron. The saturated sample was diluted 1:20 in sample treatment buffer containing 250 μl Tris–HCl 1.25 mol/L (pH: 6.8), 400 μl SDS 25%, 250 μl 2-mercaptoethanol, 500 μl glycerol 99%, 250 μl 0.2% bromophenol blue, and 3,350 μl of bidistilled water (final volume 5 ml; pH 6.8) and separated on 10% SDS-PAGE and transferred to a PVDF membrane by western blotting using standard procedures (Artuch et al. 2003). The blocked membrane was incubated with polyclonal antibodies for anti-transferrin and anti-haptoglobin glycoproteins (Dako, Germany) using an anti-rabbit peroxidase-goat conjugated as secondary antibody (Bio Rad). The visualization of the protein bands was performed using a colorimetric detection (DAB, Sigma).
IEF of Serum Transferrin (Tf-IEF)
Plasma sample from P1 (20 μl) was incubated for 30 min at room temperature with a solution of NaCl 0.9% (20 μl), 10 mM ferric citrate (2 μl), and 0.5 mM sodium hydrogen carbonate (2 μl) to saturate the transferrin with iron. After centrifugation (2,000 × g, 10 min), the supernatant was further diluted (1:5) with water.
Proteins of P2 were extracted by incubating blood spots samples for 1 h at room temperature with a solution of NaCl 0.9% (95 μl) and 10 mM ferric citrate (20 μl) and centrifugated at 3,500 × g for 10 min.
The iron-saturated serum proteins were diluted five times with water and applied to a hydrated immobiline gel (PAG plate pH 4–6.5; GE Healthcare) and separated in a Multiphore II system (GE Healthcare). Transferrin isoforms were detected after immunofixation with rabbit anti-human transferrin antibody (Dako, Germany) and Coomassie blue staining (Jaeken et al. 1984; Stibler and Jaeken 1990). The relative amounts of the transferrin isoforms were determined and quantified using the Image J 1.42q Software (Wayne Rasband National Institutes of Health, USA).
Enzymatic Analysis
Phosphomannomutase (PMM) (EC 5.4.2.8) and phosphomannose isomerase (MPI) (EC 5.3.1.8) activities were measured in leukocytes and fibroblast. The PMM and MPI activities were performed according to a procedure developed by van Schaftingen and Jaeken (1995).
High-Performance Liquid Chromatography of Transferrin
High-performance liquid chromatography of transferrin (Tf-HPLC) analysis was based on the method described by Helander et al. (2003).The HPLC system consisted of an Agilent 1100 Series liquid chromatography. Separation of the transferrin glycoforms was performed on a SOURCE 15Q PE 4.6/100 anion-exchange chromatography column (GE Healthcare) at 25°C by linear salt gradient elution at a flow rate of 1.0 ml/min. Quantification of the transferrin glycoforms relied on the selective absorbance of the iron–transferrin complex at 470 nm. The relative amount of each transferrin isoform was expressed as a percentage of the area under the curve (%AUC) (Helander et al. 2003; Quintana et al. 2009).
Intact transferrin (immunopurified as described in Sturiale et al. 2008) and its relative N-glycan pool, released by peptide-N-glycosidase F [PNGase F (EC 3.5.1.52)] treatment, were both analyzed by MALDI MS on a Voyager STR instrument (Applied Biosystems, Framingham, MA) using sinapinic acid as matrix for native protein analysis, and 2′, 4′, 6′-trihydroxyacetophenone (THAP) for glycan profiling (Sturiale et al. 2008).
Lipid-Linked Oligosaccharide Analysis
Lipid-linked oligosaccharide (LLO) analysis was measured in fibroblasts after metabolic labeling. They were size-fractionated and analyzed by HPLC. Oligosaccharides linked to dolichol were released, extracted, and analyzed by HPLC essentially as described by Denecke et al. (2005).
Molecular Studies
Genomic DNA was obtained from whole blood or fibroblasts using a commercially available kit (Qiagen, Hilden, Germany). Genomic DNA was amplified using the primers for the PMM2, MPI, ALG8, DPAGT1, and SRD5A3 genes and analyzed by direct sequencing in an AB3130 system (Applied Biosystems) (Matthijs et al. 1997, 1998; Schollen et al. 2000, 2004; Wu et al. 2003; Cantagrel et al. 2010).
Results
Biochemical Studies
Metabolic screening (amino acids, urinary organic acids, ammonia, lactate, blood pH, and very long-chain fatty acids) was normal. We found an increased serum activity of some lysosomal enzymes. Patient 1 has high levels of arylsulphatase A (EC 3.1.6.8) (53.87 nmol/h/ml) (NV: 9–30 nmol/h/ml), and patient 2 showed very high levels of β-hexosaminidase (EC 3.2.1.52) (1686,8 nmol/h/ml) (VN: 140–600 nmol/h/ml); however, they had normal levels of β-glucuronidase (EC 3.2.1.31).
During a temporary and transient hepatic problem (altered urine organic acids and plasma amino acids consistent with mild liver dysfunction), we found slightly increased levels of serum galactose, with normal enzymatic activities of galactose-1-phosphate-uridyltransferase [GALT (EC 2.7.7.12)] and galactokinase (EC 2.7.1.6), excluding classic galactosemia and galactokinase deficiency in patient 1. This increase was not observed in subsequent determinations of galactose levels in dry blood samples. Moreover, gas chromatographic analysis of galactitol and galactose in urine was normal.
Transferrin Analysis
Western blot of two serum glycoproteins (transferrin and haptoglobin) showed an altered profile similar to the CDG Type I pattern (Fig. 2b). Furthermore, Tf-IEF also showed a type I transferrin pattern in both patients (Fig. 2a), with increased asialo- and disialotransferrin isoforms and decreased tetrasialotransferrin (14% asialo-, 17% disialo-, and 25% tetrasialotransferrin; reference values: n/d, 3.8% and 46% for asialo-, disialo-, and tetrasialotransferrin, n: 24). IEF of serum Tf after neuraminidase treatment showed only one band excluding a Tf protein variant. The abnormal Tf-IEF pattern was confirmed by HPLC and MALDI-TOF MS in patient 1. She presented a clear increase in asialo- and disialotransferrin peaks: 1.43% and 3.89%, respectively [reference values: disialotransferrin: 0.28% and not detected for asialotransferrin in normal controls (n) n: 24 (Fig. 2c)]. Tf analysis by MALDI MS showed an abnormal glycosylation profile due to the presence of additional mono-glycosylated (~77.4 kDa) and a-glycosylated (~75.2 kDa) isoforms (Fig. 2d).

(a) Transferrin IEF pattern of two CDG-Ix patients: P1 serum of patient 1, P2 dried blood spot samples of patient 2, CDG Ia control of a PMM2-CDG patient, and NC a healthy control. The arrows on the right indicate tetrasialo, disialo, and asialotransferrin, carrying two, one, or no oligosaccharide chains. The comparison of transferrin isoform values (%) from patient 1 with healthy control showed an increase in the cathodic fractions (asialo- and disialo-) and a decrease in tetrasialo- of the patient respect to the healthy control. (b) SDS-PAGE profile of serum proteins from patient 1 in comparison with a healthy control (NC) and a CDG type I patient. The arrows indicate the increased bands corresponding to abnormal sialotransferrins. (c) High-performance liquid chromatography (HPLC) profile of serum transferrin of patient 1 (at the top) and of a healthy control (NC at the bottom) shows the presence of underglycosylated isoforms in the patient. The arrows indicate the increased isoforms corresponding to asialo- and disialotrasferrin. (d) MALDI-TOF mass spectrum of serum transferrin from P1 (on the right) and from a healthy control (on the left). Patient 1 showed abnormal isoforms corresponding to mono-glycosylated and a-glycosylated species
PMM2-CDG and MPI-CDG (CDG–Ib, OMIM 602579) were excluded by enzymatic analysis of leukocytes and cultured skin fibroblasts and by direct sequence analysis of PMM2 and MPI genes.
Subsequent LLO analysis of fibroblasts of patient 1 showed no abnormal accumulation of any intermediate precursor.
DNA from patient 1 has further been analyzed to identify mutations by direct gene sequencing in the context of ALG8-CDG, DPAGT1-CDG, and SRD5A3-CDG (HGNC: 23161, 2995, and 25812, respectively). No defect in those genes could be identified in this patient.
Discussion
Since glycosylation of proteins occurs in all cell types, symptoms of a glycosylation deficiency are seen in multiple organs. Previous reports indicated that the diagnosis of CDG should be considered in each patient with hypotonia, dysmorphic features, and developmental delay accompanied by cataract (Morava et al. 2008). Our patients display multisystem symptoms; however, they also showed some characteristics that are not commonly seen in these pathologies. Interestingly, during a transient hepatic problem, patient 1 presented altered urine organic acids and plasma amino acids due to a mild liver dysfunction and a slightly increased serum galactose that became normal, with normal enzymes activities. The abnormalities in the lysosomal enzymes of these patients are compatible with the protein glycosylation defects that they have, which have been described in CDG patients.
In different reviews, CDG-I patients show ophthalmological problems. Common findings are bilateral cataract, glaucoma, and optic nerve atrophy. Infantile cataract might occur in various inborn errors of metabolism including galactosemia, peroxisomal disorders, and mitochondrial disorders. Cataract has been described in PMM2-CDG adult patients, but in children it is a rare clinical finding only described in a few ALG 8-CDG (CDG-Ih), ALG2-CDG (CDG-Ii), and in CDG-Ix patients (Thiel et al. 2003; Eklund et al. 2005; Morava et al. 2008); nevertheless, cataract is an important symptom in SRD5A3-CDG (Cantagrel et al. 2010 and Kahrizi et al. 2011).
In our patients, cataracts were present very early in life; the ophthalmological examination revealed that they had affected both eyes with opacities in the central portion of the lens as well as in the lens cortex (nuclear-cortical cataracts).
At present, patient 1 presents a severe phenotype and receives anti-epileptic medication, weekly physiotherapy, and assisted therapy, as well. She has made slight progress in her motor development, muscle tone, and social interaction, and continues to have involuntary movements of the head and the upper limbs. She has visual fixation and although she seems to recognize familiar faces, her communication is limited to undifferentiated vocalization.
As we can see from the literature that the severity of the disease has no correlation with a CDG I subtype, it seems that the disease course may be influenced by different factors that improve or worsen the patient’s general condition (Morava et al. 2008). The clinical features of our patients were similar including axial hypotonia, psychomotor retardation, dysmorphic features, feeding problems, hepatomegaly, coagulopathy, and recurrent infections. Nevertheless, patient 2 was born with a severe liver involvement and recurrent neonatal infections that lead to an unfortunate early death; however, patient 1 developed a progressive neurological involvement after her first years of life.
Our patients present a similar phenotype to ALG1-CDG but not as severe as them, P1 is now 11 years old and her state is progressing. Compared with other CDG types, the phenotype in ALG1-CDG is very severe, with rapid development of microcephaly, seizures refractory to treatment, progressive stupor, and death in early infancy (Kranz et al. 2004). The hypoglycosylation of liver-derived serum glycoproteins is more profound than in other types of CDG-I. The IEF transferrin pattern in ALG1-CDG patients shows the major transferrin isoform almost missing, but P1 shows a profile that has either one or both carbohydrate side chains missing in about half of the transferrin population.
LLO analysis of fibroblasts did not identify abnormal accumulation of any dolichol-linked oligosaccharide precursors associated with some of the known forms of CDG, ruling out ALG6-CDG, ALG3-CDG, DPM1-CDG, MPDU1-CDG, ALG12-CDG, and ALG2-CDG. In this context, we screened ALG8 and DPAGT1 genes with a role in the assembly of the LLO in ER since a deficiency in those genes would also lead to normal LLO, in contrast to the other deficiencies described for the enzymes along this pathway. Finally, a normal LLO profile has recently been observed in patients with a defect in the polypropenol reductase, necessary for the synthesis of dolichol, due to mutations in the SRD5A3 gene (Cantagrel et al. 2010). SRD5A3-CDG has also been excluded in our patients.
The results observed in our patients suggest studying the defects in dolichol synthesis up to the formation of Dol-PP-GlcNAc in addition to the recently described dolichol defects named DHDDS-CDG (dehydrodolichol diphosphate synthase deficiency) and DK1-CDG (dolichol kinase deficiency).
In conclusion, we could not identify the genetic deficiency in these patients yet. Further tests will be necessary to pinpoint the defect to identify the CDG type.
Abbreviations
- CDG:
-
Congenital disorder of glycosylation
- COG:
-
Oligomeric golgi complex
- HPLC:
-
High-performance liquid chromatography
- IEF:
-
Isoelectrofocusing
- MS:
-
Mass spectrometry
- Tf:
-
Transferrin
References
Artuch R, Ferrer I, Pinedo J et al (2003) Western blotting with diaminobenzidina detection for the diagnosis of congenital disorders of glycosilation. J Neurosci Methods 125:167–171
Babovic-Vuksanovic D, O’Brien J (2007) Laboratory diagnosis of congenital disorders of glycosylation type I by analysis of transferring glycoforms. Mol Diagn Ther 11:303–311
Barone R, Sturiale L, Garozzo D (2009) Mass spectrometry in the characterization of human genetic N-glycosylation defects. Mass Spectrom Rev 28:517–542
Cantagrel V, Lefeber D, Ng BG et al (2010) SRD5A3 is required for converting polyprenol to dolichol and is mutated in a congenital glycosylation disorder. Cell 142(2):203–17
de Lonlay P, Valayannopoulos V, Dupré T, Vuillaumier-Barrot S, Seta N (2008) Congenital disorders of glycosylation (CDG). Arch Pediatr 15:602–5
Denecke J, Kranz C, Von Kleist-Retzow J et al (2005) Congenital disorder of glycosylation type Id: clinical phenotype, molecular analysis, prenatal diagnosis, and glycosylation of fetal proteins. Pediatr Res 2:248–253
Eklund EA, Sun L, Westphal V, Northrop JL, Freeze HH, Scaglia F (2005) Congenital disorder of glycosylation (CDG)-Ih patient with a severe hepato-intestinal phenotype and evolving central nervous system pathology. J Pediatr 147:847–850
Guerrera I, Kleiner O (2005) Application of mass spectrometry in proteomics. Biosci Rep 25:71–93
Helander A, Husa A, Jeppsson J (2003) Improved HPLC method for carbohydrate-deficient transferrin in serum. Clin Chem 49:1881–1890
Jaeken J (2003) Congenital disorders of glycosilation (CDG): it’s all in it! J Inherit Metab Dis 26:99–118
Jaeken J, Vanderschueren-Lodeweyckx M, Casaer P, SnoeckL CL, Eggermont E, Eeckels R (1980) Familial psychomotor retardation with markedly fluctuating serum prolactin, FSH and GH levels, partial TBG deficiency, increased serum arylsulphatase A and increased CSF protein: a new syndrome? Pediatr Res 14:179
Jaeken J, van Eijk HG, van der Heul C, Corbeel L, Eeckels R, Eggermont E (1984) Sialic acid-deficient serum and cerebrospinal fluid transferin in a newly reconized genetic syndrome. Clin Chim Acta 144:245–7
Jaeken J, Hennet T, Freeze HH, Matthijs G (2008) On the nomenclature of congenital disorders of glycosylation (CDG). J Inherit Metab Dis 31:669–672
Jaeken J, Hennet T, Matthijs G, Freeze HH (2009) CDG nomenclature: time for a change! Biochim Biophys Acta 1792:825–826
Kahrizi K, Hao Hu C, Garshasbi M et al (2011) Next generation sequencing in a family with autosomal recessive Kahrizi syndrome (OMIM 612713) reveals a homozygous frameshift mutation in SRD5A3. Eur J Hum Genet 19:115–117
Kranz C, Denecke J, Lehle L et al (2004) Congenital disorder of glycosylation type Ik (CDG-Ik): a defect of mannosyltransferase I. Am J Hum Genet 74:545–551
Lefeber D, Schonberg J, Morava E et al (2009) Deficiency of Dol-P-Man synthase subunit DPM3 bridges the congenital disorders of glycosylation with the dystroglycanopathies. Am J Hum Genet 85:76–86
Marklová E, Albahri Z (2007) Screening and diagnosis of congenital disorders of glycosylation. Clin Chim Acta 385:6–20
Matthijs G, Schollen E, Pardon E, Veiga-da-Cunha M, Jaeken J, Cassiman J-J, Van Schaftingen E (1997) Mutations in a phosphomannomutase gene, PMM2, on chromosome 16 in carbohydrate-deficient glycoprotein type I syndrome (Jaeken syndrome). Nat Genet 16:88–92, erratum: Nat Genet 16:316
Matthijs G, Schollen E, Van Schaftingen E, Cassiman JJ, Jaeken J (1998) Lack of homocygotes for the most frequent disease allele in carbohydrate-deficient glycoprotein syndrome type 1A. Am J Hum Genet 62:542–550
McKenzie F, Fietz M, Fletcher J, Smith R, Wright M, Jaeken J (2007) A previously undescribed form of congenital disorder of glycosylation with variable presentation in siblings: early fetal loss with hydrops fetalis, and infant death with hypoproteinemia. Am J Med Genet A 143A:2029–2034
Morava E, Wosik H, Karteszi J et al (2008) Congenital disorder of glycosylation type Ix: review of clinical spectrum and diagnostic steps. J Inherit Metab Dis 31:450–456
Quintana E, Navarro-Sastre A, Hernández-Pérez J et al (2009) Screening for congenital disorders of glycosylation (CDG): transferrin HPLC versus isoelectric focusing (IEF). Clin Biochem 42:408–15
Reinders J, Lewandrowski U, Moebius J, Wagner Y, Sickmann A (2004) Challenges in mass spectrometry-based proteomics. Proteomics 4:3686–3703
Schollen E, Dorland L, de Koning T et al (2000) Genomic organization of the human phosphomannose isomerase (MPI) gene and mutation analysis in patients with congenital disorders of glycosylation type Ib (CDG-Ib). Hum Mutat 16:247–252
Schollen E, Frank C, Keldermans L et al (2004) Clinical and molecular features of three patients with congenital disorders of glycosylation type Ih (CDG-Ih) (ALG8 deficiency). J Med Genet 41:550–556
Stibler H, Jaeken J (1990) Carbohydrate deficient serum transferrin in a new systemic hereditary syndrome. Arch Dis Child 65:107–111
Sturiale L, Barone R, Fuimara A et al (2005) Hypoglycosylation with increased fucosylation and branching of serum transferrin N-glycans in untrated galactosemia. Glycobiology 15:1268–76
Sturiale L, Barone R, Palmigiano A et al (2008) Multiplexed glycoproteomic analysis of glycosylation disorders by sequential yolk immunoglobulins immunoseparation and MALDI-TOF MS. Proteomics 8:3822–32
Thiel C, Schwarz M, Peng J, Grzmil M, Hasilik M, Braulke T (2003) A new type of congenital disorders of glycosylation (CDG Ii) provides new insights into the early steps of dolichol-linked oligosaccharide biosynthesis. J Biol Chem 278:22498–22505
van Schaftingen E, Jaeken J (1995) Phosphomannomutase deficiency is a cause of carbohydrate-deficient glycoprotein syndrome type I. FEBS Lett 377:318–320
Wu X, Rush JS, Karaoglu D et al (2003) Deficiency of UDP-GlcNAc: dolichol phosphate N-acetylglucosamine-1 phosphate transferase (DPAGT1) causes a novel congenital disorder of glycosylation type Ij. Hum Mutat 2:144–50
Acknowledgments
The authors are grateful to Liesbeth Keldermans from the Center for Human Genetics and Sandra Van Aerschot from the Center for Metabolic Diseases for DNA analysis and capillary zone electrophoresis. The authors would like to thank Dr. Paz Briones, Dr. Rafaél Artuch, and Dr. María Antonia Vilaseca from Barcelona (Spain). At CEMECO, this work was supported by grants from CONICET (PIP6338), FONCyT – BID PICT2350, Catholic University of Córdoba, and Argentinean National Health Ministry.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Appendices
Synopsis
This is a report of the clinical, biochemical, and molecular studies of two siblings characterized as CDG-Ix, identified in the context of a new program for the systematic identification of CDG in Argentina.
Author’s Contributions
MBBM was responsible for preparative studies in terms of isoelectrofocusing, HPLC, CDG enzymatic assays, molecular studies, data gathering, and development of the manuscript.
AD and NA contributed in the biochemical studies.
NG is a physician specialized in metabolic disease who works at CEMECO.
LS and DG contributed in the mass spectrometry analysis.
GM was responsible for the molecular studies and contributed in the development and final draft of the manuscript.
JJ provided specialized opinion in the clinical diagnosis. He contributed in the development and final draft of the manuscript.
RDK provided specialized opinion and was responsible for the evaluation and the follow-up of the patient. She coordinates the research in metabolic diseases in the Center for Study of Congenital Metabolopaties (CEMECO).
CGA is responsible for the research, conceived the idea for the chapter, and contributed to the development and final draft of the manuscript. She coordinated the CDG research program in CEMECO.
Competing Interest Statement
All authors confirm that they have no competing interests for declaration. The authors confirm independence from the sponsors. They did not receive any outside funding or grants in support of their research.
Details of Funding
This work was supported by grants from CONICET (PIP6338), (ANCyT-FONCyT) PICT2350, Catholic University of Córdoba, and Argentinean National Health Ministry. All authors confirm that they have no competing interests for declaration. The authors confirm independence from the sponsors; the content of the chapter has not been influenced by the sponsors.
Details of Ethics Approval
Written consent was obtained from the patients and their parents to participate in this study, and they allow us to submit this manuscript with images for the publication. A copy of written consent is available for review. The study was approved by the Ethics Committee of the Children’s Hospital of Córdoba (CIEIS) Act Nº 95/2007. All studies were carried out in accordance with the Word Medical Association Declaration of Helsinki.
Rights and permissions
Copyright information
© 2011 SSIEM and Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Millón, M.B.B. et al. (2011). Two Argentinean Siblings with CDG-Ix: A Novel Type of Congenital Disorder of Glycosylation?. In: JIMD Reports - Case and Research Reports, 2011/1. JIMD Reports, vol 1. Springer, Berlin, Heidelberg. https://doi.org/10.1007/8904_2011_18
Download citation
DOI: https://doi.org/10.1007/8904_2011_18
Received:
Revised:
Accepted:
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-642-17707-1
Online ISBN: 978-3-642-17708-8
eBook Packages: MedicineMedicine (R0)