
Abstract
We present a fluid-solid-growth (FSG) computational framework to simulate the mechanobiology of the arterial wall. The model utilises a realistic constitutive model that accounts for the structural arrangement of collagen fibres in the medial and adventitial layers, the natural reference configurations in which the collagen fibres are recruited to load bearing and the (normalised) mass-density of the elastinous and collagenous constituents. Growth and remodelling (G&R) of constituents is explicitly linked to mechanical stimuli: computational fluid dynamic analysis produces snapshots of the frictional forces acting on the endothelial cells; a quasi-static structural analysis is employed to quantify the cyclic deformation of the vascular cells. We apply the computational framework to simulate the evolution of a specific vascular pathology: abdominal aortic aneurysm (AAA). Two illustrative models of AAA evolution are presented. Firstly, the degradation of elastin (that is observed to accompany AAA evolution) is prescribed, and secondly, it is linked to low levels of wall shear stress (WSS). In the first example, we predict the development of tortuosity that accompanies AAA enlargement, whilst in the latter, we illustrate that linking elastin degradation to low WSS leads to enlarging fusiform AAAs. We conclude that this computational framework provides the basis for further investigating and elucidating the aetiology of AAA and other vascular diseases. Moreover, it has immediate application to tissue engineering, e.g., aiding the design and optimisation of tissue engineered vascular constructs.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Introduction
Tissue engineering offers the possibility of developing a biological substitute material in vitro with the inherent mechanical, chemical, biological, and morphological properties required in vivo, on an individual patient basis [1]. Computational modelling has an integral role to play in these ambitions. However, for in-silico modelling to realise its potential, i.e. to guide the design and optimisation of tissue engineered constructs, it must accurately represent the underlying mechanobiology. Whilst early modelling attempts are characterised by a substantial distance between computer and bench, the integration of biological experiments and simulation efforts are increasing [2].
The need for improved mechanobiological modelling is not restricted to the domain of tissue engineering, such research is necessary to guide our understanding of human physiological and pathophysiology, e.g. the evolution of vascular diseases. In this chapter, we illustrate a computational model for the evolution of abdominal aortic aneurysm (AAA). It combines a realistic micro-structural model of the arterial wall with computational fluid dynamics and structural analyses to quantify the mechanical environment that acts on the vascular cells. Growth and remodelling algorithms simulate the cells responding to mechanical stimuli and adapting the tissue structure. The model simulates a fusiform abdominal aortic aneurysm that evolves with similar mechanical, biological and morphological properties with those observed in vivo. Whilst in need of further sophistications to more accurately reflect the mechanobiology of the arterial wall, it should be readily recognisable to the reader that this computational framework has substantial potential to be applied to aid the design and optimisation of tissue engineered vascular constructs.
Abdominal Aortic Aneurysm (AAA) is characterised by a bulge in the abdominal aorta. Development is associated with dilation of the arterial wall and the possibility of rupture [3]. Prevalence rates are estimated between 1.3 and 8.9 % in men and between 1.0 and 2.2 % in women [4]. They are more common in subjects who smoke [5] and suffer from hypertension [6]. Between 80 and 90 % of ruptured AAAs will result in death [7]; rupture is responsible for 1–2 % of all deaths in the UK each year [8]. Surgery to repair the AAA is an option; however, it is a high-risk procedure with a 5 % mortality rate [9]. Intervention is recommended when the risk of rupture exceeds the risk of surgery. Statistically, this occurs when the diameter exceeds 5.5 cm [10]. However, such a criterion fails to identify small AAAs with high risk of rupture [11] and large AAAs with low risk [12]. Thus there is a pressing need for improved diagnostic criteria to aid clinical decisions: distinguishing those AAA most at risk of rupture will yield significant improvements in patient healthcare.
To understand the aetiology of AAA requires a thorough understanding of the underlying biological mechanisms that govern arterial growth and remodelling (G&R) in both healthy and diseased states [13]. This is an extremely challenging, multidisciplinary problem: not only is the function of individual components of the system elusive, their interactions remain unclear as well. In fact, modelling the mechanical response of the healthy arterial wall alone poses significant challenges in itself: it is a highly complex integrated structure [14]. However, it is envisaged that models of AAA evolution [15–18] will lead to a greater understanding of the pathogenesis of the disease and may ultimately yield improved criteria for the prediction of rupture.
A pre-requisite to modelling AAA evolution is to model the biomechanics and mechanobiology of the healthy arterial wall. Briefly, the artery, consists of three layers: intima, media and adventitia. The innermost layer is the intima, this consists of a basement membrane and a lining of endothelial cells (ECs). ECs form a permeability barrier between the blood flow, the vessel wall and surrounding tissues, and play a significant role in regulating circulatory functions. An internal elastic laminae separates the intima from the media. The media consists of a network of elastin fibres and (approximately) circumferentially orientated vascular smooth muscle cells (VSMCs) and collagen fibres. The adventitia is an outer sheath with bundles of collagen fibres, maintained by fibroblast cells [19], arranged in helical pitches around the artery. The main load bearing constituents are elastin and collagen. Collagen is considerably stiffer than elastin; however, for a healthy large elastic artery, such as the abdominal aorta, at physiological strains elastin bears most of the load [20]. This is because collagen is tortuous in nature [21, 22] and acts as a protective sheath to prevent excessive deformation of the artery.
The structure of the artery is continuously maintained by vascular cells. The morphology and functionality of the cells are intimately linked to their extracellular mechanical environment. Haemodynamic forces due to the pulsatile blood flow give rise to cyclic stretching of the extra-cellular matrix (ECM), frictional forces acting on the inner layer of the arterial wall, normal hydrostatic pressure and interstitial fluid forces due to the movement of fluid through the ECM. Mechanosensors on the cells convert the mechanical stimuli into chemical signals. Activation of second messengers (molecules that transduce the signals from the mechanoreceptors to the nucleus) follows and leads to an increase in the activity of transcription factors. Binding of the transcriptions factors to the DNA leads to activation of genes that regulate cell proliferation, apoptosis, differentiation, morphology, alignment, migration, and synthesis and secretion of various macromolecules [23]. The living response of the artery acts to ensure it functions in an optimum manner as mechanical demands change. For instance, in response to changes in flow, ECs signal VSMCs to constrict/relax to regulate the diameter of the artery to restore wall shear stress (WSS) to homeostatic levels [24].
Fibroblasts continually maintain the collagen fabric by synthesising structural proteins and matrix degrading enzymes [25]. They work on the collagen, crawling over it and tugging on it in order to compact it into sheets and draw it out into cables [26]. In doing this, the fibroblasts attach the collagen fibres to the ECM in a state of stretch. Each fibroblast can synthesise approximately 3.5 million procollagen molecules per day. However, the amount a fibroblast secretes is regulated: between 10 and 90 % of all procollagen molecules are degraded intracellularly prior to secretion [27]. This provides a mechanism for rapid adaptation of the amount of collagen secreted and thus enables the artery to rapidly adapt in response to altered environmental conditions.
During the evolution of an AAA, it is observed that there is an accompanying loss of elastin [28] due to increased elastolysis [29]. However, the highly nonlinear mechanical response of the collagen implies that the degradation of elastin alone is insufficient to explain the large dilatations that arise as the aneurysm evolves. In fact, the collagen fabric continually remodels: collagen has a half-life of 2 months [30] whereas elastin is a relatively stable protein with a long half-life (approximately 40 years [31]). Consequently, models of AAA evolution must address both the degradation of elastin and the G&R of collagen [15].
Watton et al. [15] proposed the first mathematical model of AAA evolution. The model utilises a realistic structural model for the arterial wall [32] which is adapted to incorporate variables which relate to the normalised mass density (hereon referred to as concentration) of the elastinous and collagenous constituents and the reference configurations in which the collagenous constituents are recruited to load bearing. This enables the G&R of the tissue to be simulated as an aneurysm evolves. A key assumption of the model is that collagen fibres, which are in a continual state of deposition and degradation [30], attach to the artery in a state of stretch, denoted the attachment stretch, which is independent of the current configuration of the tissue. A degradation of elastin is prescribed and differential equations are employed to evolve the reference configurations and concentrations of collagen fibres to maintain the stretch of the collagen to homeostatic levels, i.e. the attachment stretch. The model predicts evolution of AAA mechanical parameters and growth-rates consistent with clinical observations [16]. The G&R framework has subsequently been applied to consider conceptual 1D models of intracranial aneurysm (IA) evolution, i.e. enlarging (and stabilising) cylindrical and spherical membranes [33] and the evolution of saccular IAs of the internal carotid artery [34]. It has been implemented into a novel Fluid-Solid-Growth (FSG) framework to couple the G&R of aneurysmal tissue to local haemodynamic stimuli [35, 36] and applied to model patient-specific IA aneurysm evolution [37].
Watton et al. [15] and Watton and Hill [16] simulated AAA evolution by prescribing the loss of elastinous constituents. However, local aortic hemodynamic conditions may influence the risk for, and the progression of, aneurysm disease [38]. Experimental models of AAA suggest an inverse relationship between WSS and aneurysm expansion [39, 40]. In fact, expansion of AAA is believed to be related to increasing macrophage infiltration secondary to low WSS in the aneurysms recirculation region [41]. Hence, spatial and temporal changes in the WSS distribution could influence the G&R of the tissue and thus the rate at which an AAA enlarges. In this study, we apply a novel FSG framework [35] to model AAA evolution. Furthermore, we sophisticate the G&R formulation (as utilised by all previous published studies with this modelling approach [15, 16, 33–37]) to explicitly link both the growth and remodelling of the collagen fabric to cyclic deformation of the arterial wall. Two novel examples of AAA development are illustrated: firstly, we prescribe the degradation of elastin secondly, we assume that the degradation of elastin is driven by low magnitudes of WSS. In both cases, collagen remodelling and collagen growth are linked to the magnitude of the local cyclic deformation of the arterial wall.
2 Fluid-Solid-Growth Model for Aneurysm Evolution
In this section, we describe our FSG computational framework for modelling AAA evolution. Figure 1 depicts the methodology. The computational modelling cycle begins with a structural analysis of the aneurysm to solve the systolic and diastolic equilibrium deformation fields for given pressure and boundary conditions (Fig. 1i). The structural analysis quantifies the stress and stretch, and the cyclic deformation, of the ECM components and the cells (each of which may have different natural reference configurations). The geometry of the aneurysm is subsequently exported to be prepared for computational fluid dynamics (CFD) analysis (Fig. 1ii): the aneurysm geometry is integrated into a physiological geometrical domain; the domain is automatically meshed; physiological flow rate and pressure boundary conditions are applied. The flow is solved assuming rigid boundaries for the haemodynamic domain. The haemodynamic quantities of interest, for example, WSS, are then exported and interpolated onto the nodes of the structural mesh: each node of the structural mesh contains information regarding the mechanical stimuli obtained from the haemodynamic and structural analyses. G&R algorithms simulate cells responding to the mechanical stimuli and adapting the tissue (Fig. 1iii). Following G&R, the constitutive model of the aneurysmal tissue is updated and the structural analysis is re-executed to calculate the new equilibrium deformation fields. The updated geometry is exported for haemodynamic analysis. The cycle continues and as the tissue adapts an aneurysm evolves. The stages of the FSG framework, i.e. the structural modelling (Fig. 1i), CFD (Fig. 1ii) and G&R methodology (Fig. 1iii) are detailed in the subsequent subsections, i.e. Sects. 2.1, 2.2 and 2.3, respectively. Note that whilst we are considering the development of an aneurysm, we could equivalently apply such a computational framework to simulate the evolving mechanical, biological and morphological properties of a tissue engineered construct. Moreover, if the mechanobiology was clearly understood and accurately modelled, such a computational framework could guide and optimise the design of a tissue-engineered construct.

Fluid-Solid-Growth computational framework for modelling aneurysm evolution consisting of (i) structural analysis, (ii) computational fluid dynamics (CFD) analysis and (iii) G&R algorithms. Further details are provided in Sects. 2.1, 2.2 and 2.3, respectively
2.1 Structural Model of Aneurysm Evolution
A geometric nonlinear membrane theory (see, for example, [42]) is adopted to model the steady deformation of the arterial wall. The unloaded internal abdominal aorta is treated as a thin cylinder of undeformed radius \(R,\) length \(L_1,\) and thickness \(H.\) The thickness of the media \(H_M\) is assumed to be equal to 2/3 the thickness of the arterial wall, i.e. \(H_M = H/3,\) and thus the thickness of the adventitia \(H_A = H/3.\) The artery is subject to a physiological axial pre-stretch \(\lambda_z\) and pressure \(p\) which causes a circumferential stretch \(\lambda.\) A body fitted coordinate system is used to describe the cylindrical membrane with axial and azimuthal Lagrangian coordinates \(\theta_1 \in [0, L_1 ]\) and \(\theta_2 \in [0, 2 \pi R),\) respectively. Formation and development of the aneurysm is assumed to be a consequence of G&R of the material constituents of the artery. The principle of stationary potential energy is the governing equation for the steady deformation of the arterial wall. It requires that the first variation of the total potential energy vanishes,
where \(\delta\Pi_{\rm int}\) represents the variation of the internal potential energy \(\Pi_{\rm int}\) stored in the arterial wall, whilst \(\delta\Pi_{\rm ext}\) is the variation of the external potential energy \(\Pi_{\rm ext}\) caused by the normal pressure that acts on the artery. Appropriate functional forms for the spatially and temporally heterogeneous strain-energy functions (SEFs) for the media \(\Pi_M,\) and the adventitia \(\Pi_A\) must be specified so that \(\delta\Pi_{\rm int}\) can be computed. Details of the theoretical formulation to describe the deformation of the arterial wall and the numerical formulation to solve Eq. (1) can be found in [15]: the equilibrium displacement field is solved by a finite element method coded in FORTRAN 77 [43].
2.1.1 Strain-Energy Functions for Heterogeneous Aneurysmal Tissue
The arterial wall is modelled as two layers. The inner layer models the mechanical response of the media (and intima), with contributions from the elastinous constituents (ground substance, elastin fibres and passive smooth muscle cells) and a double helical pitch of collagen fibres with orientations \(\gamma_{M_p}\) to the azimuthal axis: \(p=+,\, p=-\) denote positively (\(\gamma_{M_+}>0\)) and negatively (\(\gamma_{M_-}<0\)) wound fibres, respectively. The outer layer models the mechanical response of the adventitia, which is considered to have a small elastinous contribution and a double helical pitch of collagen fibres with orientations \(\gamma_{A_p}\) (\(p=\pm\)) to the azimuthal axis. The mechanical response of each layer is modelled as the sum of a neo-Hookean strain energy function (SEF) [44] and a highly nonlinear SEF which represents the mechanical response of the collagen [32]. Spatially and temporally dependent functions are introduced for the concentration of the elastinous and collagenous constituents and the configuration in which the collagen fibres begin to be recruited to load bearing.
Recruitment stretch variables define the factor the tissue must be stretched, relative to the unloaded configuration, in the direction of a collagen fibre for it to begin to bear load. Remodelling the recruitment stretches enables the remodelling of the collagenous fabric during aneurysm evolution to be simulated. Constituent concentration variables define the ratio of the mass density of a constituent at time \(t\) to the mass density at time \(t=0\) and enable the growth/atrophy of the constituent to be simulated. For further details of the theoretical formulation to describe the G&R the interested reader is referred to [33]. The SEFs for the elastinous contributions in the medial and adventitia are multiplied by a normalised spatially and temporally dependent concentration function, denoted \(m^E (\theta_1, \theta_2, t).\) This is employed to prescribe the degradation of the elastinous constituents, where \(m^E (\theta_1,\theta_2,t=0)=1.\) Fields of spatially and temporally dependent fibre recruitment stretch \(\lambda^R_{J_p} (\theta_1, \theta_2, t)\) and concentration \(m^C_{J_p} (\theta_1, \theta_2, t)\) variables are defined throughout the midplane of the arterial wall, where the subindex \(J\) denotes the media \(M\) or the adventitia \(A.\) The fibres within each layer are orientated at an angle of \(\gamma_{J_p}\) to the azimuthal axis, where \(p\) denotes the pitch \(\pm \gamma_{J}\) relative to the azimuthal axis in the unloaded reference configuration. Hence, the SEFs are
where the material parameters for the elastinous constituent are denoted by \(K^E_J,\) whilst \(K^C_J\) and \(A^C\) are parameters that relate to the collagen fabric. The Green-Lagrange (GL) strains of the elastin, i.e. \(E_{11}, E_{22}\) and \(E_{33},\) are defined relative to the unloaded configuration; in the initial cylindrical configuration, these represent strains in the axial, azimuthal and radial directions, respectively. The GL strains in the collagen fibres are denoted by \(E^C_{J_p} (\theta_1, \theta_2, t)\) and are defined relative to the configuration in which the collagen fibres are recruited to load bearing. More specifically, the GL strains \(E^C_{J_p}\) of the collagen fibres are a function of the GL strains of the elastin resolved in the directions of the collagen fibres, denoted \(E_{J_p}.\) Thus,
where \(E^R_{J_p} = [(\lambda^R_{J_p})^2 - 1]/2,\) \(\lambda^R_{J_p}(\theta_1, \theta_2, t)\) are the recruitment stretches and \(E_{J_p} = E_{11} \sin^2 \gamma_{J_p} + E_{22} \cos^2 \gamma_{J_p} + 2E_{12} \sin \gamma_{J_p} \cos \gamma_{J_p}.\)
2.1.2 Geometry, Physiological Data and Material Parameters
The material parameters for the media and the adventitia as well as all other values which serve the basis for our subsequent computation are taken from [16] and are summarized in Table 1. In the systolic configuration, the artery has a radius \(r_s,\) an axial pre-stretch of 1.3, a circumferential stretch of 1.25 and we assume that the elastinous constituents bear 80 % of the load. We assume that the elastinous response of the adventitia is an order of magnitude weaker than that of the media, i.e. \(K^E_A = K^E_M/10\) [32] and specify the ratio of the medial and adventitial collagen material parameters to be \(K_A^C = K_M^C /4.\) The remaining three independent material parameters, namely \(K^E_M, K_M^C, A^C\) are determined so that the SEFs adequately model the mechanical behaviour of the artery (see [16]).
The Attachment Stretch \(\lambda^C_{AT}\)
Collagen fibres are in a continual state of deposition and degradation [30, 45] and they are attached to the artery in a state of stretch. Watton et al. [15] hypothesised that fibroblasts configure the collagen fibres to achieve a maximum stretch \(\lambda^C_{AT}\) during the cardiac cycle and introduced the terminology attachment stretch to denote \(\lambda^C_{AT}.\)
Magnitude of Recruitment Stretches at \(t=0\)
For a cylindrical artery at fixed axial stretch and subject to radial inflation, maximum collagen fibre stretches will occur in the systolic configuration. Hence, the initial values of the recruitment stretches are determined so that the stretch of the medial and adventitial collagen fibres at systole at \(t=0\) equal the attachment stretch, thus
2.1.3 Cyclic Deformations of Aneurysmal Tissue
To quantify the cyclic deformation we determine the geometry of the aneurysm at systolic (\(p=p_{s}\)= 16 kPa) and diastolic pressures (\(p=p_{d}\)= 10.67 kPa) as it evolves. Initially, the artery has a cylindrical configuration. It can be shown (see [15]) that the governing force-balance equation for a cylindrical membrane subject to fixed axial stretch \(\lambda_z\) and radial inflation is
where the first and second terms on the right of (5) correspond to the contribution of load bearing from the elastinous and collagenous constituents, respectively. Equation (5) is derived from the governing variational equation, i.e. (1), the functional form the of the SEFs (2) and considerations of the displacement field for a cylindrical membrane. It can be solved numerically to obtain the radius as a function of the pressure. At \(t=0,\) the diastolic and systolic displacement fields to satsify (1) are known, i.e. the artery is a cylinder of length \(\lambda_z L\) that has radius \(r_{s}\) in the systolic configuration and radius \(r_{d}\) in the diastolic configuration. As the material constituents evolve, the systolic/diastolic deformation fields to satisfy (1) are updated with a Newton-Raphson method using the systolic/diastolic deformation fields from the previous time-step as initial guesses; the positions of the boundaries of the domain (\(\theta_1 = 0, L_1\)) in the systolic/diastolic configuration are held fixed as the aneurysm evolves.
Cyclic Areal Stretch
Let \(E_{11}^D,E_{22}^D,E_{12}^D\) and \(E_{11}^S,E_{22}^S,E_{12}^S\) denote the GL strains, in the diastolic (\(D\)) and systolic (\(S\)) configurations, respectively. The cyclic areal stretch, here denoted \(A^{CS},\) is the ratio of the areal stretch at systole to the areal stretch at diastole, which expressed in terms of the Green-Lagrange strains is:
Biaxial Stretch Index \(\chi^{BSI}\)
In the initial geometrical configuration, the artery is subject to cyclic stretch that is one dimensional, i.e. the cyclic stretch is only in the circumferential direction. As the aneurysm evolves regions of the artery experience biaxial stretching. To characterise the evolution of the cyclic stretch environment, we consider a novel Biaxial Stretch Index (BSI), denoted \(\chi^{BSI}\) where \(0\le \chi^{BSI}\le 1.\) If \(\chi^{BSI}=0,\) the tissue is subject to 1D cyclic stretching whereas if \(\chi^{BSI}=1,\) the tissue is subject to equi-biaxial cyclic stretching. The index \(\chi^{BSI}\) is calculated as follows. The principal stretches \(\lambda_1, \lambda_2\) can be expressed in terms of the GL strains (see [46]), i.e.
where
We then denote the principal stretches in the diastolic configuration as \( \lambda_{\alpha}^D = \lambda_{\alpha}^D (E_{11}^D,E_{22}^D,E_{12}^D, \varphi^D)\) (\(\alpha=1,2\)) where \(\varphi^D = \varphi^D (E_{11}^D,E_{22}^D,E_{12}^D)\) is calculated using (9). We then determine the magnitude of these principal stretches (with orientations defined relative to the diastolic configuration) in the systolic configuration, i.e. \(\lambda_{\alpha}^{DS} = \lambda_{\alpha}^{DS} (E_{11}^S,E_{22}^S,E_{12}^S, \varphi^D).\) The cyclic variation of the (diastolic) principal stretches, denoted \(\lambda^{CS}_\alpha,\) are thus:
Now, let
so that \(\xi_1 +1, \xi_2 +1 \ge 1\) define the factor by which the principal stretches (with orientations defined relative to the diastolic configuration) increase during the cardiac cycle. Note the emphasis on cardiac cycle, i.e. we are not distinguishing between increases that occur between diastole and systole and increases that occur between systole and diastole. This is necessary because in certain locations, principal stretches may decrease as the pressure acting on the arterial wall increases, i.e. \(\lambda^{CS}_\alpha<1\) (\(\alpha=1\) and/or \(\alpha=2\)). In this scenario, the factor by which the principal stretches increase during the cardiac cycle is \((1/\lambda^{CS}_\alpha) >1.\) Hence to determine the factor by which the principal stretches increase during the cardiac cycle we consider \(\max \left(\lambda^{CS}_\alpha, 1/ \lambda^{CS}_\alpha \right).\)
We now define the biaxial stretch index \(\chi^{BSI}\) to be:
Suppose \((\lambda^{CS}_1 -1) = \kappa (\lambda^{CS}_2 -1),\) i.e. the cyclic (linearised) strain in the direction of the first principal stretch is a factor \(\kappa\) greater than the cyclic (linearised) strain in the direction of the second principal stretch. Then it is straightforward to deduce that \(\chi^{BSI} = 1/\kappa.\) This measure quantifies the degree of the biaxial distortion of the tissue (during the cardiac cycle) and is independent of the magnitude of the strains.
2.2 Haemodynamic Modelling
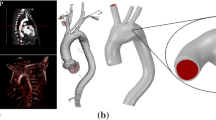
To achieve fully developed flow in the region the aneurysm develops, extensions are attached to the structural model of the artery/aneurysm. The upstream extension is taken to be a cylinder (radius \(r_S\) and length 100 mm) whilst a patient-specific illiac artery bifurcation (obtained from Magnetic Resonance Imaging) is utilised for the downstream extension [47]. Given that the real artery is not perfectly cylindrical, a connecting patch is incorporated to join the geometries together (see Fig. 2). The methodological approach to solve the haemodynamics as the aneurysm evolves proceeds as follows. The geometry of the aneurysmal section is exported (see Fig. 1) from the structural solver to the meshing suite ANSYS ICEM (ANSYS Inc, Canonsburg, PA). ANSYS ICEM automatically integrates the aneurysmal section into the larger geometrical domain, i.e. attaches the upstream and downstream extensions, and generates an unstructured tetrahedral mesh with prism layers lining the boundary in a scripted-automated manner for the fluid domain. After meshing, appropriate boundary conditions are applied (see below) and the flow is solved by ANSYS CFX (ANSYS Inc, Canonsburg, PA) which solves the incompressible Navier-Stokes equations using a finite volume formulation [48, 49]. The solver is based on a coupled approach (i.e. velocities and pressure are cast and solved as a single system) and a fully implicit time discretisation, where needed. An algebraic multigrid variant is used for convergence acceleration [50]. Blood is modelled as a Newtonian fluid [51, 52] with constant density \(\rho = 1,069\,\text{kgm}^{-3}\) [53, 54] and constant viscosity \(\eta = 0.0035\) Pa s. At the arterial wall, no slip, no-flux conditions are applied. We adopted a steady flow analysis to reduce the cost of the computational simulations. The mean flow and pressure boundary conditions are taken from a 1D model of the arterial tree [55] which has been integrated into the software suite @neufuse which was developed for the European project @neuIST [56] (www.aneurist.org: ‘Integrated Biomedical Informatics for the Management of Cerebral Aneurysms’). It solves the 1D form of the Navier-Stokes equation in a distributed model of the human systemic arteries, accounting for the ventricular-vascular interaction and wall viscoelasticity; it was recently validated through a comparison with in vivo flow measurements [57]. A flow rate of \(24 \,\text{cm}^{3} /\text{s}\) is applied at the inlet and the pressure at the illiac arteries is set to 12760 Pa.

A patient-specific iliac bifurcation artery is attached downstream of the AAA structural model; a connecting patch is created to join two geometries together. A cylindrical extension is attached upstream (not depicted)
2.3 Growth and Remodelling
To model AAA evolution, we simulate the G&R of the load bearing constituents of the arterial wall, i.e. elastin and collagen. Remodelling is associated with changes in the natural reference configurations that constituents are recruited to load bearing whereas growth is associated with changes in the mass of the constituents.
2.3.1 Elastin Degradation
To simulate the degradation of elastin that occurs during AAA evolution, two approaches are considered. Firstly, the degradation of elastin is prescribed ([15]) and secondly it is linked to the haemodynamic environment (see [35]).
-
Case (i) Prescribed elastin degradation
The degradation of elastin is prescribed over a representative timescale of 10 years (see [16]). To prescribe the loss of elastin, the following function is used:
$$ m^E (\theta_1, \theta_2, t) = 1- \left(1- m^E_{\rm min} (t) \right) \exp \left[ - \omega_1 \left({{\theta_1}\over {L_{min}}} -1 \right)^2 \right], $$(13)where \(\theta_1, \theta_2\) are the Lagrangian coordinates of the computational domain, i.e. \(\theta_1 \in \left[ 0, L_1 \right] \theta_2 \in \left[ 0, L_2 \right),\) \(m^E_{\rm min}\) represents the minimum concentration of elastin, at time \(t,\) which, by construction, is located at \((\theta_1, \theta_2) = (L_{\rm min},\theta_2).\) The parameter \(\omega_1\) controls the degree of localisation of the degradation function (we adopt \(\omega_1=20,\) see [16] for influence of this parameter). We assume that the minimum concentration decays exponentially. Thus,
$$ m^E_{\rm min} (t) = \exp \{ \ln (m^E_T) (t/T) \} = (m^E_T)^{t/T}, $$(14)where \(T=10,\, 0\le t \le T,\, m^E_{10} = 0.05\) and \(L_{\rm min} = 0.8 L_1.\) Note that at \(t=0,\, m^E_{\rm min} (t=0) =1.\)
-
Case (ii) Linking Elastin degradation to Haemodynamic Environment
Initially, the distribution of the WSS is spatially uniform. To perturb the spatial distribution of haemodynamic stimuli, a localised axisymmetric degradation of elastin is prescribed to create a small axisymmetric AAA. We assume this small aneurysm develops over several years and for numerical illustration we specifically consider a time-scale of 4 years. The functional form utilised for case (i) is adopted, i.e, (13) with \(L_{\rm min} = 0.5 L_1\) and \(m^E_4 =0.5.\) Whilst the elastin is degrading, the aneurysm continues to enlarges in size and the strains in the collagen fabric are elevated above homeostatic values, i.e. \(\lambda^C_{J} >\lambda^C_{AT}\)). We stabilise the aneurysm by switching off the degradation of elastin and allowing sufficient time for the collagen fabric to adapt to a new homeostasis, i.e. \(\lambda^C_{J_p} \rightarrow \lambda^C_{AT}\); numerically a period of 1 year or greater seems sufficient and so for \(4<t \le 5,\) we specify that no further elastin degrades (\(\forall t: 4 < t \le 5, m^E_{\rm min} (t) = m^E_4\)). Subsequent degradation of elastin (for \(t>5\)) is explicitly linked to deviations of haemodynamic stimuli from homeostatic levels (see [35] for further details on methodology). The concentration of elastin \(m^E\) evolves according to
$$ {{\partial m^E}\over {\partial t}} = - {{\fancyscript{F}}}_D D_{\rm max} m^E $$(15)where \(t\) is in years, \(D_{\rm max}\) specifies the maximum rate of degradation, and \({{\fancyscript{F}}}_D (\theta_1, \theta_2, t): 0\le {{\fancyscript{F}}}_D \le 1\) is a spatially-dependent function of the haemodynamic quantities to be linked to elastin degradation. Clearly, if \({{\fancyscript{F}}}_D = 1,\) elastin degrades at a maximum rate whilst if \({{\fancyscript{F}}}_D = 0,\) no degradation occurs.
As an illustrative example, we suppose elastin degradation is linked to low levels of WSS. More specifically, we follow [35] and assume that if the maximum value of the WSS is greater than a critical value, say \(\tau_{\rm crit},\) no degradation of elastin occurs whilst lower values of WSS give rise to degradation of elastin. Furthermore we suppose that there exists a value of WSS, say \(\tau_{X}: 0\le \tau_{X}< \tau_{\rm crit},\) at which maximum degradation occurs. We assume a simple quadratic functional form for the degradation function \({{\fancyscript{F}}}_D\) that describes the relation between the local WSS and the degree of the degradation of elastin, i.e.
$$ {{\fancyscript{F}}}_D (\tau (\theta_1, \theta_2, t)) = \left\{ \begin{array}{ll} 0, & \quad \tau \ge \tau_{\rm crit}, \\ \left({{\tau_{\rm crit} - \tau}\over {\tau_{\rm crit} - \tau_X}} \right)^2, & \quad \tau_X < \tau < \tau_{\rm crit}, \\ 1, & \quad \tau \le \tau_X. \end{array} \right. $$(16)In this study, \(D_{\rm max} = 0.5,\, \tau_{\rm crit} =0.5\) Pa and \(\tau_X = 0\) Pa.
2.3.2 Collagen Adaption
The adaption of the collagen fabric during aneurysm evolution consists of two distinct mechanisms: growth and remodelling.
2.3.3 Remodelling the Recruitment Configuration of Collagen
Evolving the reference configurations that fibres are recruited to load bearing simulates the mechanical consequences of: (i) fibre deposition and degradation in altered configurations as the aneurysm enlarges; (ii) fibroblasts configuring the collagen to achieve a maximum strain during the cardiac cycle, i.e. the attachment strain \(E^C_{AT} = ((\lambda^C_{AT})^2 -1)/2 .\) The recruitment stretches adapt so that the maximum strain in the collagen fibres remodels to \(E^C_{AT}.\) We utilise linear differential equations for the remodelling of the recruitment stretches, i.e.
where \(\alpha = 0.3\, \hbox{years}^{-1},\)
and \(E^C_{J_p} \mid_{\rm sys}\) and \(E^C_{J_p} \mid_{\rm dias}\) denote the magnitude of the collagen strains evaluated in the systolic and diastolic configurations, respectively. Note, [15] assumed that peak strains occur at systolic pressure and thus collagen remodelling is simulated by considering the steady deformations that occur in the systolic configuration. However, it was observed that for a more complex geometry, the fibres may not achieve maximum strains at systolic pressure: towards the distal and proximal ends of the AAA the axial strains increase as the pressure is reduced [15]. Hence, the subtle sophistication for the functional form for the remodelling of the recruitment stretches, i.e. (17).
2.3.4 Growth/Atrophy of the Collagen Fabric
Fibroblasts deposit collagen fibres and secrete proteases to degrade the collagenous material. They adhere to the ECM via specialised cell surface receptors, in particular integrins [58]. The integrins act as stretch sensors [59], transducing mechanical signals to the fibroblast interior, enabling fibroblasts to sense their mechanical environment. We assume that the rate of change of the concentration of the collagenous constituents is dependent on the concentration of fibroblasts \(m^F_{J_p}\) (ratio of the density of the fibroblast cells at time \(t\) to the density at time \(t=0\)) in the arterial wall and rate of synthesis and degradation of collagen. Let \({{\fancyscript{F}}}_S\) and \({{\fancyscript{F}}}_D\) be functions depicting how the rate of collagen synthesis and the secretion of matrix metalloproteases are related to the deformation of the fibroblast cell during a cardiac cycle, respectively. Rates of activity of vascular cells increase with the magnitude of the cyclic deformation [60, 61]. We assume \({\fancyscript F}_S\) and \({\fancyscript F}_D\) to be functions of the maximum strains \(E^F_{J_p} \mid_{\rm max}\) and cyclic areal stretches \(A^{CS}\) of the cells during the cardiac cycle. Under these assumptions, the rate of change of the collagen concentration is
In response to increased stretch, fibroblasts attempt to reduce their stretch and reach a new equilibrium by restructuring their cytoskeleton and ECM contacts, i.e. fibroblasts reconfigure their natural reference configuration. Hence, to simplify the mathematical analysis, we assume that the local (natural) reference configuration of a fibroblast cell is identical to that of the collagen fabric it is maintaining. Hence the GL strain of the fibroblast cell, say \(E^F_{J_p},\) is assumed to be equal to the GL strain of the collagen, i.e. \(E^F_{J_p} \equiv E^C_{J_p}.\) Furthermore, we assume that the concentration of fibroblasts in the arterial tissue is proportional to the concentration of collagenous constituents, i.e. \(m^F_{J_p} = \xi_0 m^C_{J_p}: \xi_0 >0.\) These assumptions imply that the rate of evolution of the collagen fibre concentration \(m^C_{J_p}\) is
where \({{\fancyscript{F}}}_G = \xi_0 ({{\fancyscript{F}}}_S - {{\fancyscript{F}}}_D).\) If \(E^C_{J_p} \mid_{\rm max} = E^C_{AT}\) the collagen fabric is in homeostasis, i.e. the secretion of ECM is balanced by the degradation and there is no change in concentration. Hence, for \(E^C_{J_p} \mid_{\rm max} = E^C_{AT} \) it is required that \({{\fancyscript{F}}}_G (E^C_{J_p}) = 0 .\) The exact functional form of \({{\fancyscript{F}}}_G\) is unknown. However, if the substrate is stretched, a net positive force acts on the cell and signalling to the nucleus results in an up-regulation of ECM protein expression and a down-regulation of collagenase expression. Conversely, relaxation of the substrate can trigger different signals resulting in a reversed pattern of protein expression [59], i.e. down regulation of ECM protein expression and up-regulation of collagenase expression. Although we do not explicitly model protein synthesis and enzymatic degradation, the net result is to stimulate increases/decreases in the ECM. The simplest functional form for \({{\fancyscript{F}}}_G\) that satisfies these requirements is linear,and hence we propose the following evolution equation for the collagen fibre concentration
where \(\beta (A^{CS})\) is a phenomenological growth parameter. For a healthy abdominal aorta, vascular cells experience cyclic stretching with magnitudes of approximately 10 % whilst for older, more collagenous vessels, the magnitudes may decrease to 2 % [62]. We select a functional form for \(\beta\) which increases rates of production if the cyclic stretch environment increases from its initial values for the healthy artery at \(t=0\):
This choice is somewhat arbitrary, however it acts to prevent unrealistically large cyclic deformations that can occur in regions of the tissue as the aneurysm evolves. For the analysis in this paper, we follow [37] and take \(\beta_0 = 0.7\) years\(^{-1},\) \(\beta^{CS}=3.\)
Average fibre concentration
To simplify the presentation of the results, we define the average fibre concentration \(m^C\) of the medial and adventitial layers,where
3 Physiological Growth Model Abdominal Aortic Aneurysm
We briefly review the framework of the model prior to presenting the results. Equation (2) defines the (spatially and temporally heterogeneous) SEFs for the medial and adventitial layers of the aneurysmal tissue. An AAA develops as the material constituents of the artery evolve. The variation of the total potential energy (1) governs the equilibrium displacement field, and is solved by the finite element method [15]; volume meshes of the luminal computational domain are generated and the haemodynamics are solved using finite volumes [35]. In many aneurysms, diameter enlargement is asymmetric, with primarily anterior protrusion; the posterior region is often constrained from radial expansion by the adjacent spinal column [38]. To simulate this, we follow Watton et al. [15] and model the spine as a stiff spring-backed plate [15]. Where the aneurysm wall penetrates the plate, a penalty pressure acts normal to the aneurysm. The effective pressure acting on the membrane, is given by the difference of internal physiological pressure and the penalty pressure. For further details, see [43]. Two models of AAA evolution are illustrated: case (i) prescribed elastin degradation; case (ii) the degradation of elastin is driven by low WSS. In both examples, as the geometry evolves, the collagen fabric adapts (throughout the arterial domain) to restore its strain to the homeostatic value [see (17) and (21)].
3.1 Case (i) Prescribed Elastin Degradation
First we illustrate a model of AAA evolution using a prescribed degradation of elastin. As the aneurysm evolves (see Fig. 3) it can be seen that it develops a preferential anterior bulging and becomes tortuous. Figure 3a illustrates the (prescribed) evolution of elastin concentration. In the central region of the AAA, \(m^E\) reduces to 0.05 whereas in the neck regions it is approximately 1. The average collagen concentration (see Fig. 3b) increases to compensate for loss of load borne by the elastin and the increased load acting on the wall (due to the enlargement of the geometry).

Evolution, at t = 0;2;4;6;8;10 years, of spatial distributions of: prescribed elastin concentration mE (a); average collagen concentration \(m^{C}\) (b); elastin Green-Lagrange strains E11 (c) and E22 (d); medial and adventitial collagen Green-Lagrange strain \(E_{M+}^{C}\) (e) and \(E_{A+}^{C}\) (f); cyclic areal stretch ACS (g) and Biaxial Stretch Index cBSI (h)
Figure 3c and d depict the evolution of the elastin Green-Lagrange (GL) strains \(E_{11}\) and \(E_{22},\) respectively. The strains are defined with respect to the unloaded configuration, consequently they continue to increase with the enlargement of the geometry: at \(t=0,\, E_{11} = 0.345\) and \(E_{22} =.281\) whereas at \(t=10,\) maximum values of \(E_{11} = 5.1\) and \(E_{22}=4.7\) occur in the central region of the aneurysm. Notice that \(E_{11}\) decreases in the proximal section of parent artery as the aneurysm enlarges; the axial expansion of the AAA is accentuated due to the axial retraction of the ends of the artery.
The evolution of the collagen fibre GL strains differs substantially from that of the elastin strains due to the evolution of the reference configurations that the fibres are recruited to load bearing; recall this is achieved by remodelling the fibre recruitment stretches [see (17)] which define the factor the tissue must be stretched in the direction of a fibre (relative to the unloaded reference configuration) for it to begin to bear load. Figure 3e depicts the evolution of the medial collagen GL strain \(E^C_{M+}.\) At \(t=0,\) \(E^C_{M+}\) is constant throughout the domain with a magnitude of the attachment strain, i.e. \(E^C_{M+}=E^C_{AT}=0.073.\) As the AAA enlarges, the collagen strains initially increase (at \(t=4,\) maximum values of 0.14 occur) and then reduce as the aneurysm stabilises in size and the collagen fabric remodels to a material equilibrium, i.e. \(E^C_{M+} \rightarrow E^C_{AT}\) throughout the domain. Similar results are observed for evolution of the adventitial collagen fibre GL strain \(E^C_{A+}\) (Fig. 3f).
Figure 3g illustrates the evolution of the cyclic areal stretch \(A^{CS}.\) At \(t=0,\) \(A^{CS}=1.1\) throughout the domain, i.e. the ratio of systolic to diastolic diameters is 1.1. As the elastin degrades and the collagen, which has greater mechanical nonlinearity, takes over the load bearing, the magnitude of \(A^{CS}\) decreases, i.e. to approximately 1.04 in the AAA region. However, note that \(A^{CS}\) is slightly elevated in the proximal and distal necks of the aneurysm where \(A^{CS} \approx 1.2.\) Of note, the cyclic areal stretch also decreases in the proximal section of healthy, albeit tortuous artery. This is a consequence of maximum axial strains occurring in the diastolic configuration in this section of artery, i.e. the axial strains decrease between diastole and systole. Figure 3h illustrates the evolution of the biaxial stretch index \(\chi^{BSI}.\) Initially, \(\chi^{BSI}=0\) throughout the domain: this indicates that the tissue is subject to 1D cyclic deformation (cyclic circumferential stretching). As the AAA enlarges, the distribution becomes more complex. At \(t=10,\) the anterior region of the AAA has \(\chi^{BSI} \approx 0.6\) and two regions around the aneurysm are subject to equi-biaxial stretching (\(\chi^{BSI} \approx 1\)). A more complex distribution of \(\chi^{BSI}\) evolves in the proximal section of artery due its tortuosity.
3.2 Case (ii) Elastin Degradation Linked to Low WSS
In this example, a first-stage prescribed axisymmetric degradation of elastin (with minimum elastin concentration \(m^E=0.5\) at \(t=5\)) gives rise to a small axisymmetric aneurysm. At \(t=5,\) the artery is in mechanical and material equilibrium, i.e. the collagen fabric has equilibrium strains \(E^C_{AT}\) throughout the computational domain. Consequently, if there is no further loss of elastin, there is no further adaption of the collagen fabric [via the evolution equations (17) and (21)] and the aneurysm is stable in size. However, the small prescribed axisymmetric aneurysm creates a perturbation to the WSS field (see Fig. 4a): at \(t=5\) the WSS ranges from 0.02 to 0.1 Pa within the aneurysm with lower values occurring towards the proximal end; a slight elevation of the WSS is observed distal to the aneurysm. Thus, at a second stage, subsequent degradation of elastin can be linked to the haemodynamic environment: for \(t>5,\) elastin degradation is linked to deviations of the WSS from normotensive values. Given the functional relationship between the degradation factor \({{\fancyscript{F}}}_G\) and the magnitude of WSS [see (16)], rates of degradation are elevated towards the proximal end of the aneurysm (see Fig. 4b). Hence the elastin concentration \(m^E,\) which is initially axisymmetric, develops an axial asymmetry (Fig. 4c); minimum value of elastin concentration \(m^E (t=9) =0.08.\) Notably, the aneurysm enlarges more quickly in the upstream direction. Figure 4d illustrates the evolution of the WSSG. As the aneurysm enlarges, elevated values of WSSG are observed in the proximal (200 Pa/m) and distal necks (250 Pa/m).

Evolution of wall shear stress \(\tau\) (a), degradation factor \(F^D\) (b) elastin concentration \(m^E\) (c) and WSSG (d) at \(t=0,5,6,7,8,9\) years
Figure 5a, b illustrate the evolution of the GL strains \(E_{11}\) and \(E_{22}\) of the elastin, respectively. The strains increase from \(E_{11} = 0.345\) and \(E_{22} =.281\) at \(t=0\) to \(E_{11} = 2.5\) and \(E_{22}=2.5\) at \(t=9.\) Notice that there is a slight asymmetry in the spatial distribution within the aneurysm, slightly larger magnitudes are observed towards the proximal end. Also note that as with case (i), the axial strains in the parent artery decrease as the aneurysm enlarges, i.e. the axial retraction of the ends of the artery assists the axial expansion of the AAA. Figure 5c illustrates the evolution of the GL strains of positively wound collagen fibres in the media \(E^C_{M_+}.\) At \(t=0,\) the collagen fabric is in material equilibrium, i.e. \(E^C_{J_p} = E^C_{AT}\) \((J=M,A; p =\pm)\) throughout the domain. Note that at \(t=5,\) even though the geometry has changed (and the elastin strains have increased) the collagen fabric is in homeostasis, i.e. \(E^C_{J_p} = E^C_{AT}\); the recruitment stretches and fibre concentration have evolved to restore material equilibrium in the collagen fabric. As the elastin degrades and aneurysm enlarges (\(t>5\)), the magnitude of the collagen strains increase; notice though that the increases are small relative to the large deformation that is occurring—a consequence of the remodelling of the reference configurations that the fibres are recruited to load bearing. The average collagen concentration \(m^C\) increases to compensate for the loss of load borne by the elastin (Fig. 5d).

Evolution of Elastin Green-Lagrange strains \(E_{11}\) (a) and \(E_{22}\) (b), medial collagen Green-Lagrange strain \(E^C_{M+}\) (c), average collagen concentration \(m^C\) (d), cyclic areal stretch \(A^{CS}\) (e) and the Biaxial Stretch Index \(\chi^{BSI}\) (f) at \(t=0,5,6,7,8,9\) years
Lastly, we consider the evolution of the cyclic stretch environment. Initially the cyclic areal stretch is equal to 1.1 throughout the domain. At \(t=5\) slightly elevated cyclic areal stretches (\(A^{CS}=1.12\)) are present in the proximal and distal necks of the aneurysm (see Fig. 5e). As the elastin degrades and the collagen takes over the load bearing, the cyclic areal stretch reduces to 1.04 within the aneurysm region and elevated values are observed in the aneurysm neck (1.08). The cyclic stretch environment evolves from uniaxial (\(t=0\)) to almost equi-biaxial cyclic stretching, i.e. in the aneurysm region \(\chi^{BSI}=0.7\) (see Fig. 5f). Transition regions occur in the upstream and downstream necks where the cyclic stretch environment changes from almost uniaxial (\(\chi^{BSI}=0.03\)) to equi-biaxial (\(\chi^{BSI}=0.83\)). As with case (i), as the aneurysm evolves, the proximal and distal regions of the artery experience biaxial stretching. This is a consequence of proximal/distal regions of the artery developing cyclic axial strain due to the axial expansion of the aneurysm during the cardiac cycle.
4 Discussion
We have presented a fluid-solid-growth (FSG) model for evolution of AAA. The model of the arterial wall accounts for the structural arrangement of collagen fibres in the medial and adventitial layers, the natural reference configuration that collagen fibres are recruited to load bearing and the concentrations (normalised mass-densities) of the load bearing constituents. To simulate the development of AAA we adopted two approaches: (i) we prescribed an axisymmetric degradation of elastin; (ii) we linked degradation of elastin to local haemodynamic stimuli, i.e. low WSS. In both examples, as the elastin degrades, the collagen fabric adapts (via G&R) to restore its strain to the attachment strain \(E^C_{AT}.\) The reference configurations of the collagen fibres evolve to simulate the effect of fibre deposition (with fibres attaching in a state of strain \(E^C_{AT})\) and fibre degradation in altered configurations as the aneurysm enlarges; this simulates remodelling. The concentration of collagen fibres evolves to compensate for the loss of load borne by the elastin; this simulates growth. In the first example, we illustrated a AAA that stabilises in size and develops tortuosity; to our knowledge, this is the first model of AAA evolution to predict the formation of tortuosity. In the second example, we illustrated that linking elastin degradation to low WSS predicts the evolution of enlarging fusiform aneurysms.
Computational fluid dynamic (CFD) studies of aneurysms often emphasise the role of WSS (or WSSG) on pathogenesis of the disease, sometimes extrapolating conclusions from other conditions, namely atherosclerosis However, in vivo, vascular cells are also subject to cyclic stretching due to the pulsatility of the blood pressure. Cell functionality [63] and vascular homeostasis [64, 65] are influenced by cyclic stretching. Hence, to address the G&R of the tissue that occurs during aneurysm evolution, in addition to quantifying the haemodynamic stimuli that act on the ECs, it is important to quantify the cyclic stretch environment of the vascular cells. In this paper we propose a novel FSG computational framework: rigid walls for the purpose of the CFD analysis combined with a quasi-static analysis to determine the cyclic deformation of the arterial wall. Moreover, we extended the existing G&R framework utilised to model AAA evolution [15, 16] and IA evolution [33–37, 66, 67] to link both growth and remodelling to cyclic deformation of vascular cells.
Increased tortuosity is associated with disease or vessel ageing [68]. Expanding AAAs develop specific, quantifiable shapes that can be expressed as a quantitative tortuosity index that may be relevant to their natural history [69]. Moreover, it is suggested that centreline tortuosity may become a useful addition to maximum diameter in the decision-making process of AAA treatment [70]. In fact, Shum et al. [71] analysed the tortuosity of 10 ruptured and 66 unruptured AAAs and observed that the mean tortuosity of the ruptured aneurysms (\(1.33\pm 0.12\)) was greater than the mean tortuosity of the unruptured aneurysms (\(1.23\pm 0.11\)). The mechanism for generating tortuosity is incompletely understood [72]. However, it appears to be partly attributed to failure of elastin: in an experimental study on canine carotid arteries, Dobrin et al. [73] observed that degradation of elastin caused aneurysmal dilatation and a marked decrease in longitudinal retractive force which permitted the development of tortuosity; failure of collagen causes vessels to rupture but it was observed not to facilitate the development of tortuosity. These findings are consistent with our computational model: we observed that elastin degradation gave rise to aneurysmal expansion and a reduced axial stretch of the proximal section of artery; coupling with a model of spinal contact and collagen G&R, tortuosity naturally develops. Hence, we suggest tortuosity is influenced by several factors: elastin degradation, remodelling of collagen and interaction with perivascular structures.
The structural arrangement of the cells is influenced by their local mechanical environment. For instance, ECs align with the mean direction of flow and if they are grown on a deformable substrate and subjected to cyclic uniaxial stretching, they reorient away from the stretching direction [74, 75]; in fact, it is observed that the alignment of ECs subjected to WSS appears to be significantly enhanced by the addition of cyclic stretching [76]. VSMCs align in the direction of cyclic stretching and it is observed that in 2D monolayers they align perpendicular to the applied haemodynamic WSS [77]. Fibroblasts align perpendicular to the direction of interstitial flow [78] and their orientation may be influenced by the direction of maximum principal stress or strain [79] or cyclic stretch [80]. Recently, a novel parameter was proposed to characterise the biaxial cyclic stretch environment [36], i.e. the biaxial stretch index \(\chi^{BSI}\): \(\chi^{BSI}=0\) denotes 1D cyclic stretch and \(\chi^{BSI}=1\) denotes equi-biaxial stretching. In this study, it was observed that as the AAA evolved, the BSI distribution changed significantly: within the aneurysm, the tissue experiences almost equi-biaxial cyclic stretching; however, in certain regions of the tissue, e.g. the necks, rapid transition regions were observed. If it is assumed that the AAA should evolve to achieve a particular distribution of \(\chi^{BSI}\) then analysing the evolving distributions of \(\chi^{BSI}\) could be used to test competing hypotheses for remodelling of cell/fibre alignment and dispersion. Elucidating the relationship between the mechanical environment of vascular cells and their orientation and functionality will pave the way for more sophisticated computational models.
Following Watton et al. [15] we do not explicitly remodel the collagen fibre orientations. However, it is important to appreciate that the collagen fibre orientations are defined relative to the undeformed reference configuration. Consequently, the fibres reorientate in the loaded configuration towards directions of increasing principal stretch as the aneurysm evolves; Zeinali-Davarani et al [17] use an identical approach, i.e. in the vivo configuration, newly deposited collagen fibres align along the direction of the exisiting collagen fibres. There is some justification for this approach. Fibroblasts crawl along the existing ECM matrix thus the orientation in which they deposit and degrade collagen is partly dependent on the existing ECM structure [81]. However, given that as an AAA evolves, the collagen fabric plays an increasingly important load bearing role, it is important to simulate how the collagen structure adapts to predict stress distributions. Implementing more sophisticated models to represent the remodelling of fibre alignment [82–84], collagen fibril distribution [85], fibre dispersion [86] or proteoglycan cross-bridges [87] may prove useful in this respect. Furthermore, given that VSMCs secrete connective tissue and matrix degrading enzymes [88] and are subject to apoptosis during AAA evolution [89], explicitly modelling the VSM cells with a suitable constitutive model [90–92] is needed to better understand the aetiology of AAA evolution.
We modelled the abdominal aorta as a cylindrical nonlinearly elastic membrane subject to an axial pre-stretch and uniform internal pressure. The distal and proximal ends of the abdominal aorta are fixed to simulate vascular tethering by the renal and iliac arteries. Formation and development of AAA is assumed to be a consequence of the material constituents of the artery remodelling. At physiological pressures the radius of a typical abdominal aorta is approximately 10 mm and the thickness is 1 mm. Neglecting thrombus formation, the ratio of the thickness of the wall to the diameter of the AAA will decrease as the aneurysm enlarges, therefore the deformation of the three dimensional arterial wall of the developing aneurysm is closely related to the deformation of its midplane. The residual strain that is present in the unloaded configuration gives rise to an approximately uniform strain field through the thickness of the arterial wall at physiological pressures. If it is assumed that the physiological mechanism by which collagen fibres attach to the artery is independent of both the current configuration of the artery and the radial position in the arterial wall, then the remodelling process may naturally maintain a uniform strain field (in the collagen fibres) through the thickness of the arterial wall as the AAA develops. These considerations thus support the suitability of a membrane model to model the development of a AAA at physiological pressures. Nevertheless, the G&R framework of Watton et al. [15] has recently been extended to consider transmural variations of G&R for a thick-walled model of the artery [66, 67]: the influence of transmural variations in biochemomechanical stimuli on G&R and AAA evolution will be explored in future studies.
We modelled the healthy abdominal aorta as cylindrical. However, in reality the abdominal aorta is slightly tortuous and tapers. Although, most aneurysm evolution models to date have used conceptual geometrical models for the healthy artery, recently, Zeinali-Davarani et al. [17] applied a stress-mediated constrained mixture FEM approach [93] to model AAA evolution [18] using patient-specific geometries with a nonlinear membrane formulation [65, 94]. Modelling the exact geometry of the healthy abdominal aorta would yield physiologically realistic spatial distributions of haemodynamic stimuli and thus may enable more accurate prediction of the evolution of AAA geometries. However, given that the abdominal aorta is almost cylindrical, hypotheses can be explored and insight obtained with (simpler) non-patient specific geometries. For instance, we observed that the linking elastin degradation to low WSS gives rise to enlarging fusiform aneurysms.
We adopted a steady flow analysis to reduce the cost of the computational simulations. The similarity of the spatial WSS/WSSG distributions for steady and pulsatile flow [95–97] implies that this is a reasonable approach for the purposes of investigating phenomenological hypotheses that explore the link between G&R and deviations of the WSS/WSSG from homeostatic levels [36]. However, temporal changes in WSS distribution during the cardiac cycle affect the functionality of ECs: oscillatory shear is elevated in regions of disturbed flow and is associated with proatherogenic patterns of gene expression [98, 99]. A pulsatile flow analysis yields additional quantification of the haemodynamic stimuli that act on ECs and thus is necessary to explore more sophisticated G&R hypotheses related to the haemodynamics.
Approximately 75 % of AAAs have an associated intraluminal thrombus (ILT) [100]. ILT alters the stress distribution and reduces peak wall stress in AAA. However, presence of ILT leads to regional wall weakening [101]: platelet activation, fibrin formation, binding of plasminogen and its activators, and trapping of erythrocytes and neutrophils, leads to oxidative and proteolytic injury of the arterial wall [102]. Consequently, ILT must play an important role in the aetiology of AAA and thus the biochemomechanical roles of ILT must be understood and modelled better [103]. However, incorporating a model for ILT evolution is challenging; it would require integration of a model for thrombus evolution [104–106] combined with a constitutive model describing its mechanical response [107–109]; this may merit investigation with conceptual mathematical models with simpler geometries first. We note also that our model does not include calcification of the arterial wall which are often present in AAAs [110]. Calcifications influence stress distributions [111] and thus may influence G&R and AAA evolution [112].
5 Conclusion
In order to sophisticate computational models to more accurately represent mechanobiology, guidance is needed from experiments. In return, computational models assist in the interpretation of experimental data and in the identification of questions that need to be addressed by experiments. Moreover, they can play a vital role in guiding our understanding of mechanobiology: they serve as an in silico testbed for exploration of hypotheses; enable underlying mechanisms to be evaluated with incomplete data sets and yield insight which would be impossible from in vitro/vivo experimental set-up alone. We have extended the FSG model proposed by [35] to link both growth and remodelling to cyclic deformation of vascular cells and applied it to simulate the evolution of abdominal aortic aneurysm. The model predicts an aneurysm that evolves with similar mechanical, biological and morphological properties with those observed in vivo. Whilst in need of further sophistications to more accurately reflect the underlying mechanobiology, this computational framework has clear potential to be applied to aid the design and optimisation of tissue engineered vascular constructs.
References
Barron, V., Lyons, E., Stenson-Cox, C., McHugh, P.E., Pandit, A.: Bioreactors for cardiovascular cell and tissue growth: a review. Ann. Biomed. Eng. 31, 1017–1030 (2003)
Geris, L.: In vivo, in vitro, in silico: computational tools for product and process design in tissue engineering. Springer, Heidelberg (2012)
Vorp, D.A.: Review: Biomechanics of abdominal aortic aneurysm. J. Biomech. 40, 1887–1902 (2007)
Sakalihasan, N., Kuivaniemi, H., Nusgens, B., Durieux, R., Defraigne, J.G.: Aneurysm: epidemiology aetiology and pathophysiology, biomechanics and mechanobiology of aneurysms. Springer, Heidelberg (2011)
Baxter, B.T., Terrin, M.C., Dalman, R.L.: Medical management of small abdominal aortic aneurysms. Circulation 117, 1883 (2008)
Davies, M.J.: Aortic aneurysm formation: lessons from human studies and experimental models. Circulation 98, 193–195 (1998)
Wilmink, W.B.M., Quick, C.R.G., Hubbard, C.S., Day, N.E.: The influence of screening on the incidence of ruptured abdominal aortic aneurysms. J. Vasc. Surg. 30, 203–208 (1999)
Carrell, T.W.G., Smith, A., Burnand, K.G.: Experimental techniques and models in the study of the development and treatment of abdominal aortic aneurysms. J. Surg. 86, 305–312 (1999)
Raghavan, M.L, Vorp, D.A.: Toward a biomechanical tool to evaluate rupture potential of abdominal aortic aneurysm: identification of a finite strain constitutive model and evaluation of its applicability. J. Biomech. 33, 475–482 (2000)
Powell, J.T, Brady, A.R.: Detection, management and prospects for the medical treatment of small abdominal aortic aneurysms. Arteriosclerosis Thromb. Vasc. Biol. 24, 241–245 (2004)
Powell, J.T., Gotensparre, S.M., Sweeting, M.J., Brown, L.C., Fowkes, F.G., Thompson, S.G.: Rupture rates of small abdominal aortic aneurysms: a systematic review of the literature. Eur. J. Vasc. Endovasc. Surg. 41, 2–10 (2011)
Darling, R.C., Messina, C.R., Brewster, D.C., Ottinger, L.W.: Autopsy study of unoperated abdominal aortic aneurysms: the case for early resection. Circulation 56, 161–164 (1977)
Humphrey, J.D, Taylor, C.A.: Intracranial and abdominal aortic aneurysms: Similarities, differences, and need for a new class of computational models. Ann. Rev. Biomed. Eng. 10, 221–246 (2008)
O’Connell, M.K., Murthy, S., Phan, S., Xu, C., Buchanan, J., Spilker, R., Dalman, R.L., Zarins, C.K., Denk, W., Taylor, C.A.: The three-dimensional micro- and nanostructure of the aortic medial lamellar unit measured using 3D confocal and electron microscopy imaging. Matrix Biology 27, 171–181 (2008)
Watton, P.N., Hill, N.A., Heil, M.: A mathematical model for the growth of the abdominal aortic aneurysm. Biomech. Model. Mechanobiol. 3, 98–113 (2004)
Watton, P.N., Hill, N.A.: Evolving mechanical properties of a model of abdominal aortic aneurysm. Biomech. Model. Mechanobiol. 8, 25–42 (2009)
Zeinali-Davarani, S., Sheidaei, A., Baek, S.: A finite element model of stress-mediated vascular adaptation: application to abdominal aortic aneurysms. Comput. Methods Biomech. Biomed. Eng. 14, 803–817 (2011)
Sheidaei, A., Hunley, S.C., Zeinali-Davarani, S., Raguin, L.G., Baek, S.: Simulation of abdominal aortic aneurysm growth with updating hemodynamic loads using a realistic geometry. Med. Eng. Phys. 33, 80–88 (2011)
Chiquet, M.: Regulation of extracellular matrix gene expression by mechanical stress. Matrix Biology 18, 417–426 (1999)
Armentano, R., Barra, J., Levenson, J., Simon, A., Pichel, R.: Arterial wall mechanics in conscious dogs: assessment of viscous, inertial and elastic moduli to characterize aortic wall behaviour. Circ. Res. 76, 468–478 (1995)
Shadwick, R.: Mechanical design in arteries. J. Exp. Biology 202, 3305–3313 (1999)
Raghavan, M.L., Webster, M., Vorp, D.A.: Ex-vivo bio-mechanical behavior of AAA: assessment using a new mathematical model. Ann. Biomed. Eng. 24, 573–582 (1999)
Kakisis, J.D., Liapis, C.D., Sumpio, B.E.: Effects of cyclic strain on vascular cells. Endothelium 11, 17–28 (2004)
Gleason, R.L, Humphrey, J.D.: A 2d constrained mixture model for arterial adaptations to large changes in flow, pressure and axial stretch. Math. Med. Biology 22, 347–369 (2005)
Gupta, V., Grande-Allen, K.J.: Effects of static and cyclic loading in regulating extracellular matrix synthesis by cardiovascular cells. Cardiovasc. Res. 72, 375–383 (2006)
Alberts B., Bray D., Lewis J., Raff M., Roberts K., Watson J.D., (1994) Molecular biology of the cell, 4th edn. Garland Publishing, New York
McAnulty, R.J.: Fibroblasts and myofibroblasts: their source, function and role in disease. Int. J. Biochem. Cell Biology 39, 666–671 (2007)
He, C.M, Roach, M.: The composition and mechanical properties of abdominal aortic aneurysms. J. Vasc. Surg. 20, 6–13 (1993)
Shimizu, K., Mitchell, R.N., Libby, P.: Inflammation and cellular immune responses in abdominal aortic aneurysms. Arteriosclerosis Thromb. Vasc. Biology 26, 987–994 (2006)
Nissen, R., Cardinale, G.J., Udenfriend, S.: Increased turnover of arterial collagen in hypertensive rats. Proc. Natl. Acad. Sci. U.S.A. Med. Sci. 75, 451–453 (1978)
Arribas, S.M., Hinek, A., Gonzalez, M.C.: Elastic fibres and vascular structure in hypertension. Pharm. Ther. 111, 771–791 (2006)
Holzapfel, G.A., Gasser, T.C., Ogden, R.W.: A new constitutive framework for arterial wall mechanics and a comparative study of material models. J. Elast. 61, 1–48 (2000)
Watton, P.N., Ventikos, Y., Holzapfel, G.A.: Modelling the growth and stabilisation of cerebral aneurysms. Math. Med. Biology 26, 133–164 (2009)
Watton, P.N, Ventikos, Y.: Modelling evolution of saccular cerebral aneurysms. J. Strain Anal. 44, 375–389 (2009)
Watton, P.N., Raberger, N.B., Holzapfel, G.A., Ventikos, Y.: Coupling the hemodynamic environment to the evolution of cerebral aneurysms: computational framework and numerical examples. ASME J. Biomech. Eng. 131, 101003 (2009)
Watton, P.N., Selimovic, A., Raberger, N.B., Huang, P., Holzapfel, G.A., Ventikos, Y.: Modelling evolution and the evolving mechanical environment of saccular cerebral aneurysms. Biomech. Model. Mechanobiol. 11, 109–132 (2011)
Watton, P.N., Ventikos, Y., Holzapfel, G.A.: Modelling cerebral aneurysm evolution, biomechanics and mechanobiology of aneurysms. Springer, Heidelberg (2011)
Dua, M.M, Dalman, R.L.: Hemodynamic influences on abdominal aortic aneurysm disease: application of biomechanics to aneurysm pathophysiology. Vasc. Pharm. 53, 11–21 (2010)
Nakahashi, T.K., Tsao, K.H.P.S., Sho, E., Sho, M., Karwowski, J.K., Yeh, C., Yang, R.B., Topper, J.N., Dalman, R.L.: Flow loading inducesmacrophage antioxidative gene expression in experimental aneurysms. Arteriosclerosis Thromb. Vasc. Biology 22, 2017–2022 (2002)
Hoshina, K., Sho, E., Sho, M., Nakahashi, T.K., Dalman, R.L.: Wall shear stress and strain modulate experimental aneurysm cellularity. J. Vasc. Surg. 37, 1067–1074 (2003)
Sho, E., Sho, M., Hoshina, K., et al.: Hemodynamic forces regulate mural macrophage infiltration in experimental aortic aneurysms. Exp. Mol. Pathol. 76, 108–116 (2004)
Heil, M.: The stability of cylindrical shells conveying viscous flow. J. Fluids Struct. 10, 173–196 (1996)
Watton, P.N.: Mathematical modelling of the abdominal aortic aneurysm. Ph.D. Thesis, Department of Mathematics, University of Leeds, Leeds, UK (2002)
Watton, P.N., Ventikos, Y., Holzapfel, G.A.: Modelling the mechanical response of elastin for arterial tissue. J. Biomech. 42, 1320–1325 (2009)
Humphrey, J.D.: Remodelling of a collagenous tissue at fixed lengths. J. Biomech. Eng. 121, 591–597 (1999)
Gruttmann, F., Taylor, R.L.: Theory and finite element formulation of rubberlike membrane shells using principal stretches. Int. J. Numer. Methods Eng. 35, 1111–1126 (1992)
Huang, H.: Haemodynamics in diseased arteries: effects on plaque and aneurysm progression by advanced imaging and modelling techniques. Ph.D. Thesis, Department of Engineering Science, University of Oxford, Oxford, UK (2010)
Patakar, S.V.: Numerical heat transfer and fluid flow. Hemisphere Publishing Corporation, Washington – New York – London. McGraw Hill Book Company, New York (1980)
Ferziger J.H, Peric M., (2002) Computational methods for fluid dynamics, 3rd edn. Springer, Heidelberg
Hutchinson, B.R, Raithby, G.D.: A multigrid method based on the additive correction strategy. Numer. Heat Transf. 9, 511–537 (1986)
Cebral, J.R., Castro, M.A., Appanaboyina, S., Putman, C.M., Millan, D., Frangi, A.F.: Efficient pipeline for image-based patient-specific analysis of cerebral aneurysm hemodynamics: technique and sensitivity. IEEE Trans. Med. Imaging 24, 457–467 (2005)
Fisher, C., Rossmann, J.S.: Effect of non-newtonian behavior on hemodynamics of cerebral aneurysms. ASME J. Biomech. Eng. 131, 091004 (2009)
Chatziprodromou, I., Tricoli, A., Poulikakos, D., Ventikos, Y.: Haemodynamics and wall remodelling of a growing cerebral aneurysm: a computational model. J. Biomech. 40, 412–426 (2007)
Oshima, M., Torii, R., Kobayashia, T., Taniguchic, N., Takagid, K.: Finite element simulation of blood flow in the cerebral artery. Comput. Methods Appl. Mech. Eng. 191, 661–671 (2001)
Reymond, P., Merenda, F., Perren, F., Rufenacht, D., Stergiopulos, N.: Validation of a one-dimensional model of the systemic arterial tree. Am. J. Physiol. Heart Circ. Physiol. 297, H208–H222 (2009)
Villa-Uriol M.C., Berti G., Hose D.R., Marzo A., Chiarini A., Penrose J., Pozo J., Schmidt J.G., Singh P., Lycett R., Larrabide I., Frangi A.F.: @neurist complex information processing toolchain for the integrated management of cerebral aneurysms. Interface Focus 1, 308–319 (2011)
Reymond P., Bohraus Y., Perren F., Lazeyras F., Stergiopulos N., (2011) Validation of a patient-specific one-dimensional model of the systemic arterial tree. Am. J. Physiol. Heart Circ. Physiol. 301, H1173–H1182
Wang, J.H.C, Thampaty, B.P.: An introductory review of cell mechanobiology. Biomech. Model. Mechanobiol. 5, 1–16 (2006)
Chiquet, M., Renedo, A.S., Huber, F., Flück, M.: How do fibroblasts translate mechanical signals into changes in extracellular matrix production?. Matrix Biology 22, 73–80 (2003)
Sotoudeh, M., Jalali, S., Usami, S., Shyy, J.Y., Chien, S.: A strain device imposing dynamic and uniform equi-biaxial strain to cultured cells. Ann. Biomed. Eng. 26, 181–189 (1998)
Shin, H.Y., Gerritsen, M.E., Bizios, R.: Regulation of endothelial cell proliferation and apoptosis by cyclic pressure. Ann. Biomed. Eng. 30, 297–304 (2002)
Länne, T., Sonesson, B., Bergqvist, D., Bengtsson, H., Gustafsson, D.: Diameter and compliance in the male human abdominal aorta: influence of age and aortic aneurysm. Eur. J. Vasc. Surg. 6, 178–184 (1992)
Cummins, P.M., von Offenberg Sweeney, N., Killeen, M.T., Birney, Y.A., Redmond, E.M., Cahill, P.A.: Cyclic strain-mediated matrix metalloproteinase regulation within the vascular endothelium: a force to be reckoned with. Am. J. Physiol. Heart Circ. Physiol. 292, H28–H42 (2007)
Hsiai, T.K.: Mechanical transduction coupling between endothelial and smooth muscle cells: role of hemodynamic forces. Am. J. Physiol. Cell Physiol. 294, C695–C661 (2008)
Zeinali-Davarani, S., Raguin, L.G., Baek, S.: An inverse optimization approach toward testing different hypotheses of vascular homeostasis using image-based models. Int. J. Struct. Changes Solids Mech. Appl. 3, 33–34 (2011)
Schmid, H., Watton, P.N., Maurer, M.M., Wimmer, J., Winkler, P., Wang, Y.K., Roehrle, O., Itskov, M.: Impact of transmural heterogeneities on arterial adaptation: application to aneurysm formation. Biomech. Model. Mechanobiol. 9, 295–315 (2010)
Schmid, H., Grytsan, A., Postan, E., Watton, P.N., Itskov, M.: Influence of differing material properties in media and adventitia on arterial adaption: application to aneurysm formation and rupture. Comput. Methods Biomech. Biomed. Eng. (2011). DOI:10.1080/10255842.2011.603309
Rodriguez, Z.M., Kenny, P., Gaynor, L.: Improved characterisation of aortic tortuosity. Med. Eng. Phys. 33, 1712–1719 (2011)
Pappu, S., Dardik, A., Tagare, H., Gusberg, R.J.: Beyond fusiform and saccular: a novel quantitative tortuosity index may help classify aneurysm shape and predict aneurysm rupture potential. Ann. Vasc. Surg. 22, 88–97 (2008)
Georgakarakos E., Ioannou C.V., Kamarianakis Y., Papaharilaou Y., Kostas T., Manousaki E., Katsamouris A.N.: The role of geometric parameters in the prediction of abdominal aortic aneurysm wall stress. Eur. J. Vasc. Endovasc. Surg. 39, 42–48 (2010)
Shum, J., Martufi, G., Di Martino, E., Washington, C.B., Grisafi, J., Muluk, S.C., Finol, E.A.: Quantitiatve assessment of abdominal aortic aneurysm geometry. Ann. Biomed. Eng. 39, 277–286 (2011)
Dougherty, G., Johnson, M.J.: Clinical validation of three-dimensional tortuosity metrics based on the minimum curvature of approximating polynomial splines. Med. Eng. Phys. 30, 190–198 (2008)
Dobrin, P.B., Schwarz, T.H., Baker, W.H.: Mechanisms of arterial and aneurysmal tortuosity. Surgery 104, 568–571 (1998)
Wang, J.H.C., Goldschmidt-Clermont, P., Yin, F.C.P.: Contractility affects stress fiber remodeling and reorientation of endothelial cells subjected to cyclic mechanical stretching. Ann. Biomed. Eng. 28, 1165–1171 (2000)
Owatverot, T.B., Oswald, S.J., Chen, Y., WIllie, J.J.: Effect of combined cyclic stretch and fluid shear stress on endothelial cell morphological responses. ASME J. Biomech. Eng. 127, 374–382 (2005)
Moore, J.E. Jr, Blirki, E., Sucui, A., Zhao, S., Burnier, M., Brunner, H.R., Meister, J.J.: A device for subjecting vascular endothelial cells to both fluid shear stress and circumferential cyclic stretch. Ann. Biomed. Eng. 22, 416–422 (1994)
Lee, A.A., Graham, D.A., Dela Cruz, S., Ratcliffe, A., Karlon, W.J.: Fluid shear stress-induced alignment of cultured vascular smooth muscle cells. J. Biomech. Eng. 124, 37–43 (2002)
Ng, C.P, Schwartz, M.A.: Mechanisms of interstitial flow-induced remodeling of fibroblast collagen cultures. Ann. Biomed. Eng. 34, 446–454 (2006)
Wagenseil, J.E.: Cell orientation influences the biaxial mechanical properties of fibroblast populated collagen vessels. Ann. Biomed. Eng. 32, 720–731 (2004)
Neidlinger-Wilke, C., Grood, E., Claes, L., Brand, R.: Fibroblast orientation to stretch begins within three hours. J. Orthop. Res. 20, 953–956 (2002)
Dallon, J., Sherratt, J.A.: A mathematical model for fibroblast and collagen orientation. Bulletin Math. Biology 60, 101–130 (1998)
Driessen, N.J.B., Wilson, W., Bouten, C.V.C., Baaijens, F.P.T.: A computational model for collagen fibre remodelling in the arterial wall. J. Theor. Biology 226, 53–64 (2004)
Baaijens, F., Bouten, C., Driessen, N.: Modeling collagen remodeling. J. Biomech. 43, 166–175 (2010)
Creane, A., Maher, E., Sultan, S., Hynes, N., Kelly, D.J., Lally, C.: A remodelling metric for angular fibre distributions and its application to diseased carotid bifurcations. Biomech. Model. Mechanobiol. 11(6), 869–882 (2012). doi:10.1007/s10237-011-0358-3
Zulliger, M.A., Fridez, P., Hayashi, K., Stergiopulos, N.: A strain energy function for arteries accounting for wall composition and structure. J. Biomech. 37, 989–1000 (2004)
Gasser, T.C., Ogden, R.W., Holzapfel, G.A.: Hyperelastic modelling of arterial layers with distributed collagen fibre orientations. J. Royal Soc. Interface 3, 15–35 (2006)
Gasser, T.C.: An irreversible constitutive model for fibrous soft biological tissue: a 3-d microfiber approach with demonstrative application to abdominal aortic aneurysms. Acta Biomaterialia 7, 2457–2466 (2011)
Asanuma, K., Magid, R., Johnson, C., Nerem, R.M., Galis, Z.S.: Uniaxial strain regulates matrix-degrading enzymes produced by human vascular smooth muscle cells. Am. J. Physiol. Heart Circ. Physiol. 284, H1778–H1784 (2003)
Zhang, J., Schmidt, J., Ryschich, E., Schumacher, H., Allenberg, J.R.: Increased apoptosis and decreased density of medial smooth muscle cells in human abdominal aortic aneurysms. Chin. Med. J. 116, 1549–1552 (2003)
Zulliger, M.A., Rachev, A., Stergiopulos, N.: A constitutive formulation of arterial mechanics including vascular smooth muscle tone. Am. J. Physiol. Heart Circ. Physiol. 287, H1335–1343 (2004)
Baek, S., Valentín, A., Humphrey, J.D.: Biochemomechanics of cerebral vasospasm and its resolution: II constitutive relastions and model simulations. Ann. Biomed. Eng. 35, 1498–1509 (2007)
Murtada, S., Kroon, M., Holzapfel, G.A.: A calcium-driven mechanochemical model for prediction of force generation in smooth muscle. Biomech. Model. Mechanobiol. 9, 749–762 (2010)
Baek, S., Rajagopal, K.R., Humphrey, J.D.: A theoretical model of enlarging intracranial fusiform aneurysms. J. Biomech. Eng. 128, 142–149 (2006)
Zeinali-Davarani, S., Raguin, L.G., Vorp, D.A., Baek, S.: Identification of in vivo material and geometric parameters of a human aorta: toward patient-specific modeling of abdominal aortic aneurysm. Biomech. Model. Mechanobiol. 10, 689–699 (2010)
Myers, J.G., Moore, J.A., Ojha, M., Johnston, K.W., Ethier, C.R.: Factors influencing blood flow patterns in the human right coronary artery. Ann. Biomed. Eng. 29, 109–120 (2001)
Mantha, A., Karmonik, C., Benndorf, G., Strother, C., Metcalfe, R.: Hemodynamics in a cerebral artery before and after the formation of an aneurysm. Am. J. Neuroradiol. 27, 1113–1118 (2006)
Doenitz, C.M.S.K., Zoephel, R., Brawanski, A.: A mechanism for rapid development of intracranial aneurysms: a case study. Neurosurgery 67, 1213–1221 (2010)
Chien, S.: Mechanotransduction and endothelial cell homeostasis: the wisdom of the cell. Am. J. Physiol. Heart Circ. Physiol. 292, H1209–H1224 (2007)
Chatzizisis, Y.S., Coskun, A.U., Jonas, M., Edelman, E.R., Feldman, C.L., Stone, P.H.: Role of endothelial shear stress in the natural history of coronary atherosclerosis and vascular remodeling: molecular, cellular, and vascular behavior. J. Am. Coll. Cardiol. 49, 2379–2393 (2007)
Wang, D.H.J., Makaroun, M.S., Webster, M.W., Vorp, D.A.: Effect of intraluminal thrombus on wall stress in patient-specific models of abdominal aortic aneurysms. J. Vasc. Surg. 36, 598–604 (2002)
Vorp, D.A., Lee, P.C., Wang, D.H., Makaroun, M.S., Nemoto, E.M., Ogawa, S., Webster, M.W.: Association of intraluminal thrombus in abdominal aortic aneurysm with local hypoxia and wall weakening. J. Vasc. Surg. 34, 291–299 (2001)
Michel, J.B., Martin-Ventura, J.L., Egido, J., Sakalihasan, N., Treska, V., Lindholt, J., Allaire, E., Thorsteinsdottir, U., Cockerill, G., Swedenborg, J.: Novel aspects of the pathogenesis of aneurysms of the abdominal aorta in humans. Cardiovasc. Res. 90, 18–27 (2011)
Humphrey, J.D., Holzapfel, G.A.: Mechanics, mechanobiology, and modeling of human abdominal aorta and aneurysms. J. Biomech. 45(5), 805–814 http://dx.doi.org/10.1016/j.jbiomech.2011.11.021
Xu, Z., Chen, N., Kamocka, M.M., Rosen, E.D., Alber, M.S.: Multiscale model of thrombus development. J. Royal Soc. Interface 5, 705–722 (2008)
Xu, Z., Kamocka, M., Alber, M., Rosen, E.D.: Computational approaches to studying thrombus development. Arteriosclerosis Thromb. Vasc. Biology 31, 500–505 (2011)
Biasetti, J., Hussain, F., Gasser, T.C.: Blood flow and coherent vortices in the normal and aneurysmatic aortas: a fluid dynamical approach to intra-luminal thrombus formation. J. Royal Soc. Interface 8, 1449–1461 (2011)
Vande Geest, J.P., Sacks, M.S., Vorp, D.A.: A planar biaxial constitutive relation for the luminal layer of intra-luminal thrombus in abdominal aortic aneurysms. J. Biomech. 39, 2347–2354 (2006)
van Dam, E.A., Dams, S.D., Peters, G.W.M., Rutten M., C.M., Schurink, G.W.H., Buth, J., van de Vosse, F.N.: Non-linear viscoelastic behavior of abdominal aortic aneurysm thrombus. Biomech. Model. Mechanobiol. 7, 127–137 (2008)
Tong, J., Sommer, G., Regitnig, P., Holzapfel, G.A.: Dissection properties and mechanical strength of tissue components in human carotid bifurcations. Ann. Biomed. Eng. 39, 1703–1719 (2011)
Sakalihasan, N., Michel, J.B.: Functional imaging of atherosclerosis to advance vascular biology. Eur. J. Vasc. Endovasc. Surg. 37, 728–734 (2009)
Speelman, L., Bohra, A., Bosboom, E.M., Schurink, G.W., van de Vosse, F.N., Makaroun, M.S., Vorp, D.A.: Effects of wall calcifications in patient- specific wall stress analyses of abdominal aortic aneurysms. J. Biomech. Eng. 129, 105–109 (2007)
Maier, A., Gee, M.W., Reeps, C., Eckstein, H.H., Wall, W.A.: Impact of calcifications on patient-specific wall stress analysis of abdominal aortic aneurysms. Biomech. Model. Mechanobiol. 9, 511–521 (2010)
Acknowledgments
Paul Watton is funded by The Centre of Excellence in Personalized Healthcare (funded by the Wellcome Trust and EPSRC, grant number WT 088877/Z/09/Z). This support is gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2012 Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Watton, P.N., Huang, H., Ventikos, Y. (2012). Multi-Scale Modelling of Vascular Disease: Abdominal Aortic Aneurysm Evolution. In: Geris, L. (eds) Computational Modeling in Tissue Engineering. Studies in Mechanobiology, Tissue Engineering and Biomaterials, vol 10. Springer, Berlin, Heidelberg. https://doi.org/10.1007/8415_2012_143
Download citation
DOI: https://doi.org/10.1007/8415_2012_143
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-642-32562-5
Online ISBN: 978-3-642-32563-2
eBook Packages: EngineeringEngineering (R0)