
Abstract
Infection with enteropathogenic and enterohaemorrhagic Escherichia coli (EPEC and EHEC), enteroinvasive E. coli (EIEC) and Shigella relies on the elaboration of a type III secretion system (T3SS). Few strains also encode a second T3SS, named ETT2. Through the integration of coordinated intracellular and extracellular cues, the modular T3SS is assembled within the bacterial cell wall, as well as the plasma membrane of the host cell. As such, the T3SS serves as a conduit, allowing the chaperone-regulated translocation of effector proteins directly into the host cytosol to subvert eukaryotic cell processes. Recent technological advances revealed high structural resolution of the T3SS apparatus and how it could be exploited to treat enteric disease. This chapter summarises the current knowledge of the structure and function of the E. coli T3SSs.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

1 Introduction
A healthy gastrointestinal (GI) tract encounters 9 L of fluid per day but only 100–200 ml of fluid is retained in the stools. Certain pathogenic microorganisms can alter the movement of ions and water in the gut towards net secretion, resulting in diarrhoea. With an estimated 2–4 billion incidents per year, acute diarrhoea is a significant contributor to morbidity and mortality worldwide, posing an immense burden on global health (Bern et al. 1992; Hodges and Gill 2010; Kosek et al. 2003; Snyder and Merson 1982). In low- to middle-income countries, diarrhoea is estimated to account for up to 760,000 infant deaths per annum (World Health Organisation), placing infectious diarrhoea within the five most common causes of death in children under the age of five (Liu et al. 2015).
Among bacterial agents of diarrhoeal disease, several pathotypes of Escherichia coli, Salmonella spp., Shigella spp. and Yersinia spp. depend on a specialised macromolecular syringe, the Type III Secretion System (T3SS), to cause disease. Through the T3SS, Gram-negative bacteria deliver virulence factors—also named ‘effector proteins’—into host cells or bacterial competitors. The T3SS—also known as the injectisome—spans the inner and outer bacterial membranes and punctures the host plasma membrane like a syringe to translocate the effector proteins directly from the bacteria into the cytosol of host enterocytes. This enables the pathogen to control host cell signalling pathways, creating an environmental niche in which to thrive. In some pathogenic strains of E. coli, the T3SS is responsible for the translocation of over 25 effector proteins. The roles that these effector proteins play in pathogenesis are discussed in chapter “Modulation of Host Cell Processes by T3SS Effectors”.
The term ‘Type Three Secretion System’ was coined in 1991 following the observation that Yersinia ‘Yop’ proteins were translocated into host cells in a general secretory pathway (Sec)-independent manner. Fewer than 10 years later, the Salmonella T3SS was first visualised using negative staining and electron microscopy (Kubori et al. 1998) soon followed by the E. coli T3SS (Sekiya et al. 2001). This chapter presents the contribution of this remarkable nanomachine to the pathogenesis of certain pathotypes of E. coli and reviews current knowledge on the assembly, regulation, function and importance of the T3SS. Advances in studying individual components and the impact of effector secretion on host cells have revealed fascinating complexity and sophistication to this system, indicating that our understanding of the injectisome during human infection is far from complete.
2 Enteric E. coli and the LEE-Encoded T3SS
Three currently defined pathotypes of E. coli rely on the T3SS and effector proteins to infect the human gut: enteropathogenic E. coli (EPEC), Shiga toxin-producing E. coli (STEC), of which enterohaemorrhagic E. coli (EHEC) is a subtype, and enteroinvasive E. coli (EIEC) (Clements et al. 2011; Gaytán et al. 2016). EPEC-induced diarrheoa primarily affects children under the age of 2 in low-income countries (Chen and Frankel 2005), while EHEC is also capable of causing haemorrhagic colitis and haemolytic uraemic syndrome in infected patients. EIEC shares both its virulence mechanism and clinical symptoms with Shigella spp., and it is believed that current EIEC strains represent an intermediate between E. coli and Shigella spp. The majority of T3SS components and effectors of EIEC and Shigella are encoded on the virulence plasmid pINV, while the injectisomes of EPEC and EHEC along with chaperones, the adhesin intimin, core effector proteins, a lytic transglycosilase and regulatory proteins are encoded within a genomic pathogenicity island termed the Locus of Enterocyte Effacement (LEE) (Pallen et al. 2005). Additionally, typical EPEC strains carry the large virulence EPEC adherence factor (EAF) plasmid, which encodes the bundle forming pilus (BFP) operon as well as the plasmid-encoded regulator (Per) operon, encoding PerA, PerB and PerC, which regulate LEE expression (Gomez-Duarte and Kaper 1995).
The LEE is essential for disease in these pathotypes and comprises 41 conserved genes that allow the T3SS-dependent colonisation of mammalian hosts. Its low G+C content (38% compared to 50% for the whole genome) suggests it was acquired via horizontal gene transfer. LEE islands are also found in the closely related species rabbit EPEC (REPEC), Escherichia albertii (Hyma et al. 2005) and Citrobacter rodentium, with high degrees of similarity both in terms of gene repertoire and organisation (Petty et al. 2011) (Fig. 1). The structural components of the T3SS are encoded on operons LEE1, LEE2, LEE3 and LEE4, except EscD, encoded on its own ORF. They have been demonstrated to be sufficient to assemble a functional injectisome (Ruano-Gallego et al. 2015).

The EPEC Locus of Enterocyte Effacement (LEE). This genomic island is organised into five main operons. It harbours all the necessary genes to assemble a functional T3SS, as well as regulators and core effector proteins. Minor differences are found in the EHEC and C. rodentium LEE
3 Regulation of T3SS Expression
Upon ingestion of contaminated food or water, acid resistance of the pathogens facilitates the survival of the bacteria through the low pH of the stomach (Nguyen and Sperandio 2012), and environmental signals throughout the intestine are sensed to gradually turn on the virulence factors of the pathogens (Connolly et al. 2015; Furniss and Clements 2017). These signals include temperature (Umanski et al. 2002), host signals (De Nisco et al. 2018) and microbiota signals (Carlson-Banning and Sperandio 2018).
At the site of infection, the host hormones adrenaline and noradrenaline and the quorum-sensing molecules auto-inducers 2 and 3 are produced by the gastrointestinal cells and sensed by A/E pathogens to induce the expression of the T3SS (Hughes and Sperandio 2008; Russell et al. 2007; Sperandio et al. 1999). The interplay between the host and microbiome is of significant importance to protect the intestine against infections, as A/E pathogens can sense and take advantage of an unbalanced gut environment. For example, a diet that is poor in fibre causes rapid microbiota-derived degradation of intestinal mucin and thus promotes both a more oxygenic environment and the availability of by-products like the short fatty acids succinate, butyrate and fucose (Desai et al. 2016; Pacheco et al. 2012), all of which activate expression of the T3SS.
Environmental signals are largely integrated by A/E pathogens through sensor kinases (Moreira et al. 2016) including quorum sensing that affects internal specific and global transcriptional regulators: PerC (EPEC)/PchABC (EHEC) or the histone-like nucleoid-structuring protein (H-NS) (Bustamante et al. 2001), among others (Martínez-Santos et al. 2012). The coordinate effects of these signals activate LEE transcription as well as distinct fimbrial and non-fimbrial adhesins that participate in the initial attachment to enterocytes. Most of these signals converge to regulate the transcription of the first gene encoded in operon LEE1: the LEE-encoded regulator (ler) (Bingle et al. 2014; Mellies et al. 1999). Constitutively expressed at low levels, Ler activates the transcription of operons LEE2-LEE5 counteracting the inhibitory effect of H-NS (Bustamante et al. 2001; Elliott et al. 2000; Winardhi et al. 2014), but also functions as a repressor of LEE1 itself (Berdichevsky et al. 2005; Bhat et al. 2014), creating a negative feedback loop. Extra-LEE genes that are known to be regulated by Ler in EPEC include non-LEE-encoded effectors (Nle) and espC (Mellies et al. 2001), which encodes an autotransporter extracellular serine protease that is thought to play various roles in pathogenicity (Navarro-Garcia et al. 2014; Salinger et al. 2009). In contrast, the EHEC homologue of espC, espP, is not Ler-regulated.
In addition to Ler, other LEE-encoded regulators of the T3SS expression include GrlR (the negative regulator) and GrlA (the positive regulator) (Jimenez et al. 2010; Russell et al. 2007). GrlA interacts with the promoter of LEE1 to activate its transcription, however GrlR is able to inhibit this activation by binding directly to GrlA (Padavannil et al. 2013). The action of ClpXP protease on GrlR releases GrlA under T3SS-inducing conditions (Iyoda and Watanabe 2005), but additional posttranslational modifications may be necessary for full activation (Alsharif et al. 2015).
4 Assembly of the T3SS
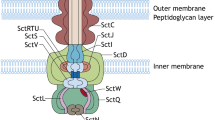
The assembly of the T3SS requires input from transcriptional regulators, chaperones, environmental cues and molecular switches. Though not yet comprehensively understood, T3SS construction can be categorised into four stages: (i) assembly of the basal body and export apparatus, (ii) assembly of the inner rod and needle, (iii) assembly of the filament and translocon and (iv) secretion of effectors. The complete E. coli injectisome is 23 nm long, 8–9 nm wide and is comprised of around 3.5 MDa of protein (Fig. 2). Throughout this chapter, E. coli nomenclature will be used, though the Salmonella, Shigella and Yersinia nomenclature can be found in Table 1.

Schematic representation of the E. coli T3SS. The T3SS consists of a number of coaxial ring-like structures. It comprises an export apparatus (red) a base complex and a needle filament (blue) through which unfolded effectors are channelled. At the distal end of the injectisome is a pore that inserts into the host membrane. The cytoplasmic ATPase complex (green) powers injectisome assembly as well as the translocation of effector proteins
5 Assembly of the Basal Body and Export Apparatus
There are four major components of the injectisome: the needle, the basal body, the export apparatus and the cytoplasmic protein complexes. The order of construction is disputed within the field and between species, which may represent divergence in the assembly pathway. An ‘outside-in’ model has been proposed for Yersinia beginning with the outer membrane ring and working inwards towards the export apparatus (Diepold et al. 2010, 2011) and the reverse ‘inside-out’ model has been described for Salmonella and E. coli (Wagner et al. 2010). This ‘inside-out’ model begins with nucleation of the inner membrane export apparatus components EscR, EscS and EscT forming a stable complex. EscRST recruits the export gate EscV oligomeric ring and the autoprotease EscU, completing the 5-member export apparatus EscRSTUV (Diepold et al. 2011). Autoproteolytic cleavage of EscU is an important signal for the union of the basal body and the export apparatus. The basal body ring components are exported via the Sec pathway, inserted within the bacterial membranes and come together with the export apparatus. A homo-oligomeric EscD ring at the inner membrane encircles the EscJ ring, which does not possess a transmembrane region but is instead tethered to the inner membrane. In the periplasm, EscD interacts with the outer membrane ring of EscC (Creasey et al. 2003a; Ogino et al. 2006) and is presumed to bind the EscJ ring. EscC belongs to the secretin family of proteins found in several bacterial secretion systems. Unlike other T3SS where the insertion and oligomerisation of the secretin are directed by pilotin, a lipoprotein not identified in A/E pathogens, EscC is believed to be regulated by other T3SS components (Gauthier and Finlay 2003). Assembly and coupling of the membrane rings to the export complex are followed by the formation of the EscQ cytosolic ring sorting complex, which recruits EscN and EscL, two parts of the three-component ATPase complex (Biemans-Oldehinkel et al. 2011). The ATPase complex is only activated upon the conformational change in EscL induced by the binding of EscO to EscN (Romo-Castillo et al. 2014). The function of the ATPase complex will be discussed in a later section.
Formation of the basal body is the only stage of assembly where components are directed by the Sec pathway. Once assembled, the estimated dimensions of the EPEC T3SS basal body are 16.7 ± 1.9 nm wide at the outer membrane ring, 18.1 ± 2.5 nm wide at the inner membrane ring and 31.4 ± 4.3 nm tall (Sekiya et al. 2001). This immature structure can secrete the so-called early- and mid-substrates required for construction of the inner rod, needle, tip and translocon.
6 The C-Ring/Sorting Complex
Multiple EscQ subunits make up the C-ring/sorting complex that sits at the foot of the basal body (Biemans-Oldehinkel et al. 2011; Pallen et al. 2005). In the flagellar T3SS, the C-ring is a three-component structure that has been attributed to generating flagellar torque and rotational switching, though whether this translates to the injectisome is debated. However, based on data from the Salmonella T3SS, the C-ring has been proposed to form a so-called ‘sorting platform’ (Lara-Tejero et al. 2011). Co-immunoprecipitation experiments show that the Salmonella EscQ homologue is predominantly associated with translocon proteins, but after deletion of translocator substrates is instead occupied by late effector proteins. These experiments also showed its interaction with the ATPase complex subunit homologues of EscN and EscL (Biemans-Oldehinkel et al. 2011). Most EscQ homologues have an internal translation site, giving rise to both the full length protein and a truncated C-terminal product, homologous to two of the three flagellar C-ring components (Bzymek et al. 2012; Lorenz et al. 2012; Notti et al. 2015). However, it is not clear whether EscQ expressed by A/E pathogens is produced in this way, and it is currently thought that the sorting platform is composed of EscQ, EscL and EscK, a crucial protein for substrate secretion (Soto et al. 2017).
7 The ATPase Complex
EscN, EscL and EscO together constitute the T3SS ATPase complex. It is located at the base of the export apparatus and has structural similarity to both the flagellar and the F1 ATPase. At the core of this complex is the multifunctional, multi-domain protein EscN. The EscN N-terminus facilitates its hexameric self-oligomerisation, while the central domain harbours a conserved ATPase domain and the C-terminus is the proposed recognition site for T3SS substrates. Aside from energising the secretion process, EscN is thought to serve as a docking site for the chaperone-substrate complex (Gauthier and Finlay 2003; Thomas et al. 2004), enabling the ATP hydrolysis-dependent uncoupling of these complexes, and unfolding of the protein which is to be secreted. As the needle pore is 2–3 nm wide this unfolding is essential for T3SS assembly. Binding of EscO to EscN promotes a change of conformation in EscL that activates EscN (Biemans-Oldehinkel et al. 2011; Romo-Castillo et al. 2014). Due to the structural similarity to the F1 ATPase, it is assumed that effector translocation is powered by a combination of ATP hydrolysis and proton motive force (Ibuki et al. 2011; Imada et al. 2007; Romo-Castillo et al. 2014; Zarivach et al. 2007). There is currently not enough data to confirm that EscN is the sole energiser of the T3SS, and due to a lack of high quality structural data, the mechanics of energising is still poorly understood.
8 Inner Rod and Needle Assembly
Once the basal body and the export apparatus join, EscI monomers assemble to create a hollow inner rod. EscI is also thought to have a role in substrate regulation, a theory supported by the fact that EscI interacts with EscU and EscP, components that contribute to substrate recognition and regulation. As the T3SS must penetrate the bacterial peptidoglycan layer, the specialised lytic transglycosilase EtgA, a peptidoglycan degrading enzyme, is required for this step of the assembly (García-Gómez et al. 2011). EtgA interacts directly with EscI, a relationship shown to enhance EtgA enzymatic activity (Burkinshaw et al. 2015). The EscF polymer, which forms the needle of the T3SS, is believed to be generated simultaneously with EscI (Gaytán et al. 2016). To prevent premature self-polymerisation of EscF, co-chaperones EscE and EscG bind EscF in the bacterial cytoplasm (Sal-Man et al. 2013).
At 23 nm long and 8–9 nm wide (Ogino et al. 2006; Sekiya et al. 2001), the EPEC injectisome is the shortest T3SS, compared to Salmonella enterica (25–80 nm), Shigella flexneri (45–50 nm), Yersinia pestis (41 nm) and Yersinia enterocolitica (58 nm). Despite this variation between bacterial genera, needle length tends to be conserved within species, perhaps reflecting its role in determining host tropism. Needle completion also controls the switch from the secretion of early substrates to middle and late substrates for the assembly of the filament and translocon structures (Buttner 2012; Minamino et al. 2004). In EPEC, the specificity switch occurs when EscP interacts with EscU, causing a conformational change in EscU that ultimately signals a substrate switch from injectisome components to translocated proteins (Feria et al. 2012). There are several suggested mechanisms for this switch. One hypothesis, named the infrequent ruler model, proposes that EscP is occasionally secreted in an elongated form and its passage through the needle is hindered by EscF subunits. Thus, as the needle grows, the chance of EscP interacting with EscU in the cytosol is increased (Feria et al. 2012). This is an adaptation of the ruler model first published in 2003, wherein EscP is anchored to both the tip of the growing filament and the basal body: once EscP is stretched to its maximum capacity, EscF is no longer incorporated into the needle and the substrate switches (Journet et al. 2003). In both models, the length of EscP determines substrate switching, hence the name ‘ruler protein’. In an alternative model, the inner rod regulates the needle length and timing of the substrate-switching event. Overexpression of the Yersinia inner rod protein results in shorter needles, while mutations presumed to slow inner rod assembly cause an elongated needle (Wood et al. 2008). Accordingly, EPEC EscI has also been shown to interact with EscP and EscU (Creasey et al. 2003a; Sal-Man et al. 2012). It is likely that needle length and substrate switching is in fact controlled by a combination of these models.
9 Filament and Translocon Assembly
The T3SS EspA filament is an extension of the EscF needle structure that is polymorphous among EPEC and EHEC isolates (Daniell et al. 2001a; Neves et al. 2003a). After translocation through the T3SS, EspA subunits self-polymerise via their C-terminal coiled-coil domains, with the completed filament averaging 90 nm in length (Daniell et al. 2001b; Delahay et al. 1999; Knutton et al. 1998). These coiled-coil domains are not only important for filament assembly, but also for prohibiting the immature polymerisation of EspA in the bacterial cytosol, where they are bound by the chaperone CesAB (Yip et al. 2005). Unlike the needle, the filament displays a more variable length seemingly dependent on the availability of EspA subunits, and can be up to 700 nm long (Crepin et al. 2005; Sekiya et al. 2001). The filament has proposed roles in bacterial adhesion and sensing of mammalian cells (Cleary et al. 2004). Interestingly, once intimate attachment has been achieved the EspA filament is disassembled by an unknown mechanism, and is absent from the mature A/E lesions (Dahan et al. 2004; Knutton et al. 1998).
The translocon is assembled by hetero-oligomerisation of EspB and EspD with 6–8 subunits, and has a pore size of 3–5 nm (Ide et al. 2001). It is responsible for puncturing the mammalian cell membrane, with both components predicted to possess transmembrane domains. Additionally, EspD comprises a C-terminal coiled-coil domain, which is necessary for A/E lesion formation (Daniell et al. 2001b). It was recently shown that host cells may be able to sense this puncturing, as contact with the EPEC injectisome was sufficient to induce activation of NF-κB, which in turn is subverted by anti-inflammatory effectors (Litvak et al. 2017).
10 The Roles of Chaperones in T3SS Assembly and Secretion
Chaperones are required in the bacterial cytoplasm throughout the assembly of the T3SS, and for the translocation of late effectors during the infection of host cells. They tend to be small acidic proteins and hold roles in delivering subunits and effectors to the export complex, preventing homo- and hetero-oligomerisation or degradation of substrates in the cytoplasm. Chaperones are also thought to play a role in defining the substrate hierarchy. Eight chaperones have been characterised in the LEE, and are classified according to their substrate: those that are specific for one effector protein (Class IA: CesF and CesL), several effectors (Class IB: CesT), translocators (Class II: CesAB, CesD and CesD2), and needle subunits (Class III: EscE and EscG) (reviewed in Gaytán et al. 2016; Izoré et al. 2011).
Together, class II chaperones control the secretion of translocators EspA, EspB, EspD. With the exception of CesD2, which only has one substrate (EspD) (Neves et al. 2003b), these chaperones work in coordination. Secretion of EspB and EspD requires CesD (Wainwright and Kaper 1998). EspB also interacts with CesAB, as does EspA, prior to secretion (Creasey et al. 2003b; Yip et al. 2005). In addition to CesAB, EspA is also chaperoned by CesA2 (Su et al. 2008). Both class III chaperones, EscG and EscE, are important for EscF secretion, as discussed above (Sal-Man et al. 2013). EscF is sometimes referred to as an ‘early substrate’, while the translocators EspA, EspB and EspD are ‘mid-substrates’ (Table 1).
Upon the completion of T3SS assembly, a disputed external signal leads to a second substrate switch from translocators to effectors, which are injected into host cells via the completed T3SS. This event involves the regulatory SepD-SepL-CesL complex. SepD and SepL, dubbed the gatekeeper proteins, simultaneously promote translocator secretion while preventing the premature translocation of late effectors (Deng et al. 2004, 2015; O’Connell et al. 2004). While the mechanism of promoting translocator secretion is unclear, it is thought that the interaction between SepL and the effector protein Tir is responsible for preventing effector secretion. Under the established effector hierarchy, Tir is the first effector to be translocated, and thus its interaction with SepL prevents translocation of any other effectors (Thomas et al. 2007; Wang et al. 2008). The Tir binding site of SepL is shared with the inner membrane ring protein EscD, perhaps playing a role in relief from the suppression of late effector secretion (Wang et al. 2008). Furthermore, in A/E pathogens removal of calcium from growth medium switches the secretion specificity from translocators to effectors (Deng et al. 2005; Gaytán et al. 2017; Ide et al. 2003; Kenny et al. 1997); under normal calcium conditions, the ruler protein EscP binds extracellular calcium flowing into the incomplete injectisome, stabilising its interaction with SepL and inhibiting effector secretion (Shaulov et al. 2017). However, contact between EspA/B/D and the host membrane does not play a role in specificity switching, as previously thought (Gaytán et al. 2017).
11 The ETT2: A Second T3SS?
In 2001, analysis of the EHEC O157:H7 genome revealed a second type III secretion system cluster, designated the E. coli Type Three Secretion System 2 (ETT2). The ETT2 locus appears to have been inserted into the E. coli genome immediately upstream tRNA glyU locus, spanning 27.5 kb and 35 open reading frames (ORFs) (Ren et al. 2004). It was initially noted that the ETT2 is remarkably homologous to the S. enterica serovar Typhimurium T3SS, SPI-1, with some genes sharing up to 64% identity with their Salmonella counterparts. This homology guided further analysis of each ORF, leading to the discovery that several genes in the ETT2 contain frameshift mutations, rendering it non-functional. However, fragments of the ETT2 have been identified in a variety of enteric E. coli isolates from several mammalian and avian sources (Osawa et al. 2006; Zhou et al. 2014). The isoform of each ETT2 identified is now categorised from ‘type A’ to ‘type K’ per its completeness, where type A encodes all 35 ORFs (Cheng et al. 2012). Interestingly, the ETT2 is considered intact in the emerging enteropathogen E. albertii, which gives a more specific idea of when the locus was acquired (Ooka et al. 2015). As such, ETT2 classification may provide an insight into the phylogenetic origin of E. coli isolates.
A secretion-competent ETT2 has never been identified, therefore the question of its function remains unsolved. In earlier works, it was suggested that proteins encoded by the ETT2 could complement the function of the LEE-encoded T3SS This now seems unlikely, given the mutational attrition within ORFs in the ETT2 and the lack of ETT2-specific effectors. Interestingly, deletion of ETT2 genes impacts several aspects of bacterial virulence (Ideses et al. 2005), although the mechanistic details behind these observations remain unclear. Two ETT2 encoded regulators, EtrA and EivF, repress LEE expression and therefore reduce bacterial adherence (Zhang et al. 2004). This could explain why an ΔetrA mutant was also deficient in intracellular survival (Wang et al. 2017). A third regulator, EtrB, was shown to activate LEE expression by direct interaction with Ler, the master regulator of the LEE pathogenicity island (Luzader et al. 2016). More recently, Wang et al. showed that deletion of the ETT2 putative ATPase EivC inhibited the flagellar motility of the avian pathogenic E. coli strain APCE94. The lack of motility was attributed to downregulation of the flagellum and upregulation of fimbrial genes. Additionally, the ΔeivC strain had significantly decreased intracellular survival compared to WT APCE94 (Wang et al. 2016). Together, these observations suggest that the ETT2 is not just an artefact, as it was once assumed.
12 In Vitro and in Vivo Tools to Study the E. coli T3SS
Significant strides have been made delineating the molecular mechanisms that underpin T3SS-dependent virulence in enteric E. coli, aided by technical advances in fluorescence microscopy, electron cryotomography and single-cell super-resolution techniques. Our structural understanding of T3SS architecture is becoming increasingly clear as crystal structures of components are being solved, and defined functions during infection are being assigned to T3SS components and effectors. Numerous studies in vivo have demonstrated the importance of the T3SS for A/E virulence, including a study with human volunteers that confirmed the requirement for structural T3SS components (EspB and EspA) for the development of diarrhoea (Donnenberg et al. 1993; Tacket et al. 2000). However, furthering our understanding of the T3SS requires robust in vitro and in vivo models that do not rely on the availability of human samples. Intestinal biopsies have historically been used for in vitro organ culture (IVOC) models to demonstrate how A/E pathogens attach to the apical portion of intestinal crypts (Shaw et al. 2005). However, for biochemical assays, in vitro studies typically use cultured cells (such as HeLa cells or polarised Caco-2 cells) (Wong et al. 2011).
In vivo models of A/E pathogenesis allow studies of the complex interplay between the pathogen and its host as well as the intestinal microbiota, which is known to play a vital role in disease development. A variety of surrogate species have been infected with EPEC and EHEC, including Caenorhabditis elegans, pigs, baboons, macaques, infant rabbits, ferrets and cows (Ritchie 2014; Law et al. 2013), but these techniques inevitably suffer from the bacterium not representing a natural pathogen. Therefore, closely related animal-specific A/E pathogens serve as a better proxy for human infection: the use of REPEC is limited as it causes high mortality (Milon et al. 1999), but C. rodentium is an indispensable tool for studying A/E pathogenesis. C. rodentium causes transmissible murine colonic hyperplasia with A/E lesions indistinguishable from those caused by EPEC and EHEC (Collins et al. 2014; Schauer et al. 1995). In general, murine models benefit from the availability of inbred or genetically manipulated mice that can be maintained under germ-free or controlled pathogenic conditions. The C. rodentium model continues to give invaluable insights into the physiological outcomes of the translocation of effector proteins in vivo and can be combined with biochemical techniques such as mass spectrometry to uncover strategies used by A/E pathogens to circumvent immune responses and cause disease.
13 Exploiting the T3SS to Treat Disease
The genetic variety of pathogenic E. coli strains underpins their success but makes treatment of disease difficult. Rehydration therapy is currently the most effective way to manage symptoms of E. coli infection, which can become dangerous if it is not self-limiting. The traditional treatment for unidentified bacterial infections associated with diarrhoea is often antibiotics. Broad-spectrum antibiotics may kill the offending bacterium but will also affect commensals. Additionally, some antibiotics can exacerbate expression of Shiga toxins if they are present (Freedman et al. 2016), which can lead to HUS and ultimately renal failure. These problems could be bypassed by targeting the T3SS: the injectisome is not required for growth, so its inhibition should not enforce selective pressure on targeted bacteria. Further, the T3SS is only conserved in pathogenic bacteria, so commensals will not be affected by treatment, and the development of anti-T3SS drugs for E. coli could be adapted to target other T3SS-expressing pathogens.
There are several elements of type III secretion that can be targeted. Aside from inhibiting the basal body, vaccines can be developed against the translocon and needle, LEE regulatory elements can be manipulated, effectors can be counteracted and secretion can be inhibited (Charro and Mota 2015). One of the most studied classes of anti-T3SS compounds is salicylidene acylhydrazides (SAHs), which subvert secretion in several ways. First, SAHs interact with the inner membrane proteins of the basal body to restrict the passage of effectors. Owing to its homology with the T3SS basal body, SAHs have also been shown to block the flagellar apparatus, effectively reducing bacterial motility. Lastly, SAHs have been shown to interact with proteins involved in E. coli metabolism, resulting in the downregulation of T3SS gene expression (Mcshan and Guzman 2015).
Although other classes of compounds have been shown to inhibit the T3SS, their development as drugs is hindered by their toxicity to eukaryotic cells, their similarity to existing drugs against which pathogens have gained resistance, difficulty introducing the compound into the gut, or lack of a clear bacterial target for their action. Owing to the intricacy of LEE regulation, our incomplete knowledge of T3SS effectors and the problems with anti-T3SS compounds described above, vaccines against the bacterial surface structures represent the most promising treatment candidates (O’Ryan et al. 2015). For example, self-polymerisation of EspA can be inhibited by treatment with synthetic EspA-like coiled-coil domains, which effectively outcompete endogenous EspA interactions to inhibit the secretion of effectors and A/E lesion formation (Larzábal et al. 2010). Additionally, when produced either in planta or in vitro and administered orally to ruminants, EspA itself is able to act as a vaccine to reduce bacterial shedding (Miletic et al. 2017; Potter et al. 2004). Indeed, many other injectisome proteins and pathogenic E. coli virulence factors are highly immunogenic in ruminants, including flagellin, intimin, Tir, EspB and EspD. Antibodies against these proteins have been identified in bovine and human colostrum and offer protection for both calves and human infants against EHEC colonisation (Loureiro et al. 1998; Vilte et al. 2008), often by reducing ruminants’ shedding (McNeilly et al. 2015).
Although the complexity of the T3SS presents an obstacle for inhibiting its action, it is a targeted membrane-specific nanomachine capable of secreting cargo, and this itself is an exploitable property (González-Prieto and Lesser 2018). Recently, all of the structural components of the EPEC injectisome were inserted in five independent transcriptional units, or engineered LEEs (eLEE), into the genome of the commensal strain E. coli K-12. Rational design of each eLEEs meant their expression can be controlled, avoiding the intricate regulation found in the A/E pathogens. The resulting strain, named synthetic injector E. coli (SIEC), was demonstrated to assemble functional injectisomes and efficiently translocate T3-substrate proteins (Ruano-Gallego et al. 2015). Indeed, several studies have also demonstrated its use as a drug delivery system by engineering a signal sequence from an effector onto the protein of interest. This includes delivery of a nuclear-targeted recombinase for editing the genome of induced pluripotent stem cells (Bichsel et al. 2011), delivery of antigens for immunotherapy (Le Gouëllec et al. 2013) and delivery of angiogenic inhibitors for the shrinkage of tumours (Shi et al. 2016).
A current challenge in the field is shifting from studying T3SS effector proteins and injectisome components in isolation to applying a holistic approach that considers the role of effectors in context of the full effector repertoire, while also considering spatio-temporal regulatory mechanisms. Many mechanistic details on the assembly and regulation of the T3SS must still be elucidated, including fully defining the signal that triggers the translocation of effector proteins upon contact of host cells. Answering these questions will help design strategies to interfere with the system to help relieve the global burden of enteric E. coli infections on human health.
References
Alsharif G, Ahmad S, Islam MS, Shah R, Busby SJ, Krachler AM (2015) Host attachment and fluid shear are integrated into a mechanical signal regulating virulence in Escherichia coli O157:H7. Proc Natl Acad Sci U S A 112:5503–5508
Berdichevsky T, Friedberg D, Nadler C, Rokney A, Oppenheim A, Rosenshine I (2005) Ler is a negative autoregulator of the LEE1 operon in enteropathogenic Escherichia coli. J Bacteriol 187:349–357
Bern C, Martines J, de Zoysa I, Glass RI (1992) The magnitude of the global problem of diarrhoeal disease: a ten-year update. Bull World Health Organ 70:705–714
Bhat A, Shin M, Jeong J-H, Kim H-J, Lim H-J, Rhee JH, Paik S-Y, Takeyasu K, Tobe T, Yen H et al (2014) DNA looping-dependent autorepression of LEE1 P1 promoters by Ler in enteropathogenic Escherichia coli (EPEC). Proc Natl Acad Sci U S A 111:E2586–E2595
Bichsel C, Neeld DK, Hamazaki T, Wu D, Chang LJ, Yang L, Terada N, Jin S (2011) Bacterial delivery of nuclear proteins into pluripotent and differentiated cells. PLoS ONE 6
Biemans-Oldehinkel E, Sal-Man N, Deng W, Foster LJ, Finlay BB (2011) Quantitative proteomic analysis reveals formation of an EscL-EscQ-EscN type III complex in enteropathogenic Escherichia coli. J Bacteriol 193:5514–5519
Bingle LEH, Constantinidou C, Shaw RK, Islam MS, Patel M, Snyder LAS, Lee DJ, Penn CW, Busby SJW, Pallen MJ (2014) Microarray analysis of the Ler regulon in enteropathogenic and enterohaemorrhagic Escherichia coli strains. PLoS ONE 9:1–12
Burkinshaw BJ, Deng W, Lameignère E, Wasney GA, Zhu H, Worrall LJ, Finlay BB, Strynadka NCJ (2015) Structural analysis of a specialized type III secretion system peptidoglycan-cleaving enzyme. J Biol Chem 290:10406–10417
Bustamante VH, Santana FJ, Calva E, Puente JL (2001) Transcriptional regulation of type III secretion genes in enteropathogenic Escherichia coli: ler antagonizes H-NS-dependent repression. Mol Microbiol 39:664–678
Buttner D (2012) Protein export according to schedule: architecture, assembly, and regulation of type III secretion systems from plant- and animal-pathogenic bacteria. Microbiol Mol Biol Rev 76:262–310
Bzymek KP, Hamaoka BY, Ghosh P (2012) Two translation products of Yersinia yscQ assemble to form a complex essential to type III secretion. Biochemistry 51:1669–1677
Carlson-Banning KM, Sperandio V (2018) Enterohemorrhagic Escherichia coli outwits hosts through sensing small molecules. Curr Opin Microbiol 41:83–88
Charro N, Mota LJ (2015) Approaches targeting the type III secretion system to treat or prevent bacterial infections. Expert Opin Drug Discov 10:373–387
Chen HD, Frankel G (2005) Enteropathogenic Escherichia coli: unravelling pathogenesis. FEMS Microbiol Rev 29:83–98
Cheng D, Zhu S, Su Z, Zuo W, Lu H (2012) Prevalence and isoforms of the pathogenicity island ETT2 among Escherichia coli isolates from colibacillosis in pigs and mastitis in cows. Curr Microbiol 64:43–49
Cleary J, Lai L-C, Shaw RK, Straatman-Iwanowska A, Donnenberg MS, Frankel G, Knutton S (2004) Enteropathogenic Escherichia coli (EPEC) adhesion to intestinal epithelial cells: role of bundle-forming pili (BFP), EspA filaments and intimin. Microbiology 150:527–538
Clements A, Young JC, Constantinou N, Frankel G (2011) Infection strategies of enteric pathogenic Escherichia coli. Gut Microbes 3:71–87
Collins JW, Keeney KM, Crepin VF, Rathinam VAK, Fitzgerald KA, Finlay BB, Frankel G (2014) Citrobacter rodentium: infection, inflammation and the microbiota. Nat Rev Microbiol 12:612–623
Connolly JPR, Brett Finlay B, Roe AJ (2015) From ingestion to colonization: the influence of the host environment on regulation of the LEE encoded type III secretion system in enterohaemorrhagic Escherichia coli. Front Microbiol 6:1–15
Creasey EA, Delahay RM, Daniell SJ, Frankel G (2003a) Yeast two-hybrid system survey of interactions between LEE-encoded proteins of enteropathogenic Escherichia coli. Microbiology 149:2093–2106
Creasey EA, Friedberg D, Shaw RK, Umanski T, Knutton S, Rosenshine I, Frankel G (2003b) CesAB is an enteropathogenic Escherichia coli chaperone for the type-III translocator proteins EspA and EspB. Microbiology 149:3639–3647
Crepin V, Shaw R, Abe C, Knutton S, Frankel G (2005) Polarity of enteropathogenic Escherichia coli EspA filament assembly and protein secretion. J Bacteriol 187:2881–2889
Dahan S, Knutton S, Shaw RK, Crepin VF, Dougan G, Frankel G (2004) Transcriptome of enterohemorrhagic Escherichia coli O157 adhering to eukaryotic plasma membranes. Infect Immun 72:5452–5459
Daniell SJ, Takahashi N, Wilson R, Friedberg D, Rosenshine I, Booy FP, Shaw RK, Knutton S, Frankel G, Aizawa S (2001a) The filamentous type III secretion translocon of enteropathogenic Escherichia coli. Cell Microbiol 3:865–871
Daniell SJ, Delahay RM, Shaw RK, Hartland EL, Pallen MJ, Booy F, Ebel F, Knutton S, Frankel G (2001b) Coiled-coil domain of enteropathogenic Escherichia coli type III secreted protein EspD is involved in EspA filament-mediated cell attachment and hemolysis. Infect Immun 69:4055–4064
De Nisco NJ, Rivera-Cancel G, Orth K (2018) The biochemistry of sensing: enteric pathogens regulate type III secretion in response to environmental and host cues. MBio 9:e02122-17
Delahay RM, Knutton S, Shaw RK, Hartland EL, Pallen MJ, Frankel G (1999) The coiled-coil domain of EspA is essential for the assembly of the type III secretion translocon on the surface of enteropathogenic Escherichia coli. J Biol Chem 274:35969–35974
Deng W, Puente JL, Gruenheid S, Li Y, Vallance BA, Vázquez A, Barba J, Ibarra JA, O’Donnell P, Metalnikov P et al (2004) Dissecting virulence: systematic and functional analyses of a pathogenicity island. Proc Natl Acad Sci 101:3597–3602
Deng W, Li Y, Hardwidge PR, Frey EA, Pfuetzner RA, Lee S, Gruenheid S, Strynakda NCJ, Puente JL, Finlay BB (2005) Regulation of type III secretion hierarchy of translocators and effectors in attaching and effacing bacterial pathogens. Infect Immun 73:2135–2146
Deng W, Yu HB, Li Y, Finlay BB (2015) SepD/SepL-dependent secretion signals of the type III secretion system translocator proteins in enteropathogenic Escherichia coli. J Bacteriol 197:1263–1275
Desai MS, Seekatz AM, Koropatkin NM, Kamada N, Hickey CA, Wolter M, Pudlo NA, Kitamoto S, Terrapon N, Muller A et al (2016) A dietary fiber-deprived gut microbiota degrades the colonic mucus barrier and enhances pathogen susceptibility. Cell 167(1339–1353):e21
Diepold A, Amstutz M, Abel S, Sorg I, Jenal U, Cornelis GR (2010) Deciphering the assembly of the Yersinia type III secretion injectisome. EMBO J 29:1928–1940
Diepold A, Wiesand U, Cornelis GR (2011) The assembly of the export apparatus (YscR, S, T, U, V) of the Yersinia type III secretion apparatus occurs independently of other structural components and involves the formation of an YscV oligomer. Mol Microbiol 82:502–514
Donnenberg MS, Tacket CO, James SP, Losonsky G, Nataro JP, Wasserman SS, Kaper JB, Levine MM (1993) Role of the eaeA gene in experimental enteropathogenic Escherichia coli infection. J Clin Invest 92:1412–1417
Elliott SJ, Sperandio V, Girón JA, Shin S, Mellies JL, Wainwright L, Hutcheson SW, McDaniel TK, Kaper JB (2000) The locus of enterocyte effacement (LEE)-encoded regulator controls expression of both LEE- and non-LEE-encoded virulence factors in enteropathogenic and enterohemorrhagic Escherichia coli. Infect Immun 68:6115–6126
Feria JM, García-Gómez E, Espinosa N, Minamino T, Namba K, González-Pedrajo B (2012) Role of escp (Orf16) in injectisome biogenesis and regulation of type III protein secretion in enteropathogenic Escherichia coli. J Bacteriol 194:6029–6045
Freedman SB, Xie J, Neufeld MS, Hamilton WL, Hartling L, Tarr PI (2016) Shiga toxin-producing Escherichia coli infection, antibiotics, and risk of developing hemolytic uremic syndrome: a meta-analysis. Clin Infect Dis 62:1251–1258
Furniss RCD, Clements A (2017) Regulation of the locus of enterocyte effacement in attaching and effacing pathogens. J Bacteriol JB.00336-17
García-Gómez E, Espinosa N, de la Mora J, Dreyfus G, González-Pedrajo B (2011) The muramidase EtgA from enteropathogenic Escherichia coli is required for efficient type III secretion. Microbiology 157:1145–1160
Gauthier A, Finlay BB (2003) Translocated intimin receptor and its chaperone interact with ATPase of the type III secretion apparatus of enteropathogenic Escherichia coli. J Bacteriol 185:6747–6755
Gaytán MO, Martínez-Santos VI, Soto E, González-Pedrajo B (2016) Type three secretion system in attaching and effacing pathogens. Front Cell Infect Microbiol 6:129
Gaytán MO, Monjarás Feria J, Soto E, Espinosa N, Benítez JM, Georgellis D, González-Pedrajo B (2017) Novel insights into the mechanism of SepL-mediated control of effector secretion in enteropathogenic Escherichia coli. Microbiologyopen e00571
Gomez-Duarte OG, Kaper JB (1995) A plasmid-encoded regulatory region activates chromosomal eaeA expression in enteropathogenic Escherichia coli. Infect Immun 63:1767–1776
González-Prieto C, Lesser CF (2018) Rationale redesign of type III secretion systems: toward the development of non-pathogenic E. coli for in vivo delivery of therapeutic payloads. Curr Opin Microbiol 41:1–7
Hodges K, Gill R (2010) Infectious diarrhea: cellular and molecular mechanisms. Gut Microbes 1:4–21
Hughes DT, Sperandio V (2008) Inter-kingdom signalling: communication between bacteria and their hosts. Nat Rev Microbiol 6:111–120
Hyma KE, Lacher DW, Nelson AM, Bumbaugh AC, Janda JM, Strockbine NA, Young VB, Whittam TS (2005) Evolutionary genetics of a new pathogenic Escherichia species: Escherichia albertii and related Shigella boydii strains. J Bacteriol 187:619–628
Ibuki T, Imada K, Minamino T, Kato T, Miyata T, Namba K (2011) Common architecture of the flagellar type III protein export apparatus and F- and V-type ATPases. Nat Struct Mol Biol 18:277–282
Ide T, Laarmann S, Greune L, Schillers H, Oberleithner H, Schmidt MA (2001) Characterization of translocation pores inserted into plasma membranes by type III-secreted Esp proteins of enteropathogenic Escherichia coli. Cell Microbiol 3:669–679
Ide T, Michgehl S, Knappstein S, Heusipp G, Schmidt MA (2003) Differential modulation by Ca2+ of type III secretion of diffusely adhering enteropathogenic Escherichia coli. Infect Immun 71:1725–1732
Ideses D, Gophna U, Paitan Y, Chaudhuri RR, Pallen MJ, Ron EZ (2005) A degenerate type III secretion system from septicemic Escherichia coli contributes to pathogenesis. J Bacteriol 187:8164–8171
Imada K, Minamino T, Tahara A, Namba K (2007) Structural similarity between the flagellar type III ATPase FliI and F1-ATPase subunits. Proc Natl Acad Sci U S A 104:485–490
Iyoda S, Watanabe H (2005) ClpXP protease controls expression of the type III protein secretion system through regulation of RpoS and GrlR levels in enterohemorrhagic Escherichia coli. J Bacteriol 187:4086–4094
Izoré T, Job V, Dessen A (2011) Biogenesis, regulation, and targeting of the type III secretion system. Structure 19:603–612
Jimenez R, Cruz-Migoni SB, Huerta-Saquero A, Bustamante VH, Puente JL (2010) Molecular characterization of GrlA, a specific positive regulator of ler expression in enteropathogenic Escherichia coli. J Bacteriol 192:4627–4642
Journet L, Agrain C, Broz P, Cornelis GR (2003) The needle length of bacterial injectisomes is determined by a molecular ruler. Science (80-) 302:1757–1760
Kenny B, Abe A, Stein M, Finlay BB (1997) Enteropathogenic Escherichia coli protein secretion is induced in response to conditions similar to those in the gastrointestinal tract. Infect Immun 65:2606–2612
Knutton S, Rosenshine I, Pallen MJ, Nisan I, Neves BC, Bain C, Wolff C, Dougan G (1998) A novel EspA-associated surface organelle of enteropathogenic Escherichia coli involved in protein translocation into epithelial cells. EMBO J 17:2166–2176
Kosek M, Bern C, Guerrant RL (2003) The global burden of diarrhoeal disease, as estimated from studies published between 1992 and 2000. Bull World Health Organ 81:197–204
Kubori T, Matsushima Y, Nakamura D, Uralil J, Lara-Tejero M, Sukhan A, Galán JE, Aizawa SI (1998) Supramolecular structure of the Salmonella typhimurium type III protein secretion system. Science 280:602–605
Lara-Tejero M, Kato J, Wagner S, Liu X, Galán JE (2011) A sorting platform determines the order of protein secretion in bacterial type III systems. Science 331:1188–1191
Larzábal M, Mercado EC, Vilte DA, Salazar-González H, Cataldi A, Navarro-Garcia F (2010) Designed coiled-coil peptides inhibit the type three secretion system of enteropathogenic Escherichia coli. PLoS ONE 5
Law RJ, Gur-Arie L, Rosenshine I, Brett Finlay B (2013) In vitro and in vivo model systems for studying enteropathogenic Escherichia coli infections. Cold Spring Harb Perspect Med 3:a009977
Le Gouëllec A, Chauchet X, Laurin D, Aspord C, Verove J, Wang Y, Genestet C, Trocme C, Ahmadi M, Martin S et al (2013) A safe bacterial microsyringe for in vivo antigen delivery and immunotherapy. Mol Ther 21:1076–1086
Litvak Y, Sharon S, Hyams M, Zhang L, Kobi S, Katsowich N, Dishon S, Nussbaum G, Dong N, Shao F et al (2017) Epithelial cells detect functional type III secretion system of enteropathogenic Escherichia coli through a novel NF-κB signaling pathway. PLoS Pathog 13:e1006472
Liu L, Oza S, Hogan D, Perin J, Rudan I, Lawn JE, Cousens S, Mathers C, Black RE (2015) Global, regional, and national causes of child mortality in 2000–13, with projections to inform post-2015 priorities: an updated systematic analysis. Lancet 385:430–440
Lorenz C, Hausner J, Büttner D (2012) HrcQ provides a docking site for early and late type III secretion substrates from Xanthomonas. PLoS ONE 7
Loureiro I, Frankel G, Adu-Bobie J, Dougan G, Trabulsi LR, Carneiro-Sampaio MM (1998) Human colostrum contains IgA antibodies reactive to enteropathogenic Escherichia coli virulence-associated proteins: intimin, BfpA, EspA, and EspB. J Pediatr Gastroenterol Nutr 27:166–171
Luzader DH, Willsey GG, Wargo MJ, Kendall MM (2016) The ETT2-encoded regulator EtrB modulates enterohemorrhagic Escherichia coli virulence gene expression. Infect Immun 84:2555–2565
Martínez-Santos VI, Medrano-López A, Saldaña Z, Girón JA, Puente JL (2012) Transcriptional regulation of the ecp operon by EcpR, IHF, and H-NS in attaching and effacing Escherichia coli. J Bacteriol 194:5020–5033
McNeilly TN, Mitchell MC, Corbishley A, Nath M, Simmonds H, McAteer SP, Mahajan A, Low JC, Smith DGE, Huntley JF et al (2015) Optimizing the protection of cattle against Escherichia coli O157:H7 colonization through immunization with different combinations of H7 flagellin, Tir, intimin-531 or EspA. PLoS ONE 10:1–19
Mcshan AC, Guzman RN De (2015) The bacterial type III secretion system as a target for developing new antibiotics. Chem Biol Drug Des 85:30–42
Mellies JL, Elliott SJ, Sperandio V, Donnenberg MS, Kaper JB (1999) The Per regulon of enteropathogenic Escherichia coli: identification of a regulatory cascade and a novel transcriptional activator, the locus of enterocyte effacement (LEE)-encoded regulator (Ler). Mol Microbiol 33:296–306
Mellies JL, Navarro-Garcia F, Okeke I, Frederickson J, Nataro JP, Kaper JB (2001) espC pathogenicity island of enteropathogenic Escherichia coli encodes an enterotoxin. Infect Immun 69:315–324
Miletic S, Hünerberg M, Kaldis A, MacDonald J, Leuthreau A, McAllister T, Menassa R (2017) A plant-produced candidate subunit vaccine reduces shedding of enterohemorrhagic Escherichia coli in ruminants. Biotechnol J 12:1–9
Milon A, Oswald E, De Rycke J (1999) Rabbit EPEC: a model for the study of enteropathogenic Escherichia coli. Vet Res 30:203–219
Minamino T, Saijo-Hamano Y, Furukawa Y, González-Pedrajo B, Macnab RM, Namba K (2004) Domain organization and function of Salmonella FliK, a flagellar hook-length control protein. J Mol Biol 341:491–502
Moreira CG, Russell R, Mishra AA, Narayanan S, Ritchie JM, Waldor MK, Curtis MM, Winter SE, Weinshenker D, Sperandio V (2016) Bacterial adrenergic sensors regulate virulence of enteric pathogens in the gut. MBio 7:e00826-16
Navarro-Garcia F, Serapio-Palacios A, Vidal JE, Isabel Salazar M, Tapia-Pastrana G (2014) EspC promotes epithelial cell detachment by enteropathogenic Escherichia coli via sequential cleavages of a cytoskeletal protein and then focal adhesion proteins. Infect Immun 82:2255–2265
Neves BC, Shaw RK, Frankel G, Knutton S (2003a) Polymorphisms within Espa filaments of enteropathogenic and enterohemorrhagic Escherichia coli. Infect Immun 71:2262–2265
Neves BC, Mundy R, Petrovska L, Dougan G, Knutton S, Frankel G (2003b) CesD2 of enteropathogenic Escherichia coli is a second chaperone for the type III secretion translocator protein EspD. Infect Immun 71:2130–2141
Nguyen Y, Sperandio V (2012) Enterohemorrhagic E. coli (EHEC) pathogenesis. Front Cell Infect Microbiol 2:1–7
Notti RQ, Bhattacharya S, Lilic M, Stebbins CE (2015) A common assembly module in injectisome and flagellar type III secretion sorting platforms. Nat Commun 6:7125
O’Connell CB, Creasey EA, Knutton S, Elliott S, Crowther LJ, Luo W, John Albert M, Kaper JB, Frankel G, Donnenberg MS (2004) SepL, a protein required for enteropathogenic Escherichia coli type III translocation, interacts with secretion component SepD. Mol Microbiol 52:1613–1625
Ogino T, Ohno R, Sekiya K, Kuwae A, Matsuzawa T, Nonaka T, Fukuda H, Imajoh-ohmi S, Abe A (2006) Assembly of the type III secretion apparatus of enteropathogenic Escherichia coli. J Bacteriol 188:2801–2811
Ooka T, Ogura Y, Katsura K, Seto K, Kobayashi H, Kawano K, Tokuoka E, Furukawa M, Harada S, Yoshino S et al (2015) Defining the genome features of Escherichia albertii, an emerging enteropathogen closely related to Escherichia coli. Genome Biol Evol 7:3170–3179
O’Ryan M, Vidal R, del Canto F, Salazar JC, Montero D (2015) Vaccines for viral and bacterial pathogens causing acute gastroenteritis: Part I: Overview, vaccines for enteric viruses and Vibrio cholerae. Hum Vaccin Immunother 11:584–600
Osawa K, Shibata M, Nishiyama Y, Kurokawa M, Yamamoto G, Kinoshita S, Kataoka N (2006) Identification of the ETT2 locus in human diarrheagenic Escherichia coli by multiplex PCR. J Infect Chemother 12:157–159
Pacheco AR, Curtis MM, Ritchie JM, Munera D, Waldor MK, Moreira CG, Sperandio V (2012) Fucose sensing regulates bacterial intestinal colonization. Nature 492:113–117
Padavannil A, Jobichen C, Mills E, Velazquez-Campoy A, Li M, Leung KY, Mok YK, Rosenshine I, Sivaraman J (2013) Structure of GrlR–GrlA complex that prevents GrlA activation of virulence genes. Nat Commun 4:2–11
Pallen MJ, Beatson SA, Bailey CM (2005) Bioinformatics analysis of the locus for enterocyte effacement provides novel insights into type-III secretion. BMC Microbiol 5:9
Petty NK, Feltwell T, Pickard D, Clare S, Toribio AL, Fookes M, Roberts K, Monson R, Nair S, Kingsley RA et al (2011) Citrobacter rodentium is an unstable pathogen showing evidence of significant genomic flux. PLoS Pathog 7
Potter AA, Klashinsky S, Li Y, Frey E, Townsend H, Rogan D, Erickson G, Hinkley S, Klopfenstein T, Moxley RA et al (2004) Decreased shedding of Escherichia coli O157:H7 by cattle following vaccination with type III secreted proteins. Vaccine 22:362–369
Ren CP, Chaudhuri RR, Fivian A, Bailey CM, Antonio M, Barnes WM, Pallen MJ (2004) The ETT2 gene cluster, encoding a second type III secretion system from Escherichia coli, is present in the majority of strains but has undergone widespread mutational attrition. J Bacteriol 186:3547–3560
Ritchie JM (2014) Animal models of enterohemorrhagic Escherichia coli infection. Microbiol, Spectr, p 2
Romo-Castillo M, Andrade A, Espinosa N, Feria JM, Soto E, Díaz-Guerrero M, González-Pedrajo B (2014) EscO, a functional and structural analog of the flagellar fliJ protein, is a positive regulator of EscN ATPase activity of the enteropathogenic Escherichia coli injectisome. J Bacteriol 196:2227–2241
Ruano-Gallego D, Álvarez B, Fernández LÁ (2015) Engineering the controlled assembly of filamentous injectisomes in E. coli K-12 for protein translocation into mammalian cells. ACS Synth Biol 4:1030–1041
Russell RM, Sharp FC, Rasko DA, Sperandio V (2007) QseA and GrlR/GrlA regulation of the locus of enterocyte effacement genes in enterohemorrhagic Escherichia coli. J Bacteriol 189:5387–5392
Salinger N, Kokona B, Fairman R, Okeke IN (2009) The plasmid-encoded regulator activates factors conferring lysozyme resistance on enteropathogenic Escherichia coli strains. Appl Environ Microbiol 75:275–280
Sal-Man N, Deng W, Finlay BB (2012) EscI: a crucial component of the type III secretion system forms the inner rod structure in enteropathogenic Escherichia coli. Biochem J 442:119–125
Sal-Man N, Setiaputra D, Scholz R, Deng W, Yu ACY, Strynadka NCJ, Finlay BB (2013) EscE and EscG are cochaperones for the type III needle protein EscF of enteropathogenic Escherichia coli. J Bacteriol 195:2481–2489
Schauer DB, Zabel BA, Pedraza IF, O’Hara CM, Steigerwalt AG, Brenner DJ (1995) Genetic and biochemical characterization of Citrobacter rodentium sp. nov. J Clin Microbiol 33:2064–2068
Sekiya K, Ohishi M, Ogino T, Tamano K, Sasakawa C, Abe A (2001) Supermolecular structure of the enteropathogenic Escherichia coli type III secretion system and its direct interaction with the EspA-sheath-like structure. Proc Natl Acad Sci U S A 98:11638–11643
Shaulov L, Gershberg J, Deng W, Finlay BB, Sal-Man N (2017) The ruler protein EscP of the enteropathogenic Escherichia coli type III secretion system is involved in calcium sensing and secretion hierarchy regulation by interacting with the gatekeeper protein SepL. MBio 8:1–15
Shaw RK, Cleary J, Murphy MS, Frankel G, Knutton S (2005) Interaction of enteropathogenic Escherichia coli with human intestinal mucosa: role of effector proteins in brush border remodeling and formation of attaching and effacing lesions. Infect Immun 73(2):1243–1251. https://doi.org/10.1128/IAI.73.2.1243-1251.2005
Shi L, Yu B, Cai C-H, Huang J-D (2016) Angiogenic inhibitors delivered by the type III secretion system of tumor-targeting Salmonella typhimurium safely shrink tumors in mice. AMB Express 6:56
Snyder JD, Merson MH (1982) The magnitude of the global problem of acute diarrhoeal disease: a review of active surveillance data. Bull World Health Organ 60:605–613
Soto E, Espinosa N, Díaz-Guerrero M, Gaytán MO, Puente JL, González-Pedrajo B (2017) Functional characterization of EscK (Orf4), a sorting platform component of the enteropathogenic Escherichia coli injectisome. J Bacteriol 199
Sperandio V, Mellies JL, Nguyen W, Shin S, Kaper JB (1999) Quorum sensing controls expression of the type III secretion gene transcription and protein secretion in enterohemorrhagic and enteropathogenic Escherichia coli. Proc Natl Acad Sci U S A 96:15196–15201
Su MSW, Kao HC, Lin CN, Syu WJ (2008) Gene l0017 encodes a second chaperone for EspA of enterohaemorrhagic Escherichia coli O157: H7. Microbiology 154:1094–1103
Tacket CO, Sztein MB, Losonsky G, Abe A, Finlay BB, McNamara BP, Fantry GT, James SP, Nataro JP, Levine MM, Donnenberg MS (2000) Role of EspB in experimental human enteropathogenic Escherichia coli infection. Infect Immun 68(6):3689–3695
Thomas J, Stafford GP, Hughes C (2004) Docking of cytosolic chaperone-substrate complexes at the membrane ATPase during flagellar type III protein export. Proc Natl Acad Sci U S A 101:3945–3950
Thomas NA, Deng W, Baker N, Puente J, Finlay BB (2007) Hierarchical delivery of an essential host colonization factor in enteropathogenic Escherichia coli. J Biol Chem 282:29634–29645
Umanski T, Rosenshine I, Friedberg D (2002) Thermoregulated expression of virulence genes in enteropathogenic Escherichia coli. Microbiology 148:2735–2744
Vilte DA, Larzábal M, Cataldi ÁA, Mercado EC (2008) Bovine colostrum contains immunoglobulin G antibodies against intimin, EspA, and EspB and inhibits hemolytic activity mediated by the type three secretion system of attaching and effacing Escherichia coli. Clin Vaccine Immunol 15:1208–1213
Wagner S, Königsmaier L, Lara-Tejero M, Lefebre M, Marlovits TC, Galán JE (2010) Organization and coordinated assembly of the type III secretion export apparatus. Proc Natl Acad Sci U S A 107:17745–17750
Wainwright LA, Kaper JB (1998) EspB and EspD require a specific chaperone for proper secretion from enteropathogenic Escherichia coli. Mol Microbiol 27:1247–1260
Wang D, Roe AJ, McAteer S, Shipston MJ, Gally DL (2008) Hierarchal type III secretion of translocators and effectors from Escherichia coli O157:H7 requires the carboxy terminus of SepL that binds to Tir. Mol Microbiol 69:1499–1512
Wang S, Liu X, Xu X, Yang D, Wang D, Han X, Shi Y, Tian M, Ding C, Peng D et al (2016) Escherichia coli type III secretion system 2 ATPase EivC Is involved in the motility and virulence of avian pathogenic Escherichia coli. Front Microbiol 7:1–14
Wang S, Xu X, Liu X, Wang D, Liang H, Wu X, Tian M, Ding C, Wang G, Yu S (2017) Escherichia coli type III secretion system 2 regulator EtrA promotes virulence of avian pathogenic Escherichia coli. Microbiology (United Kingdom) 163:1515–1524
Winardhi RS, Gulvady R, Mellies JL, Yan J (2014) Locus of enterocyte effacement-encoded regulator (Ler) of pathogenic Escherichia coli competes off histone-like nucleoid-structuring protein (H-NS) through noncooperative DNA binding. J Biol Chem 289:13739–13750
Wong AR, Pearson JS, Bright MD, Munera D, Robinson KS, Lee SF, Frankel G, Hartland EL (2011) Enteropathogenic and enterohaemorrhagic Escherichia coli: even more subversive elements. Mol Microbiol 80(6):1420–1438. https://doi.org/10.1111/j.1365-2958.2011.07661.x. Epub 2011 May 5
Wood SE, Jin J, Lloyd SA (2008) YscP and YscU switch the substrate specificity of the Yersinia type III secretion system by regulating export of the inner rod protein YscI. J Bacteriol 190:4252–4262
Yip CK, Finlay BB, Strynadka NCJ (2005) Structural characterization of a type III secretion system filament protein in complex with its chaperone. Nat Struct Mol Biol 12:75–81
Zarivach R, Vuckovic M, Deng W, Finlay BB, Strynadka NCJ (2007) Structural analysis of a prototypical ATPase from the type III secretion system. Nat Struct Mol Biol 14:131–137
Zhang L, Chaudhuri RR, Hobman JL, Patel MD, Antony C, Sarti D, Roe AJ, Vlisidou I, Shaw RK, Falciani F et al (2004) Regulators encoded in the Escherichia coli type III secretion system 2 gene cluster influence exression of genes withing the locus of enterocyte effacement in Enterohemorrhagic E. coli O157:H7. Infect Immun 72:7282–7293
Zhou M, Guo Z, Duan Q, Hardwidge PR, Zhu G (2014) Escherichia coli type III secretion system 2: a new kind of T3SS? Vet Res 45:32
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Slater, S.L., Sågfors, A.M., Pollard, D.J., Ruano-Gallego, D., Frankel, G. (2018). The Type III Secretion System of Pathogenic Escherichia coli. In: Frankel, G., Ron, E. (eds) Escherichia coli, a Versatile Pathogen. Current Topics in Microbiology and Immunology, vol 416. Springer, Cham. https://doi.org/10.1007/82_2018_116
Download citation
DOI: https://doi.org/10.1007/82_2018_116
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-99663-9
Online ISBN: 978-3-319-99664-6
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)