
Abstract
Acute myelogenous leukemia (AML) is a bone marrow disease in which the leukemic cells show constitutive release of a wide range of CCL and CXCL chemokines and express several chemokine receptors. The AML cell release of various chemokines is often correlated and three release clusters have been identified: CCL2–4/CXCL1/8, CCL5/CXCL9–11, and CCL13/17/22/24/CXCL5. CXCL8 is the chemokine usually released at highest levels. Based on their overall constitutive release profile, patients can be classified into distinct subsets that differ in their T cell chemotaxis towards the leukemic cells. The release profile is modified by hypoxia, differentiation status, pharmacological interventions, and T cell cytokine responses. The best investigated single chemokine in AML is CXCL12 that binds to CXCR4. CXCL12/CXCR4 is important in leukemogenesis through regulation of AML cell migration, and CXCR4 expression is an adverse prognostic factor for patient survival after chemotherapy. Even though AML cells usually release high levels of several chemokines, there is no general increase of serum chemokine levels in these patients and the levels are also influenced by patient age, disease status, chemotherapy regimen, and complicating infections. However, serum CXCL8 levels seem to partly reflect the leukemic cell burden in AML. Specific chemokine inhibitors are currently being developed, although redundancy and pleiotropy of the chemokine system are obstacles in drug development.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Treg Cell
- Acute Myelogenous Leukemia
- Bone Marrow Stromal Cell
- Allogeneic Stem Cell Transplantation
- CXCR4 Expression
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Introduction
Chemokines are involved in the regulation of cell survival, proliferation, and trafficking (Bendall 2005; Tanaka et al. 2005; Balkwill 2004; Rosenkilde and Schwartz 2004; Allavena et al. 2005). All these processes are important in the development of acute myelogenous leukemia (AML), an aggressive bone marrow malignancy, and the AML patients are often subclassified according to their prognosis, that is, risk of primary therapy resistance or later disease relapse (Estey and Döhner 2006; Harris et al. 1999). Primary human AML cells usually show constitutive release of a wide range of chemokines and have several chemokine receptors on the cell’s surface. These chemokine/chemokine receptor expression patterns are probably important for both disease development (i.e., leukemogenesis) and chemosensitivity (i.e., response to therapy).
2 Primary Human AML Cells Often Show Constitutive Chemokine Release
The AML cells as well as their neighboring bone marrow stromal cells produce survival- and growth-regulatory cytokines, including chemokines belonging to both the CCL and CXCL subclasses (Bruserud et al. 2007; Balkwill 2004). The remaining normal hematopoietic cells and bone marrow infiltrating immunocompetent cells also release chemokines and express a wide range of chemokine receptors (Bruserud et al. 2007; Laurence 2006; Moser and Loetscher 2002; Christopherson and Hromas 2001; Mantovani et al. 2004; Broxmeyer 2008; Homey et al. 2002; Qin et al. 1998; Kim 2006; Honczarenko et al. 2006; Cignetti et al. 2003; Jin et al. 2007). For example, the stromal cells (1) release CCL2, CCL4, CCL5, CCL20, CXCL8, CXCL12, and CX3CL1 and (2) express CCR1, CCR7, CCR9, CXCR4, CXCR5, and CXCR6 (Honczarenko et al. 2006). The chemokines thereby constitute a bidirectional interacting network between leukemic and nonleukemic cells.
2.1 Constitutive Chemokine Release by Primary Human AML Cells
A broad constitutive chemokine release profile is often detected in AML, but the profile shows both qualitative and quantitative differences between individual patients (Bruserud et al. 2007). The release of different chemokines is often correlated so that distinct release clusters can be identified: (1) CCL2–4/CXCL1/8, (2) CCL5/CXCL9–11 (possibly also CCL23), and (3) CCL13/17/22/24/CXCL5 (possibly also CXCL6). This means that individual patients usually show either high or low release for all chemokines within the same cluster; the molecular mechanisms behind this coordinated release are not known, but common transcriptional regulation seems to be important for at least the CCL2–4/CXCL1/8 cluster. It should be emphasized that there is a wide variation in the release of each chemokine between individual patients, and this is illustrated by the summary of the overall results presented in Table 1 (Bruserud et al. 2007). For many patients, additional chemokines are also released. Individual AML patients can therefore be subclassified based on their overall chemokine release profile (Bruserud et al. 2007):
-
A relatively large group (approximately 20–30% of patients) shows undetectable or low levels of most chemokines with decreased in vitro chemotaxis of immunocompetent cells towards the AML cells
-
The majority of the other patients shows relatively high release for the CCL2–4/CXCL1/8 chemokine cluster eventually in combination with other single chemokines
-
The remaining minority shows high CCL2–4/CXCL1/8 levels and in addition high levels of the CCL13/17/22/24/CXCL5 and CCL5/CXCL9–11 clusters
No single chemokine or chemokine cluster showed any correlations with clinical or biological AML cell characteristics (i.e., morphology, membrane molecule expression, genetic abnormalities) in this study (Bruserud et al. 2007). Taken together, these observations therefore suggest that the chemokine release profile rather than single chemokines should be examined in biological studies of human AML.
2.2 Modulation of the Constitutive Chemokine Release
Even though the constitutive chemokine release by primary human AML cells seems to be carefully controlled and appears in clusters (see Sect. 2.1), several factors can modulate the release profile. However, this modulation will often be similar for chemokines within the same cluster.
2.2.1 Differentiation Induction
Cytokines, chemotherapeutics, all-trans retinoic acid (ATRA), and vitamin D3 can induce differentiation of AML blasts towards a dendritic cell phenotype (Bruserud and Gjertsen 2000). This phenotype includes altered chemokine levels with high release of CCL17 and CCL22 similar to normal dendritic cells but usually without effects on other chemokines in the CCL13/17/22/24/CXCL5 cluster (Olsnes et al. 2008).
2.2.2 Tissue Oxygenation
The oxygen pressure (pO2) in human bone marrow is decreased and is estimated to be 50–55 mmHg (atmospheric pO2 corresponding to 140–160 mmHg) (Harrison et al. 2002; Cummins and Taylor 2005). The most important hypoxia-responsive transcription factor is HIF-1, which is known to directly regulate CXCL12 and CXCR4 expression and increase the expression of proangiogenic CXCL8 (Wenger et al. 2005; Hirota and Semenza 2006; Lisy and Peet 2008). Exposure of primary human AML cells to hypoxia increases HIF-1 levels and the release of several other chemokines especially within the CCL2–4/CXCL1/8 cluster (Hatfield, unpublished data).
2.2.3 Pharmacological Interventions
NF-κB is important for transcriptional regulation of several chemokines and can be targeted by specific inhibitors and by the proteasomal inhibitor bortezomib. NF-κB expression by primary AML cells correlates with mRNA and protein levels of the CCL2–4/CXCL1/8 release cluster, an observation further supporting that common transcriptional regulation is important for this clustering (Bruserud et al. 2007). The specific inhibitor BMS345541 decreases the release of these chemokines, and bortezomib also decreases these chemokines, except for CXCL8, which is increased (Bruserud et al. 2007; Olsnes et al. 2009). The most likely explanation for the CXCL8 discrepancy between these two drugs is that bortezomib has additional effects and not only inhibits NF-κB.
The protein kinase C δ agonist PEP005 induces growth inhibition and apoptosis of primary human AML cells together with increased release of several T cell chemotactic chemokines, especially chemokines within the CCL2–4/CXCL1/8 and CCL5/CXCL9–11 clusters (Olsnes et al. 2009). Such a combination of direct antileukemic effects and immunostimulation through increased local T cell recruitment is uncommon and may result in synergistic antileukemic effects.
The drug JTE-607 inhibits the release of several cytokines. In a murine AML model, it had an antileukemic effect comparable to the maximum tolerable dose of cytarabine and was associated with decreased CXCL8 levels (Uesato et al. 2006). These decreased CXCL8 levels may be caused by decreased constitutive AML cell release, but it is not known whether the antileukemic activity depends on this effect. Furthermore, all-trans retinoic acid (ATRA) is mandatory in the treatment of acute promyelocytic leukemia (APL) (Bruserud and Gjertsen 2000), and it is also tried in the treatment of other AML variants (Bruserud et al. 2006). In vitro studies have shown that ATRA or vitamin D3 derivatives can increase CXCR1 expression (Zahn et al. 1997) as well as decrease CXCL8 release by myeloid leukemia cells (Dubois et al. 1994; Srivastava and Ambrus 2004). The overall chemokine release profiles were not characterized in these pharmacological studies, and it is not known whether other chemokines within the CCL2–4/CXCL1/8 cluster also are affected. It is not known whether such effects contribute to the disease-stabilization observed for a subset of AML patients receiving ATRA-based palliative therapy (Bruserud et al. 2006).
2.2.4 Nonleukemic Stromal Cells
The bidirectional crosstalk between primary human AML cells and their neighboring nonleukemic stromal cells alters AML cell release of both CCL and CXCL chemokines. This has been observed both for fibroblasts, osteoblasts, and endothelial cells, but the wide variation in chemokine release between individual patients is maintained even in the presence of stromal cells (Bruserud et al. 2004; Olsnes et al. 2008; Glenjen et al. 2003, 2004; Hatfield et al. 2006, 2009).
Leukemic cells from most AML patients show a high constitutive release of CXCL8 (Bruserud et al. 2007). The cytokine crosstalk between AML cells and microvascular endothelial cells, fibroblasts, or osteoblasts increases the local CXCL8 levels and the proliferation of these nonleukemic stromal cells (Bruserud et al. 2004; Hatfield et al. 2006, 2008, 2009; Ryningen et al. 2005). Furthermore, CXCL8-binding receptors are expressed both by AML cells and endothelial cells (Bruserud et al. 2007; Tobler et al. 1993; Strieter et al. 1995; Xie 2001); AML-derived CXCL8 may therefore be involved both in autocrine and paracrine circuits in the bone marrow microenvironment.
The high levels of proangiogenic CXCL8 may contribute to the increased microvessel density in AML bone marrow (de Bont et al. 2001; Hatfield et al. 2005). Primary AML cells show constitutive release of several additional proangiogenic mediators, although there are both qualitative and quantitative differences between individual patients (Bruserud et al. 2007; Lee et al. 2007a). Among these proangiogenic nonchemokine mediators are angiopoietin-1 (Ang-1), Ang-2, hepatocyte growth factor (HGF), vascular endothelial growth factor (VEGF), platelet-derived growth factor (PDGF), interleukin-6 (IL-6), matrix metalloproteases (MMPs), and IL-1. The AML cells also show constitutive release of antiangiogenic molecules, including CXCL9–11, IL-12, and thrombospondin (Bruserud et al. 2004; Hatfield et al. 2005). However, the constitutive release of antiangiogenic CXCL9–11 is lower than the CXCL8 release (Bruserud et al. 2007), showing that at least for the angioregulatory chemokines the balance is in favor of angiogenesis.
2.2.5 Cellular Immune Responses
Leukemia-directed T cell reactivity is important for the antileukemic effect of allogeneic stem cell transplantation (Ersvaer et al. 2007a, b; Paczesny et al. 2010; Engelhardt and Crowe 2010; Kittan and Hildebrandt 2010; Löffler et al. 2010), and antileukemic immune effects may also be important in patients receiving conventional chemotherapy (Ersvær et al. 2007b). IFN-γ is released at high levels by activated T cells derived from healthy individuals (Bruserud et al. 1993), patients receiving allogeneic (Bruserud et al. 1993) and autologous (Wendelbo et al. 2004a) stem cell transplantation, and patients with severe chemotherapy-induced cytopenia (Wendelbo et al. 2004b); IFN-γ reduces the constitutive release of proangiogenic CXCL8 and increases antiangiogenic CXCL9–11 by primary human AML cells (Ersvaer et al. 2007a). Antiangiogenic effects may thereby become a part of antileukemic T cell reactivity.
3 Chemokine Receptors on Primary Human AML Cells
We previously examined CCR1–5 and CXCR1–4 expression at the protein level by primary human AML cells (Bruserud et al. 2007). These nine receptors can bind 18 CCL (CCL2–5, 7, 8, 11–17, 22–24, 26, 28) and 11 CXCL chemokines (CXCL1–3,5–12) (Bendall 2005; Tanaka et al. 2005; Balkwill 2004; Rosenkilde and Schwartz 2004; Allavena et al. 2005). When comparing the expression for the total AML cell populations, the chemokine receptor expression varied considerably: (1) CCR3 and CXCR1 showed low levels for all patients; (2) CCR5, CXCR2, and CXCR3A generally showed intermediate expression; and (3) CCR1, CCR2, CCR4, and CXCR4 showed relatively high expression (Bruserud et al. 2007).
We have now analyzed the associations among the expression of these nine chemokine receptors, genetic abnormalities, and differentiation status for the patients included in our previous study (Bruserud et al. 2007). No clustering of receptor expression was observed similar to the chemokine release. Surprisingly, these additional studies demonstrated that Flt3-internal tandem duplication (ITD) was associated with decreased CCR1 and CXCR4 expression in these relatively old patients with severe leukemization (Fig. 1). Furthermore, high CCR1 and CCR2 expression among total AML cells was also associated with morphological signs of monocytic differentiation (Fig. 1), and the expression of these two receptors was also inversely correlated with expression of the CD34 stem cell marker (data not shown).

Chemokine receptor expression by primary human AML cells (Bruserud et al. 2007). Receptor expression was analyzed by flow cytometry and the results are presented as the percentage of positive cells. Morphological signs of differentiation was classified according to the French–American–British classification, and monocytic differentiation is then classified as M4/M5. The figure shows that primary AML cells with morphological signs of monocytic differentiation have an increased expression of CCR1 (Mann-Whitney, U-test, p = 0.03) and CCR2 (p = 0.023) (upper part). Furthermore, Flt3-ITD, which is a genetic abnormality associated with adverse prognosis, is associated with decreased expression of CCR1 (p = 0.048) and CXCR4 (p = 0.047) (lower part)
The chemokine receptor expression varies within the AML cell population in each patient (Bruserud et al. 2007). We observed increased expression of several receptors by the leukemic CD34+ subset (often only a minority) compared with the CD34− cell subset in the same patient (Bruserud et al. 2007). This was most clearly seen for CCR5 and CXCR3A and also for CCR1, CCR2, and CCR4, and this difference was not altered by in vitro exposure to hematopoietic growth factors.
Our previous studies have demonstrated that primary human AML cells show constitutive release of several chemokines, and as described earlier, the leukemic cells also express the receptors for these chemokines. Even though autocrine circuits are formed thereby, this receptor/ligand expression is not associated with autocrine proliferation. Furthermore, for most patients exogenous chemokines do not affect spontaneous or cytokine-dependent AML cell proliferation either, although altered proliferation is observed for a minority of patients, with growth enhancement being most common. For these exceptional patients, altered proliferation was observed also for the more immature clonogenic cells. Thus, most chemokines have only minor direct effects on growth regulation in the AML cells.
4 The CXCL12/CXCR4 System in Human AML
4.1 CXCR4 and CXCL12 Expression in AML Bone Marrow
CXCR4 expression is detectable at the mRNA level for the large majority of patients (Cignetti et al. 2003), and studies at the protein level have confirmed this (Bruserud et al. 2007; Möhle et al. 1998) with an average percentage of CXCR4+ cells comparable to normal CD34+ hematopoietic cells (Möhle et al. 1998). However, the variation between patients is much wider than the variation between normal CD34+ cells from healthy individuals (Möhle et al. 1998). Some studies suggest that CXCR4 expression is strongest for AML cells with a monocytic phenotype and in APL (Löffler et al. 2010; Cignetti et al. 2003; Möhle et al. 2000); in case of monocytic differentiation, the increased CXCR4 expression seems to be a part of a more complex phenotype with increased expression of other chemokine receptors (CCR1, CCR2), costimulatory molecules (CD40, CD86), death receptors (TNFR1, TNFR2, Fas), and several adhesion molecules (Burger and Kipps 2006; Brouwer et al. 2001).
Detectable release of CXCL12, the only CXCR4 ligand, by primary human AML cells is seen only for a minority of patients (Bruserud et al. 2007; Cignetti et al. 2003). Less than half of the patients show detectable mRNA expression (Cignetti et al. 2003), and when investigating CXCL12 release by in vitro cultured AML cells, low but detectable levels were seen only for ten out of 68 patients (Table 1) (Bruserud et al. 2007). Thus, autocrine CXCR4/CXCL12 loops are probably uncommon in human AML. The major source of CXCL12 in AML bone marrow seems to be the constitutive release by various stromal cells (Brouwer et al. 2001), including osteoblasts in endosteal stem cell niches and endothelial cells in vascular niches. At these sites, CXCL12 may facilitate survival, self-renewal, and localization of normal stem cells and possibly also leukemic cells (Broxmeyer 2008).
4.2 Biological Effects of CXCR4-Initiated Signaling in AML
CXCR4 cooperates with the very late antigen (VLA)-4 and other integrins, the hyaluronan receptor CD44, and possibly also the surface sialomucin podocalyxin in the regulation of AML cell adhesion and migration (Burger 2009; Burger and Bürkle 2007; Burger et al. 2003; Riccioni et al. 2006; Voermans et al. 2002; Jin et al. 2006; Tavernier-Tardy et al. 2009). Both CXCR4 and VLA-4 seem to mediate resistance to cytarabine-induced apoptosis through these interactions (Burger et al. 2003). CXCR4 is thereby a part of a larger functional entity that seems important for anchoring AML cells to the bone marrow and for possibly facilitating their migration to stem cell niches, with the maintenance of their immature phenotype (Rombouts et al. 2004). Finally, the hypoxic bone marrow microenvironment causes upregulation of CXCR4 expression, failure to internalize CXCR4 in response to CXCL12 ligation, and altered shedding of soluble CXCR4 (Fiegl et al. 2009). Thus, CXCR4 is not only important for migration and differentiation but also for the adaption to the hypoxic microenvironment.
Cellular microparticles are submicron vesicles that are shed from the plasma membrane, and CXCR4+ microparticles are detected both in the peripheral blood and bone marrow plasma of healthy individuals as well as AML patients (Kalinkovich et al. 2006). CXCR4+ microparticles are increased in AML and express CD45, whereas most microparticles in healthy individuals express CD41. In vitro studies have demonstrated that these microparticles can transfer biologically active CXCL12 to AML cells.
Whether CXCR4 is important for AML cell migration outside the human bone marrow remains controversial. One study described an association between the CXCR4 G801A gene polymorphism and extramedullary disease (Dommange et al. 2006), but this association was not observed in another study (Ponziani et al. 2008). Other chemokines may also influence extramedullary AML cell trafficking since another study described a correlation between extramedullary AML and coexpression of CCL2/CCR2 (Cignetti et al. 2003). Finally, two relatively small studies including only 11 and 21 patients, respectively, showed conflicting results with regard to whether CXCR4 is important for engraftment of human AML cells in NOD/SCID mice (Tavor et al. 2004; Monaco et al. 2004a, b).
4.3 CXCR4/CXCL12 Has a Prognostic Impact in Human AML
CXCR4 expression is significantly increased in AML cells derived from patients with Flt3-ITD (Rombouts et al. 2004) (Fig. 1). Therefore, to investigate the prognostic impact of CXCR4 expression independent of the Flt3-ITD effect, Konoplev et al. (Konoplev et al. 2007) analysed survival after chemotherapy for patients with normal karyotype and no Flt3-ITD. CXCR4 was expressed by the AML cells for 70% of the patients. The initial complete remission rate did not differ, but patients with CXCR4+ leukemic cells had decreased event-free and overall long-term survival. This was later confirmed by others (Spoo et al. 2007). Taken together, these results suggest that high CXCR4 expression has an adverse prognostic impact independent of Flt3-ITD.
A small study investigated the prognostic impact of CXCR4, VLA-4, and focal adhesion kinase (FAK) in AML (Tavernier-Tardy et al. 2009). CXCR4 cooperates with VLA-4 in AML cell migration (Burger et al. 2003), and FAK is also important in cell adhesion by regulating multiple signal-transduction pathways (Sieg et al. 2000). The expression of each single molecule was associated with decreased overall survival, but the strongest impact was observed for patients showing combined expression of at least two or all three markers. These observations suggest that the adverse prognosis associated with CXCR4 reflects the impact of a more complex phenotype.
4.4 CXCR4 as a Possible Therapeutic Target in Human AML
Several CXCR4 inhibitors have been developed (Zeng et al. 2006, 2009; Tavor et al. 2008; Liesveld et al. 2007; Nervi et al. 2009; Li et al. 2008), and clinical studies have demonstrated that CXCR4 inhibition can be used for mobilization of peripheral blood stem cells (Calandra et al. 2010). However, the use of CXCR4 inhibitors in AML therapy is also supported by several experimental observations:
-
CXCR4 inhibitors decrease chemotaxis of human AML cell lines against CXCL12 or bone marrow stromal cells (Zeng et al. 2009; Tavor et al. 2008; Li et al. 2008) and inhibit transmigration of AML cells through stromal and endothelial cell monolayers (Liesveld et al. 2007)
-
Bone marrow stromal cells have a protective effect against chemotherapy-induced apoptosis in primary human AML cells, and CXCR4-antagonists decrease this protection and enhance the proapoptotic effects of the cytotoxic drug cytarabine (Zeng et al. 2009; Tavor et al. 2008). This effect is possibly mediated through inhibition of CXCL12-mediated activation of ERK and AKT (Zeng et al. 2009; Tavor et al. 2008). The chemosensitizing effect has also been detected in vivo in murine AML models (Nervi et al. 2009)
-
Flt3-ITD activates CXCR4 signaling, and CXCR4 inhibition then increases the sensitivity of Flt3-ITD+ leukemic cells to proapoptotic Flt3 inhibitors (Zeng et al. 2009)
-
CXCR4 inhibition induces differentiation and proliferation arrest in U937 AML cells, possibly through inhibition of CXCL12-dependent elastase that is constitutively expressed (Tavor et al. 2008)
-
Studies in murine models have shown that CXCR4 inhibitors decrease bone marrow homing and thereby mobilize both normal and leukemic cells from the bone marrow to the blood (Zeng et al. 2009; Nervi et al. 2009)
These effects were observed with the inhibitory polypeptide RCP168 or the second-generation small molecule reversible CXCR4 inhibitors AMD3465 or AMD3100. Similar effects can also be induced by berberine, an isoquinoline derivative that inhibits stromal cell release of CXCL12 (Li et al. 2008).
5 Leukemogenesis Through Transcriptional Regulation in the Chemokine System
MEIS1 is a HOX cofactor that contributes to leukemogenesis in AML (Bruserud et al. 2006). Results from an animal AML model demonstrated that MEIS1 upregulated Flt3 and occupied regulatory sequences of the Flt3 as well as the CCL3, CCL4, and CXCL4 genes (Argiropoulos et al. 2008). CCL3 was then important for the marrow-repopulating activity of AML cells, suggesting that altered chemokine expression is involved in leukemogenesis.
The NF-κB transcription factor is another regulator of chemokine expression in AML cells (Bruserud et al. 2007), and it is also regarded as important in leukemogenesis (Olsnes et al. 2009; Reikvam et al. 2009). Furthermore, the histone acetyltransferase Monocyte zinc finger (MOZ) increases CXCL8 release through a direct interaction with the p65 subunit of the NF-κB complex; MOZ can also be rearranged in human AML, and the fusion protein formed with the coactivator CREB binding protein (CBP) is then important in leukemogenesis (Bruserud et al. 2006). Thus, AML-associated genetic abnormalities that are regarded as important contributors in leukemogenesis may mediate their leukemogenic effects through the chemokine regulator NF-κB and thereby increase expression of CXCL8 and possibly also other NF-κB regulated chemokines (Bruserud et al. 2007). The same mechanism may be operative for translocations involving the RUNX1 or AML-1 transcription factor because MOZ also interacts with this transcription factor and thereby increase CCL3 expression (Mrózek et al. 2004; Bristow and Shore 2003), another member of the CCL2–4/CXCL1/8 release cluster (see Sect. 2.1).
Even though the molecular details behind transcriptional regulation of chemokine expression in primary human AML cells are largely unknown, the regulation seems to involve several transcription factors (NF-κB, MOZ, RUNX1) that can be involved in AML-associated genetic abnormalities. These results suggest that several chemokines and not only CXCL12/CXCR4 may contribute in leukemogenesis.
6 Chemokine Serum Levels in AML
Even though AML cells show constitutive release of several chemokines, there is no general increase in the serum levels of these mediators in untreated patients. However, increased CXCL8 serum levels are detected for patients with untreated disease and especially for patients with monocyte AML variants (Hsu et al. 2002; Liu et al. 1999; Negaard et al. 2009). These levels normalize when patients achieve complete hematological remission (Hsu et al. 2002), but increased levels can later be detected as a part of the acute phase reaction during febrile neutropenia and especially in patients with septicemia or septic shock (Ostermann et al. 1994; Bruserud et al. 1996; Schönbohn et al. 1995). Furthermore, increased levels of CCL2, CXCL10 (only younger patients), and CXCL12 have also been detected in patients with untreated disease (Kalinkovich et al. 2006; Mazur et al. 2007; Olsnes et al. 2006). The increased levels of total CXCL12 are then accompanied by decreased levels of the functional noncleaved form (Kalinkovich et al. 2006). Neither CCL2 nor CXCL10 levels are affected by chemotherapy (Mazur et al. 2007; Olsnes et al. 2006), and increased CXCL10 levels persist even after induction of hematological remission (Olsnes et al. 2006). Finally, CCL17 levels are decreased and CCL18 levels are not altered in patients with untreated disease, and CCL17 levels will decrease further following intensive chemotherapy and during febrile neutropenia (Olsnes et al. 2006; Struyf et al. 2003). We therefore conclude that systemic chemokine levels in patients with untreated AML are determined by several factors and not only by the constitutive AML cell release.
7 Chemotaxis of Immunocompetent Cells in Human AML
7.1 T Cell Chemotaxis
Experimental studies have demonstrated that T cells are able to migrate towards primary AML cells, but the T cell chemotaxis varies between patients and is decreased for those patients who do not show constitutive chemokine release (see Sect. 2.1) (Bruserud et al. 2007). CCL5 and CXCL10 contribute to the chemotaxis but it is likely that other chemokines are also involved because AML cells often show constitutive release of several T cell chemotactic chemokines, including CCL1–5/7/11/13/17/20–22 and CXCL6/8–12 (Bruserud et al. 2007; Olsnes et al. 2006). Especially, CXCL8 is usually released at high levels for most patients and normal CD4+ as well as CD8+ T cells migrate after stimulation with CXCL8 (Ward et al. 1998). However, the T cell population in untreated AML patients is abnormal with increased numbers of circulating T cells, cytotoxic CD3+56+ T cells are frequently oligoclonal and in a higher state of activation with abnormal gene expression profiles, and these T cells are unable to form effective immune synapses with autologous AML cells (Le Dieu et al. 2009). The T cell population normalizes after remission induction, but it is not known whether chemotaxis towards AML cells is abnormal in untreated AML or after achievement of complete hematological remission.
7.2 Chemotaxis of Regulatory T Cells
T lymphocytes generally express several chemokine receptors (Ward et al. 1998; Campbell et al. 2003; Muller et al. 2002), and regulatory T (Treg) cells show a distinct expression profile and seem to be highly attracted by CCR4 ligation (CCL17, CCL22) and by ligation of CCR8 (CCL1) that seems to be more selectively expressed on Treg cells (Engelhardt and Crowe 2010; Iellem et al. 2001). Primary AML cells can be induced to differentiate towards AML-dendritic cells with high release of CCL17 and CCL22 (Olsnes et al. 2008; Köhler et al. 2000), and several normal immunocompetent T cell subsets (CD4+ and CD8+ T cells, Treg cells) show increased migration towards such cells (Olsnes et al. 2008). However, even in the presence of CCL17/CCL22-neutralizing antibodies, the number of migrating cells was higher than for primary AML cells, an observation clearly demonstrating that other chemokines are also involved.
Treg cells seem to have a stronger migration towards dendritic AML cells than other T cell subsets (Olsnes et al. 2008). Animal studies suggest that circulating Treg cells express CXCR4, CCR2, CCR5, CCR6, and CCR9 in addition to CCR 4 and CCR8 (see above) (Lee et al. 2007b; Yi et al. 2006). This expression profile shows that Treg chemotaxis will depend on the overall local chemokine network, although it is known that certain chemokines may have a predominant role in certain clinical situations (Haas et al. 2007; Olkhanud et al. 2009).
The frequency of circulating Treg cells is increased in patients with untreated AML (Szczepansky et al. 2009). The increased levels persist after remission induction, an observation suggesting that this is a disease-induced and chemoresistant immunomodulation with a biological impact even after achievement of disease control. The constitutive AML cell release of several Treg-recruiting chemokines may lead to colocalization of leukemic and Treg cells. This may explain the adverse prognostic impact of high pretherapy levels of circulating Treg cells (Szczepansky et al. 2009).
7.3 Chemotaxis of Monocytes
Previous in vitro studies have shown that the migration of normal monocytes towards primary human AML cells differs between patients (Legdeur et al. 1997, 2001). For a minority of patients this migration is low, but for most patients a high degree of migration is observed and CCL2 (a ligand of the CCR2 receptor) is the most important single chemotactic chemokine. These observations are also consistent with the studies of constitutive chemokine release by primary human AML cells (see Sect. 2.1); CCL2 is released at relatively high levels for most patients but often together with other CCR2 ligands or monocyte-chemotactic chemokines (Bruserud et al. 2007). The high monocyte migration towards AML cells is therefore expected. Furthermore, CD40 ligation of AML cells will increase the release of chemotactic chemokines, including CCL5 and CXCL8, and thereby increase monocyte as well as T and NK cell chemotaxis (Costello et al. 2000). The recruited monocytes may then have cytotoxic effects against AML cells, but the CCL2 effect is limited to monocyte migration without any effect on the antileukemic cytotoxicity of the recruited cells (Legdeur et al. 1997, 2001). Alternatively, the recruited monocytes may represent an AML-stimulating mechanism through their release of proangiogenic mediators (Dimberg 2010).
7.4 Chemotaxis of Natural Killer Cells
Natural killer (NK) cells can also mediate antileukemic activity, and they express several chemokine receptors (ligands given in parenthesis), including CCR1 (CCL2/3/5/7/14–16/23), CCR4 (CCL17/22), CCR6 (CCL20), CCR7 (CCL19/21) CXCR1 (CXCL6/7/8), CXCR3 (CXCL9–11), CXCR4 (CXCL12), CXCR6 (CXCL16), and CX3CR1 (CX3CL1) (Maghasachi 2010). The expression of the individual receptors may differ between various NK cell subsets and may also be dependent on the activation status of the cells, as described in detail by Maghasachi (Maghasachi 2010). However, it can be seen that many of these ligands are constitutively released by AML cells (see Table 1 and Sect. 2.1), including the chemokines within the CCL2–4/CXCL1/8 cluster that are released for most patients. One would therefore expect NK cells to migrate towards primary human AML cells.
8 Chemokine-Mediated Suppression of Normal Hematopoiesis
AML is a bone marrow disease, and leukemia-induced bone marrow failure is an important clinical characteristic (Estey and Döhner 2006). Previous experimental studies have shown that several chemokines have direct or indirect effects on normal hematopoiesis. First, several chemokines seem to directly inhibit normal hematopoiesis, including CCL3, CXCL4, CXCL5, and CXCL8 (Dimberg 2010; Lambert et al. 2007). For CCL chemokines, the suppression seems to be linked to a specific molecular motif that was identified in the inhibitory CCL3 but not in the noninhibitory CCL5 (Ottersbach et al. 2006). Residues within this region probably contribute to the binding of other inhibitory chemokines to their receptors (Bondue et al. 2002; Lecomte-Raclet et al. 2000), and based on the comparison of CCL3 and CCL5, the inhibition is probably mediated through the formation of a helical turn preceding the first β-strand in CCL3 (Ottersbach et al. 2006). Second, CCL18 and CCL2 seem to stimulate hematopoiesis (Broxmeyer 2008), but this is probably an indirect effect mediated through growth factor release from neighboring monocytes (Wimmer et al. 2006). Finally, injection of chemokines into mice has demonstrated that several of these mediators affect normal hematopoiesis, but it is not known whether direct or indirect effects are most important. Dose-dependent in vivo suppression has then been demonstrated for CCL2, CCL3, CCL19, CCL20, CXCL4, CXCL5, CXCL8, CXCL9, and XCL1 (Broxmeyer et al. 2006). Several chemokine combinations showed synergistic inhibitory effects, and suppression of hematopoiesis was associated with accelerated recovery in response to the toxic effects of cytarabine.
Taken together, these observations demonstrate that chemokine-induced suppression of hematopoiesis seems more common than stimulation, and Table 1 shows that several of the suppressing chemokines are constitutively released at high levels by primary human AML cells, especially the cluster I chemokines CCL2–4/CXCL5/8. Constitutive chemokine release may thereby contribute to the disease-associated bone marrow failure in AML. Chemokine effects on normal hematopoiesis thus differ from leukemic hematopoiesis where most chemokines either have no or weak enhancing effects on AML cell proliferation (see Sect. 3). Finally, enhancement of chemokine-mediated myelosuppression may represent a possible therapeutic strategy for myeloprotection in patients receiving intensive anticancer therapy, and this may be achieved through pharmacological inhibition of the chemokine-degrading decoy receptors (Bonecchi et al. 2010).
9 Concluding Remarks
Hanahan et al. (Hanahan and Weinberg 2000) suggested that malignant diseases have six fundamental hallmarks, and Mantovani et al. (Mantovani 2009) later suggested that cancer-associated inflammation is a seventh hallmark. The local chemokine network can affect all these hallmarks in human AML (discussed in detail earlier):
-
The three characteristics associated with cancer cell proliferation are limitless replicative potential, self-sufficiency in growth signals, and insensitivity to antigrowth signals. Chemokines can affect the growth of primary human AML cells directly, but in our available in vitro models, this is observed only for a minority of patients (Bruserud et al. 2007) and paracrine mechanisms are more likely to be involved. The importance of paracrine circuits is also supported by experimental studies describing expression of several chemokine receptors, by bone marrow stromal cells, including CCR1, CCR7, CCR9, CXCR4–6 (Honczarenko et al. 2006). Among these receptors, CCR1 binds at least three of the chemokines secreted by AML cells, namely CCL3, CCL5, and CCL13. When cultured in serum-free medium, the stromal cells release several chemokines that can bind to receptors expressed by the AML cells, including CCL2, CCL4, CCL5, CCL20, CXCL8, CXCL12, and CXC3L1 (Honczarenko et al. 2006).
-
Evading apoptosis. Inhibition of chemokine signaling (i.e., CXCR4 antagonists) potentiates proapoptotic chemotherapy effects (Fig. 2).
-
Sustained angiogenesis. Several proangiogenic chemokines are constitutively released by the AML cells at high levels for almost all patients, while antiangiogenic chemokines are released at lower levels (Bruserud et al. 2007; Dimberg 2010).
-
Tissue evasion and metastasis. The CXCL12/CXCR4 system and CCL2/CCR2 are important for AML cell migration and thereby for bone marrow infiltration.
-
Inflammatory microenvironment. The clinical importance of an inflammatory microenvironment in AML is generally accepted only for patients receiving allogeneic stem cell transplantation, and for these patients chemokine-targeting therapy is now considered as an immunomodulatory treatment. However, the balance between various immunocompetent cells may then be of particular importance (Szczepansky et al. 2009), especially in patients treated with allogeneic stem cell transplantation where this balance is essential for induction of antileukemic T cell reactivity vs. the risk of GVHD due to excess proinflammatory reactivity towards host antigens (Kittan and Hildebrandt 2010).
Thus, therapeutic targeting of the chemokine system would interfere with fundamental cancer cell characteristics or important paracrine mechanisms. This therapeutic targeting of the chemokine system can include specific agents directed against the chemokines or their receptors (Fig. 2). However, downstream intracellular signaling involves several pathways, and specific inhibitors of intracellular mediators are now considered for cancer treatment and would then be expected to modulate chemokine effects on the malignant cells. Modulation of the chemokine network may also become useful in patients receiving allogeneic stem cell transplantation and possibly also when immunotherapy is tried in combination with conventional chemotherapy. Finally, analysis of AML-associated chemokine mRNA expression in bone marrow may become useful in monitoring of treatment responses and detection of minimal residual disease. A recent study described that mRNA expression of CCL23 together with six other disease markers could be used for early detection of AML relapse (Steinbach et al. 2006). Thus, a better understanding of the chemokine system in human AML will probably lead to the development of new diagnostic tools as well as new therapeutic strategies.

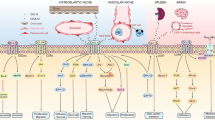
Pharmacological targeting of the chemokine system, a general overview of possible strategies (for additional references see (Bruserud et al. 2007; Olsnes et al. 2009; Zebisch et al. 2007; Hatfield et al. 2005; Tavor et al. 2008; Calandra et al. 2010)). First, specific inhibition can be achieved through specific targeting of chemokines or chemokine receptors. Monoclonal antibodies can then be used either to neutralize chemokines or to inhibit chemokine receptors. Small molecule inhibitors can also effectively target specific chemokine receptors, and nonfunctional chemokines can inhibit chemokine oligomerization or interfere with the binding of chemokines to the extracellular matrix or cell surfaces. Second, chemokine-induced signaling downstream of the chemokine receptors can be altered by specific inhibitors; this last strategy will not be specific for chemokine-initiated signaling because other receptors may also affect the same pathways. Various pathways can then be involved in the intracellular signaling downstream of the receptor, and these are coupled to heterotrimeric G-proteins (subunits α, β, and γ, only the two last functional units being presented in the figure). Specific inhibitors have been developed against several of these mediators, as indicated in the figure, including protein kinase inhibitors, farnesyl transferase inhibitors, and proteasome inhibitors. Here we show only two of the possible signaling pathways that can be activated after receptor ligation and activation of the functional G-protein unit (the βγ-dimer). These inhibitors represent experimental tools, but several of them are also used in clinical therapy, for example, bortezomib and farnesyl transferase inhibitors. (βγ, the βγ subunit of the heterotrimeric G-protein; PI3K, phosphoinositide 3-kinase; Akt, serine/threonine-specific protein kinase and also known as protein kinase B; NF-κB, nuclear factor-κB; Ras, small GTPase; MEK, MAPK/ERK kinase and also known as MAPK kinase; ERK, extracellular-signal regulated kinase and also known as mitogen-activated protein kinase or MAPK)
Abbreviations
- AML:
-
Acute myelogenous leukemia
- ATRA:
-
All-trans retinoic acid
- FAK:
-
Focal adhesion kinase
- HIF:
-
Hypoxia inducible factor
- IL:
-
Interleukin
- ITD:
-
Internal tandem duplication
- MMP:
-
Matrix metalloproteases
- MOZ:
-
Monocyte zinc finger
- NK:
-
Natural killer
- TNF:
-
Tumor necrosis factor
- VLA:
-
Very late antigen
References
Allavena P, Marchesi F, Mantovani A (2005) The role of chemokines and their receptors in tumor progression and invasion: potential new targets of biological therapy. Curr Cancer Ther Rev 1:81–92
Argiropoulos B, Palmqvist L, Yung E, Kuchenbauer F, Heuser M, Sly LM, Wan A, Krystal G, Humphries RK (2008) Linkage of Meis1 leukemogenic activity to multiple downstream effectors including Trib2 and Ccl3. Exp Hematol 36:845–859
Balkwill F (2004) Cancer and the chemokine network. Nat Rev Cancer 4:540–550
Bendall L (2005) Chemokines and their receptors in disease. Histol Histopathol 20:907–926
Bondue A, Jao SC, Blanpain C, Parmentier M, LiWang PJ (2002) Characterization of the role of the N-loop of MIP-1 beta in CCR5 binding. Biochemistry 41:13548–13555
Bonecchi R, Savino B, Borroni EM, Mantovani A, Locati M (2010) Chemokine decoy receptors: structure-function and biological properties. Curr Top Microbiol Immunol, DOI 10.1007/82_2010_19
Bristow CA, Shore P (2003) Transcriptional regulation of the human MIP-1alpha promoter by RUNX1 and MOZ. Nucleic Acids Res 31:2735–2744
Brouwer RE, Hoefnagel J, Borger van Der Burg B, Jedema I, Zwinderman KH, Starrenburg IC, Kluin-Nelemans HC, Barge RM, Willemze R, Falkenburg JH (2001) Expression of co-stimulatory and adhesion molecules and chemokine or apoptosis receptors on acute myeloid leukaemia: high CD40 and CD11a expression correlates with poor prognosis. Br J Haematol 115:298–308
Broxmeyer HE (2008) Chemokines in hematopoiesis. Curr Opin Hematol 15:49–58
Broxmeyer HE, Pelus LM, Kim CH, Hangoc G, Cooper S, Hromas R (2006) Synergistic inhibition in vivo of bone marrow myeloid progenitors by myelosuppressive chemokines and chemokine-accelerated recovery of progenitors after treatment of mice with Ara-C. Exp Hematol 34:1069–1077
Bruserud Ø, Gjertsen BT (2000) New strategies for the treatment of acute myelogenous leukemia: differentiation induction – present use and future possibilities. Stem Cells 18:157–165
Bruserud Ø, Hamann W, Patel S, Ehninger G, Schmidt H, Pawelec G (1993) IFN-gamma and TNF-alpha secretion by CD4+ and CD8+ TCR αβ+ T-cell clones derived early after allogeneic bone marrow transplantation. Eur J Haematol 51:73–79
Bruserud Ø, Halstensen A, Peen E, Solberg CO (1996) Serum levels of adhesion molecules and cytokines in patients with acute leukaemia. Leuk Lymphoma 23:423–430
Bruserud Ø, Ryningen A, Wergeland L, Glenjen NI, Gjertsen BT (2004) Osteoblasts increase proliferation and release of pro-angiogenic interleukin 8 by native human acute myelogenous leukemia blasts. Haematologica 89:391–402
Bruserud Ø, Stapnes C, Tronstad KJ, Ryningen A, Anensen N, Gjertsen BT (2006) Protein lysine acetylation in normal and leukaemic haematopoiesis: HDACs as possible therapeutic targets in adult AML. Expert Opin Ther Targets 10:51–68
Bruserud Ø, Ryningen A, Olsnes AM, Stordrange L, Øyan AM, Kalland KH, Gjertsen BT (2007) Subclassification of patients with acute myelogenous leukemia based on chemokine responsiveness and constitutive chemokine release by their leukemic cells. Haematologica 92:332–341
Burger JA (2009) CXCR4 in acute myelogenous leukemia (AML): when too much attraction is bad for you. Leuk Res 33:747–748
Burger JA, Bürkle A (2007) The CXCR4 chemokine receptor in acute and chronic leukaemia: a marrow homing receptor and potential therapeutic target. Br J Haematol 137:288–296
Burger JA, Kipps TJ (2006) CXCR4: a key receptor in the crosstalk between tumor cells and their microenvironment. Blood 107:1761–1767
Burger JA, Spoo A, Dwenger A, Burger M, Behringer D (2003) CXCR4 chemokine receptors (CD184) and α4β1 integrins mediate spontaneous migration of human CD34+ progenitors and acute myeloid leukaemia cells beneath marrow stromal cells (pseudoemperipolesis). Br J Haematol 122:579–589
Calandra G, Bridger G, Fricker S (2010) CXCR4 in clinical hematology. Curr Top Microbiol Immunol, DOI 10.1007/82_2010_26
Campbell DJ, Kim CH, Butcher EC (2003) Chemokines in the systemic organization of immunity. Immunol Rev 195:58–71
Christopherson K, Hromas R (2001) Chemokine regulation of normal and pathologic immune responses. Stem Cells 19:388–396
Cignetti A, Vallario A, Roato I, Circosta P, Strola G, Scielzo C, Allione B, Garetto L, Caligaris-Cappio F, Ghia P (2003) The characterization of chemokine production and chemokine receptor expression reveals possible functional cross-talks in AML blasts with monocytic differentiation. Exp Hematol 31:495–503
Costello RT, Mallet F, Chambost H, Sainty D, Arnoulet C, Gastaut JA, Olive D (2000) Acute myeloid leukaemia triggering via CD40 induces leukocyte chemoattraction and cytotoxicity against allogenic or autologous leukemic targets. Leukemia 14:123–128
Cummins EP, Taylor CT (2005) Hypoxia-responsive transcription factors. Pflugers Arch 450:363–371
de Bont ES, Vellenga E, Molema G, van Wering E, de Leij LF, Kamps WA (2001) A possible role for spontaneous interleukin-8 production by acute myeloid leukemic cells in angiogenesis related processes: work in progress. Med Pediatr Oncol 37:511–517
Dimberg A (2010) Chemokines in angiogenesis. Curr Top Microbiol Immunol, DOI 10.1007/82_2010_21
Dommange F, Cartron G, Espanel C, Gallay N, Domenech J, Benboubker L, Ohresser M, Colombat P, Binet C, Watier H, Herault O, GOELAMS Study Group (2006) CXCL12 polymorphism and malignant cell dissemination/tissue infiltration in acute myeloid leukemia. FASEB J 20:1913–1915
Dubois C, Schlageter MH, de Gentile A, Balitrand N, Toubert ME, Krawice I, Fenaux P, Castaigne S, Najean Y, Degos L (1994) Modulation of IL-8, IL-1 beta, and G-CSF secretion by all-trans retinoic acid in acute promyelocytic leukemia. Leukemia 8:1750–1757
Engelhardt BE, Crowe JE (2010) Homing in an acute graft-versus-host disease: Tissue-specific T regulatory and Th 17 cells. Curr Top Microbiol Immunol, DOI 10.1007/82_2010_24
Ersvaer E, Skavland J, Ulvestad E, Gjertsen BT, Bruserud Ø (2007a) Effects of interferon gamma on native human acute myelogenous leukaemia cells. Cancer Immunol Immunother 56:13–24
Ersvær E, Olsnes AM, Bruserud Ø (2007b) The immunological dilemma: cellular innate and adaptive immune response versus acute myelogenous leukemia. Open Hematol J 1:1–14
Estey E, Döhner H (2006) Acute myeloid leukaemia. Lancet 368:1894–1907
Fiegl M, Samudio I, Clise-Dwyer K, Burks JK, Mnjoyan Z, Andreeff M (2009) CXCR4 expression and biologic activity in acute myeloid leukemia are dependent on oxygen partial pressure. Blood 113:1504–1512
Glenjen N, Hovland R, Wergeland L, Wendelbo Ø, Ernst P, Bruserud Ø (2003) The angioregulatory phenotype of native human acute myelogenous leukemia cells: influence of karyotype, Flt3 abnormalities and differentiation status. Eur J Haematol 71:163–173
Glenjen N, Ersvaer E, Ryningen A, Bruserud Ø (2004) In vitro effects of native human acute myelogenous leukemia blasts on fibroblasts and osteoblasts. Int J Cancer 111:858–867
Haas J, Schopp L, Storch-Hagenlocher B, Fritzsching B, Jacobi C, Milkova L, Fritz B, Schwarz A, Suri-Payer E, Hensel M, Wildemann B (2007) Specific recruitment of regulatory T-cells into the CSF in lymphomatous and carcinomatous meningitis. Blood 111:761–766
Hanahan D, Weinberg RA (2000) The hallmarks of cancer. Cell 100:57–70
Harris NL, Jaffe ES, Diebold J, Flandrin G, Muller-Hermelink HK, Vardiman J, Lister TA, Bloomfield CD (1999) World Health Organization classification of neoplastic diseases of the hematopoietic and lymphoid tissues: report of the Clinical Advisory Committee meeting-Airlie House, Virginia, November 1997. J Clin Oncol 17:3835–3849
Harrison JS, Rameshwar P, Chang V, Bandari P (2002) Oxygen saturation in the bone marrow of healthy volunteers. Blood 99:394
Hatfield KJ, Olsnes AM, Gjertsen BT, Bruserud Ø (2005) Antiangiogenic therapy in acute myelogenous leukemia: targeting of vascular endothelial growth factor and interleukin 8 as possible antileukemic strategies. Curr Cancer Drug Targets 5:229–248
Hatfield K, Ryningen A, Corbascio M, Bruserud Ø (2006) Microvascular endothelial cells increase proliferation and inhibit apoptosis of native human acute myelogenous leukemia blasts. Int J Cancer 119:2313–2321
Hatfield KJ, Hovland R, Øyan AM, Kalland KH, Ryningen A, Gjertsen BT, Bruserud Ø (2008) Release of angiopoietin-1 by primary human acute myelogenous leukemia cells is associated with mutations of nucleophosmin, increased by bone marrow stromal cells and possibly antagonized by high systemic angiopoietin-2 levels. Leukemia 22:287–293
Hatfield K, Øyan AM, Ersvaer E, Kalland KH, Lassalle P, Gjertsen BT, Bruserud Ø (2009) Primary human acute myeloid leukaemia cells increase the proliferation of microvascular endothelial cells through the release of soluble mediators. Br J Haematol 144:53–68
Hirota K, Semenza GL (2006) Regulation of angiogenesis by hypoxia-inducible factor 1. Crit Rev Oncol Hematol 59:15–26
Homey B, Muller A, Zlotnik A (2002) Chemokines: agents for the immunotherapy of cancer? Nat Rev Immunol 2:175–184
Honczarenko M, Le Y, Swierkowski M, Ghiran I, Glodek AM, Silberstein LE (2006) Human bone marrow stromal cells express a distinct set of biologically functional chemokine receptors. Stem Cells 24:1030–1041
Hsu HC, Lee YM, Tsai WH, Jiang ML, Ho CH, Ho CK, Wang SY (2002) Circulating levels of thrombopoietic and inflammatory cytokines in patients with acute myeloblastic leukemia and myelodysplastic syndrome. Oncology 63:64–69
Iellem A, Mariani M, Lang R, Recalde H, Panina-Bordignon P, Sinigaglia F, D’Ambrosio D (2001) Unique chemotactic response profile and specific expression of chemokine receptors CCR4 and CCR8 by CD4+CD25+ regulatory T cells. J Exp Med 194:847–853
Jin L, Hope KJ, Zhai Q, Smadja-Joffe F, Dick JE (2006) Targeting of CD44 eradicates human acute myeloid leukemic stem cells. Nat Med 12:1167–1174
Jin JO, Park HY, Kim JW, Park JI, Hong YS, Min do S, Kwak JY (2007) Phosphatidic acid induces the differentiation of human acute promyelocytic leukemic cells into dendritic cell-like. J Cell Biochem 100:191–203
Kalinkovich A, Tavor S, Avigdor A, Kahn J, Brill A, Petit I, Goichberg P, Tesio M, Netzer N, Naparstek E, Hardan I, Nagler A, Resnick I, Tsimanis A, Lapidot T (2006) Functional CXCR4-expressing microparticles and SDF-1 correlate with circulating acute myelogenous leukemia cells. Cancer Res 66:11013–11020
Kim CH (2006) Migration and function of FoxP3+ regulatory T cells in the hematolymphoid system. Exp Hematol 34:1033–1040
Kittan NA, Hildebrandt GC (2010) The chemokine system – a possible therapeutic target in acute graft versus host disease. Curr Top Microbiol Immunol, DOI 10.1007/82_2010_23
Köhler T, Plettig R, Wetzstein W, Schmitz M, Ritter M, Mohr B, Schaekel U, Ehninger G, Bornhäuser M (2000) Cytokine-driven differentiation of blasts from patients with acute myelogenous and lymphoblastic leukemia into dendritic cells. Stem Cells 18:139–147
Konoplev S, Rassidakis GZ, Estey E, Kantarjian H, Liakou CI, Huang X, Xiao L, Andreeff M, Konopleva M, Medeiros LJ (2007) Overexpression of CXCR4 predicts adverse overall and event-free survival in patients with unmutated FLT3 acute myeloid leukemia with normal karyotype. Cancer 109:1152–1156
Lambert MP, Rauova L, Bailey M, Sola-Visner MC, Kowalska MA, Poncz M (2007) Platelet factor 4 is a negative autocrine in vivo regulator of megakaryopoiesis: clinical and therapeutic implications. Blood 110:1153–1160
Laurence AD (2006) Location, movement and survival: the role of chemokines in haematopoiesis and malignancy. Br J Haematol 132:255–267
Le Dieu R, Taussig DC, Ramsay AG, Mitter R, Miraki-Moud F, Fatah R, Lee AM, Lister TA, Gribben JG (2009) Peripheral blood T cells in AML patients at diagnosis have abnormal phenotype and genotype and form defective immune synapses with AML blasts. Blood 114:3909–3916
Lecomte-Raclet L, Rholam M, Alemany M, Lazar N, Simenel C, Delepierre M, Han ZC, Cohen P, Caen JP (2000) Dual structural requirements for multilineage hematopoietic-suppressive activity of chemokine-derived peptides. Biochemistry 39:9612–9622
Lee CY, Tien HF, Hu CY, Chou WC, Lin LI (2007a) Marrow angiogenesis-associated factors as prognostic biomarkers in patients with acute myelogenous leukaemia. Br J Cancer 97:877–882
Lee JH, Kang SG, Kim CH (2007b) FoxP3+ T cells undergo conventional first switch to lymphoid tissue homing receptors in thymus but accelerated second switch to nonlymphoid tissue homing receptors in secondary lymphoid tissues. J Immunol 178:301–311
Legdeur MC, Beelen RH, Schuurhuis GJ, Broekhoven MG, van de Loosdrecht AA, Tekstra J, Langenhuijsen MM, Ossenkoppele GJ (1997) A functional study on the migration of human monocytes to human leukemic cell lines and the role of monocyte chemoattractant protein-1. Leukemia 11:1904–1908
Legdeur MC, Broekhoven MG, Schuurhuis GJ, Beelen RH, Ossenkoppele GJ (2001) Monocyte-chemoattractant-protein-1-mediated migration of human monocytes towards blasts from patients with acute myeloid leukemia. Cancer Immunol Immunother 50:16–22
Li H, Guo L, Jie S, Liu W, Zhu J, Du W, Fan L, Wang X, Fu B, Huang S (2008) Berberine inhibits SDF-1-induced AML cells and leukemic stem cells migration via regulation of SDF-1 level in bone marrow stromal cells. Biomed Pharmacother 62:573–578
Liesveld JL, Bechelli J, Rosell K, Lu C, Bridger G, Phillips G 2nd, Abboud CN (2007) Effects of AMD3100 on transmigration and survival of acute myelogenous leukemia cells. Leuk Res 31:1553–1563
Lisy K, Peet DJ (2008) Turn me on: regulating HIF transcriptional activity. Cell Death Differ 15:642–649
Liu J, Zeng H, Zhang Y (1999) Study on the expression of interleukin-8 and its receptors in acute leukemia. Zhonghua Xue Ye Xue Za Zhi 20:24–26
Löffler J, Mezger M, Ok M, Oliver Morton C, Einsele H (2010) Genetic polymorphisms in the cytokine and chemokine system – their possible importance in allogeneic stem cell transplantation. Curr Top Microbiol Immunol, DOI 10.1007/82_2010_22
Maghasachi AA (2010) Role of chemokines in the biology of natural killer cells. Curr Top Microbiol Immunol, DOI 10.1007/82_2010_20
Mantovani A (2009) Cancer: inflaming metastasis. Nature 457:36–37
Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M (2004) The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol 25:677–686
Mazur G, Wróbel T, Butrym A, Kapelko-Słowik K, Poreba R, Kuliczkowski K (2007) Increased monocyte chemoattractant protein 1 (MCP-1/CCL-2) serum level in acute myeloid leukemia. Neoplasma 54:285–289
Möhle R, Bautz F, Rafii S, Moore MA, Brugger W, Kanz L (1998) The chemokine receptor CXCR-4 is expressed on CD34+ hematopoietic progenitors and leukemic cells and mediates transendothelial migration induced by stromal cell-derived factor-1. Blood 91:4523–4530
Möhle R, Schittenhelm M, Failenschmid C, Bautz F, Kratz-Albers K, Serve H, Brugger W, Kanz L (2000) Functional response of leukaemic blasts to stromal cell-derived factor-1 correlates with preferential expression of the chemokine receptor CXCR4 in acute myelomonocytic and lymphoblastic leukaemia. Br J Haematol 110:563–572
Monaco G, Belmont JW, Konopleva M, Andreeff M, Tavor S, Petit I, Kollet O, Lapidot T (2004a) Correlation between CXCR4 and homing or engraftment of acute myelogenous leukemia. Cancer Res 64:6832
Monaco G, Konopleva M, Munsell M, Leysath C, Wang RY, Jackson CE, Korbling M, Estey E, Belmont J, Andreeff M (2004b) Engraftment of acute myeloid leukemia in NOD/SCID mice is independent of CXCR4 and predicts poor patient survival. Stem Cells 22:188–201
Moser B, Loetscher P (2002) Lymphocyte traffic control by chemokines. Nat Immunol 2:123–128
Mrózek K, Heerema NA, Bloomfield CD (2004) Cytogenetics in acute leukemia. Blood Rev 18:115–136
Muller G, Hopken UE, Stein H, Lipp M (2002) Systemic immunoregulatory and pathogenic functions of homeostatic chemokine receptors. J Leukoc Biol 72:1–8
Negaard HF, Iversen N, Bowitz-Lothe IM, Sandset PM, Steinsvik B, Østenstad B, Iversen PO (2009) Increased bone marrow microvascular density in haematological malignancies is associated with differential regulation of angiogenic factors. Leukemia 23:162–169
Nervi B, Ramirez P, Rettig MP, Uy GL, Holt MS, Ritchey JK, Prior JL, Piwnica-Worms D, Bridger G, Ley TJ, DiPersio JF (2009) Chemosensitization of acute myeloid leukemia (AML) following mobilization by the CXCR4 antagonist AMD3100. Blood 113:6206–6214
Olkhanud PB, Baatar D, Bodogai M, Hakim F, Gress R, Anderson RL, Deng J, Xu M, Briest S, Biragyn A (2009) Breast cancer lung metastasis requires expression of chemokine receptor CCR4 and regulatory T cells. Cancer Res 69:5996–6004
Olsnes AM, Motorin D, Ryningen A, Zaritskey AY, Bruserud Ø (2006) T lymphocyte chemotactic chemokines in acute myelogenous leukemia (AML): local release by native human AML blasts and systemic levels of CXCL10 (IP-10), CCL5 (RANTES) and CCL17 (TARC). Cancer Immunol Immunother 55:830–840
Olsnes AM, Ryningen A, Ersvaer E, Bruserud Ø (2008) In vitro induction of a dendritic cell phenotype in primary human acute myelogenous leukemia (AML) blasts alters the chemokine release profile and increases the levels of T cell chemotactic CCL17 and CCL22. J Interferon Cytokine Res 28:297–310
Olsnes AM, Ersvær E, Ryningen A, Paulsen K, Hampson P, Lord JM, Gjertsen BT, Kristoffersen EK, Bruserud Ø (2009) The protein kinase C agonist PEP005 increases NF-kappaB expression, induces differentiation and increases constitutive chemokine release by primary acute myeloid leukaemia cells. Br J Haematol 145:761–774
Ostermann H, Rothenburger M, Mesters RM, van de Loo J, Kienast J (1994) Cytokine response to infection in patients with acute myelogenous leukaemia following intensive chemotherapy. Br J Haematol 88:332–337
Ottersbach K, McLean J, Isaacs NW, Graham GJ (2006) A310 helical turn is essential for the proliferation-inhibiting properties of macrophage inflammatory protein-1 alpha (CCL3). Blood 107:1284–1291
Paczesny S, Hanauer D, Sun Y, Reddy P (2010) New perspectives on the biology of acute GVHD. Bone Marrow Transplant 15:1–11
Ponziani V, Mannelli F, Bartalucci N, Gianfaldoni G, Leoni F, Antonioli E, Guglielmelli P, Ciolli S, Bosi A, Vannucchi AM (2008) No role for CXCL12-G801A polymorphism in the development of extramedullary disease in acute myeloid leukemia. Leukemia 22:669–671
Qin S, Rottman JB, Myers P, Kassam N, Weinblatt M, Loetscher M, Koch AE, Moser B, Mackay CR (1998) The chemokine receptors CXCR3 and CCR5 mark subsets of T cells associated with certain inflammatory reactions. J Clin Invest 101:746–754
Reikvam H, Olsnes AM, Gjertsen BT, Ersvaer E, Bruserud Ø (2009) Nuclear factor-κB signaling – a contributor in leukemogenesis and a target for pharmacological intervention in human acute myelogenous leukemia. Crit Rev Oncog 15:1–41
Riccioni R, Calzolari A, Biffoni M, Senese M, Riti V, Petrucci E, Pasquini L, Cedrone M, Lo-Coco F, Diverio D, Foà R, Peschle C, Testa U (2006) Podocalyxin is expressed in normal and leukemic monocytes. Blood Cells Mol Dis 37:218–225
Rombouts EJ, Pavic B, Löwenberg B, Ploemacher RE (2004) Relation between CXCR-4 expression, Flt3 mutations, and unfavorable prognosis of adult acute myeloid leukemia. Blood 104:550–557
Rosenkilde MM, Schwartz TW (2004) The chemokine system – a major regulator of angiogenesis in health and disease. APMIS 112:481–495
Ryningen A, Wergeland L, Glenjen N, Gjertsen BT, Bruserud Ø (2005) In vitro crosstalk between fibroblasts and native human acute myelogenous leukemia (AML) blasts via local cytokine networks results in increased proliferation and decreased apoptosis of AML cells as well as increased levels of proangiogenic Interleukin 8. Leuk Res 29:185–196
Schönbohn H, Schuler M, Kolbe K, Peschel C, Huber C, Bemb W, Aulitzky WE (1995) Plasma levels of IL-1, TNF alpha, IL-6, IL-8, G-CSF, and IL1-RA during febrile neutropenia: results of a prospective study in patients undergoing chemotherapy for acute myelogenous leukemia. Ann Hematol 71:161–168
Sieg DJ, Hauck CR, Ilic D, Klingbeil CK, Schaefer E, Damsky CH, Schlaepfer DD (2000) FAK integrates growth-factor and integrin signals to promote cell migration. Nat Cell Biol 2:249–256
Spoo AC, Lübbert M, Wierda WG, Burger JA (2007) CXCR4 is a prognostic marker in acute myelogenous leukemia. Blood 109:786–791
Srivastava MD, Ambrus JL (2004) Effect of 1, 25(OH)2 vitamin D3 analogs on differentiation induction and cytokine modulation in blasts from acute myeloid leukemia patients. Leuk Lymphoma 45:2119–2126
Steinbach D, Schramm A, Eggert A, Onda M, Dawczynski K, Rump A, Pastan I, Wittig S, Pfaffendorf N, Voigt A, Zintl F, Gruhn B (2006) Identification of a set of seven genes for the monitoring of minimal residual disease in pediatric acute myeloid leukemia. Clin Cancer Res 12:2434–2441
Strieter RM, Polverini PJ, Kunkel SL, Arenberg DA, Burdick MD, Kasper J, Dzuiba J, Van Damme J, Walz A, Marriott D, Chan S-Y, Roczniak S, Shanafelt AB (1995) The functional role of the ELR motif in CXC chemokine-mediated angiogenesis. J Biol Chem 270:27348–27357
Struyf S, Schutyser E, Gouwy M, Gijsbers K, Proost P, Benoit Y, Opdenakker G, Van Damme J, Laureys G (2003) PARC/CCL18 is a plasma CC chemokine with increased levels in childhood acute lymphoblastic leukemia. Am J Pathol 163:2065–2075
Szczepansky MJ, Szajnik M, Czystowska M, Mandapathil M, Strauss L, Welsh A, Foon KA, Whiteside TL, Boyiadzis M (2009) Increased frequency and suppression by regulatory T cells in patients with acute myelogenous leukemia. Clin Cancer Res 15:3325–3332
Tanaka T, Bai Z, Srinoulprasert Y, Yang BG, Hayasaka H, Miyasaka M (2005) Chemokines in tumor progression and metastasis. Cancer Sci 96:317–322
Tavernier-Tardy E, Cornillon J, Campos L, Flandrin P, Duval A, Nadal N, Guyotat D (2009) Prognostic value of CXCR4 and FAK expression in acute myelogenous leukemia. Leuk Res 33:764–768
Tavor S, Petit I, Porozov S, Avigdor A, Dar A, Leider-Trejo L, Shemtov N, Deutsch V, Naparstek E, Nagler A, Lapidot T (2004) CXCR4 regulates migration and development of human acute myelogenous leukemia stem cells in transplanted NOD/SCID mice. Cancer Res 64:2817–2824
Tavor S, Eisenbach M, Jacob-Hirsch J, Golan T, Petit I, Benzion K, Kay S, Baron S, Amariglio N, Deutsch V, Naparstek E, Rechavi G (2008) The CXCR4 antagonist AMD3100 impairs survival of human AML cells and induces their differentiation. Leukemia 22:2151–2158
Tobler A, Moser B, Dewald B, Geiser T, Studer H, Baggiolini M, Fey MF (1993) Constitutive expression of interleukin-8 and its receptor in human myeloid and lymphoid leukemia. Blood 82:2517–2525
Uesato N, Fukui K, Maruhashi J, Tojo A, Tajima N (2006) JTE-607, a multiple cytokine production inhibitor, ameliorates disease in a SCID mouse xenograft acute myeloid leukemia model. Exp Hematol 34:1385–1392
Voermans C, van Heese WP, de Jong I, Gerritsen WR, van Der Schoot CE (2002) Migratory behavior of leukemic cells from acute myeloid leukemia patients. Leukemia 16:650–657
Ward SG, Bacon K, Westwick J (1998) Chemokines and T lymphocytes: more than an attraction. Immunity 9:1–11
Wendelbo Ø, Nesthus I, Sjo M, Ernst P, Bruserud Ø (2004a) Cellular immune responses in multiple myeloma patients with treatment-induced cytopenia early after high-dose chemotherapy and autologous peripheral blood stem cell transplantation. Leuk Res 28:461–468
Wendelbo Ø, Nesthus I, Sjo M, Paulsen K, Ernst P, Bruserud Ø (2004b) Functional characterization of T lymphocytes derived from patients with acute myelogenous leukemia and chemotherapy-induced leukopenia. Cancer Immunol Immunother 53:740–747
Wenger RH, Stiehl DP, Camenisch G (2005) Integration of oxygen signaling at the consensus HRE. Sci STKE 306:re12
Wimmer A, Khaldoyanidi SK, Judex M, Serobyan N, Discipio RG, Schraufstatter IU (2006) CCL18/PARC stimulates hematopoiesis in long-term bone marrow cultures indirectly through its effect on monocytes. Blood 108:3722–3729
Xie K (2001) Interleukin-8 and human cancer biology. Cytokine Growth Factor Rev 12:375–391
Yi H, Zhen Y, Jiang L, Zheng J, Zhao Y (2006) The phenotypic characterization of naturally occurring regulatory CD4+CD25+ T cells. Cell Mol Immunol 3:189–195
Zahn S, Zwirner J, Spengler HP, Götze O (1997) Chemoattractant receptors for interleukin-8 and C5a: expression on peripheral blood leukocytes and differential regulation on HL-60 and AML-193 cells by vitamin D3 and all-trans retinoic acid. Eur J Immunol 27:935–940
Zebisch A, Czernilofsky AP, Keri G, Smigelskaite J, Sill H, Troppmair J (2007) Signaling through RAS-RAF-MEK-ERK: from basics to bedside. Curr Med Chem 14:601–623
Zeng Z, Samudio IJ, Munsell M, An J, Huang Z, Estey E, Andreeff M, Konopleva M (2006) Inhibition of CXCR4 with the novel RCP168 peptide overcomes stroma-mediated chemoresistance in chronic and acute leukemias. Mol Cancer Ther 5:3113–3121
Zeng Z, Shi YX, Samudio IJ, Wang RY, Ling X, Frolova O, Levis M, Rubin JB, Negrin RR, Estey EH, Konoplev S, Andreeff M, Konopleva M (2009) Targeting the leukemia microenvironment by CXCR4 inhibition overcomes resistance to kinase inhibitors and chemotherapy in AML. Blood 113:6215–6224
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2010 Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Kittang, A.O., Hatfield, K., Sand, K., Reikvam, H., Bruserud, Ø. (2010). The Chemokine Network in Acute Myelogenous Leukemia: Molecular Mechanisms Involved in Leukemogenesis and Therapeutic Implications. In: Bruserud, O. (eds) The Chemokine System in Experimental and Clinical Hematology. Current Topics in Microbiology and Immunology, vol 341. Springer, Berlin, Heidelberg. https://doi.org/10.1007/82_2010_25
Download citation
DOI: https://doi.org/10.1007/82_2010_25
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-642-12638-3
Online ISBN: 978-3-642-12639-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)