
Abstract
Bipolar disorders are severe and have a high prevalence; despite this, the neurobiological mechanisms are far from being elucidated, and this limits the development of new treatments. Although the aetiology of bipolar disorders is not yet fully understood, it is accepted that the disorder(s) may result from the interaction between genetic factors that cause susceptibility and predisposing, precipitating and perpetuating environmental factors, such as stress and traumatic events. A pathophysiological formulation of the disease suggests that dysfunctions in intracellular biochemical cascades, oxidative stress and mitochondrial dysfunction impair the processes linked to neuronal plasticity, leading to cell damage and the consequent loss of brain tissue that has been identified in post-mortem and neuroimaging studies. The data we have reviewed suggests that peripheral biomarkers related to hormones, inflammation, oxidative stress and neurotrophins are altered in bipolar disorders, especially during acute mood episodes. Together, these changes have been associated with a systemic toxicity of the disease and the damage resulting from multiple episodes. Systemic toxicity related to recurrent episodes in bipolar disorder may influence brain anatomical changes associated with the progression of stress and neuroplasticity in bipolar disorder and the response to treatment.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
Bipolar affective disorder is a severe and chronic mental disorder, which often puts the lives of those affected at risk. Despite this severity and high prevalence, the pathophysiological mechanisms are far from being elucidated, which limits the development of new treatments.
Bipolar disorder (BD) is considered a clinical syndrome, as the current state of the evidence is insufficient to allow its characterization based on aetiology or pathophysiology. Also, BD has a complex clinical course, involving manic, depressive and/or mixed episodes, which makes study a challenge for researchers in the field.
Conceptually, it is clear that BD is complex and multifactorial. Although the aetiology of BD is not yet defined (Bobo 2017), it is accepted that the disorder may result from the interaction between genetic factors that cause susceptibility and predisposing, precipitating and perpetuating environmental factors, such as stress and traumatic events (Caspi and Moffitt 2006; Barnett and Smoller 2009).
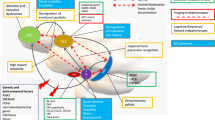
A research approach that describes reliable neurobiological findings based on psychopathological syndrome will be more solid contrasted to a non-aetiologic system of classification. Integrative approaches to understanding complex health issues can transcend disciplinary and knowledge boundaries and provide opportunities to view phenomena from diverse perspectives. A future diagnostic criteria system in which aetiology and pathophysiology are essential in diagnostic decision-making would bring psychiatry closer to other specialties of medicine. The precision medicine might deconstruct traditional symptom-based categories. Patients with a range of mood disorders may be studied across several platforms to parse current heterogenous syndromes into homogenous clusters (see Fig. 1).

Precision medicines in mood disorders integrate platforms to parse current heterogenous syndromes into homogenous clusters. Adapted from Cuthbert and Insel (2013). Illustrations courtesy of Romayne Gadelrab
Although bipolar disorder (BD) is not a typical neurodegenerative disease, several studies have associated BD with stress and death of glial cells and neurons (Machado-Vieira et al. 2009). In an attempt to understand the changes present in BD, initially, the focus of the research was the dysregulation of neurotransmitters, and then it became intracellular signalling (Hahn and Friedman 1999; Manji et al. 2011); more recently, mitochondrial energy metabolism, oxidative stress and neurogenesis have been the focus of studies in BD (Berk et al. 2011). The hypothesis was raised that the loss of neurotrophic support, oxidative stress and inflammation are associated with more advanced stages of BD, occurring progressively (Berk et al. 2014). In this view, mitochondrial function and neuroprotection may be particularly relevant in BD.
One of the strategies most used by researchers is to study the biochemical targets of therapeutically relevant drugs, verifying changes in these targets in both manic and depressive episodes. Biochemical and molecular targets believed to be involved in the neurobiology/pathophysiology of this disorder are neurotransmitter systems (serotonergic, dopaminergic and glutamatergic), neurotrophic factors, PKC, GSK-3, mitochondria and oxidative stress (Zarate et al. 2006).
To explain the changes in brain cells in BD, different hypotheses have been considered, including the presence of greater oxidative stress in the CNS of patients with BD (Machado-Vieira et al. 2007; Wang et al. 2009). Reactive oxygen species (ROS) are toxic substances produced in all cells of the human body that have the potential to cause molecular damage and impair cell function. It is necessary that the cells have detoxification systems, which, under normal physiology, is accomplished by the action of antioxidant substances. However, there may be situations in which the production of ROS exceeds the competence of the antioxidant system and oxidative degradation of molecules may occur, characterized as a state of oxidative stress. It is possible to investigate the presence of oxidative stress in cells by detecting the final products of oxidative reactions and studying the activity of antioxidant enzymes (Andreazza et al. 2008).
2 Neurotransmitters
Studies have described neurochemical changes in BD by evaluating various hormones, neurotransmitters and their metabolites, second messengers and neurotrophic and gene factors, in plasma, cerebrospinal fluid, platelets and brain slices. Concerning changes in neurotransmission systems associated with the disease, studies have described changes in the regulation of biogenic amines in BD (Young et al. 1994). These studies have shown changes in the regulation of noradrenergic, serotonergic, dopaminergic and cholinergic systems. These biogenic amines are widely distributed in the limbic system, which is involved in the modulation of sleep-wakefulness, appetite, endocrine functions and behavioural states, such as irritability, anxiety, euphoria, anhedonia and fear.
It has also been suggested that changes related in monoaminergic neurotransmitters may occur in BD due to changes in the sensitivity of their receptors. It is worth remembering that, as it is a multifactorial and polygenic disease from the biological point of view, these findings related to changes in neurotransmission systems should not be evaluated in isolation. Future studies that seek to bring new knowledge about the metabolic interrelation between the different neurotransmission systems supposedly altered in BD may provide a better assessment of the importance of these findings. The study of the possible relationship between the abnormalities observed in these neurotransmission systems and the clinical and cognitive manifestation of BD will also assist in the evaluation of the influence of specific biochemical changes in the modulation of both mood and cognitive and neurovegetative functions.
2.1 Serotoninergic System
Serotonin (5-HT) modulates different neuronal activities and several physiological and behavioural functions, such as impulse control, aggressiveness and suicidal tendencies (Shiah and Yatham 2000). Decreased 5-HT release and activity may be associated with some abnormalities such as suicidal ideation, suicide attempts, aggression and sleep disorders, which are frequent findings in bipolar disorders (Ackenheil 2001).
Since the 1970s, many authors (e.g. Prange et al. 1974) have suggested the involvement of 5-HT in the pathophysiology of BD, formulating the permissive hypothesis, in which a deficit in central serotonergic neurotransmission would allow expression of both the manic phase and the depressive; however, such phases would differ to central catecholamine (norepinephrine and dopamine) levels, which would be elevated in mania and decreased in depression. Also, decreased levels of 5-hydroxy indole acetic acid (5-HIAA), the major metabolite of serotonin, have been demonstrated in the CSF of manic and depressed patients compared to healthy controls, suggesting that both mania and depression are associated with a reduction in function central serotoninergic. A post-mortem study of the brains of patients with BD also found significantly lower levels of 5-HIAA in the frontal and parietal cortex, compared with controls, providing further evidence for the hypothesis of decreased central serotonergic activity in bipolar disorders (Baumann et al. 1999). Neuroendocrine challenge studies (Juruena 2014), when analysed together, suggest that the presynaptic activity of serotonin in the CNS is decreased, whereas the sensitivity of receptors postsynaptic is increased in mania (Shiah and Yatham 2000).
2.2 Dopaminergic System
Dopamine is a catecholaminergic neurotransmitter present in the central and peripheral nervous systems. Dopamine is synthesized in the brain from L-dihydroxyphenylalanine (L-Dopa) and has five distinct receptors, which have different affinities for dopamine. Type 1 dopamine receptors include D1 and D5 receptors, while type 2 receptors include D2, D3 and D4 receptors (Beaulieu and Gainetdinov 2011). Type D1 receptors are coupled to the Gαs protein, which activates adenylate cyclase, leading to increase in cyclic adenosine monophosphate (cyclic AMP), while D2 receptors are coupled to the Gαi protein, which inhibits adenylate cyclase, reducing the cytoplasmic concentration of cyclic AMP. Dopamine is involved in physiological functions such as blood pressure, kidney function, glucose homeostasis, voluntary movements, cognition, reward system, sleep and memory. Therefore, some researchers suggest that the dopaminergic system is involved in the pathophysiology of BD and that excessive dopaminergic stimulation is related to and perhaps triggers manic symptoms (Beaulieu and Gainetdinov 2011).
One of the most consistent findings about the role of dopamine in the neurobiology of BD is the fact that dopaminergic agonists, both direct and indirect, simulate episodes of mania or hypomania in patients with bipolar disorder with an underlying or predisposition (Brunello and Tascedda 2003). Ackenheil (2001) suggested that although the results were not consistent, a higher dopaminergic activity induced by increased release, decreased buffering capacity by vesicles synaptic disorders or the higher sensitivity of the dopaminergic may be associated with the development of manic symptoms, while decreased activity dopaminergic is associated with depression.
Pioneering studies on dopaminergic changes in BD had demonstrated high levels of dopamine in the urine of BD patients during manic episodes (Joyce et al. 1995). Also, a clinical study has shown that the administration of amphetamine (AMPH), an agonist substance in the dopaminergic system, triggers manic symptoms in both BD patients and healthy volunteers, with behavioural changes being more intense in those with BD (Anand et al. 2011). Studies suggest that the pathophysiology of BD may be linked to changes in the dopaminergic system and that the therapeutic effects of mood stabilizers may be linked to the modulation of this system (Bunney and Garland 1982). Therefore, the decrease in the expression or activity of the dopamine transporter (DAT) can lead to an increase in extracellular dopamine, suggesting that changes in this protein may contribute to the clinical manifestations of BD (Berk et al. 2007).
Therefore, it is believed that defects in the homeostatic mechanisms that respond to a hyperdopaminergic state, in the manic phase, may result in an excessive reduction in dopaminergic function, leading to a hypodopaminergic state and depression. In contrast, a weak regulatory response to a hypodopaminergic state may lead to a manic state. Dopamine is associated with induction of oxidative stress state in the brain and is metabolized via (a) enzymatic processes by the monoamine oxidase (MAO), which generates H2O2, a ROS, and dihydroxyphenylacetic acid, or via (b) autocatalysis by reaction with Fe2+ and H2O2 forming 6-hydroxydopamine (Obata 2002). Further studies are indeed necessary to understand how MAO contributes to oxidative damage in BD. Such studies would contribute a crucial piece of information – how oxidative stress modulates neurotransmitter levels.
2.3 Norepinephrinergic System
Studies have described the function of this system in depressive states. In these states, lower norepinephrine deficits and lower a2 receptor sensitivity have been reported, in contrast to a tendency toward higher norepinephrine activity in manic states (Ackenheil 2001). Following this logic, Baumann et al. (1999) observed that individuals with BD present higher numbers of pigmented cells in the locus coeruleus than do unipolar patients. Also, Shiah and Yatham (2000) suggested that diminished central 5-HT function concomitant with increased noradrenergic function may be involved in the genesis of mania.
Studies – evaluating norepinephrinergic metabolism in manic patients compared to healthy controls – describe that the results for plasma levels of MHPG may reflect the pathophysiology of BD from the manic state to the depressive state more than the plasma levels of HVA or BDNF. These data suggest that peripheral MHPG, which is associated with norepinephrine levels in the brain, could be used as a biomarker for whole mood states in BD. The MHPG levels, reflecting the norepinephrine levels in the brain, are likely to reflect the clinical characteristics of the switching process in BD and to have prognostic significance for the treatment of manic and depressive states (Kurita et al. 2014). When lithium was used, a significant decrease in these markers was observed in manic patients. However, on the other hand, the level of MHPG can be very different from individual to individual. Therefore, a patient’s MHPG level must be known because it can change over time (Kurita et al. 2014).
2.4 GABAergic System
The monoaminergic hypothesis is not able to explain the pathophysiology of mood disorders fully. Moreover, the balance between excitatory and inhibitory impulses may be essential for processing information and preserving cognitive functions. Furthermore, new therapies have been developed to modulate the neurotransmission of gamma-aminobutyric acid (GABA) and glutamate, the main inhibitory and excitatory neurotransmitters, respectively (Wilkinson and Sanacora 2019). The dysregulation of the levels of these leads to an alteration of activity and of the neural resting state, which, chronically, might result in a maladaptation of these neuronal systems contributing to the appearance of mood symptoms (Fee et al. 2017).
Clinical data indicate that decreased GABAergic activity is associated with manic and depressive states and that GABAergic agonists have mood-stabilizing properties. Low levels of GABAergic activity have been found in the plasma of bipolar patients during both depressive and manic episodes (Petty et al. 1993).
The ratio between GABA and glutamate appears to be altered in patients with MDD (Sanacora et al. 2004). There is still no consensus about either peripheral or central levels of these neurotransmitters in patients with bipolar disorders due to the difficulty of studies with regard to controlling the effects of medication and post-mortem changes (Brady et al. 2013; Sanacora et al. 2008). Even so, patients who present a reduction in cortical GABA appear to have more significant cognitive impairments, especially inhibitory control (Bhagwagar et al. 2008; Luscher et al. 2011).
2.5 Glutamatergic System
Glutamate is the most important excitatory neurotransmitter in the brain. The role of this amino acid is to generate excitatory postsynaptic potential. However, its performance in brain functioning is much more diverse and complex (Rahn et al. 2012). In brain development, glutamate acts on migration, differentiation and the neuron’s survivability (Luján et al. 2005). In mature brains, it is also inherently involved with neuroplasticity and neurotoxicity (Dong et al. 2009). The involvement of glutamate in excitatory synaptic transmission, neurotoxicity and neuroplasticity processes has led to growing interest in its role in the pathophysiology of psychiatric disorders, particularly in BD.
Investigation of the glutamatergic system in BD has been carried out using different approaches. Studies that measured glutamate in the plasma of patients with BD have found conflicting results and have been done on small samples of both patients and controls. Altamura et al. (1993) and Hoekstra et al. (2006) found increased levels of glutamate in patients in depression and mania, respectively. Although this results from the first line of evidence that the glutamatergic system is altered in BD, plasma glutamate levels may not adequately represent brain levels, given that the glutamate present in the brain is produced locally since that from outside the CNS does not cross the blood-brain barrier.
A greater number of studies have investigated glutamate in BD in post-mortem brain tissue (Law and Deakin 2001; McCullumsmith et al. 2007). Hashimoto et al. (2007) measured glutamate levels in the brain and found increased levels in the frontal cortex of patients with BD. Other studies investigated the protein or RNA expression of receptor subunits or other molecules belonging to the glutamatergic system and found abnormalities in areas of the brain associated with BD, including the dorsolateral prefrontal cortex (DLPFC) (Beneyto and Meador-Woodruff 2006), the hippocampus (Law and Deakin 2001; McCullumsmith et al. 2007), the striatum (Kristiansen and Meador-Woodruff 2005) and the thalamus (Clinton and Meador-Woodruff 2004). The results are consistent, showing a decrease in the expression of several molecules linked to glutamatergic transmission in the brains of patients with BD. These findings have supported the hypothesis of hyperglutamatergic state in the brain of patients with BD, leading to a subsensitization of receptors, as a compensation mechanism to modulate excessive glutamatergic activity (Rao et al. 2007). In fact, chronic administration of N-methyl-D-aspartate (NMDA), a glutamate analogue from which the NMDA receptor is named, causes a decrease in the expression of NR-1 and NR-3A subunits of this receptor in cell cultures neurons (Rao et al. 2007).
Further evidence that the glutamatergic system is altered in BD comes from studies demonstrating the action of mood stabilizers in this system. Chronic treatment with lithium or valproic acid decreases the synaptic levels of glutamate through several mechanisms (Sanacora et al. 2008). Both drugs have the effect of decreasing the expression of the GluR1 subunit of alpha-amino-3-hydroxy-methyl-5-4-isoxazolpropionic (AMPA) receptors in the hippocampus with prolonged treatment at clinically relevant doses (Du et al. 2003).
More support comes from recent studies indicating that AMPA receptor antagonists attenuate several manifest behaviours produced by the administration of amphetamine to rats (Bäckström and Hyytiä 2003). Lithium appears to increase neuronal excitability in the hippocampal CA1 synapses (Colino et al. 1998), increasing the effectiveness of these pathways. This appears to be due to the capacity of lithium to potentiate currents through AMPA receivers, through the selective increase in the opening of these channels (Gebhardt and Cull-Candy 2010). Lamotrigine, a drug used to prevent and treat depressive episodes in BD, inhibits the release of glutamate in the hippocampus of rats (Leach et al. 1986). Riluzole, another substance that inhibits the release of glutamate at the synapse, has shown antidepressant effect in preliminary studies (Zarate et al. 2005; Sanacora et al. 2007).
The above findings contribute to the hypothesis that glutamate levels are increased in BD and some of the effectiveness of mood stabilizers may come from an effect on glutamatergic neurotransmission.
In summary, the most current studies on the pathophysiology of BD have identified several macroscopic changes in brain areas and circuits, in addition to histopathological and chemical changes, at the tissue, intracellular and synaptic levels associated with the disease. A pathophysiological hypothesis of the disease suggests that dysfunctions in intracellular biochemical cascades, oxidative stress and mitochondrial dysfunction impair the processes linked to neuronal plasticity, leading to cell damage and the consequent loss of brain tissue that has been identified in post-mortem and neuroimaging. The investigation of the glutamatergic system in BD has been carried out using different methodological approaches. Studies that measured glutamate in the plasma of patients with BD have found conflicting results and have been done on small samples from patients and controls. Altamura et al. (1993) and Hoekstra et al. (2006) found increased levels of glutamate in patients in depression and mania, respectively. Although this results from the first line of evidence that the glutamatergic system is altered in BD, plasma glutamate levels may not adequately represent brain levels, given that the glutamate present in the brain is produced locally, since that from outside the CNS does not cross the blood-brain barrier.
A greater number of studies have investigated glutamate in BD in post-mortem brain tissue (Law and Deakin 2001; McCullumsmith et al. 2007). Hashimoto et al. (2007) measured glutamate levels in the brain and found increased levels in the frontal cortex of patients with BD. Other studies investigated the protein or RNA expression of receptor subunits or other molecules belonging to the glutamatergic system and found abnormalities in areas of the brain associated with BD, including the dorsolateral prefrontal cortex (DLPFC) (Beneyto and Meador-Woodruff 2006), the hippocampus (Law and Deakin 2001; McCullumsmith et al. 2007), the striatum (Kristiansen and Meador-Woodruff 2005) and the thalamus (Clinton and Meador-Woodruff 2004). The results are consistent, showing a decrease in the expression of several molecules linked to glutamatergic transmission in the brains of patients with BD. These findings have supported the hypothesis of a hyperglutamatergic state in the brain of patients with BD, leading to a subsensitization of receptors, as a compensatory mechanism to modulate excessive glutamatergic activity (Rao et al. 2007). In fact, chronic administration of N-methyl-D-aspartate (NMDA), a glutamate analogue from which the NMDA receptor is named, causes a decrease in the expression of NR-1 and NR-3A subunits of this receptor in cell cultures neurons (Rao et al. 2007).
Other evidence that the glutamatergic system is altered in BD comes from studies demonstrating the action of mood stabilizers in this system. Chronic treatment with lithium or valproic acid decreases the synaptic levels of glutamate through several mechanisms (Sanacora et al. 2008). Both drugs have the effect of decreasing the expression of the GluR1 subunit of alpha-amino-3-hydroxy-methyl-5-4-isoxazolpropionic (AMPA) receptors in the hippocampus with prolonged treatment and at clinically relevant doses (Du et al. 2003).
Additional support for this evidence comes from recent studies indicating that AMPA receptor antagonists attenuate several manifest behaviours produced by the administration of amphetamine to rats (Bäckström and Hyytiä 2003). Lithium appears to increase neuronal excitability in the hippocampal CA1 synapses (Colino et al. 1998), increasing the effectiveness of these pathways. This appears to be due to the capacity of lithium to potentiate currents through AMPA receivers, through the selective increase in the opening of these channels (Gebhardt and Cull-Candy 2010). Lamotrigine, a drug used to prevent and treat depressive episodes in BD, inhibits the release of glutamate in the hippocampus of rats (Leach et al. 1986). Riluzole, another substance that inhibits the release of glutamate at the synapse, has shown antidepressant effect in preliminary studies (Zarate et al. 2005; Sanacora et al. 2007).
In summary, the above findings contribute to the hypothesis that glutamate levels are increased in BD and some of the effectiveness of mood stabilizers comes from an effect on glutamatergic neurotransmission.
Most current studies on the pathophysiology of BD have identified several macroscopic changes in brain areas and circuits, in addition to histopathological and chemical changes, at the tissue, intracellular and synaptic levels associated with the disease. A pathophysiological hypothesis of the disease suggests that dysfunctions in intracellular biochemical cascades, oxidative stress and mitochondrial dysfunction impair the processes linked to neuronal plasticity, leading to cell damage and the consequent loss of brain tissue that has been identified in post-mortem and neuroimaging. The metabolites studied provide important clues about the processes linked to cellular energy metabolism, the intracellular pathway of phosphatidylinositol, neurotransmission and glutamatergic metabolism, in addition to providing measures of cellular vulnerability.
3 Intracellular Signalling
Most current studies on the pathophysiology of BD have identified several macroscopic changes in brain areas and circuits, in addition to histopathological and chemical changes, at the tissue, intracellular and synaptic levels associated with the disease. A pathophysiological hypothesis of the disease suggests that dysfunctions in intracellular biochemical cascades, oxidative stress and mitochondrial dysfunction impair the processes linked to neuronal plasticity, leading to cell damage and the consequent loss of brain tissue that has been identified in post-mortem and neuroimaging. Therefore, the metabolites studied provide essential clues about the processes linked to cellular energy metabolism, the intracellular pathway of phosphatidylinositol, neurotransmission and glutamatergic metabolism, in addition to providing measures of cellular vulnerability (Zarate et al. 2006).
In the case of oxidative stress, changes in the main antioxidant enzymes have already been described, such as the increased activity of superoxide dismutase (SOD, EC 1.15.1.1) in patients experiencing an episode of mania or depression and decreased catalase (CAT, EC 1.11.1.6) in medicated euthymic patients and an increase in non-medicated manic patients (Steckert et al. 2010). Among several markers of oxidative stress already evaluated in bipolar disorder, a meta-analysis showed that levels of lipid peroxidation, nitric oxide and DNA/RNA damage are significantly elevated in patients when compared to controls (Brown et al. 2014).
Bioenergetic changes in BD have been described in the last decades. Several studies have presented robust evidence to support the involvement of mitochondrial dysfunction in the pathophysiology of BD, through changes involving oxidative phosphorylation, the glycolytic pathway, phospholipid metabolism, decreased total energy production and/or availability of substrates, as well as abnormalities in the morphology and intracellular distribution of mitochondria. In fact, a recent integrative literature review, involving neuroimaging studies, has demonstrated increased levels of lactate in various brain regions and cerebrospinal fluid in patients with the disorder, which indicates an increase in anaerobic and extramitochondrial glucose metabolism, consistent with a weakened mitochondrial metabolism in BD. This fact is supported by data demonstrating that BD patients also have significantly lower brain levels of phosphocreatine (PCr, a high-energy compound), as well as a reduction in N-acetyl-aspartate (NAA) levels, along with correlation negative between NAA/creatine + PCr levels or NAA levels and disease duration. These indicate neurodevelopmental changes and provide indirect evidence that mitochondrial dysfunction may play a role in disease progression (Scaini et al. 2016). Also, it has been shown that patients with this disorder have reduced levels of inorganic phosphate, a regulator of oxidative phosphorylation, and Na+/K+-ATPase activity, as well as a significant decrease in the levels of adenosine diphosphate (ADP), but not in ATP concentrations. These suggest that the reduction may be driven by increased adenylate kinase activity, providing ATP at the cost of ADP. Added to this hypothesis is the fact that, during the process of visual stimulation, healthy individuals show significant reductions in PCr levels, but not in ATP levels, as expected since PCr is being used for the synthesis of ATP from the creatine kinase reaction. However, patients with BD have a different profile, since significant reductions in ATP levels were observed, but not in PCr levels. Also, a recent study demonstrated a significant reduction in the rate of direct reaction effect of creatine kinase in the absence of changes in concentration in ATP and PCr in patients with BD during the first episode. These data corroborating the previously presented data, according to which patients with the disorder have a baseline and normal concentrations of ATP and PCr, but disability to replenish ATP concentrations in the brain during periods of high energy demand (Du et al. 2018).
Corroborating the involvement of mitochondrial dysfunction in the pathophysiology of BD, a recent study demonstrated that patients with the disorder have an imbalance between the processes of fusion and mitochondrial fission, observed by increased levels in fission protein (Fis-1) and decreased levels of protein of fusion proteins (Mfn-2 and Opa-1), suggesting that the process of mitochondrial dynamics in patients with BD is dysfunctional, which may increase mitochondrial fragmentation (Scaini et al. 2017). In fact, morphological abnormalities have been described (more mitochondria in a smaller size) and an abnormal pattern of agglomeration and marginalization in the intracellular distribution of mitochondria in neurons in the prefrontal cortex of the post-mortem brain and peripheral cells of patients with BD (Cataldo et al. 2010). Furthermore, it is known that morphological and process changes in mitochondrial dynamics can directly activate the apoptotic pathway. This fact was evidenced by an increase in the protein levels of the active form of caspase-3, which was shown to be negatively correlated with the protein levels of Mfn-2 and Opa-1, as well as by a decrease in anti-apoptotic factors. In addition, the same study indicated that the changes observed peripherally were directly correlated with functional decline in patients with BD (Scaini et al. 2017).
As previously described, mitochondrial dynamics involves not only the processes of fission and fusion but also the movement of mitochondria through neurons, a process influenced by the concentration of ATP and Ca + 2. In addition to changes in ATP levels, as already mentioned, studies have pointed out that BD patients have changes in intracellular Ca + 2 signalling, causing an increase in cytosolic Ca + 2 concentrations, related to excitotoxicity, decreased mitochondrial viability and, for the end, cell death (Duchen 2000).
In addition to providing most of the cellular ATP, mitochondria also play a central role in a wide variety of metabolic pathways and cellular functions. Since the brain uses about 20% of the body’s total ATP, mitochondrial dysfunction significantly impacts brain functions. Although many questions remain open, many studies have shown that mitochondrial dysfunction, at both peripheral and CNS levels, is related to the pathophysiology of bipolar disorders.
4 Adenylate Cyclase Signalling Pathway
Intracellular ATP plays a central role in energy homeostasis, being the main product of reactions such as photophosphorylation, respiration and fermentation, acting as an energetic donor in most endergonic biosynthetic processes that support cell survival, proliferation and motility. The cytoplasm of most mammalian cells contains an ATP concentration in the range of 5–10 mM, and even higher concentrations are stored in the form of vesicles at synaptic terminals of neurons (Adinolfi et al. 2010).
Once in the extracellular environment, ATP has a signalling role, as in proliferation, mitogenesis and cell differentiation (Burnstock and Verkhratsky 2010), and in high concentrations, ATP extracellular cell causes cytotoxic effects (Lemmens et al. 1996). Specifically, in the central nervous system (CNS), extracellular ATP can act both as a fast-exciting neurotransmitter, by supporting Ca+2 waves between astrocytes, and as a neuromodulator (Fam et al. 2000). Since the extracellular concentration of ATP is in the nanomolar range in physiological situations, even a small release of ATP can generate a very significant signal, due to this low background (Adinolfi et al. 2010). In this context, studies demonstrate that ATP can activate microglial cells to release cytokines (Sanz and Di Virgilio 2000) and, therefore, seems to act in a pro-inflammatory way, while adenosine, a product of the hydrolysis of this nucleotide, may have an anti-inflammatory effect (Bours et al. 2006, Di Virgilio et al. 2009). Adenosine is also a neuromodulator capable of mediating neuroprotection by decreasing membrane excitability, limiting the influx of calcium and exerting modulating effects on glial cells (Ribeiro et al. 2003). Also, it is well established as a presynaptic inhibitor, reducing the release of neurotransmitters such as glutamate, dopamine, serotonin and acetylcholine (Brundege and Dunwiddie 1997).
There is essential adaptive plasticity of the ectonucleotidase pathway, with the main function in the CNS to protect the cells of this noble organ, avoiding accumulations, absence and exacerbated stimuli of these signalling molecules, when necessary. In general, in the sense of a decrease in ATP levels, and an increase in adenosine levels, since this is a neuroprotective molecule (Bonan 2012).
Associated with changes in neurotrophins, other factors linked to the homeostasis of the central nervous system may change during episodes, such as the increase in free radicals (oxidative stress). The imbalance in the production and control of reactive oxygen species, demonstrated, among other results.
5 Neurotrophins and Neurogenesis
Biochemical studies from peripheral samples from patients have been important in clarifying the pathophysiology of bipolar disorder. In this context, a growing body of evidence suggests changes in molecules associated with neuroplasticity (Scola and Andreazza 2015). Among these molecules, the role of neurotrophins stands out, which comprise a class of highly abundant proteins in the nervous system with fundamental functions in neuronal survival, growth and plasticity (Huang and Reichardt 2001).
In this perspective, the neurotrophins are proteins responsible for mediating neuronal survival and differentiation, modulation and transmission of synaptic plasticity. BDNF (brain-derived neurotrophic factor) is the most abundant neurotrophic factor in the central nervous system of adult humans and is highly expressed in brain areas known to regulate complex cognitive functions (Post 2007). It is known that there is evidence of changes in BDNF in neuropsychiatric diseases such as multiple sclerosis, Alzheimer’s disease and Parkinson’s disease (Duman and Monteggia 2006).
The evidence suggests a role for BDNF in the pathogenesis of BD. Blood levels of BDNF have been described as decreased in BD patients during manic, depressed and even euthymic episodes (Palomino et al. 2006; Machado-Vieira et al. 2007; Monteleone et al. 2008; de Oliveira et al. 2009). But these results have not been replicated in other studies (Yoshimura et al. 2006; Mackin et al. 2007). In one study, it was described that BD patients had lower BDNF levels than healthy controls (Lin 2009). Taking into account the different affective poles, the results showed statistically significant differences in BDNF only among manic patients or depressed state and controls and not between patients in euthymic state and controls. Also, BDNF levels increased significantly after pharmacological treatment of the manic state. These findings indicate that BDNF levels are abnormally reduced in the manic and depressed states of BD and that the reduced level in the manic state increases after pharmacological treatment. This suggests a potential role in the level of BDNF in the blood as a state-dependent BD biomarker (Hashimoto 2010).
Also, studies also indicate changes in other neurotrophins and trophic factors in bipolar patients, including neurotrophin-3, neurotrophin-4/5, glia-derived neurotrophic factor (GDNF) and neural (NGF) and endothelial growth factors vascular (VEGF), reinforcing the hypothesis that impairments in neuroplasticity are involved in the pathophysiology of bipolar disorder (Scola and Andreazza 2015).
6 Neuroendocrine
There is evidence that in bipolar disorder, dysfunction of the hypothalamic-pituitary-adrenal (HPA) axis is associated with neuroprogression, which can induce persistent changes in the HPA axis that lead to depression and more refractory bipolar episodes in adults. It is believed that disturbance of the hormonal stress system can cause problems of thought and memory and worsen depressive symptoms in bipolar disorder (Watson et al. 2004). Hypoactivation of the HPA axis has been observed in depression with atypical characteristics, but also in other depression subtypes, in manic/hypomanic episodes. These abnormalities are related to the feedback of glucocorticoids, on the HPA axis, and their binding to glucocorticoid receptors (GR and MR). These abnormalities seem to be related to changes in the ability to circulate glucocorticoids to exert their negative feedback on the secretion of hormones from the HPA axis by binding to mineralocorticoid (MR) and glucocorticoid (GR) receptors in HPA tissues (Juruena et al. 2015). Bipolar disorder is multifactorial concerning the possible factors that contribute to their aetiology – biological and environmental factors (Juruena 2014, see Fig. 2).

The impact on neuroprogression, adapted from Moylan et al. (2013). Illustrations courtesy of Romayne Gadelrab
Regarding neurobiological markers, cortisol is considered one of the main mediators of allostatic load (McEwen 2006). Several studies in bipolar disorder – and major depressive disorder – demonstrate dysregulation of the HPA axis, with the maintenance of high levels of cortisol even in stages of remission of the disease, due to possible negative impaired feedback in the HPA axis (Watson et al. 2004; Ellenbogen et al. 2010; Juruena et al. 2010). Also, abnormalities in axis regulation may predict depressive (Juruena et al. 2009) and manic relapses (Maripuu et al. 2016). It can be considered that the reduction in resilience with the repetition of episodes and the increase in reactivity to stressors in the progression of the disease may be related to the deregulation of the HPA axis (Juruena 2014).
Bipolar disorder has also been associated with impaired cell resilience. Changes in cell signalling pathways, that is, in the cell’s ability to deal with a given stimulus and the response to it, may be associated with greater vulnerability to stressful events, and this includes changes in neurotransmitters, trophic cascade, calcium, anti-apoptotic factors and pathway survival (Belvederi Murri et al. 2016).
HPA axis and renin-angiotensin-aldosterone system (RAAS) impairment may be some of the intrinsic aetiological factors (Juruena et al. 2015; Heuser et al. 2000; Keller et al. 2017; Murck et al. 2019). MR and GR receptors in the brain are involved in the regulation of stress hormone secretion and complex behaviour, such as emotion, memory and sleep. In the pathophysiology of stress-related psychiatric disorders, these receptors have not been sufficiently characterized, but HPA axis and RAAS interact with each other, and both are mediated by the limbic system (de Kloet and Joëls 2017; Harris et al. 2013; Murck et al. 2012).
Aldosterone and cortisol bind to mineralocorticoid receptors (MR) and glucocorticoid receptors (GR), respectively, to exert their actions on RAAS and HPA axis. MR/GR imbalance and inadequate levels of aldosterone and cortisol may impair resilience capacity of subjects, raising their vulnerable phenotype to disorders related to stress exposition (de Kloet and Joëls 2017; Gomez-Sanchez and Gomez-Sanchez 2014). As HPA axis and RAAS adequate functioning also depend on their receptors’ well-functioning, their gene receptors’ polymorphisms are important biological factors to be considered in bipolar patients. Some of MR and GR gene polymorphisms have been shown in the literature associated with the imbalanced functioning HPA axis and RAAS (DeRijk et al. 2008; van Leeuwen et al. 2011).
7 Conclusion
The data we have reviewed suggests that peripheral biomarkers related to hormones, inflammation, oxidative stress and neurotrophins are altered in the bipolar disorder, especially during acute mood episodes. Together, these changes have been associated with a systemic toxicity of the disease and the damage resulting from multiple episodes.
Systemic toxicity related to recurrent episodes in bipolar disorder can influence brain anatomical changes associated with the progression of bipolar disorder and the response to stress and neuroplasticity.
References
Ackenheil M (2001) Neurotransmitters and signal transduction processes in bipolar affective disorders: a synopsis. J Affect Disord 62(1–2):101–111
Adinolfi E, Cirillo M, Woltersdorf R, Falzoni S, Chiozzi P, Pellegatti P, Callegari MG, Sandonà D, Markwardt F, Schmalzing G, Di Virgilio F (2010) Trophic activity of a naturally occurring truncated isoform of the P2X7 receptor. FASEB J 24(9):3393–3404
Altamura C, Mauri M, Ferrara A, Moro A, D'Andrea G, Zamberlan F (1993) Plasma and platelet excitatory amino acids in psychiatric disorders. Am J Psychiatry 150:1731–1733
Anand A, Barkay G, Dzemidzic M, Albrecht D, Karne H, Zheng QH, Hutchins GD, Normandin MD, Yoder KK (2011) Striatal dopamine transporter availability in unmedicated bipolar disorder. Bipolar Disord 13(4):406–413
Andreazza AC, Kauer-Sant'anna M, Frey BN, Bond DJ, Kapczinski F, Young LT, Yatham LN (2008) Oxidative stress markers in bipolar disorder: a meta-analysis. J Affect Disord 111(2-3):135–144
Bäckström P, Hyytiä P (2003) Attenuation of cocaine-seeking behaviour by the AMPA/kainate receptor antagonist CNQX in rats. Psychopharmacology (Berl) 166:69–76
Barnett JH, Smoller JW (2009) The genetics of bipolar disorder. Neuroscience 164(1):331–343
Baumann B, Danos P, Krell D, Diekmann S, Wurthmann C, Bielau H et al (1999) Unipolar-bipolar dichotomy of mood disorders is supported by noradrenergic brainstem system morphology. J Affect Disord 54(1–2):217–224
Beaulieu JM, Gainetdinov RR (2011) The physiology, signalling, and pharmacology of dopamine receptors. Pharmacol Rev 63(1):182–217
Belvederi Murri M, Prestia D, Mondelli V et al (2016) The HPA axis in bipolar disorder: systematic review and metaanalysis. Psychoneuroendocrinology 63:327–342
Beneyto M, Meador-Woodruff J (2006) Lamina-specific abnormalities of AMPA receptor trafficking and signaling molecule transcripts in the prefrontal cortex in schizophrenia. Synapse 60:585–598
Berk M, Dodd S, Kauer-Sant'anna M, Malhi GS, Bourin M, Kapczinski F, Norman T (2007) Dopamine dysregulation syndrome: implications for a dopamine hypothesis of bipolar disorder. Acta Psychiatr Scand Suppl 434:41–49
Berk M, Kapczinski F, Andreazza AC, Dean OM, Giorlando F, Maes M, Yücel M, Gama CS, Dodd S, Dean B, Magalhães PV, Amminger P, McGorry P, Malhi GS (2011) Pathways underlying neuroprogression in bipolar disorder: focus on inflammation, oxidative stress and neurotrophic factors. Neurosci Biobehav Rev 35(3):804–817
Berk M, Berk L, Dodd S, Cotton S, Macneil C, Daglas R, Conus P, Bechdolf A, Moylan S, Malhi GS (2014) Stage managing bipolar disorder. Bipolar Disord 16(5):471–477
Bhagwagar Z et al (2008) Low GABA concentrations in occipital cortex and anterior cingulate cortex in medication-free, recovered depressed patients. Int J Neuropsychopharmacol 11(2):255–260
Bobo WV (2017) The diagnosis and management of bipolar I and II disorders: clinical practice update. Mayo Clin Proc 92(10):1532–1551
Bonan CD (2012) Ectonucleotidases and nucleotide/nucleoside transporters as pharmacological targets for neurological disorders. CNS Neurol Disord Drug Targets 11(6):739–750
Bosaipo NB, Borges VF, Juruena MF (2017) Bipolar disorder: a review of conceptual and clinical aspects. Medicina 50(Suppl 1):72–84
Bours MJ, Swennen EL, Di Virgilio F, Cronstein BN, Dagnelie PC (2006) Adenosine 5′-triphosphate and adenosine as endogenous signaling molecules in immunity and inflammation. Pharmacol Ther 112(2):358–404
Brady RO et al (2013) Brain gamma-aminobutyric acid (GABA) abnormalities in bipolar disorder. Bipolar Disord 15(4):434–439
Brown NC, Andreazza AC, Young LT (2014) An updated meta-analysis of oxidative stress markers in bipolar disorder. Psychiatry Res 218(1–2):61–68
Brundege JM, Dunwiddie TV (1997) Role of adenosine as a modulator of synaptic activity in the central nervous system. Adv Pharmacol 39:353–391
Brunello N, Tascedda F (2003) Cellular mechanisms and second messengers: relevance to the psychopharmacology of bipolar disorders. Int J Neuropsychopharmacol 6(2):181–189
Bunney WE Jr, Garland BL (1982) A second generation catecholamine hypothesis. Pharmacopsychiatria 15(4):111–115
Burnstock G, Verkhratsky A (2010) Long-term (trophic) purinergic signalling: Purinoceptors control cell proliferation, differentiation and death. Cell Death Dis 1:e9
Caspi A, Moffitt TE (2006) Gene-environment interactions in psychiatry: joining forces with neuroscience. Nat Rev Neurosci 7(7):583–590
Cataldo AM, McPhie DL, Lange NT, Punzell S, Elmiligy S, Ye NZ et al (2010) Abnormalities in mitochondrial structure in cells from patients with bipolar disorder. Am J Pathol 177(2):575–585
Clinton SM, Meador-Woodruff JH (2004) Abnormalities of the NMDA receptor and associated intracellular molecules in the thalamus in schizophrenia and bipolar disorder. Neuropsychopharmacology 29:1353–1362
Colino A, García-Seoane JJ, Valentín A (1998) Action potential broadening induced by lithium may cause a presynaptic enhancement of excitatory synaptic transmission in neonatal rat hippocampus. Eur J Neurosci 10:2433–2443
Cuthbert BN, Insel TR (2013) Toward the future of psychiatric diagnosis: the seven pillars of RDoC. BMC Med 11:126
de Kloet ER, Joëls M (2017) Brain mineralocorticoid receptor function in control of salt balance and stress-adaptation. Physiol Behav 178:13–20
de Oliveira GS, Ceresér KM, Fernandes BS et al (2009) Decreased brain-derived neurotrophic factor in medicated and drug-free bipolar patients. J Psychiatr Res 43:1171–1174
Derijk RH, van Leeuwen N, Klok MD, Zitman FG (2008) Corticosteroid receptor-gene variants: modulators of the stress-response and implications for mental health. Eur J Pharmacol 585(2–3):492–501
Di Virgilio F, Ceruti S, Bramanti P, Abbracchio MP (2009) Purinergic signalling in inflammation of the central nervous system. Trends Neurosci 32(2):79–87
Dong XX, Wang Y, Qin ZH (2009) Molecular mechanisms of excitotoxicity and their relevance to pathogenesis of neurodegenerative diseases. Acta Pharmacol Sin 30:379–387
Du J, Gray NA, Falke C, Yuan P, Szabo S, Manji HK (2003) Structurally dissimilar antimanic agents modulate synaptic plasticity by regulating AMPA glutamate receptor subunit GluR1 synaptic expression. Ann N Y Acad Sci 1003:378–380
Du F, Yuksel C, Chouinard VA, Huynh P, Ryan K, Cohen BM, Öngür D (2018) Abnormalities in high-energy phosphate metabolism in first-episode bipolar disorder measured using 31P-magnetic resonance spectroscopy. Biol Psychiatry 84(11):797–802
Duchen MR (2000) Mitochondria and Ca(2+)in cell physiology and pathophysiology. Cell Calcium 28(5–6):339–348
Duman RS, Monteggia LM (2006) A neurotrophic model for stress-related mood disorders. Biol Psychiatry 59(12):1116–1127
Ellenbogen MA, Santo JB, Linnen AM, Walker CD, Hodgins S (2010) High cortisol levels in the offspring of parents with bipolar disorder during two weeks of daily sampling. Bipolar Disord 12:77–86
Fam SR, Gallagher CJ, Salter MW (2000) P2Y(1) purinoceptor-mediated Ca(2+) signaling and Ca(2+) wave propagation in dorsal spinal cord astrocytes. J Neurosci 20(8):2800–2808
Fee C, Banasr M, Sibille E (2017) Somatostatin-positive GABA Interneuron deficits in depression: cortical microcircuit and therapeutic perspectives. Biol Psychiatry 82(8):549–559
Gebhardt C, Cull-Candy SG (2010) Lithium acts as a potentiator of AMPAR currents in hippocampal CA1 cells by selectively increasing channel open probability. J Physiol 588:3933–3941
Gomez-Sanchez E, Gomez-Sanchez CE (2014) The multifaceted mineralocorticoid receptor. Compr Physiol 4(3):965–994
Hahn CG, Friedman E (1999) Abnormalities in protein kinase C signaling and the pathophysiology of bipolar disorder. Bipolar Disord 1(2):81–86
Harris AP, Holmes MC, de Kloet ER, Chapman KE, Seckl JR (2013) Mineralocorticoid and glucocorticoid receptor balance in control of HPA axis and behaviour. Psychoneuroendocrinology 38(5):648–658
Hashimoto K (2010) Brain-derived neurotrophic factor as a biomarker for mood disorders: an historical overview and future directions. Psychiatry Clin Neurosci 64(4):341–357
Hashimoto K, Sawa A, Iyo M (2007) Increased levels of glutamate in brains from patients with mood disorders. Biol Psychiatry 62:1310–1316
Heuser I, Deuschle M, Weber B, Stalla GK, Holsboer F (2000) Increased activity of the hypothalamus-pituitary-adrenal system after treatment with the mineralocorticoid receptor antagonist spironolactone. Psychoneuroendocrinology 25(5):513–518
Hoekstra R, Fekkes D, Loonen A, Pepplinkhuizen L, Tuinier S, Verhoeven W (2006) Bipolar mania and plasma amino acids: increased levels of glycine. Eur Neuropsychopharmacol 16:71–77
Huang EJ, Reichardt LF (2001) Neurotrophins: roles in neuronal development and function. Annu Rev Neurosci 24:677–736. Review
Joyce PR, Fegursson DM, Woollard G, Abbott RM, Horwood LJ, Upton J (1995) Urinary catecholamines and plasma hormones predict mood state in rapid cycling bipolar affective disorder. J Affect Disord 33(4):233–243
Juruena MF, Cleare AJ, Papadopoulos AS, Poon L, Lightman S, Pariante CM (2010) The prednisolone suppression test in depression: dose-response and changes with antidepressant treatment. Psychoneuroendocrinology 35(10):1486–1491
Juruena MF (2014) Early-life stress and HPA axis trigger recurrent adulthood depression. Epilepsy Behav 38:148–159
Juruena MF, Pariante CM, Papadopoulos AS, Poon L, Lightman S, Cleare AJ (2009) Prednisolone suppression test in depression: prospective study of the role of HPA axis dysfunction in treatment resistance. Br J Psychiatry 194:342–349
Juruena MF, Baes CW, Menezes IC, Graeff FG (2015) Early life stress in depressive patients: role of glucocorticoid and mineralocorticoid receptors and of hypothalamic-pituitary-adrenal axis activity. Curr Pharm Des 21(11):1369–1378
Keller J, Gomez R, Williams G, Lembke A, Lazzeroni L, Murphy GM et al (2017) HPA Axis in major depression: cortisol, clinical symptomatology, and genetic variation predict cognition. Mol Psychiatry 22(4):527–536
Kristiansen L, Meador-Woodruff J (2005) Abnormal striatal expression of transcripts encoding NMDA interacting PSD proteins in schizophrenia, bipolar disorder and major depression. Schizophr Res 78:87–93
Kurita M, Nishino S, Numata Y, Okubo Y, Sato T (2014) The noradrenaline metabolite MHPG is a candidate biomarker from the manic to the remission state in bipolar disorder I: a clinical naturalistic study. PLoS One 9(6):e100634
Law AJ, Deakin JF (2001) Asymmetrical reductions of hippocampal NMDAR1 glutamate receptor mRNA in the psychoses. Neuroreport 12:2971–2974
Leach MJ, Marden CM, Miller AA (1986) Pharmacological studies on lamotrigine, a novel potential antiepileptic drug: II. Neurochemical studies on the mechanism of action. Epilepsia 27:490–497
Lemmens R, Vanduffel L, Teuchy H, Culic O (1996) Regulation of proliferation of LLC-MK2 cells by nucleosides and nucleotides: the role of ectoenzymes. Biochem J 316(Pt 2):551–557
Lin PY (2009) State-dependent decrease in levels of brain-derived neurotrophic factor in bipolar disorder: a meta-analytic study. Neurosci Lett 466:139–143
Luján R, Shigemoto R, López-Bendito G (2005) Glutamate and GABA receptor signalling in the developing brain. Neuroscience 130:567–580
Luscher B, Shen Q, Sahir N (2011) The GABAergic deficit hypothesis of major depressive disorder. Mol Psychiatry 16(4):383–406
Machado-Vieira R, Dietrich MO, Leke R et al (2007) Decreased plasma brain derived neurotrophic factor levels in unmedicated bipolar patients during manic episode. Biol Psychiatry 61:142–144
Machado-Vieira R, Manji HK, Zarate CA Jr (2009) The role of lithium in the treatment of bipolar disorder: convergent evidence for neurotrophic effects as a unifying hypothesis. Bipolar Disord 11(Suppl 2):92–109
Mackin P, Gallagher P, Watson S, Young AH, Ferrier IN (2007) Changes in brain-derived neurotrophic factor following treatment with mifepristone in bipolar disorder and schizophrenia. Aust N Z J Psychiatry 41:321–326
Manji HK, Henter ID, Zarate CA Jr (2011) Bipolar disorder: a neurobiological synthesis. Curr Top Behav Neurosci 5:331–340
Maripuu M, Wikgren M, Karling P, Adolfsson R, Norrback KF (2016) Relative hypocortisolism is associated with obesity and the metabolic syndrome in recurrent affective disorders. J Affect Disord 204:187–196
Mazer AK, Cleare AJ, Young AH, Juruena MF (2019) Bipolar affective disorder and borderline personality disorder: differentiation based on the history of early life stress and psychoneuroendocrine measures. Behav Brain Res 357–358:48–56
McCullumsmith R, Kristiansen L, Beneyto M, Scarr E, Dean B, Meador-Woodruff J (2007) Decreased NR1, NR2A, and SAP102 transcript expression in the hippocampus in bipolar disorder. Brain Res 1127:108–118
McEwen B (2006) Dialogues Clin Neurosci 8(4)
Monteleone P, Serritella C, Martiadis V, Maj M (2008) Decreased levels of serum brain-derived neurotrophic factor in both depressed and euthymic patients with unipolar depression and in euthymic patients with bipolar I and II disorders. Bipolar Disord 10:95–100
Moylan S, Maes M, Wray N, Berk M (2013) The neuroprogressive nature of major depressive disorder: pathways to disease evolution and resistance, and therapeutic implications. Mol Psychiatry 18:595–606
Murck H, Schüssler P, Steiger A (2012) Renin-angiotensin-aldosterone system: the forgotten stress hormone system: relationship to depression and sleep. Pharmacopsychiatry 45(3):83–95
Murck H, Braunisch MC, Konrad C, Jezova D, Kircher T (2019) Markers of mineralocorticoid receptor function: changes over time and relationship to response in patients with major depression. Int Clin Psychopharmacol 34(1):18–26
Obata T (2002) Dopamine efflux by MPTP and hydroxyl radical generation. J Neural Transm (Vienna) 109(9):1159–1180
Palomino A, Vallejo-Illarramendi A, González-Pinto A et al (2006) Decreased levels of plasma BDNF in first-episode schizophrenia and bipolar disorder patients. Schizophr Res 86:321–322
Petty F, Kramer GL, Fulton M, Moeller FG, Rush AJ (1993) Low plasma GABA is a marker of a characteristic similar to bipolar disorder. Neuropsychopharmacology 9(2):125–132
Post RM (2007) Role of BDNF in bipolar and unipolar disorder: clinical and theoretical implications. J Psychiatr Res 41(12):979–990
Prange AJ Jr, Wilson IC, Lynn CW, Alltop LB, Stikeleather RA (1974) L-tryptophan in mania. Contribution to a permissive hypothesis of affective disorders. Arch Gen Psychiatry 30(1):56–62
Rahn KA, Slusher BS, Kaplin AI (2012) Glutamate in CNS neurodegeneration and cognition and its regulation by GCPII inhibition. Curr Med Chem 19:1335–1345
Rao J, Ertley R, Rapoport S, Bazinet R, Lee H (2007) Chronic NMDA administration to rats up-regulates frontal cortex cytosolic phospholipase A2 and its transcription factor, activator protein-2. J Neurochem 102:1918–1927
Ribeiro JA, Sebastiao AM, de Mendonca A (2003) Participation of adenosine receptors in neuroprotection. Drug News Perspect 16(2):80–86
Sanacora G et al (2004) Subtype-specific alterations of gamma-aminobutyric acid and glutamate in patients with major depression. Arch Gen Psychiatry 61(7):705–713
Sanacora G, Kendell SF, Levin Y, Simen AA, Fenton LR, Coric V, Krystal JH (2007) Preliminary evidence of riluzole efficacy in antidepressant-treated patients with residual depressive symptoms. Biol Psychiatry 61:822–825
Sanacora G, Zarate CA, Krystal JH, Manji HK (2008) Targeting the glutamatergic system to develop novel, improved therapeutics for mood disorders. Nat Rev Drug Discov 7:426–437
Sanz JM, Di Virgilio F (2000) Kinetics and mechanism of ATP-dependent IL-1 beta release from microglial cells. J Immunol 164(9):4893–4898
Scaini G, Rezin GT, Carvalho AF, Streck EL, Berk M, Quevedo J (2016) Mitochondrial dysfunction in bipolar disorder: evidence, pathophysiology and translational implications. Neurosci Biobehav Rev 68:694–713
Scaini G, Fries GR, Valvassori SS, Zeni CP, Zunta-Soares G, Berk M et al (2017) Perturbations in the apoptotic pathway and mitochondrial network dynamics in peripheral blood mononuclear cells from bipolar disorder patients. Transl Psychiatry 7(5):e1111
Scola G, Andreazza AC (2015) The role of neurotrophins in bipolar disorder. Prog Neuropsychopharmacol Biol Psychiatry 56:122–128
Shiah IS, Yatham LN (2000) Serotonin in mania and in the mechanism of action of mood stabilizers: a review of clinical studies. Bipolar Disord 2(2):77–92
Steckert AV, Valvassori SS, Moretti M, Dal-Pizzol F, Quevedo J (2010) Role of oxidative stress in the pathophysiology of bipolar disorder. Neurochem Res 35(9):1295–1301
van Leeuwen N, Bellingrath S, de Kloet ER, Zitman FG, DeRijk RH, Kudielka BM, Wüst S (2011) Human mineralocorticoid receptor (MR) gene haplotypes modulate MR expression and transactivation: implication for the stress response. Psychoneuroendocrinology 36(5):699–709
Wang JF, Shao L, Sun X, Young LT (2009) Increased oxidative stress in the anterior cingulate cortex of subjects with bipolar disorder and schizophrenia. Bipolar Disord 11:523–529
Watson S, Gallagher P, Ritchie JC, Ferrier IN, Young AH (2004) Hypothalamic-pituitary-adrenal axis function in patients with bipolar disorder. Br J Psychiatry 184:496–502
Wilkinson ST, Sanacora G (2019) A new generation of antidepressants: an update on the pharmaceutical pipeline for novel and rapid-acting therapeutics in mood disorders based on glutamate/GABA neurotransmitter systems. Drug Discov Today 24(2):606–615
Yoshimura R, Nakano Y, Hori A et al (2006) Effect of risperidone on plasma catecholamine metabolites and brain-derived neurotrophic factor in patients with bipolar disorders. Hum Psychopharmacol 21:433–438
Young LT, Warsh JJ, Kish SJ et al (1994) Reduced brain 5-HT and elevated NE turnover and metabolites in bipolar affective disorder. Biol Psychiatry 35:121–127
Zarate CA Jr, Quiroz JA, Singh JB, Denicoff KD, De Jesus G, Luckenbaugh DA, Charney DS, Manji HK (2005) An open-label trial of the glutamate-modulating agent riluzole in combination with lithium for the treatment of bipolar depression. Biol Psychiatry 57(4):430–432
Zarate CJ, Singh J, Manji H (2006) Cellular plasticity cascades: targets for the development of novel therapeutics for bipolar disorder. Biol Psychiatry 59:1006–1020
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Young, A.H., Juruena, M.F. (2020). The Neurobiology of Bipolar Disorder. In: Young, A.H., Juruena, M.F. (eds) Bipolar Disorder: From Neuroscience to Treatment. Current Topics in Behavioral Neurosciences, vol 48. Springer, Cham. https://doi.org/10.1007/7854_2020_179
Download citation
DOI: https://doi.org/10.1007/7854_2020_179
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-72142-8
Online ISBN: 978-3-030-72143-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)