
Abstract
Despite the extensive research in the field, the precise etiology of bipolar disorder (BD) is not clear; neither, then, is our understanding of the pathogenesis of bipolar depression. What we do know is largely gleaned from investigations of patients with BD irrespective of their mood state. The most consistent neuropathological findings include structural, cellular, and functional changes in cortical and limbic regions, and most explanations for the illness involve pathways that ultimately result in cerebral atrophy and cell loss. Though many theories have been proposed, here, the focus is on the current leading hypotheses of mitochondrial involvement, oxidative stress, the role of inflammation, and neurotrophic factors. Future research is required with a specific focus on the depressive phase of BD in addition to the study of biomarkers to aid clinicians in the diagnosis and targeted treatment of bipolar depression.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Understanding the Neurobiology of Bipolar Depression
Despite extensive research in the field, the precise etiology of bipolar disorder (BD) is not clear. Neither, then, do we have a fulsome understanding of the neurobiology of bipolar depression. Some insights have been gained from knowledge of the mechanisms associated with major depressive disorder (MDD ); however, it is largely believed that bipolar depression is unique because of its differential response to conventional antidepressant treatment as well as its divergent prognosis and course of illness. Our understanding of the pathogenesis of bipolar depression comes from the investigation of patients with BD irrespective of their mood state. Few studies focus exclusively on bipolar depression. Subsequently, the existing literature exploring the neurobiology of BD is robust but can be piecemeal with respect to the depressive state. This chapter will feature key areas of research into the pathophysiology of BD, taking the opportunity to highlight depression-specific findings where available.
Though many theories have been proposed, here, the focus is on the current leading hypotheses of mitochondrial involvement, oxidative stress, the role of inflammation, and neurotrophic factors. Other hypotheses have implicated the serotonergic system and neurotransmitter dysfunction (see Wang and Young (2009) for review) but will not be discussed here in detail. The preceding chapter provides an in-depth discussion of genetic factors implicated in bipolar depression and so will not be reviewed again here.
We begin with a review of the neuropathological features of BD since most explanations for the illness involve pathways that ultimately result in cerebral atrophy and cell loss. Finally, we attend to the future of research in this field, highlighting the need for biomarkers to guide diagnosis and treatment of this complex illness.
2 Neuropathological Studies Reveal Cellular Loss in Cerebral Cortical and Limbic Regions
Structural, cellular, and functional changes have long been observed in the brains of patients with BD. These changes are often classified based on region, allowing researchers to postulate relationships between neuroanatomy and behavioral correlates of BD. The neural basis of affective states includes the subcortical limbic system as well as cortical regions like the anterior cingulate cortex (ACC ) and dorsolateral prefrontal cortex (dlPFC ). The limbic system comprises primitive structures (e.g., amygdala, hippocampus, thalamus) essential to one’s ability to perceive, process, and create memories about emotions and experiences with emotional valence (Janak and Tye 2015). The cerebral cortex is implicated in emotion through its extensive connections to the limbic system and other brain regions. In the cortex, complex sensory information is processed and, through its connections, the cortex is believed to modulate one’s experience of emotion (Salzman and Fusi 2010). In BD, neuroanatomical abnormalities are observed in both limbic and cortical areas, and these changes are fundamental to understanding the neurobiology of the illness.
The amygdala is widely understood to be the seat of our emotions and is necessarily implicated in BD, with its prominent affective states. Using MRI to estimate amygdala volume, Hartberg and colleagues (2015) recently reported a decrease in left amygdala volume in patients with BD not treated with lithium. These researchers note that lithium-treated patients displayed larger amygdalar volumes, suggesting a neuroprotective effect for lithium. This study corroborates other evidence linking amygdala structure to BD (Foland-Ross et al. 2012; Inal-Emiroglu et al. 2015; Kittel-Schneider et al. 2015). Previous reports investigating volumes and neurons in amygdala of postmortem specimens highlight a decrease in the size of neuronal cell bodies as well as decreased density of neurons in certain nuclei of the amygdala.
The hippocampus is known for its role in memory, and the thalamus is known as a relay station between cortical and subcortical regions; however, both regions play a key role in regulating emotion and have been implicated in the neurobiology of BD. Postmortem brain samples from patients with BD have smaller CA1 pyramidal hippocampal neurons compared to a control population (Liu et al. 2007). Some reports support a reduction in thalamic volume and support cells in BD patients (Bielau et al. 2005; Byne et al. 2008). In contrast, there is compelling evidence for decreased hippocampal volume in MDD coming from both imaging and postmortem brain studies (Campbell and Macqueen 2004). This potentially highlights different pathophysiological mechanisms in MDD vs. BD.
The ACC is thought to be a hub in the network between our cortical cognitive capacity and our subcortical limbic emotional experiences, and the subgenual region of the ACC (sgACC) is particularly implicated in emotion regulation (Ghaznavi and Deckersbach 2012). In patients with bipolar depression, gray matter volume, blood flow, and metabolism in the sgACC are decreased. Early investigation of postmortem ACC specimens from BD patients revealed a deficit in both the number and density of glia cells, i.e., the nonneuronal cells that play a supportive function in the central nervous system facilitating neuronal growth and differentiation (Wake et al. 2013). Notably, the structural changes observed in the sgACC were present in patients with a family history of BD at the outset of their illness (Hirayasu et al. 1999). Further, recent imaging analyses highlight the functional connectivity between brain regions and note that the children of patients with BD are more likely to exhibit atypical connection patterns between cortical neurons (including the sgACC) and subcortical areas (Singh et al. 2014). Taken together, these findings suggest that pathophysiologic changes in patients with this disorder are not entirely due to the damaging effect of repeated mood episodes but can be present at the outset of the illness. Here again, lithium treatment is associated with a reversal in the sgACC gray matter volume deficits observed in patients with BD (Moore et al. 2009). In addition, response to ketamine—whose novel therapeutic use shows efficacy in treatment-resistant depression—is linked to sgACC activity in patients with bipolar depression (Nugent et al. 2014) (Fig. 6.1).

Areas where regional metabolic rate of glucose (rMRGlu) following placebo infusion was significantly correlated with percent improvement in Montgomery–Åsberg Depression Rating Scale (MADRS) score following ketamine administration. Crosshair is centered on the finding in the subgenual anterior cingulate cortex (sgACC), but the cluster with the peak voxel is in the dorsal cingulate. Image threshold at p < 0.05 uncorrected. The extent threshold was set such that only the clusters remaining significant after correction for multiple comparisons (p corrected < 0.01) are shown (Nugent et al. 2014). Reprinted with permission from Nugent et al. (2014), Fig. 2
The dlPFC is implicated in attention and executive functioning—patients with BD have long been known to have decreased performance on tasks that require these cognitive capacities (Rubinsztein et al. 2006; Taylor and Abrams 1987). The dlPFC has also been identified as a central locus in the cortical emotional processing network that plays a role in cognitive reappraisals of emotionally salient events (Morawetz et al. 2016). Thus, it is expected that the dlPFC is a region fundamental to the pathophysiology of BD where maladaptive affect regulation and extreme appraisals of affective states are key deficits (Palmier-Claus et al. 2015). Indeed, patients with BD display deficient connections between the dlPFC and subcortical limbic regions, suggesting a possible mechanism for emotion dysregulation in BD (Radaelli et al. 2015).
Examination of the cellular architecture of the dlPFC found decreased density of glial cells in certain cortical layers of brain specimens of BD patients. Studies also observed a significant reduction in the number of pyramidal neurons with a trend towards decreased neuron size. Subsequent studies inspecting postmortem samples corroborated these results. MRI quantifying volume of gray matter showed decreased dlPFC volumes in children and adolescents with BD (Adleman et al. 2012), suggesting that these structural changes may underlie the pathogenesis of BD.
These above-noted structural abnormalities are widely accepted as fundamental to the pathology of BD, though the mechanisms that underlie these changes remain unclear. In the next section, we describe some of the potential models that lead to cell loss including work from our own laboratory.
3 Proposed Mechanisms Underlying Cortical and Limbic Cell Loss
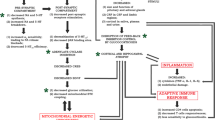
The proposed mechanisms thought to underlie these structural brain changes fall loosely into two categories: (1) processes that increase destruction of neuronal cells or (2) processes with dysfunctional neuroprotective mechanisms. We begin by examining the role of mitochondrial dysfunction and the destructive impact of oxidative stress and then explore the role of inflammation before concluding with a discussion of neurotrophic factors.
3.1 The Role of Mitochondrial Dysfunction
The past 20 years have seen a growing literature supporting the concept of dysfunctional mitochondrial energy metabolism in the pathogenesis of BD. Mitochondria are ubiquitous organelles providing cellular energy in the form of adenosine triphosphate (ATP ). Energy is created via the process of oxidative phosphorylation, i.e., the transfer of electrons via mitochondrial membrane proteins (the mitochondrial electron transport chain (mETC )) and creation of a proton gradient. The mETC is a series of protein complexes (I–V) situated in the inner mitochondrial membrane. Protons are shuttled across these complexes and the resulting electrochemical gradient allows for the transfer of electrons to an oxygen molecule (i.e., reduction of oxygen) and the creation of ATP.
Reactive oxygen species (ROS ) are formed as a byproduct of oxidative phosphorylation. These compounds have both physiological and pathological functions. In our normal physiological state, we utilize endogenous antioxidants to counteract the pathological or harmful effects of a buildup of ROS . Human brain tissue is specifically susceptible to ROS damage because of its high oxygen content and ROS overproduction; alternately, ROS that overwhelm the endogenous antioxidants can create neuronal damage specifically targeting lipids (key in neuronal membranes), proteins, and DNA (Moniczewski et al. 2015). The pathological effects of ROS , or oxidative stress, are reflected via a number of peripheral and central indicators and are associated with the pathophysiology of BD. ROS levels are elevated in BD, and it has been suggested that this likely reflects deficient energy metabolism resulting from mitochondrial dysfunction (Wang 2007). Many lines of evidence highlight a role for mitochondrial dysfunction in BD; for instance, the mitochondria of patients with BD exhibit morphological changes and distributional abnormalities within cells (Cataldo et al. 2010).
Patients differentially express genes related to energy metabolism with decreased expression of genes encoding for certain mitochondrial subunits, and downregulation of mitochondrial genes has been observed (Kato 2007; Naydenov et al. 2007; Sun et al. 2006; Washizuka et al. 2005). In depressed patients with BD, there is evidence for an increase in the expression of genes encoding for the mitochondrial ETC that is hypothesized to reflect an increase in turnover of mitochondria in this population (Beech et al. 2010).
Increased lactate levels have been found in brain tissue and cerebrospinal fluid from patients (Kato et al. 1992; Regenold et al. 2009). Earlier research by Kato and colleagues identified decreased intracellular pH in patients with BD, while a significant increase in pH was observed in patients during a depressive state (Kato et al. 1992). Low pH is associated with an increase in lactate—the byproduct of anaerobic glycolysis, which is the alternative energy-generating process arising when the function of mitochondria is reduced. The process of oxidative phosphorylation in mitochondria is meant to be the main source of cerebral energy, but in conditions of mitochondrial dysfunction, there can be a shift to anaerobic glycolysis and, subsequently, an accumulation of lactate. There is some evidence to suggest that it is the increase in lactate—following a state of mitochondrial dysfunction—that accounts for the reduced pH in patients with BD. It has been suggested that the increase in intracellular pH observed during the depressive state might reflect the brain’s attempt to correct pH imbalance (Kato et al. 1992; Stork and Renshaw 2005).
Phosphocreatine (PCr ) is created from creatinine and ATP and functions as an energy store for use during acute neuronal activity (MacDonald et al. 2006). Chronically decreased levels of PCr are indicative of mitochondrial dysfunction as evidenced by low brain PCr levels in other known mitochondrial disorders (Barbiroli et al. 1993; Chaturvedi and Flint Beal 2013; Eleff et al. 1990). Reductions in PCr have long been noted in BD, with research identifying low focal levels of PCr in the left frontal lobe of patients in the depressed state (Kato et al. 1994, 1995; Moore et al. 1997).
In all, there is impressive evidence supporting a role for mitochondrial dysfunction in the pathophysiology of BD. Oxidative stress is implicated as a proposed mechanism generating the brain atrophy and decreased cell density expressed in the population. Though data describing associations of oxidative stress and BD are abundant, the direction of the association remains unknown—does BD create oxidative stress or does oxidative stress create a milieu in which BD can develop?
3.2 Mitochondrial Dysfunction Leads to Oxidative Stress
Lipid peroxidation refers to the specific oxidative stress damage to lipids. This damage is key in CNS pathology because of the high content of lipids in white matter in the brain. Thiobarbituric acid reactive substances (TBARS ) are byproducts of lipid peroxidation that can be quantified from serum samples. Elevated serum levels of TBARS were quantified from a population of BD patients in various affective states (i.e., mania, depression, and euthymia (Andreazza et al. 2007a; Kunz et al. 2008)). Marked elevations were noted in manic patients, though levels in depressed patients were also increased (Kunz et al. 2008). Bannerjee and colleagues investigated the effects of lithium on TBARS and found that BD patients had elevated serum TBARS and that lithium treatment significantly reduced markers of lipid peroxidation (Banerjee et al. 2012).
4-Hydroxy-2-nonenal (4-HNE ), another marker of lipid peroxidation, is significantly increased in the postmortem brain specimens of patients with BD (Wang et al. 2009). Notably, the 4-HNE was sampled from cells of the ACC —a region with noted structural deficits and highlighted as key in the pathogenesis of BD (discussed above in Sect. 6.1.1). This postmortem sample included patients who had received pharmacological treatment for BD; however, subsequent analysis suggest that the differences identified were indicative of the pathological process in BD rather than changes induced by chronic medication (Wang et al. 2009). Malondialdehyde (MDA ) is created when ROS degrade cellular lipid components, and peripheral serum levels of MDA are increased in patients with BD compared to nonpsychiatric controls (Can et al. 2011). Interestingly, post hoc analyses that stratified patients by medication subtype found that those on a combination of antidepressant and antipsychotic medication (a popular combination in bipolar depression) exhibited decreased serum MDA values. In addition, the impact of antipsychotics and antidepressants suggest that they modulate antioxidant capacity to combat oxidative stress (Tang and Wang 2013). Recent evidence links lipid peroxidation measured peripherally to changes in white matter tracts, which may underscore the importance of this process to the pathophysiology of BD (Versace et al. 2014). These findings also identify a potential biomarker to be measured in blood and might specifically reflect changes in brain in this disorder (Fig. 6.2).


Panel (a). The posterior distribution of each white matter (WM) tract is displayed in isosurface mode. The forceps minor and major are represented in red; the anterior thalamic radiation in yellow; the angular bundle of the cingulum in light green; the cingulate gyrus of the cingulum in emerald green; the cortico-spinal tract in purple; the inferior longitudinal fasciculus in orange; the arcuate bundle of the superior longitudinal fasciculus in aquamarine; the III bundle of the superior longitudinal fasciculus in gray; and the uncinate fasciculus in blue. All fibers were thresholded at 20 % of their maximum. The background image depicts the fractional anisotropy (FA) image in color-code convention in one of our participants: voxels with a red color define left-to-right oriented fibers; voxels with a blue color define inferior-to-superior oriented fibers; and voxels with a green color define anterior-to-posterior oriented fibers. The isosurface superimposed on the sagittal view of the colored FA image shows the characteristic anterior–posterior alignment of the cingulum (green voxels) and the origin of the forceps minor and major (red voxels in the genu and splenium of the corpus callosum). Panel (b). Error bar graphs depict the between-group differences in central measures (top; FA in the left corner and radial diffusivity (RD) in the right corner) across all WM tracts and in peripheral measures (bottom; lipid hydroperoxides (LPH) in the left corner and 4-HNE in the right corner) in 24 currently euthymic patients with bipolar disorder (BD) and 19 gender-matched healthy controls (CONT). Panel (c). Scatter plot graphs represent the linear relationship between mean FA (left) and mean RD (right) and LPH across all study participants in the forceps minor. Reprinted with permission from Versace et al. (2014), Fig. 1
Similarly, proteins are vulnerable to oxidative stress damage that can be measured by protein carbonylation, and when ROS attack amino acid side chains of proteins, 3-nitrotyrosine (3-NT) is formed. Increased levels of protein carbonylation (i.e., oxidative stress damage of proteins) have been detected in BD patients in a number of studies. Kapczinski and colleagues quantified serum levels of 3-NT and found a positive correlation with BD patients in a depressive state (Kapczinski et al. 2011).
8-Hydroxy-2-deoxyguanosine (8-OHdG ) is measured to detect ROS -induced DNA damage. Levels of 8-OHdG are elevated in patients with BD and correlate positively to number of previous manic episodes (Soeiro-de-Souza et al. 2013). Longitudinal observation of 8-OHdG levels across the affective states in BD found elevations in 8-OHdG at baseline, six months, and 12 months, independent of symptomatology (Munkholm et al. 2015). Other studies corroborate the finding of increased oxidative damage to nucleic acids of DNA and RNA (D’Addario et al. 2012; Dell’Osso et al. 2014; Huzayyin et al. 2014).
There is strong evidence to support the notion of increased oxidative damage in BD. Many findings are generally applicable to the illness while others show selective changes for the depressed state.
3.3 Decreased Antioxidant Capacity Facilitates Oxidative Stress Damage
Oxidative stress damage can be magnified by deficient antioxidant capacity. Some of our endogenous antioxidants include superoxide dismutase (SOD ), catalase, and glutathione (Soczynska et al. 2008), and abnormalities in antioxidant activity have been reported in patients with BD. Though it is reasonable to expect deficient antioxidant activity if oxidative stress damage were to underlie the pathogenesis of bipolar depression, some research demonstrates an increase in antioxidant activity. Andreazza and colleagues identified increases in SOD activity in BD patients during affective states (both depressed and manic) though not during euthymia (Andreazza et al. 2007a). Similar results have been replicated (Kuloglu et al. 2002; Kunz et al. 2008). The authors conceptualize this increase in SOD activity as an overall deficient state of antioxidant capacity responding to the increased oxidative stress during an affective (e.g., depressive) episode. Other studies have reported lower levels of SOD in bipolar depression. Selek and colleagues found that SOD levels were significantly lower in patients with bipolar depression compared to controls, and though these levels improved over the course of pharmacological treatment, they did not reach the baseline levels in the control group (Selek et al. 2008). As the brain has limited levels of SOD and catalase, glutathione plays a primary role as an antioxidant, and literature supports abnormalities of this antioxidant in BD patients (Bengesser et al. 2015; Gawryluk et al. 2011).
It follows, then, that bolstering antioxidant capacity provides a potential treatment option for bipolar depression. N-acetylcysteine (NAC ) is a compound that creates the precursor to glutathione, and administration of NAC can be used to increase physiological stores of this antioxidant. In one study, patients with bipolar depression had their maintenance medication augmented with NAC for eight weeks (Berk et al. 2008) and displayed a significant reduction in depressive symptoms. These patients were then followed in a randomized, placebo-controlled trial divided into two groups: one receiving NAC daily for six months and the other placebo. The NAC group had a prolonged reduction in depressive symptoms (Berk et al. 2011). Another small study of BD patients supports these data; higher remission rates for affective symptoms were observed in the NAC -treated patients vs. their control group (Magalhães et al. 2011).
3.4 Overactive Inflammatory Pathways Are Implicated in BD
We now turn our focus to the role of inflammation in BD . This theory is salient in bipolar depression specifically because the link between markers of inflammation and clinical states of depression has long been known. There has been extensive research investigating the role of inflammation and cytokines in MDD (see Zunszain et al. (2011) for a review), with more recent interest in BD. It is not yet clear if inflammation has a causative role in the pathogenesis of BD or if manic and depressive episodes chronically activate the immune system, which then gets identified by alterations in central and peripheral inflammatory markers. Regardless, the role of inflammation is receiving considerable attention in the field, as it is linked with affective episodes, oxidative stress, and the medical comorbidities common in patients with BD. Given the growing research indicating the importance of the inflammatory process, we take the opportunity to explore the literature here.
C-reactive protein (CRP) is an acute phase reactant protein created in the liver and released into the bloodstream after acute injury, infection, or other systemic inflammation (Dargel et al. 2015). Clinically, serum CRP is measured to detect an acute inflammatory response or to monitor the condition of someone with a chronic inflammatory illness. Overall, the literature robustly supports elevations of CRP in BD. Most data highlight a positive correlation between serum CRP and manic episodes, and there are reports of a similar association in the depressive phase of the illness (Cunha et al. 2008; Dargel et al. 2015; De Berardis et al. 2008; Dickerson et al. 2007, 2013; Goldstein et al. 2011).
Cytokines are another class of proteins that are implicated in inflammatory states. Every cell has the capacity to generate this heterogeneous group of proteins though specific immune-related cells can enhance their production (e.g., lymphocytes) (Müller and Schwarz 2007). Cytokines can have varied effects depending on their physiological targets, and these potent molecules tend to be classified as proinflammatory (i.e., inducing inflammatory responses) or anti-inflammatory. Alterations in circulating cytokines have been recorded in both manic and depressed patients with BD with literature supporting a positive association between proinflammatory cytokines and mood episodes (Goldstein et al. 2011; Modabbernia et al. 2013; Rao et al. 2010; Su et al. 2011). A comprehensive meta-analysis of research quantifying peripheral levels of proinflammatory markers found evidence for elevated levels of cytokines, including tumor necrosis factor alpha (TNF-α) and interleukin-4 (IL-4) (Munkholm et al. 2013). The studies appraised included manic, depressed, and euthymic patients. Other data support differences between these markers based on affective valence, where CRP and soluble IL-2 receptor (sIL-2R) a re consistently elevated in manic episodes, and IL-6 and TNF-α tend to be higher in depressive states (Cunha et al. 2008; De Berardis et al. 2008; Ortiz-Domínguez et al. 2007).
Pharmacological treatments have varied effects on these inflammatory markers. Lithium treatment has been implicated as anti-inflammatory, given that a comparison between treated and untreated patients revealed lower levels of cytokines in the lithium group (Boufidou et al. 2004). Interestingly, in vitro models using cells from a normal control sample revealed a proinflammatory effect of lithium treatment (Knijff et al. 2007; Petersein et al. 2015) though other preclinical models identified a reduction in cytokines with valproic acid (Himmerich et al. 2014).
The role of inflammation in BD is strengthened by acknowledgement of the high prevalence of certain medical conditions in this population. BD is highly comorbid with cardiovascular disease and type 2 diabetes mellitus, at rates exceeding those found in MDD (Angst et al. 2002; Birkenaes et al. 2007). This morbidity, and subsequent increased mortality, is not immediately related to the factors common in severe and persistent mental illness (e.g., suicide, exposure to medications, low income, physical inactivity, obesity, etc.). Inflammation is implicated in the pathogenesis of both cardiovascular disease and insulin resistance and, increasingly, research is linking these altered inflammatory networks to those that might underlie BD (Goldstein and Young 2013).
In terms of mechanism, recent research links the overactivation of inflammatory pathways to oxidative stress. Studies suggest that the overproduction of ROS by the dysfunctional mitochondrial membrane protein, complex I, can stimulate inflammatory networks and increase levels of proinflammatory cytokines. The nod-like receptor pyrin domain containing 3 (NLRP3) inflammasome is hypothesized as one mechanism for this activation. The NLRP3 inflammasome is a protein complex that can gauge oxidative species. In the presence of ROS , NLRP3 activates downstream inflammatory processes via the enzyme, caspase-1 (López-Armada et al. 2013). When ROS production is blocked, there is no activation of the NLRP3 inflammasome (Zhou et al. 2011). New research from our lab identified increases in NLRP3 components specific to mitochondrial samples from patients with BD but not in other brain or cytoplasmic samples (Kim, HK et al., in preparation). These findings implicate both oxidative stress and increased activation of the inflammasome in the pathophysiology of BD.
3.5 Oxidative Stress and Inflammation Can Accelerate Apoptosis
Growing literature highlights the process of apoptosis as central to the morphologic changes in the brains of patients with BD and oxidative stress. Its subsequent activation of inflammation has a significant impact on this critical cellular pathway. Apoptosis is the programmed process of cell death involving an ordered process with many steps, including cell shrinkage, dissociation of nuclear components, labeling the cell for destruction, and removal by phagocytosis. Dysregulated apoptotic pathways have been postulated as the mechanism leading to the cell loss and brain atrophy seen in the brains of patients with BD (Gigante et al. 2011). Reductions in nuclear size and DNA fragmentation have been observed at higher rates in patient samples than control populations (Buttner et al. 2007; Munkholm et al. 2015; Uranova et al. 2001). Further, the amount of DNA damage observed was positively correlated with depressive symptoms (Andreazza et al. 2007b).
A number of proteins regulate apoptosis. B-cell lymphoma 2 (Bcl-2) denotes one family of these proteins, and Bcl-2 proteins are considered antiapoptotic—thus protective of neuronal cells. Brain-derived neurotrophic factor (BDNF ) , a neurotrophin, is also considered an antiapoptotic factor. Proapoptotic compounds include caspase-3, caspase-9, Bcl-2-associated death promotor (BAD) , and Bcl-2-associated X protein (BAX) . Analysis of prefrontal cortical samples from brains of patients with BD revealed abnormal apoptotic pathways (Kim et al. 2010); specifically, the authors identified a decrease in protective, antiapoptotic factors (i.e., Bcl-2 and BDNF ) and an increase in the expression of proapoptotic factors (i.e., BAX, BAD, caspase-3, and 9).
The link between BD and medical comorbidities was introduced in the section above. Here, we highlight the association because of the role of apoptosis in cellular aging. Telomere length is one indication of cellular aging as telomeres are regions on genetic material that shorten with each cell division in the life cycle; abnormal apoptotic pathways, as well as oxidative stress, are known to induce pathological shortening of telomeres (Lindqvist et al. 2015). BD patients have notably shortened telomeres (Lima et al. 2015), and one study provides evidence for a negative correlation between telomere length and number of depressive episodes in BD-II patients (Elvsåshagen et al. 2011). The concept of apoptosis-induced cellular aging synthesizes the morphological changes, mitochondrial dysfunction, inflammatory associations, and clinical comorbidities seen in BD. Future research will have to investigate and integrate knowledge about the impact of affective states within this model.
3.6 Growth Factors Are Implicated in Both Oxidative Stress and Inflammatory Models of BD
Dysfunctional neurotrophic factors are another potential mechanism by which cellular atrophy may occur. In general, neurotrophic factors are proteins responsible for the growth and maintenance of neurons and their support cells. They act by binding to tyrosine kinase receptors and initiating a cascade of events that inhibits apoptotic pathways. This suggests an alternative or potentially synergistic mechanism underlying the cell loss and brain atrophy observed in the brains of patients with BD. The neurotrophins of the nerve growth family are heterogenous and include BDNF , neurotrophin-3 (NT-3), and NT-4/5. Glial cell line-derived neurotrophic factor (GDNF ) and vascular endothelial growth factor (VEGF ) are other growth factors connected to the progression of mood disorders. To date, the bulk of the research has focused on BDNF in BD, though evidence does implicate a role for other growth factors.
BDNF is the most widely studied neurotrophin, and is implicated in the neurobiology of BD via a number of proposed mechanisms. BDNF is a key protein in regulating the survival of neurons in both the central and peripheral nervous system. As a growth factor, BDNF plays a role in promoting the development and differentiation of neurons and synapses. It is also negatively correlated with measures of oxidative stress (Kapczinski et al. 2008). The concept of neuronal growth and survival is fundamental and underlies the idea that low levels of BDNF create an inability to protect the survival or promote the growth of neurons in the brain areas implicated in BD (Hashimoto 2010). Patients with the BDNF polymorphism gene (resulting in deficient BDNF activity) had the smallest hippocampal volumes compared to patients homozygous for the gene and nonpsychiatric controls (Chepenik et al. 2009). Other data demonstrate decreased volume of the ACC in patients carrying the polymorphism gene vs. homozygotes (Matsuo et al. 2009). It has also been suggested that dysfunctional BDNF activity prevents neuroplasticity and malformation of specific cortical and subcortical regions, given that the BDNF polymorphism gene is associated with childhood, adolescent, and early onset BD (Inal-Emiroglu et al. 2015). Decreased peripheral BDNF levels have been observed during both episodes of depression and mania (de Oliveira et al. 2009; Fernandes et al. 2009, 2011; Machado-Vieira et al. 2007). Finally, there exist data to support the notion that BDNF levels improve with pharmacotherapy (Dwivedi and Zhang 2015; Yasuda et al. 2009). Of interest, ECT—one of the few known effective options for treatment resistant depression in BD—increases BDNF in multiple brain areas and, further, chronic convulsive stimuli can generate regrowth of neurons.
Levels of NT-3 and NT-4/5 are increased in manic and depressive episodes (Loch et al. 2015; Rybakowski et al. 2013; Walz et al. 2007). Loch and colleagues specifically investigated depressed patients with BD who were medication free at the time of study entry. Their baseline levels of NT-3 and NT-4/5 were elevated compared to healthy controls, and these levels did not change significantly after six weeks of lithium treatment (Loch et al. 2015).
GDNF has been investigated in BD with mixed results. Some studies reported a decrease of this neurotrophin in BD patients during manic and depressive phases (Takebayashi et al. 2006; Zhang et al. 2010), while others reported an increase or no difference compared to euthymic patients or healthy controls (Otsuki et al. 2008; Rosa et al. 2006; Rybakowski et al. 2013; Tunca et al. 2015).
VEGF is not a neurotrophic factor, but a growth factor involved in the development and proliferation of blood vessels. It is believed that VEGF is implicated in CNS diseases through its impact on cerebral vasculature (Scola and Andreazza 2015). In BD, there is evidence to support increased peripheral levels of VEGF (Lee and Kim 2012; Shibata et al. 2013) and a role for VEGF in response to treatments specific to bipolar depression. The association of VEGF and BD can be linked to our earlier discussion of inflammation and cardiovascular comorbidity. Vasculogenesis and endothelial dysfunction are implicated in the pathophysiology of cardiovascular disease (with a role for both VEGF and BDNF ), and the hope is that future research will elucidate how these changes in growth factors are involved in the pathogenesis of both these physical and mental illnesses (Goldstein and Young 2013).
4 Neurobiology of BD: The Past, Present, and Future
Over 20 years ago, Post proposed the “kindling” hypothesis of BD (Post 1992). He acknowledged that psychosocial stressors are important as triggers for mood episodes in early BD and theorized that subsequent neuropathological and genetic changes could sensitize the brain, resulting in autonomously triggered mood episodes later in the illness. This work integrated biopsychosocial evidence available at the time and presented a comprehensive theory for the neurobiology of BD.
In the intervening years, however, advances in research techniques both in psychiatry and other fields have given rise to the staging model of BD (Kapczinski et al. 2014). In a similar fashion, this model aims to integrate the many dimensions of BD and outlines a series of progressive stages from a latent phase to the significantly symptomatic Stage IV, where patients have substantial functional, clinical, and cognitive impairments (Fig. 6.3). The model is presented as a means to target and predict response to treatment. Indeed, one common criticism in the field of psychiatry is that our diagnostic structure relies largely on patients’ accounts of their subjective experience and collateral interpretations of behavior change, as well as our own clinical observations. This diagnostic process can lead to misdiagnosis and inefficient or inappropriate treatment. Undertreating bipolar depression has a significant impact on both patients and their support networks as sustained depressive symptoms can be incredibly debilitating.

Reprinted with permission from Kapczinski et al. (2009), Table 1
Peripheral biomarkers are fundamental to the staging model, as the hope is that they can be used to identify the neuroprogression at different stages of BD. The concept of biomarkers is at the forefront of research now with a global acknowledgement of a need for biologically valid markers that can aid in early identification, diagnosis, and treatment of those with mental illness. The neurobiological pathways described in this chapter highlight some of the most promising areas for future development. Markers of oxidative stress, lipid peroxidation, protein carbonylation, inflammation, apoptosis, and neurotrophin status are all potential areas for investigation.
5 Summary
Knowledge about the neurobiology of bipolar depression is gleaned largely from what is known about the pathophysiology of BD in general. Cell loss and brain atrophy in cortical and limbic areas are consistent findings and specific to BD. Though the mechanisms underlying these morphological changes are not fully understood, promising hypotheses include oxidative stress induced by mitochondrial dysfunction and dysregulated inflammatory processes . These potential mechanisms are thought to lead to an increase in apoptosis. Neurotrophins are also implicated in the pathophysiology of BD with links to both oxidative stress and inflammation. The future of this field will depend on research with a specific focus on the depressive phase of BD as well as the study of biomarkers to aid clinicians in the diagnosis and targeted treatment of bipolar depression.
References
Adleman NE, Fromm SJ, Razdan V, Kayser R, Dickstein DP, Brotman MA, Pine DS, Leibenluft E (2012) Cross-sectional and longitudinal abnormalities in brain structure in children with severe mood dysregulation or bipolar disorder. J Child Psychol Psychiatry 53(11):1149–1156
Andreazza AC, Cassini C, Rosa AR, Leite MC, de Almeida LMV, Nardin P, Cunha ABN, Cereser KM, Santin A, Gottfried C, Salvador M, Kapczinski F, Goncalves CA (2007a) Serum S100B and antioxidant enzymes in bipolar patients. J Psychiatr Res 41(6):523–529
Andreazza AC, Noronha Frey B, Erdtmann B, Salvador M, Rombaldi F, Santin A, Goncalves CA, Kapczinski F (2007b) DNA damage in bipolar disorder. Psychiatry Res 153(1):27–32
Angst F, Stassen HH, Clayton PJ, Angst J (2002) Mortality of patients with mood disorders: follow-up over 34–38 years. J Affect Dis 68(2–3):167–181
Banerjee U, Dasgupta A, Rout JK, Singh OP (2012) Effects of lithium therapy on Na+–K+-ATPase activity and lipid peroxidation in bipolar disorder. Prog Neuropsychopharmacol Biol Psychiatry 37(1):56–61
Barbiroli B, Montagna P, Martinelli P, Lodi R, Iotti S, Cortelli P, Funicello R, Zaniol P (1993) Defective brain energy metabolism shown by in vivo 31P MR spectroscopy in 28 patients with mitochondrial cytopathies. J Cereb Blood Flow Metab 13(3):469–474
Beech RD, Lowthert L, Leffert JJ, Mason PN, Taylor MM, Umlauf S, Lin A, Lee JY, Maloney K, Muralidharan A, Lorberg B, Zhao H, Newton SS, Mane S, Epperson CN, Sinha R, Blumberg H, Bhagwagar Z (2010) Increased peripheral blood expression of electron transport chain genes in bipolar depression. Bipolar Disord 12(8):813–824
Bengesser SA, Lackner N, Birner A, Fellendorf FT (2015) Peripheral markers of oxidative stress and antioxidative defense in euthymia of bipolar disorder—Gender and obesity effects. J Affect Disord 172:367–374
Berk M, Copolov DL, Dean O, Lu K, Jeavons S, Schapkaitz I, Anderson-Hunt M, Bush AI (2008) N-acetyl cysteine for depressive symptoms in bipolar disorder—a double-blind randomized placebo-controlled trial. Biol Psychiatry 64(6):468–475
Berk M, Dean O, Cotton SM, Gama CS, Kapczinski F, Fernandes BS, Kohlmann K, Jeavons S, Hewitt K, Allwang C, Cobb H, Bush AI, Schapkaitz I, Dodd S, Malhi GS (2011) The efficacy of N-acetylcysteine as an adjunctive treatment in bipolar depression: an open label trial. J Affect Disord 135(1–3):389–394
Bielau H, Trübner K, Krell D, Agelink MW, Bernstein HG, Stauch R, Mawrin C, Danos P, Gerhard L, Bogerts B, Baumann B (2005) Volume deficits of subcortical nuclei in mood disorders. Eur Arch Psychiatry Clin Neurosci 255(6):401–412
Birkenaes AB, Opjordsmoen S, Brunborg C, Engh JA, Jonsdottir H, Ringen PA, Simonsen C, Vaskinn A, Birkeland KI, Friis S, Sundet K, Andreassen OA (2007) The level of cardiovascular risk factors in bipolar disorder equals that of schizophrenia: a comparative study. J Clin Psychiatry 68(6):917–923
Boufidou F, Nikolaou C, Alevizos B, Liappas IA, Christodoulou GN (2004) Cytokine production in bipolar affective disorder patients under lithium treatment. J Affect Disord 82(2):309–313
Buttner N, Bhattacharyya S, Walsh J, Benes FM (2007) DNA fragmentation is increased in non-GABAergic neurons in bipolar disorder but not in schizophrenia. Schizophr Res 93(1–3):33–41
Byne W, Tatusov A, Yiannoulos G, Vong GS, Marcus S (2008) Effects of mental illness and aging in two thalamic nuclei. Schizophr Res 106(2–3):172–181
Campbell S, Macqueen G (2004) The role of the hippocampus in the pathophysiology of major depression. J Psychiatry Neurosci 29(6):417–426
Can M, Güven B, Atik L, Konuk N (2011) Lipid peroxidation and serum antioxidant enzymes activity in patients with bipolar and major depressive disorders. J Mood Disord 1(1):14
Cataldo AM, McPhie DL, Lange NT, Punzell S, Elmiligy S, Ye NZ, Froimowitz MP, Hassinger LC, Menesale EB, Sargent LW, Logan DJ, Carpenter AE, Cohen BM (2010) Abnormalities in mitochondrial structure in cells from patients with bipolar disorder. Am J Pathol 177(2):575–585
Chaturvedi RK, Flint Beal M (2013) Mitochondrial diseases of the brain. Free Radic Biol Med 63:1–29
Chepenik LG, Fredericks C, Papademetris X, Spencer L, Lacadie C, Wang F, Pittman B, Duncan JS, Staib LH, Duman RS, Gelernter J, Blumberg HP (2009) Effects of the brain-derived neurotrophic growth factor val66met variation on hippocampus morphology in bipolar disorder. Neuropsychopharmacology 34(4):944–951
Cunha ÂB, Andreazza AC, Gomes FA, Frey BN, Silveira LE, Gonçalves CA, Kapczinski F (2008) Investigation of serum high-sensitive C-reactive protein levels across all mood states in bipolar disorder. Eur Arch Psychiatry Clin Neurosci 258(5):300–304
D’Addario C, Dell’Osso B, Palazzo MC, Benatti B, Lietti L, Cattaneo E, Galimberti D, Fenoglio C, Cortini F, Scarpini E, Arosio B, Di Francesco A, Di Benedetto M, Romualdi P, Candeletti S, Mari D, Bergamaschini L, Bresolin N, Maccarrone M, Altamura AC (2012) Selective DNA methylation of BDNF promoter in bipolar disorder: differences among patients with BDI and BDII. Neuropsychopharmacology 37(7):1647–1655
Dargel AA, Godin O, Kapczinski F, Kupfer DJ, Leboyer M (2015) C-reactive protein alterations in bipolar disorder: a meta-analysis. J Clin Psychiatry 76(2):142–150
De Berardis D, Conti CM, Campanella D, Carano A, Scali M, Valchera A, Serroni N, Pizzorno AM, D’Albenzio A, Fulcheri M, Gambi F, La Rovere R, Cotellessa C, Salerno RM, Ferro FM (2008) Evaluation of C-reactive protein and total serum cholesterol in adult patients with bipolar disorder. Int J Immunopathol Pharmacol 21(2):319–324
de Oliveira GS, Ceresér KM, Fernandes BS, Kauer-Sant’Anna M, Fries GR, Stertz L, Aguiar B, Pfaffenseller B, Kapczinski F (2009) Decreased brain-derived neurotrophic factor in medicated and drug-free bipolar patients. J Psychiatr Res 43(14):1171–1174
Dell’Osso B, D’Addario C, Carlotta Palazzo M, Benatti B, Camuri G, Galimberti D, Fenoglio C, Scarpini E, Di Francesco A, Maccarrone M, Altamura AC (2014) Epigenetic modulation of BDNF gene: differences in DNA methylation between unipolar and bipolar patients. J Affect Disord 166:330–333
Dickerson F, Stallings C, Origoni A, Boronow J, Yolken R (2007) Elevated serum levels of C-reactive protein are associated with mania symptoms in outpatients with bipolar disorder. Prog Neuropsychopharmacol Biol Psychiatry 31(4):952–955
Dickerson F, Stallings C, Origoni A, Vaughan C, Khushalani S, Yolken R (2013) Elevated C-reactive protein and cognitive deficits in individuals with bipolar disorder. J Affect Disord 150(2):456–459
Dwivedi T, Zhang H (2015) Lithium-induced neuroprotection is associated with epigenetic modification of specific BDNF gene promoter and altered expression of apoptotic-regulatory proteins. Front Neurosci 8:457
Eleff SM, Barker PB, Blackband SJ, Chatham JC, Lutz NW, Johns DR, Bryan RN, Hurko O (1990) Phosphorus magnetic resonance spectroscopy of patients with mitochondrial cytopathies demonstrates decreased levels of brain phosphocreatine. Ann Neurol 27(6):626–630
Elvsåshagen T, Vera E, Bøen E, Bratlie J, Andreassen OA, Josefsen D, Malt UF, Blasco MA, Boye B (2011) The load of short telomeres is increased and associated with lifetime number of depressive episodes in bipolar II disorder. J Affect Disord 135(1–3):43–50
Fernandes BS, Gama CS, Kauer-Sant’Anna M, Lobato MI, Belmonte-de-Abreu P, Kapczinski F (2009) Serum brain-derived neurotrophic factor in bipolar and unipolar depression: a potential adjunctive tool for differential diagnosis. J Psychiatr Res 43(15):1200–1204
Fernandes BS, Gama CS, Maria Ceresér K, Yatham LN, Fries GR, Colpo G, de Lucena D, Kunz M, Gomes FA, Kapczinski F (2011) Brain-derived neurotrophic factor as a state-marker of mood episodes in bipolar disorders: a systematic review and meta-regression analysis. J Psychiatr Res 45(8):995–1004
Foland-Ross LC, Brooks JO, Mintz J, Bartzokis G, Townsend J, Thompson PM, Altshuler LL (2012) Mood-state effects on amygdala volume in bipolar disorder. J Affect Disord 139(3):298–301
Gawryluk JW, Wang J-F, Andreazza AC, Shao L, Young LT (2011) Decreased levels of glutathione, the major brain antioxidant, in post-mortem prefrontal cortex from patients with psychiatric disorders. Int J Neuropsychopharmacol 14(1):123–130
Ghaznavi S, Deckersbach T (2012) Rumination in bipolar disorder: evidence for an unquiet mind. Biol Mood Anxiety Disord 2(1):2
Gigante AD, Young LT, Yatham LN, Andreazza AC, Nery FG, Grinberg LT, Heinsen H, Lafer B (2011) Morphometric post-mortem studies in bipolar disorder: possible association with oxidative stress and apoptosis. Int J Neuropsychopharmacol 14(8):1075–1089
Goldstein B, Young LT (2013) Toward clinically applicable biomarkers in bipolar disorder: focus on BDNF, inflammatory markers, and endothelial function. Curr Psychiatry Rep 15(12):1–7
Goldstein BI, Collinger KA, Lotrich F, Marsland AL, Gill M-K, Axelson DA, Birmaher B (2011) Preliminary findings regarding proinflammatory markers and brain-derived neurotrophic factor among adolescents with bipolar spectrum disorders. J Child Adolesc Psychopharmacol 21(5):479–484
Hartberg CB, Jørgensen KN, Haukvik UK, Westlye LT, Melle I, Andreassen OA, Agartz I (2015) Lithium treatment and hippocampal subfields and amygdala volumes in bipolar disorder. Bipolar Disord 17(5):496–506
Hashimoto K (2010) Brain-derived neurotrophic factor as a biomarker for mood disorders: an historical overview and future direction. Psychiatry Clin Neurosci 64(4):341–357
Himmerich H, Bartsch S, Hamer H, Mergl R, Schonherr J, Petersein C, Munzer A, Kirkby KC, Bauer K, Sack U (2014) Modulation of cytokine production by drugs with antiepileptic or mood stabilizer properties in anti-CD3- and anti-Cd40-stimulated blood in vitro. Oxid Med Cell Longev 2014:806162
Hirayasu Y, Shenton ME, Salisbury DF, Kwon JS et al (1999) Subgenual cingulate cortex volume in first-episode psychosis. Am J Psychiatry 156(7):1091–1093
Huzayyin AA, Andreazza AC, Turecki G, Cruceanu C, Rouleau GA, Alda M, Young LT (2014) Decreased global methylation in patients with bipolar disorder who respond to lithium. Int J Neuropsychopharmacol 17(4):561–569
Inal-Emiroglu FN, Karabay N, Resmi H, Guleryuz H (2015) Correlations between amygdala volumes and serum levels of BDNF and NGF as a neurobiological marker in adolescents with bipolar disorder. J Affect Disord 182:50–56
Janak PH, Tye KM (2015) From circuits to behaviour in the amygdala. Nature 517(7534):284–292
Kapczinski F, Frey BN, Andreazza AC, Kauer-Sant’Anna M, Cunha AB, Post RM (2008) Increased oxidative stress as a mechanism for decreased BDNF levels in acute manic episodes. Rev Bras Psiquiatr 30(3):243–245
Kapczinski F, Dias VV, Kauer-Sant’Anna M, Frey BN, Grassi-Oliveira R, Colom F, Berk M (2009) Clinical implications of a staging model for bipolar disorders. Expert Rev Neurother 9(7):957–966
Kapczinski F, Dal-Pizzol F, Teixeira AL, Magalhaes PVS, Kauer-Sant’Anna M, Klamt F, Moreira JCF, Augusto de Bittencourt Pasquali M, Fries GR, Quevedo J, Gama CS, Post R (2011) Peripheral biomarkers and illness activity in bipolar disorder. J Psychiatr Res 45(2):156–161
Kapczinski F, Magalhães PVS, Balanzá-Martinez V, Dias VV, Frangou S, Gama CS, Gonzalez-Pinto A, Grande I, Ha K, Kauer-Sant’Anna M, Kunz M, Kupka R, Leboyer M, Lopez-Jaramillo C, Post RM, Rybakowski JK, Scott J, Strejilevitch S, Tohen M, Vazquez G, Yatham L, Vieta E, Berk M (2014) Staging systems in bipolar disorder: an International Society for Bipolar Disorders Task Force Report. Acta Psychiatr Scand 130(5):354–363
Kato T (2007) Mitochondrial dysfunction as the molecular basis of bipolar disorder: therapeutic implications. CNS Drugs 21(1):1–11
Kato T, Takahashi S, Shioiri T, Inubushi T (1992) Brain phosphorous metabolism in depressive disorders detected by phosphorus-31 magnetic resonance spectroscopy. J Affect Disord 26(4):223–230
Kato T, Takahashi S, Shioiri T, Murashita J, Hamakawa H, Inubushi T (1994) Reduction of brain phosphocreatine in bipolar II disorder detected by phosphorus-31 magnetic resonance spectroscopy. J Affect Disord 31(2):125–133
Kato T, Shioiri T, Murashita J, Hamakawa H, Takahashi Y, Inubushi T, Takahashi S (1995) Lateralized abnormality of high energy phosphate metabolism in the frontal lobes of patients with bipolar disorder detected by phase-encoded 31P-MRS. Psychol Med 25(03):557–566
Kim H-W, Rapoport SI, Rao JS (2010) Altered expression of apoptotic factors and synaptic markers in postmortem brain from bipolar disorder patients. Neurobiol Dis 37(3):596–603
Kittel-Schneider S, Wobrock T, Scherk H, Schneider-Axmann T, Trost S, Zilles D, Wolf C, Schmitt A, Malchow B, Hasan A, Backens M, Reith W, Falkai P, Gruber O, Reif A (2015) Influence of DGKH variants on amygdala volume in patients with bipolar affective disorder and schizophrenia. Eur Arch Psychiatry Clin Neurosci 265(2):127–136
Knijff EM, Nadine Breunis M, Kupka RW, de Wit HJ, Ruwhof C, Akkerhuis GW, Nolen WA, Drexhage HA (2007) An imbalance in the production of IL-1ß and IL-6 by monocytes of bipolar patients: restoration by lithium treatment. Bipolar Disord 9(7):743–753
Kuloglu M, Ustundag B, Atmaca M, Canatan H, Tezcan AE, Cinkilinc N (2002) Lipid peroxidation and antioxidant enzyme levels in patients with schizophrenia and bipolar disorder. Cell Biochem Funct 20(2):171–175
Kunz M, Gama CS, Andreazza AC, Salvador M, Ceresér KM, Gomes FA, Belmonte-de-Abreu PS, Berk M, Kapczinski F (2008) Elevated serum superoxide dismutase and thiobarbituric acid reactive substances in different phases of bipolar disorder and in schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry 32(7):1677–1681
Lee B-H, Kim Y-K (2012) Increased plasma VEGF levels in major depressive or manic episodes in patients with mood disorders. J Affect Disord 136(1–2):181–184
Lima IMM, Barros A, Rosa DV, Albuquerque M (2015) Analysis of telomere attrition in bipolar disorder. J Affect Disord 172:43–47
Lindqvist D, Epel ES, Mellon SH, Penninx BW, Révész D, Verhoeven JE, Reus VI, Lin J, Mahan L, Hough CM, Rosser R, Saverio Bersani F, Blackburn EH, Wolkowitz OM (2015) Psychiatric disorders and leukocyte telomere length: underlying mechanisms linking mental illness with cellular aging. Neurosci Biobehav Rev 55:333–364
Liu L, Schulz SC, Lee S, Reutiman TJ, Fatemi SH (2007) Hippocampal CA1 pyramidal cell size is reduced in bipolar disorder. Cell Mol Neurobiol 27(3):351–358
Loch AA, Zanetti MV, de Sousa RT, Chaim TM, Serpa MH, Gattaz WF, Teixeira AL, Machado-Vieira R (2015) Elevated neurotrophin-3 and neurotrophin 4/5 levels in unmedicated bipolar depression and the effects of lithium. Prog Neuropsychopharmacol Biol Psychiatry 56:243–246
López-Armada MJ, Riveiro-Naveira RR, Vaamonde-García C, Valcárcel-Ares MN (2013) Mitochondrial dysfunction and the inflammatory response. Mitochondrion 13:106–118
MacDonald ML, Naydenov A, Chu M, Matzilevich D, Konradi C (2006) Decrease in creatine kinase messenger RNA expression in the hippocampus and dorsolateral prefrontal cortex in bipolar disorder. Bipolar Disord 8(3):255–264
Machado-Vieira R, Dietrich MO, Leke R, Cereser VH, Zanatto V, Kapczinski F, Souza DO, Portela LV, Gentil V (2007) Decreased plasma brain derived neurotrophic factor levels in unmedicated bipolar patients during manic episode. Biol Psychiatry 61(2):142–144
Magalhães PV, Dean OM, Bush AI, Copolov DL, Malhi GS, Kohlmann K, Jeavons S, Schapkaitz I, Anderson-Hunt M, Berk M (2011) N-acetyl cysteine add-on treatment for bipolar II disorder: a subgroup analysis of a randomized placebo-controlled trial. J Affect Disord 129(1–3):317–320
Matsuo K, Walss-bass C, Nery FG, Nicoletti MA, Hatch JP, Frey BN, Monkul ES, Zunta-soares GB, Bowden CL, Escamilla MA, Soares JC (2009) Neuronal correlates of brain-derived neurotrophic factor Val66Met polymorphism and morphometric abnormalities in bipolar disorder. Neuropsychopharmacology 34(8):1904–1913
Modabbernia A, Taslimi S, Brietzke E, Ashrafi M (2013) Cytokine alterations in bipolar disorder: a meta-analysis of 30 studies. Biol Psychiatry 74(1):15–25
Moniczewski A, Gawlik M, Smaga I, Niedzielska E, Krzek J, Przegaliński E, Pera J, Filip M (2015) Oxidative stress as an etiological factor and a potential treatment target of psychiatric disorders. Part 1. Chemical aspects and biological sources of oxidative stress in the brain. Pharmacol Rep 67(3):560–568
Moore CM, Christensen JD, Lafer B, Fava M et al (1997) Lower levels of nucleoside triphosphate in the basal ganglia of depressed subjects: a phosphorous-31 magnetic resonance spectroscopy study. Am J Psychiatry 154(1):116–118
Moore GJ, Cortese BM, Glitz DA, Zajac-Benitez C, Quiroz JA, Uhde TW, Drevets WC, Manji HK (2009) A longitudinal study of the effects of lithium treatment on prefrontal and subgenual prefrontal gray matter volume in treatment-responsive bipolar disorder patients. J Clin Psychiatry 70(5):699–705
Morawetz C, Bode S, Baudewig J, Kirilina E, Heekeren HR (2016) Changes in effective connectivity between dorsal and ventral prefrontal regions moderate emotion regulation. Cereb Cortex 26(5):1923–1937
Müller N, Schwarz MJ (2007) The immune-mediated alteration of serotonin and glutamate: towards an integrated view of depression. Mol Psychiatry 12(11):988–1000
Munkholm K, Braüner JV, Kessing LV, Vinberg M (2013) Cytokines in bipolar disorder vs. healthy control subjects: a systematic review and meta-analysis. J Psychiatr Res 47(9):1119–1133
Munkholm K, Poulsen HE, Kessing LV, Vinberg M (2015) Elevated levels of urinary markers of oxidatively generated DNA and RNA damage in bipolar disorder. Bipolar Disord 17(3):257–268
Naydenov AV, MacDonald ML, Ongur D, Konradi C (2007) Differences in lymphocyte electron transport gene expression levels between subjects with bipolar disorder and normal controls in response to glucose deprivation stress. Arch Gen Psychiatry 64(5):555–564
Nugent AC, Diazgranados N, Carlson PJ, Ibrahim L, Luckenbaugh DA, Brutsche N, Herscovitch P, Drevets WC, Zarate CA (2014) Neural correlates of rapid antidepressant response to ketamine in bipolar disorder. Bipolar Disord 16(2):119–128
Ortiz-Domínguez A, Hernández ME, Berlanga C, Gutiérrez-Mora D, Moreno J, Heinze G, Pavón L (2007) Immune variations in bipolar disorder: phasic differences. Bipolar Disord 9(6):596–602
Otsuki K, Uchida S, Watanuki T, Wakabayashi Y, Fujimoto M, Matsubara T, Funato H, Watanabe Y (2008) Altered expression of neurotrophic factors in patients with major depression. J Psychiatr Res 42(14):1145–1153
Palmier-Claus JE, Dodd A, Tai S, Emsley R, Mansell W (2015) Appraisals to affect: testing the integrative cognitive model of bipolar disorder. Br J Clin Psychol 2015
Petersein C, Sack U, Mergl R, Schonherr J, Schmidt F, Lichtblau N, Kirkby K, Bauer K, Himmerich H (2015) Impact of lithium alone and in combination with antidepressants on cytokine production in vitro. J Neural Transm 122(1):109–122
Post RM (1992) Transduction of psychosocial stress into the neurobiology of recurrent affective disorder. Am J Psychiatry 149(8):999–1010
Radaelli D, Sferrazza Papa G, Vai B, Poletti S, Smeraldi E, Colombo C, Benedetti F (2015) Fronto-limbic disconnection in bipolar disorder. Eur Psychiatry 30(1):82–88
Rao JS, Harry GJ, Rapoport SI, Kim HW (2010) Increased excitotoxicity and neuroinflammatory markers in postmortem frontal cortex from bipolar disorder patients. Mol Psychiatry 15(4):384–392
Regenold WT, Phatak P, Marano CM, Sassan A, Conley RR, Kling MA (2009) Elevated cerebrospinal fluid lactate concentrations in patients with bipolar disorder and schizophrenia: implications for the mitochondrial dysfunction hypothesis. Biol Psychiatry 65(6):489–494
Rosa AR, Frey BN, Andreazza AC, Cereser KM, Cunha ABM, Quevedo J, Santin A, Gottfried C, Goncalves CA, Vieta E, Kapczinski F (2006) Increased serum glial cell line-derived neurotrophic factor immunocontent during manic and depressive episodes in individuals with bipolar disorder. Neurosci Lett 407(2):146–150
Rubinsztein JS, Michael A, Underwood BR, Tempest M, Sahakian BJ (2006) Impaired cognition and decision-making in bipolar depression but no ‘affective bias’ evident. Psychol Med 36(5):629–639
Rybakowski JK, Permoda-Osip A, Skibinska M, Adamski R, Bartkowska-Sniatkowska A (2013) Single ketamine infusion in bipolar depression resistant to antidepressants: are neurotrophins involved? Hum Psychopharmacol 28(1):87–90
Salzman CD, Fusi S (2010) Emotion, cognition, and mental state representation in amygdala and prefrontal cortex. Ann Rev Neurosci 33:173–202
Scola G, Andreazza AC (2015) The role of neurotrophins in bipolar disorder. Prog Neuropsychopharmacol Biol Psychiatry 56:122–128
Selek S, Savas HA, Gergerlioglu HS, Bulbul F, Uz E, Yumru M (2008) The course of nitric oxide and superoxide dismutase during treatment of bipolar depressive episode. J Affect Disord 107(1–3):89–94
Shibata T, Yamagata H, Uchida S, Otsuki K, Hobara T, Higuchi F, Abe N, Watanabe Y (2013) The alteration of hypoxia inducible factor-1 (HIF-1) and its target genes in mood disorder patients. Prog Neuropsychopharmacol Biol Psychiatry 43:222–229
Singh MK, Chang KD, Kelley RG, Saggar M, Reiss AL, Gotlib IH (2014) Early signs of anomalous neural functional connectivity in healthy offspring of parents with bipolar disorder. Bipolar Disord 16(7):678–689
Soczynska JK, Kennedy SH, Chow CSM, Woldeyohannes HO, Konarski JZ, McIntyre RS (2008) Acetyl-L-carnitine and a-lipoic acid: possible neurotherapeutic agents for mood disorders? Expert Opin Investig Drugs 17(6):827–843
Soeiro-de-Souza MG, Andreazza AC, Carvalho AF, Machado-Vieira R, Young LT, Moreno RA (2013) Number of manic episodes is associated with elevated DNA oxidation in bipolar I disorder. Int J Neuropsychopharmacol 16(7):1505–1512
Stork C, Renshaw PF (2005) Mitochondrial dysfunction in bipolar disorder: evidence from magnetic resonance spectroscopy research. Mol Psychiatry 10(10):900–919
Su SC, Sun MT, Wen MJ, Lin CJ, Chen YC, Hung YJ (2011) Brain-derived neurotrophic factor, adiponectin, and proinflammatory markers in various subtypes of depression in young men. Int J Psychiatry Med 42(3):211–226
Sun X, Jun-Feng W, Tseng M, Young LT (2006) Downregulation in components of the mitochondrial electron transport chain in the postmortem frontal cortex of subjects with bipolar disorder. J Psychiatr Neurosci 31(3):189–196
Takebayashi M, Hisaoka K, Nishida A, Tsuchioka M, Miyoshi I, Kozuru T, Hikasa S, Okamoto Y, Shinno H, Morinobu S, Yamawaki S (2006) Decreased levels of whole blood glial cell line-derived neurotrophic factor (GDNF) in remitted patients with mood disorders. Int J Neuropsychopharmacol 9(5):607–612
Tang V, Wang J-F (2013) Oxidative stress in bipolar disorder. Biochem Anal Biochem 2(2):1–8
Taylor MA, Abrams R (1987) Cognitive impairment patterns in schizophrenia and affective disorder. J Neurol Neurosurg Psychiatry 50(7):895–899
Tunca Z, Kivircik Akdede B, Özerdem A, Alkin T, Polat S, Ceylan D, Bayin M, Cengizçetin Kocuk N, Simsek S, Resmi H, Akan P (2015) Diverse glial cell line-derived neurotrophic factor (GDNF) support between mania and schizophrenia: a comparative study in four major psychiatric disorders. Eur Psychiatry 30(2):198–204
Uranova N, Orlovskaya D, Vikhreva O, Zimina I, Kolomeets N, Vostrikov V, Rachmanova V (2001) Electron microscopy of oligodendroglia in severe mental illness. Brain Res Bull 55(5):597–610
Versace A, Andreazza AC, Young LT, Fournier JC, Almeida JR, Stiffler RS, Lockovich JC, Aslam HA, Pollock MH, Park H, Nimgaonkar VL, Kupfer DJ, Phillips ML (2014) Elevated serum measures of lipid peroxidation and abnormal prefrontal white matter in euthymic bipolar adults: toward peripheral biomarkers of bipolar disorder. Mol Psychiatry 19(2):200–208
Wake H, Moorhouse AJ, Miyamoto A, Nabekura J (2013) Microglia: actively surveying and shaping neuronal circuit structure and function. Trends Neurosci 36(4):209–217
Walz JC, Andreazza AC, Frey BN, Cacilhas AA, Cereser KMM, Cunha ABM, Weyne F, Stertz L, Santin A, Goncalves CA, Kapczinski F (2007) Serum neurotrophin-3 is increased during manic and depressive episodes in bipolar disorder. Neurosci Lett 415(1):87–89
Wang JF (2007) Defects of mitochondrial electron transport chain in bipolar disorder: implications for mood-stabilizing treatment. Can J Psychiatry 52(12):753–762
Wang JF, Young LT (2009) Understanding the neurobiology of bipolar depression. In: Zarate CA, Manji HK (eds) Bipolar depression: molecular neurobiology, clinical diagnosis and pharmacotherapy. Milestones in drug therapy. Birkhäuser Verlag, Basel, pp 77–94
Wang JF, Shao L, Sun X, Young LT (2009) Increased oxidative stress in the anterior cingulate cortex of subjects with bipolar disorder and schizophrenia. Bipolar Disord 11(5):523–529
Washizuka S, Kakiuchi C, Mori K, Tajima O, Akiyama T, Kato T (2005) Expression of mitochondria-related genes in lymphoblastoid cells from patients with bipolar disorder. Bipolar Disord 7(2):146–152
Yasuda S, Liang MH, Marinova Z, Yahyavi A, Chuang DM (2009) The mood stabilizers lithium and valproate selectively activate the promoter IV of brain-derived neurotrophic factor in neurons. Mol Psychiatry 14(1):51–59
Zhang X, Zhang Z, Sha W, Xie C, Xi G, Zhou H, Zhang Y (2010) Effect of treatment on serum glial cell line-derived neurotrophic factor in bipolar patients. J Affect Disord 126(1–2):326–329
Zhou R, Yazdi AS, Menu P, Tschopp J (2011) A role for mitochondria in NLRP3 inflammasome activation. Nature 469(7329):221–225
Zunszain PA, Anacker C, Cattaneo A, Carvalho LA, Pariante CM (2011) Glucocorticoids, cytokines and brain abnormalities in depression. Prog Neuropsychopharmacol Biol Psychiatry 35(3):722–729
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland (outside the USA)
About this chapter
Cite this chapter
Chintoh, A.F., Young, L.T. (2016). Understanding the Neurobiology of Bipolar Depression. In: Zarate Jr., C., Manji, H. (eds) Bipolar Depression: Molecular Neurobiology, Clinical Diagnosis, and Pharmacotherapy. Milestones in Drug Therapy. Springer, Cham. https://doi.org/10.1007/978-3-319-31689-5_6
Download citation
DOI: https://doi.org/10.1007/978-3-319-31689-5_6
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-31687-1
Online ISBN: 978-3-319-31689-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)