
Abstract
Mood disorders are associated with persistently high rates of morbidity and mortality, despite the widespread availability of antidepressant treatments. One limitation to extant therapeutic options has been that nearly all approved antidepressant pharmacotherapies exert a similar primary action of blocking monoamine transporters, and few options exist for transitioning treatment resistant patients to alternatives with distinct mechanisms. An emerging area of science that promises novel pathways to antidepressant and mood-stabilizing therapies has followed from evidence that immunological factors play major roles in the pathophysiology of at least some mood disorder subtypes. Here we review evidence that the compounds that reduce the release or signaling of neuroactive cytokines, particularly IL-1β, IL-6, and TNF-α, can exert antidepressant effects in subgroups of depressed patients who are identified by blood-based biomarkers associated with inflammation. Within this context we discuss the role of microglia in central neuroinflammation, and the interaction between the peripheral immune system and the central synaptic microenvironment during and after neuroinflammation. Finally we review data using preclinical neuroinflammation models that produce depression-like behaviors in experimental animals to guide the discovery of novel neuro-immune drug targets.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

Keywords
Mood disorders constitute clinically pleomorphic syndromes consisting of behavioral and experiential changes in the emotional, cognitive, visceral, and appetitive domains that show moderate to high heritability, but remain idiopathic with respect to etiology. The main mood disorders, major depressive disorder (MDD) and bipolar disorder (BD) show relatively high lifetime prevalence rates [1] and despite the availability of many antidepressant drugs, MDD is ranked by the World Health Organization (WHO) as the highest global cause of “years of life lived with disability” for all age groups. The persistence of this global public health problem partly reflects the limited efficacy of extant therapies, as about one third of MDD patients do not achieve remission despite multiple trials using different treatments, while another third experience illness relapse and recurrence despite continued adherence to initially effective treatments [2–4]. One limitation of extant antidepressant pharmacotherapies is that they essentially all target biogenic amine based mechanisms, so that for patients who do not respond to such mechanisms, therapeutic options with distinct mechanisms have been largely unavailable.
Notably, the results of studies that compared depressed patients who respond to monoamine reuptake inhibiting agents versus those who do not consistently have shown that the non-responders manifest abnormal elevations in a variety of pro-inflammatory immunological markers [5–9]. These data converge with evidence that factors within the innate and the adaptive immune system play roles in the pathophysiology of MDD and BD, potentially thereby illuminating new targets for novel therapeutics in mood disorders [10, 11]. As reviewed below the findings that administration of pro-inflammatory cytokines such as interferon-alpha or low dose endotoxin can induce depressive symptoms in a subset of humans who have not previously been depressed [12], along with the implication of immune pathway dysregulation by genome-wide association studies (GWAS) of primary mood disorders, suggest that some individuals have a biological diathesis to manifest depressive symptoms under immune challenge [13, 14]. Such conclusions have been corroborated pre-clinically by similar phenomena, specifically by showing that immune activation produces depression-like behaviors in repeatedly stressed animals and that these behaviors can be prevented or reversed by anti-inflammatory treatments [15]. Similarly, an emerging clinical literature provides evidence that some types of anti-inflammatory treatments can produce antidepressant effects in depressed patients with peripheral blood evidence of inflammation [16].
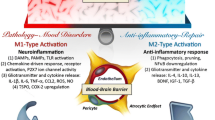
1 Interplay of the Immune System and the Central Nervous System (CNS)
The emerging neuro-immunological literature suggests that immune cells in the periphery and/or the brain interact with neurons in the CNS to play roles in the pathophysiology of mood disorders [11, 17]. These data point to the existence of a bi-directional immune-connectivity between the peripheral and central compartments [18–20]. The interplay of the immune system and the CNS involving pro-inflammatory cytokines, chemokines, and related molecular processes that lead to microglial activation and astrogliosis is referred to as neuroinflammation. However, in the CNS the biological concomitants of an inflammatory state differ in many respects from conventional inflammation involving peripheral immune cells [21]. Thus the neuroimmunology field has broadened in perspective to also encompass the mechanisms by which the peripheral immune system modulates central neurophysiology. In contrast, the neuroinflammatory changes in microglia, astrocytes, and oligodendrocytes that putatively contribute to the causal mechanisms underlying multiple sclerosis, Parkinson’s disease, and epilepsy are generally absent in mood disorders. For example, post mortem studies of glial cell function, structure and density do not show the astrocytosis and amoeboid microglial morphology that is manifest in multiple sclerosis, trauma, or neurodegeneration. Instead such studies have demonstrated reductions in oligodendroglia, impaired astroglial function, and intermediate morphologies of activated microglia [22]. One exception to this general set of findings in post mortem studies of mood disorders involves elderly patients characterized by a late age of depression onset; such patients show clinical and neuropathological evidence for a pathophysiological process mediated via cerebrovascular disease, including astrogliosis, inflammation, and other histopathological correlates of ischemic disease [23]. Nevertheless, debate remains whether neuroinflammatory processes play pathological or adaptive/compensatory roles in the pathophysiology underlying early onset mood disorders, which instead have been associated with a combination of genetic and environmental (e.g., early life trauma) risk factors [24–26].
In the CNS, bone marrow derived immune cells have a restricted access due to an intact blood–brain barrier (BBB) and blood–CSF barrier. During an injury or infection when this barrier is compromised, peripheral immune cells can penetrate the CNS causing neuroinflammation. Nevertheless, other conditions exist in which macrophages and monocytes from the periphery can migrate into the CNS [27, 28] and the CNS lymphatics may serve as conduits of peripheral to central cellular migration [29, 30]. In addition, and probably more pertinent to the topic of neuroinflammation, microglia constitute the critical cell types that change from a “surveillance” mode to a “response” mode during injury and disease pathology. Resting microglia manifest a distinct “ramified” morphology whose function is to sense the local environment and maintain homeostasis among the neurons, astrocytes, and oligodendrocytes that participate in synaptic function [31] and transmission. During pathology associated with neuroinflammation, microglia respond by adopting an amoeboid morphology and release gliotransmitters such as pro-inflammatory cytokines (IL-1β, IL-6, TNF-α, IFN-γ), chemokines, glutamate, ATP, nitric oxide, and reactive oxygen and nitrogen species that alter the neuro-glia functional interactions [32]. Microglial activation also involves up-regulation of cellular markers, increased microglial proliferation and migration, and a shift in structure and function towards an “M1” pro-inflammatory phenotype. The M1 phenotype occurs in response to tissue injury, stress, and infection as part of the adaptive immune response which ultimately leads to reparative processes. The repair is mediated by microglia, predominantly of the anti-inflammatory (M2) phenotype, which are more phagocytic in nature. Nevertheless, microglia more commonly exist in a range of phenotypes that are intermediate in morphology between the M1 and M2 [33–35]; the differential roles of M1/M2 microglia and their role in CNS (patho)physiology are reviewed elsewhere [36, 37]. Notably, post mortem assessment of brain tissue from patients with MDD and BD revealed that the microglia manifest such an intermediate “activated” morphology associated with greater quinolinic acid expression (implying pro-inflammatory activation of the kynurenine pathway) in the subgenual anterior cingulate cortex area consistently implicated in the pathophysiology of mood disorders [38].
One type of phagocytic activity performed by the microglia involves synaptic pruning that regulates interneuronal connectivity and restores the optimal multipartite synaptic function from the altered states that arise during neuroinflammatory states [39–41]. Thus in mood disorders it has remained unclear whether microglial activation manifests as a reparative response or instead comprises a pathological mechanism that initiates disruption of normative neurophysiology [42]. In other neuropathological conditions, the extant data suggest that neuroinflammation can play pathological roles under some conditions and adaptive/restorative roles in others, with both roles potentially co-existing within the context of a particular CNS disease states. Nevertheless, chronic and/or dysregulated neuroinflammation eventually contributes to a pathological phenotype within the CNS. For example, the extant preclinical and emerging clinical data suggest that glial factors released from microglia and astrocytes during neuroinflammation modulate synaptic plasticity and neurogenesis and impact the neurocircuitry in a manner that can manifest behaviorally in much of the symptomology that defines mood disorders.
Peripheral immune cells also play roles in CNS function that include supporting learning and memory [43–48], protecting against pathogens (e.g., as evidenced by IFN-γ-mediated control of Toxoplasma gondii [49], and inducing neuropathology (e.g., in multiple sclerosis). The peripheral immune system also can play a beneficial or healing role in CNS pathology. For example, a controlled amplification of the autoimmune response was associated with improved neuronal survival in rodent models of acute CNS injury [50] and chronic neurodegeneration [51, 52]. The complex interplay between the peripheral immune system and the CNS in mediating beneficial components of the immune response to CNS pathology is only beginning to be elucidated.
Conversely, changes in peripheral immune cell populations have also been associated with CNS pathologies that do not feature clear penetration of the blood–brain or blood–CSF barrier by circulating cells. Pro-inflammatory cytokines released during peripheral infection are associated with behavioral correlates of depressive mood—termed sickness behavior [53]. Links between sickness behavior and a tryptophan metabolizing enzyme, namely indoleamine 2,3 dioxygenase (IDO), have been demonstrated [54], establishing a potential association between cytokines and monoamine deficiency.
Moreover, post-traumatic stress disorder (PTSD), a syndrome characterized by chronic anxiety, depression, and hyperarousal arising in the aftermath of traumatic stress constitutes a condition that links CNS pathology, stress, and immune system dysregulation at the level of inflammatory cascades and gene networks. For example, in soldiers studied before and after deployment in areas of active conflict, CD14+ monocyte-associated factors were differentially regulated in PTSD sufferers [55]. Notably monocytes are primary producers of the neuroactive cytokines IL-6, IL-1β, and TNF-α, which have been linked to mood disorders in preclinical studies and clinical populations (reviewed elsewhere in this volume). CD14+ monocytes are mobilized into circulation primarily by CCL2, a chemokine produced by glial [56] and blood–brain barrier cells [57] during neuroinflammation. These findings implicate a link between glial activation and loss of peripheral immune homeostasis leading to chronic feedback between the CNS and periphery in PTSD.
2 Abnormalities in Immunological Factors in a Subset of Patients with Mood Disorders Suggest Novel Antidepressant Targets for Such Subgroups
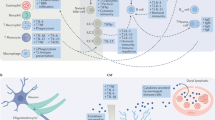
A rapidly expanding scientific literature suggests that alterations in immune system function and neuroinflammation play major roles in the pathophysiology of at least some subtypes of mood disorders [17, 58–61]. This evidence has encouraged targeted and rational drug discovery efforts with a view to intervene using immune modulating treatments for mood disorders [16]. Immune mediators for which the mean concentrations are increased in the blood and cerebrospinal fluid (CSF) of patients with mood disorders versus healthy controls, both when assessed at baseline and after exposure to stressors, include IL-6, IL-1β, IFN-α, TNF-α, prostaglandin E2, and the chemokine CCL2 [62–66]. The mRNA transcripts for these cytokines and for other related innate immune system genes have also been elevated in peripheral blood cells in patients with mood disorders relative to healthy controls matched for age, BMI, smoking, and comorbid medical conditions [6, 22, 67–70]. The clinical significance of these findings is supported by evidence that the elevations of these cytokines in the plasma or CSF of patients with MDD or BD relative to controls are correlated with illness severity and/or suicidality ratings (reviewed elsewhere in this volume). Moreover, successful response to conventional antidepressant drugs is associated with reductions in these cytokine levels in depressed patients, although non-response to conventional antidepressants is predicted by higher IL1-β, IL-6, and CRP levels in the pre-treatment baseline [6, 9, 71]. The preliminary evidence reviewed below suggests that in depressed patients who manifest both resistance to monoamine reuptake inhibitor antidepressant agents and elevation in pro-inflammatory cytokines/acute phase proteins, certain classes of anti-inflammatory agents can produce antidepressant effects.
The relationship between immune challenge and the development of “sickness behavior” as well as other more clearly pathological depressive symptomatology is instructive in considering the etiology of mood disorders. In one of the clearest examples indicating that elevated cytokine signaling can cause depressive symptoms, immune challenge with interferon-α (IFN-α) during the treatment of hepatitis C or other medical conditions reliably has induced the major depressive syndrome (and less commonly manic symptoms) in 30–40% of previously non-depressed humans [64]. This neuropsychiatric sequelae of IFN-α converges with other types of evidence to suggest that elevated signaling of some neuroactive cytokines can play a causal role in inducing depressive symptoms [72]. Within the days following IFN-α administration, previously non-depressed patients show a behavioral complex that includes anorexia, fatigue, lower mood, reduced social interaction, and reduced engagement in pleasurable activities, a symptom complex referred to in the research literature as “sickness behavior” [73]. Notably, in the subset of patients who receive IFN-α who go on to develop major depressive episodes, more specific depressive symptoms such as pessimism, anxiety, and suicide ideation arise later than the initial appearance of sickness behavior, and the likelihood of developing an MDE continues to rise with longer time spent receiving IFN-α [74]. Thus the symptoms of the major depressive episode differ from those of sickness behavior by magnitude in some cases (e.g., more severely depressed mood and pervasive anhedonia in MDE) and by quality in others (e.g., pathological anxiety and suicide ideation). In addition there is also evidence that IFN-α induced depression differs from depression arising in medically healthy MDD subjects by the presence of pathological guilt in the latter, but not in the former condition [75, 76]. These data are notable within the context of evidence that primary MDD is heterogeneous, with subgroups that are distinguishable on the basis of clinical symptomatology as well as immunological markers [58], as reviewed below.
The prevailing hypothesis holds that IFN-α induced major depressive episodes constitute an example of a “pro-inflammatory state induced mood disorder” that manifests in some individuals exposed to IFN-α on the basis of a biological predisposition. A corollary to this hypothesis posits that a subset of the “primary” MDD population also manifests depressive symptoms due to the influence of elevated neuroactive cytokine signaling caused via other etiologies. An example of the existence of a biological predisposition toward IFN-α induced depression was provided by the report of a single nucleotide polymorphism (SNP) in the IL-6 receptor gene that resulted in lower IL-6 expression, and also was associated with decreased susceptibility to the development of depressive symptoms during IFN-α treatment [77].
Notably, some patients who manifest IFN-α induced major depressive episodes improve during SSRI treatment, leading physicians to prophylactically initiate SSRIs in the weeks prior to initiating IFN-α for some patients [78]. Of the symptom domains affected by IFN-α, however, the depressed mood symptom dimension is most responsive, whereas the anxiety, cognitive and neurovegetative symptoms appear less responsive (or unresponsive) to prophylactic SSRI treatment [79]. These observations hold intriguing therapeutic implications, because in patients with primary mood disorders, higher blood levels of proinflammatory cytokines or their mRNA transcripts predict non-response to treatment with SSRIs or other conventional antidepressants [6–9]. It is conceivable that depressed patients who both manifest chronic inflammation and prove nonresponsive to conventional antidepressant drug treatment may benefit from immune modulating treatments.
The hypothesis that anti-inflammatory agents may exert antidepressant effects in depressed patients has been tested both in patients who have primary MDD and in patients with autoimmune disorders who manifest clinically significant depressive symptoms [16]. For example, patients with psoriasis who received the anti-TNF-α agent etanercept showed significant improvement in depressive symptoms in response to drug versus placebo as assessed using conventional depression rating scales, and this difference was evident earlier than the associated changes in pain or skin lesions [80], implying the antidepressant response occurred independently of psychological benefits related to the improvement in the skin lesions per se. In this study patients treated with etanercept also had significant improvements in fatigue. Notably, while the improvements in fatigue correlated with decreasing joint pain, the improvements in depression were less correlated with objective measures of skin clearance or joint pain.
3 Immunological Biomarker Data from Mood Disordered Samples Shows Heterogeneity That May Hold Therapeutic Implications
In studies of primary MDD patients treated using anti-inflammatory agents, the extant data suggest that subgroups characterized by high levels of pro-inflammatory biomarkers are most likely to benefit (see below). This observation appears intuitive when the findings that mean concentrations of cytokine levels are elevated between depressed and control samples are considered in further detail. The distribution of the immunological data from these studies suggests that the differences reported in mean values are attributable to a subset(s) of the depressed patients. This observation appears consistent with accumulating evidence of biological and genetic differences between subtypes of depressed subjects with MDD, who otherwise appear phenotypically homogeneous in many aspects of symptom presentation. For example, from the Netherlands Study of Depression and Anxiety database, Lamers et al. [58] used subgroups defined initially using cluster analysis of depressive signs and symptoms, and then further differentiated these subtypes based on serum protein profiles. The identified analytes consisted largely of inflammatory (e.g., CRP) and metabolic markers (e.g., insulin), supporting the conceptualization of a subtype(s) characterized by metabolic disturbances and inflammation. These researchers [81] also showed that these subgroups appeared stable across time, with patients moving between different levels of severity, but not between subtypes, during longitudinal follow-up. In another example, data from the Mood Inflame Consortium identified three MDD subtypes: one manifest in MDD patients aged ≥28 years that was characterized by increased expression of monocyte genes and decreased expression of glucocorticoid receptor (GR) α versus β subunit ratio, a second in MDD patients <28 years of age who showed a severe course of depression (characterized by recurrent type, illness onset <15 years of age, history of childhood trauma, and prominent panic/arousal symptoms) but monocyte gene expression similar to healthy controls, and a third also manifest in MDD patients <28 years of age characterized by a milder illness course (most with first episode of depression, age at onset ≥15 years, and absent panic symptoms) that exhibited a strongly reduced inflammatory monocyte activation compared to controls [82].
Within the bipolar spectrum of mood disorders, another study from the Mood Inflame Consortium identified a biomarker signature composed of multiple immunological factors that discriminated the majority of BD patients from healthy controls. Using whole-genome expression profiling of RNA obtained from purified CD14+ monocytes, Padmos and colleagues reported elevated mRNAs of inflammatory (e.g., TNF, PDE4B, IL-1β, IL6, TNFAIP3), trafficking, survival (e.g., BCL2A), and mitogen-activated protein kinase pathway (e.g., MAPK6, ATF3) genes in BD subjects in various illness phases, as well as in affected offspring of BD parents [69]. Notably, in peripheral blood mononuclear cells (PBMC) from the same subjects assessed via fluorescence-activated cell sorting (FACS) analysis, the percentages of anti-inflammatory CD4+CD25highFoxP3+ regulatory T cells were higher in BD patients <40 years of age, while percentages of Th1, Th2, and Th17 cells were normal. Together these results thus showed enhancement of both pro-inflammatory monocyte and anti-inflammatory T cell mediators in BD [83].
4 Novel Drug Targets at the Crossroads of Neuroimmunology and Mood Disorders
With continued and refined understanding of the role of immune cells and their mediators in the periphery and the CNS, it is anticipated that new mechanisms will be discovered that can exert antidepressant and mood-stabilizing effects in primary mood disorders. Several comprehensive reviews have highlighted potential drug targets in neuroimmunology for mood disorders [11, 21, 84]. In this chapter, we summarize evidence that highlights TNF-α, IL-6, and IL-1β signaling in the pathophysiology of mood disorders.
TNF-α: TNF-α signaling appears to play a major role in mood disorders [85]. In meta-analyses of clinical studies, plasma TNF-α correlated with depression severity and level of resistance to conventional antidepressants [62]. A causal relationship between TNF-α elevation and depressive symptoms was suggested by observations that in patients with immunological diseases such as rheumatoid arthritis and psoriasis, anti-TNF-α treatment alleviates depressed mood; as reviewed above, these antidepressant effects do not appear attributable simply to improvement in sickness symptoms, such as fatigue, or in the underlying autoimmune disorder [86]. Consistent with these observations, the TNF-α receptor 1, TNF-α receptor 2, and TNF-α knockout mouse models all show antidepressant-like phenotypes [87, 88]. Likewise, systemic administration of antibodies targeting TNF-α in chronic models of stress reversed the anhedonic behaviors, suggesting that TNF-α signaling contributes to depressogenic behaviors in rodents [89, 90].
Nevertheless, a clinical study of the efficacy of infliximab (a monoclonal antibody against TNF-α) in depressed patients generated negative results on depressive symptoms rating using a conventional depression rating scale [91]. A post hoc investigation of data from this study, however, revealed a significant positive correlation between clinical improvement and pre-treatment levels of the nonspecific inflammation marker, CRP, raising the possibility that antidepressant effects may be limited to individuals who manifest a pro-inflammatory diathesis. Nevertheless, because the test of the a priori hypothesis in this study was negative, the question has remained whether targeting TNF-α via large molecules introduced in the periphery alone can produce an antidepressant effect (since very low proportions of peripherally administered monoclonal antibodies enter the brain following acute treatment), or whether therapies that reduce TNF-α signaling must instead directly engage targets in the CNS.
IL-6: In studies of MDD or BD one of the more highly replicated biomarker abnormalities has been an elevation in peripheral blood IL-6 concentrations [92]. Notably during IFN-α treatment the magnitude of the increase in plasma and CSF IL-6 levels correlates positively with depressive symptom severity. Conversely, the above-mentioned functional polymorphism in the promoter region of the IL-6 gene (rs1800795) that results in decreased IL-6 expression is associated with a significantly lower risk for developing major depressive episodes during IFN-α treatment [77]. The relationship to IL-6 function is compatible with findings that, in patients with primary mood disorders higher IL-6 levels in the CSF correlated with suicidality, and elevated IL-6 levels in the plasma correlated with non-responsiveness to conventional antidepressant drugs [93]. In contrast, during the euthymic (i.e., asymptomatic) phase of BD, the CSF concentration of IL-6 was decreased with respect to healthy controls, despite the same BD subjects showing an abnormal elevation in the CSF levels of IL-1β [65].
Although IL-6 can be released by immune cells in the CNS as well as in the periphery, preclinical evidence suggests that elevated IL-6 release from peripheral immune cells is sufficient to induce depressive behaviors, irrespective of central immune system activation. In studies conducted by Hodes and colleagues [15] to elucidate the biological basis of susceptibility to depression-like behaviors under stress, mice that developed a persistent depression-like phenotype in response to social defeat stress (SDS) were compared to genetically identical mice that did not develop depression-like behaviors under SDS. The susceptible animals differed from the resilient animals by showing elevated basal IL-6 levels in the pre-SDS condition and higher IL-6 release in response to the stressed condition. In addition, white blood cells sampled pre-SDS from susceptible mice showed higher LPS-induced IL-6 release ex vivo compared to cells from resilient mice. Crucially, the susceptibility to the depression-like phenotype could be altered toward either susceptibility or resilience by generating bone marrow chimeras that had hemopoetic stem cells transplanted from high IL-6 expressing mice or IL-6 knockout mice, respectively. The bone marrow recipients in these studies had received radiation to their bodies while the head was shielded, so the hemopoetic stem cells in periphery conferred the susceptibility to the depression-like phenotype under stress.
IL-1β: In contrast to the therapeutic potential offered by neutralizing IL-6 predominantly in the periphery, the extant data suggest that for the pro-inflammatory cytokine IL-1β, reducing signaling in the brain may prove critical to achieving antidepressant effects. IL-1β is probably the most potent pro-inflammatory cytokine released from microglia in the brain. Clinical studies found that IL-1β is present at abnormally higher levels in plasma, CSF, and postmortem brain tissue of individuals with mood disorders, and that IL-1β levels correlated positively with depression severity [63, 65]. Anisman and colleagues reported increased IL-1β production from lymphocytes in patients with dysthymic disorder and a modest correlation existed between the cytokine and depressive symptoms [94]. In studies of primary mood disordered subgroups, IL-1β has been linked with both geriatric depression and postpartum depression [95, 96].
In animal models of stress-induced depression-like behaviors, several groups showed that IL-1β signaling is critical to the acquisition of the depression-like phenotype [97, 98]. The development of the depressive behavioral phenotype during chronic stress can be blocked by IL-1 receptor antagonists, and is absent in IL-1R receptor knockout mice [99]. In addition, manipulation of central IL-1β, either by exogenous administration of IL-1β directly into the brain, or by selective ablation of signaling via pharmacology or genetics, produced behavioral analogues of depression when IL-1β was increased, or antidepressant-like effects when IL-1β was decreased [100]. The IL-1β driven changes in the brain resulted in decreased neurogenesis in the hippocampus [99] and increased corticosterone response to stress in the periphery [101], suggesting an interplay between stress-induced-IL-1β release and HPA axis function.
Recently, it was shown that both acute and chronic stress increase brain IL-1β release [102] [100]. Stress-induced IL-1β release appears to be driven by ATP-induced activation of the P2X purinoceptor 7 ion channel (P2X7), and genetic deletion of P2X7 receptors results in antidepressant-like reversal of stress-induced depressogenic behaviors in rodents [102, 103]. Other experimental evidence has similarly demonstrated that P2X7 activation causes release of IL-1β ([104]). The initiation of transcription and translation of the pro-form of IL-1β is induced by activation of Toll-like receptor (TLR), but it is the second signal from P2X7 (due to ATP activating the ion channel) that results in maturation and release of the pro-inflammatory IL-1β cytokine; this process has been referred as a “two-hit” model of IL-1β release. Priming of the TLRs is achieved by factors such as cellular debris, by endotoxins, by damage- and pathogen-associated molecular pattern molecules (DAMPs and PAMPs, respectively). Since P2X7 is abundantly expressed in blood cells, IL-1β release in the blood has been used as a biomarker of P2X7 activity in both preclinical and clinical assessment of target engagement.
Based on robust microglial expression of P2X7, and IL-1β signaling leading to neuroinflammation, CNS penetrable P2X7 antagonists would be potentially beneficial for treating mood disorders, and there is growing evidence that strengthens the role of P2X7 in MDD and BD. Several human genetic studies have associated the highly polymorphic P2RX7 gene with the risk for developing both BD and MDD, and some of these mutations have been linked to a modulation of P2X7 channel function in vitro [105, 106]. The rs2230912-G allele exhibits a gain-of-function and human monocytes expressing this variant secreted more IL-1β in response to activation of P2X7 than monocytes expressing a wild-type variant [107]. It is conceivable that such a variant in P2X7 receptors based in human microglia would lead to enhanced IL-1β release (or production), leading to neuroinflammation over time. Nevertheless, several other GWAS studies have not confirmed the association between P2RX7 variants and the risk for mood disorders [108], so the relationship between the variation in P2RX7 and depression is not yet established. The lack of clarity for a genetic association of P2RX7 variation in the risk for mood disorders (or any disease phenotype) is perhaps not surprising as the underlying factors of such pathologies are often a result of interplay between genetic (often many genes), environmental, and developmental factors.
In addition to the human genetic literature, several laboratories have demonstrated that P2X7 knockout mice manifest a protective phenotype in models of depression and mania, strengthening the hypothesis that P2X7 antagonism may be therapeutically beneficial in mood disorders. Consistent with the antidepressant phenotype observed in P2X7 knockout mice, emerging data suggest that P2X7 antagonists can reverse depressogenic behaviors in animal models. For example, pharmacological antagonism of P2X7 (by AZ-10606120 and A-804598) restored the deficit observed in the preference for a sucrose solution (a putative behavioral analogue of anhedonia) induced by either chronic stress or systemic administration of lipopolysaccharide (LPS) administration [103]. Recently, it was shown that a P2X7 selective, brain-penetrant antagonist was efficacious in chronic stress models in rats [102]. In addition, a large corpus of evidence suggests that manipulation of central IL-1β (by either exogenous administration or selective ablation of signaling by pharmacological or genetic manipulation) results in depression-like behaviors when IL-1β is increased, or in resilience against the development of depression-like behaviors when IL-1β is decreased [97, 99, 109]. These observations appear consistent with the above-mentioned findings that IL-1β levels are abnormally elevated in the plasma, cerebrospinal fluid (CSF), and postmortem brain tissue obtained from MDD and BD patients [63, 65, 110].
Preclinical data also suggest that P2X7 antagonism may produce anti-manic or mood-stabilizing effects in BD [111]. For example, P2X7 antagonism produced attenuation of amphetamine-induced sensitization of hyperactivity [112, 113], a putative rodent model of mania-like behavior, and similar phenotypes were observed in P2X7 knockout mice [111] suggesting a potential therapeutic role of P2X7 antagonism in the manic phase of BD. Taken together, it remains plausible that a selective and brain-penetrant P2X7 antagonist may be therapeutically beneficial in mood disorders, especially targeting treatment resistant patient populations.
5 Conclusion
Neuroimmunology stands at the interface of emerging biology and breakthrough therapeutics for mood disorders. Taken together, the extant data support the hypothesis that elevated cytokine levels contribute to the pathophysiology of depression and the neurobiological mechanisms underlying resistance to conventional antidepressant drugs, in at least a subpopulation of depressed patients. They also suggest that specific cytokines (such as TNF-α, IL-6, and IL-1β) and their effectors and regulators (such as P2X7) may constitute novel therapeutic targets for depression. However, the extant postmortem data also indicate that mood disorders are not associated with classical neuroinflammation, and in vivo blood-based biomarker studies suggest that not all patients suffering from mood disorders manifest an inflammatory component. Consequently, for clinical proof-of-concept studies with compounds that target signaling of microglia, astrocytes, or cytokines/chemokines, it may prove necessary to discriminate the patient population suffering from concomitant depression and neuroinflammation through the aid of immunological biomarkers.
References
Kessler RC, Berglund P, Demler O, Jin R, Koretz D, Merikangas KR, Rush AJ, Walters EE, Wang PS, National Comorbidity Survey Replication (2003) The epidemiology of major depressive disorder: results from the National Comorbidity Survey Replication (NCS-R). JAMA 289:3095–3105
O’Leary OF, Dinan TG, Cryan JF (2015) Faster, better, stronger: towards new antidepressant therapeutic strategies. Eur J Pharmacol 753:32–50
Papakostas GI, Ionescu DF (2015) Towards new mechanisms: an update on therapeutics for treatment-resistant major depressive disorder. Mol Psychiatry 20:1142–1150
Rush AJ, Trivedi MH, Wisniewski SR, Nierenberg AA, Stewart JW, Warden D, Niederehe G, Thase ME, Lavori PW, Lebowitz BD, McGrath PJ, Rosenbaum JF, Sackeim HA, Kupfer DJ, Luther J, Fava M (2006) Acute and longer-term outcomes in depressed outpatients requiring one or several treatment steps: a STAR*D report. Am J Psychiatry 163:1905–1917
Carvalho LA, Torre JP, Papadopoulos AS, Poon L, Juruena MF, Markopoulou K, Cleare AJ, Pariante CM (2013) Lack of clinical therapeutic benefit of antidepressants is associated overall activation of the inflammatory system. J Affect Disord 148:136–140
Cattaneo A, Gennarelli M, Uher R, Breen G, Farmer A, Aitchison KJ, Craig IW, Anacker C, Zunsztain PA, McGuffin P, Pariante CM (2013) Candidate genes expression profile associated with antidepressants response in the GENDEP study: differentiating between baseline ‘predictors’ and longitudinal ‘targets’. Neuropsychopharmacology 38:377–385
O’Brien SM, Scully P, Fitzgerald P, Scott LV, Dinan TG (2007) Plasma cytokine profiles in depressed patients who fail to respond to selective serotonin reuptake inhibitor therapy. J Psychiatric Res 41:326–331
Uher R, Tansey KE, Dew T, Maier W, Mors O, Hauser J, Dernovsek MZ, Henigsberg N, Souery D, Farmer A, McGuffin P (2014) An inflammatory biomarker as a differential predictor of outcome of depression treatment with escitalopram and nortriptyline. Am J Psychiatry 171:1278–1286
Yoshimura R, Hori H, Ikenouchi-Sugita A, Umene-Nakano W, Ueda N, Nakamura J (2009) Higher plasma interleukin-6 (IL-6) level is associated with SSRI- or SNRI-refractory depression. Prog Neuropsychopharmacol Biol Psychiatry 33:722–726
Pariante CM (2016) Neuroscience, mental health and the immune system: overcoming the brain-mind-body trichotomy. Epidemiol Psychiatr Sci 25(2):101–105
Wohleb ES, Franklin T, Iwata M, Duman RS (2016) Integrating neuroimmune systems in the neurobiology of depression. Nat Rev Neurosci 17:497–511
Sandiego CM, Gallezot JD, Pittman B, Nabulsi N, Lim K, Lin SF, Matuskey D, Lee JY, O’Connor KC, Huang Y, Carson RE, Hannestad J, Cosgrove KP (2015) Imaging robust microglial activation after lipopolysaccharide administration in humans with PET. Proc Natl Acad Sci U S A 112:12468–12473
Haapakoski R, Ebmeier KP, Alenius H, Kivimaki M (2016) Innate and adaptive immunity in the development of depression: an update on current knowledge and technological advances. Prog Neuropsychopharmacol Biol Psychiatry 66:63–72
Morris G, Berk M (2015) The many roads to mitochondrial dysfunction in neuroimmune and neuropsychiatric disorders. BMC Med 13:68
Hodes GE, Pfau ML, Leboeuf M, Golden SA, Christoffel DJ, Bregman D, Rebusi N, Heshmati M, Aleyasin H, Warren BL, Lebonte B, Horn S, Lapidus KA, Stelzhammer V, Wong EH, Bahn S, Krishnan V, Bolanos-Guzman CA, Murrough JW, Merad M, Russo SJ (2014) Individual differences in the peripheral immune system promote resilience versus susceptibility to social stress. Proc Natl Acad Sci U S A 111:16136–16141
Kohler O, Benros ME, Krogh J (2015) Anti-inflammatory intervention in depression--reply. JAMA Psychiatry 72:512–513
Najjar S, Pearlman DM, Alper K, Najjar A, Devinsky O (2013) Neuroinflammation and psychiatric illness. J Neuroinflammation 10:43
Ben-Shaanan TL, Azulay-Debby H, Dubovik T, Starosvetsky E, Korin B, Schiller M, Green NL, Admon Y, Hakim F, Shen-Orr SS, Rolls A (2016) Activation of the reward system boosts innate and adaptive immunity. Nat Med 22:940–944
Louveau A, Harris TH, Kipnis J (2015) Revisiting the mechanisms of CNS immune privilege. Trends Immunol 36:569–577
Yirmiya R, Goshen I (2011) Immune modulation of learning, memory, neural plasticity and neurogenesis. Brain Behav Immun 25:181–213
Bhattacharya A, Derecki NC, Lovenberg TW, Drevets WC (2016) Role of neuro-immunological factors in the pathophysiology of mood disorders. Psychopharmacology (Berl) 233:1623–1636
Savitz J, Frank MB, Victor T, Bebak M, Marino JH, Bellgowan PS, McKinney BA, Bodurka J, Kent Teague T, Drevets WC (2013) Inflammation and neurological disease-related genes are differentially expressed in depressed patients with mood disorders and correlate with morphometric and functional imaging abnormalities. Brain Behav Immun 31:161–171
Savitz J, Drevets WC (2009) Bipolar and major depressive disorder: neuroimaging the developmental-degenerative divide. Neurosci Biobehav Rev 33:699–771
Craddock N, Sklar P (2013) Genetics of bipolar disorder. Lancet 381:1654–1662
Edvardsen J, Torgersen S, Roysamb E, Lygren S, Skre I, Onstad S, Oien PA (2009) Unipolar depressive disorders have a common genotype. J Affect Disord 117:30–41
Milaneschi Y, Lamers F, Peyrot WJ, Abdellaoui A, Willemsen G, Hottenga JJ, Jansen R, Mbarek H, Dehghan A, Lu C, Group Ciw, Boomsma DI, Penninx BW (2016) Polygenic dissection of major depression clinical heterogeneity. Mol Psychiatry 21:516–522
Capuron L, Miller AH (2011) Immune system to brain signaling: neuropsychopharmacological implications. Pharmacol Ther 130:226–238
Wohleb ES, McKim DB, Sheridan JF, Godbout JP (2014) Monocyte trafficking to the brain with stress and inflammation: a novel axis of immune-to-brain communication that influences mood and behavior. Front Neurosci 8:447
Louveau A, Smirnov I, Keyes TJ, Eccles JD, Rouhani SJ, Peske JD, Derecki NC, Castle D, Mandell JW, Lee KS, Harris TH, Kipnis J (2015) Structural and functional features of central nervous system lymphatic vessels. Nature 523:337–341
Raper D, Louveau A, Kipnis J (2016) How do meningeal lymphatic vessels drain the CNS? Trends Neurosci
Wu Y, Dissing-Olesen L, MacVicar BA, Stevens B (2015) Microglia: dynamic mediators of synapse development and plasticity. Trends Immunol 36:605–613
Ben Achour S, Pascual O (2010) Glia: the many ways to modulate synaptic plasticity. Neurochem Int 57:440–445
Orihuela R, McPherson CA, Harry GJ (2016) Microglial M1/M2 polarization and metabolic states. Br J Pharmacol 173(4):649–665
Ransohoff RM (2016) A polarizing question: do M1 and M2 microglia exist? Nat Neurosci 19:987–991
Tang Y, Le W (2015) Differential roles of M1 and M2 microglia in neurodegenerative diseases. Mol Neurobiol 53(2):1181–1194
Cherry JD, Olschowka JA, O’Banion MK (2014) Neuroinflammation and M2 microglia: the good, the bad, and the inflamed. J Neuroinflammation 11:98
Prinz M, Priller J (2014) Microglia and brain macrophages in the molecular age: from origin to neuropsychiatric disease. Nat Rev Neurosci 15:300–312
Steiner J, Walter M, Gos T, Guillemin GJ, Bernstein HG, Sarnyai Z, Mawrin C, Brisch R, Bielau H, Meyer zu Schwabedissen L, Bogerts B, Myint AM (2011) Severe depression is associated with increased microglial quinolinic acid in subregions of the anterior cingulate gyrus: evidence for an immune-modulated glutamatergic neurotransmission? J Neuroinflammation 8:94
Aguzzi A, Barres BA, Bennett ML (2013) Microglia: scapegoat, saboteur, or something else? Science 339:156–161
Chung WS, Barres BA (2012) The role of glial cells in synapse elimination. Curr Opin Neurobiol 22:438–445
Zhan Y, Paolicelli RC, Sforazzini F, Weinhard L, Bolasco G, Pagani F, Vyssotski AL, Bifone A, Gozzi A, Ragozzino D, Gross CT (2014) Deficient neuron-microglia signaling results in impaired functional brain connectivity and social behavior. Nat Neurosci 17:400–406
Yirmiya R, Rimmerman N, Reshef R (2015) Depression as a microglial disease. Trends Neurosci 38:637–658
Brynskikh A, Warren T, Zhu J, Kipnis J (2008) Adaptive immunity affects learning behavior in mice. Brain Behav Immun 22:861–869
Derecki NC, Cardani AN, Yang CH, Quinnies KM, Crihfield A, Lynch KR, Kipnis J (2010) Regulation of learning and memory by meningeal immunity: a key role for IL-4. J Exp Med 207:1067–1080
Jiang NM, Tofail F, Moonah SN, Scharf RJ, Taniuchi M, Ma JZ, Hamadani JD, Gurley ES, Houpt ER, Azziz-Baumgartner E, Haque R, Petri WA Jr (2014) Febrile illness and pro-inflammatory cytokines are associated with lower neurodevelopmental scores in Bangladeshi infants living in poverty. BMC Pediatr 14:50
Kipnis J, Cohen H, Cardon M, Ziv Y, Schwartz M (2004) T cell deficiency leads to cognitive dysfunction: implications for therapeutic vaccination for schizophrenia and other psychiatric conditions. Proc Natl Acad Sci U S A 101:8180–8185
Wolf SA, Steiner B, Akpinarli A, Kammertoens T, Nassenstein C, Braun A, Blankenstein T, Kempermann G (2009) CD4-positive T lymphocytes provide a neuroimmunological link in the control of adult hippocampal neurogenesis. J Immunol 182:3979–3984
Ziv Y, Ron N, Butovsky O, Landa G, Sudai E, Greenberg N, Cohen H, Kipnis J, Schwartz M (2006) Immune cells contribute to the maintenance of neurogenesis and spatial learning abilities in adulthood. Nat Neurosci 9:268–275
Yarovinsky F (2014) Innate immunity to Toxoplasma gondii infection. Nat Rev Immunol 14:109–121
Moalem G, Leibowitz-Amit R, Yoles E, Mor F, Cohen IR, Schwartz M (1999) Autoimmune T cells protect neurons from secondary degeneration after central nervous system axotomy. Nat Med 5:49–55
Hofstetter HH, Sewell DL, Liu F, Sandor M, Forsthuber T, Lehmann PV, Fabry Z (2003) Autoreactive T cells promote post-traumatic healing in the central nervous system. J Neuroimmunol 134:25–34
Kipnis J, Mizrahi T, Yoles E, Ben-Nun A, Schwartz M (2002) Myelin specific Th1 cells are necessary for post-traumatic protective autoimmunity. J Neuroimmunol 130:78–85
Kelley KW, Bluthe RM, Dantzer R, Zhou JH, Shen WH, Johnson RW, Broussard SR (2003) Cytokine-induced sickness behavior. Brain Behav Immun 17(Suppl 1):S112–S118
Dantzer R (2016) Role of the Kynurenine metabolism pathway in inflammation-induced depression: preclinical approaches. Curr Top Behav Neurosci. doi:10.1007/7854_2016_6
Breen MS, Maihofer AX, Glatt SJ, Tylee DS, Chandler SD, Tsuang MT, Risbrough VB, Baker DG, O’Connor DT, Nievergelt CM, Woelk CH (2015) Gene networks specific for innate immunity define post-traumatic stress disorder. Mol Psychiatry 20(12):1538–1545
Glabinski AR, Balasingam V, Tani M, Kunkel SL, Strieter RM, Yong VW, Ransohoff RM (1996) Chemokine monocyte chemoattractant protein-1 is expressed by astrocytes after mechanical injury to the brain. J Immunol 156:4363–4368
Andjelkovic AV, Pachter JS (2000) Characterization of binding sites for chemokines MCP-1 and MIP-1alpha on human brain microvessels. J Neurochem 75:1898–1906
Lamers F, Bot M, Jansen R, Chan MK, Cooper JD, Bahn S, Penninx BW (2016) Serum proteomic profiles of depressive subtypes. Transl Psychiatry 6:e851
Noto C, Rizzo LB, Mansur RB, McIntyre RS, Maes M, Brietzke E (2014) Targeting the inflammatory pathway as a therapeutic tool for major depression. Neuroimmunomodulation 21:131–139
Rosenblat JD, Cha DS, Mansur RB, McIntyre RS (2014) Inflamed moods: a review of the interactions between inflammation and mood disorders. Prog Neuropsychopharmacol Biol Psychiatry 53:23–34
Walker AK, Kavelaars A, Heijnen CJ, Dantzer R (2014) Neuroinflammation and comorbidity of pain and depression. Pharmacol Rev 66:80–101
Dowlati Y, Herrmann N, Swardfager W, Liu H, Sham L, Reim EK, Lanctot KL (2010) A meta-analysis of cytokines in major depression. Biol Psychiatry 67:446–457
Jones KA, Thomsen C (2013) The role of the innate immune system in psychiatric disorders. Mol Cell Neurosci 53:52–62
Pace TW, Miller AH (2009) Cytokines and glucocorticoid receptor signaling. Relevance to major depression. Ann N Y Acad Sci 1179:86–105
Soderlund J, Olsson SK, Samuelsson M, Walther-Jallow L, Johansson C, Erhardt S, Landen M, Engberg G (2011) Elevation of cerebrospinal fluid interleukin-1ss in bipolar disorder. J Psychiatry Neurosci 36:114–118
Young JJ, Bruno D, Pomara N (2014) A review of the relationship between proinflammatory cytokines and major depressive disorder. J Affect Disord 169:15–20
Carvalho LA, Bergink V, Sumaski L, Wijkhuijs J, Hoogendijk WJ, Birkenhager TK, Drexhage HA (2014) Inflammatory activation is associated with a reduced glucocorticoid receptor alpha/beta expression ratio in monocytes of inpatients with melancholic major depressive disorder. Transl Psychiatry 4:e344
Jansen R, Penninx BW, Madar V, Xia K, Milaneschi Y, Hottenga JJ, Hammerschlag AR, Beekman A, van der Wee N, Smit JH, Brooks AI, Tischfield J, Posthuma D, Schoevers R, van Grootheest G, Willemsen G, de Geus EJ, Boomsma DI, Wright FA, Zou F, Sun W, Sullivan PF (2016) Gene expression in major depressive disorder. Mol Psychiatry 21(3):339–347
Padmos RC, Hillegers MH, Knijff EM, Vonk R, Bouvy A, Staal FJ, de Ridder D, Kupka RW, Nolen WA, Drexhage HA (2008) A discriminating messenger RNA signature for bipolar disorder formed by an aberrant expression of inflammatory genes in monocytes. Arch Gen Psychiatry 65:395–407
Powell TR, McGuffin P, D’Souza UM, Cohen-Woods S, Hosang GM, Martin C, Matthews K, Day RK, Farmer AE, Tansey KE, Schalkwyk LC (2014) Putative transcriptomic biomarkers in the inflammatory cytokine pathway differentiate major depressive disorder patients from control subjects and bipolar disorder patients. PLoS One 9:e91076
Hannestad J, DellaGioia N, Bloch M (2011) The effect of antidepressant medication treatment on serum levels of inflammatory cytokines: a meta-analysis. Neuropsychopharmacology 36:2452–2459
Baraldi S, Hepgul N, Mondelli V, Pariante CM (2012) Symptomatic treatment of interferon-alpha-induced depression in hepatitis C: a systematic review. J Clin Psychopharmacol 32:531–543
Dantzer R, Bluthe RM, Laye S, Bret-Dibat JL, Parnet P, Kelley KW (1998) Cytokines and sickness behavior. Ann N Y Acad Sci 840:586–590
Miller AH, Maletic V, Raison CL (2009) Inflammation and its discontents: the role of cytokines in the pathophysiology of major depression. Biol Psychiatry 65:732–741
Capuron L, Fornwalt FB, Knight BT, Harvey PD, Ninan PT, Miller AH (2009) Does cytokine-induced depression differ from idiopathic major depression in medically healthy individuals? J Affect Disord 119:181–185
Pasquini M, Speca A, Mastroeni S, Delle Chiaie R, Sternberg CN, Biondi M (2008) Differences in depressive thoughts between major depressive disorder, IFN-alpha-induced depression, and depressive disorders among cancer patients. J Psychosom Res 65:153–156
Bull SJ, Huezo-Diaz P, Binder EB, Cubells JF, Ranjith G, Maddock C, Miyazaki C, Alexander N, Hotopf M, Cleare AJ, Norris S, Cassidy E, Aitchison KJ, Miller AH, Pariante CM (2009) Functional polymorphisms in the interleukin-6 and serotonin transporter genes, and depression and fatigue induced by interferon-alpha and ribavirin treatment. Mol Psychiatry 14:1095–1104
Udina M, Hidalgo D, Navines R, Forns X, Sola R, Farre M, Capuron L, Vieta E, Martin-Santos R (2014) Prophylactic antidepressant treatment of interferon-induced depression in chronic hepatitis C: a systematic review and meta-analysis. J Clin Psychiatry 75:e1113–e1121
McNutt MD, Liu S, Manatunga A, Royster EB, Raison CL, Woolwine BJ, Demetrashvili MF, Miller AH, Musselman DL (2012) Neurobehavioral effects of interferon-alpha in patients with hepatitis-C: symptom dimensions and responsiveness to paroxetine. Neuropsychopharmacology 37:1444–1454
Tyring S, Gottlieb A, Papp K, Gordon K, Leonardi C, Wang A, Lalla D, Woolley M, Jahreis A, Zitnik R, Cella D, Krishnan R (2006) Etanercept and clinical outcomes, fatigue, and depression in psoriasis: double-blind placebo-controlled randomised phase III trial. Lancet 367:29–35
Lamers F, Rhebergen D, Merikangas KR, de Jonge P, Beekman AT, Penninx BW (2012) Stability and transitions of depressive subtypes over a 2-year follow-up. Psychol Med 42:2083–2093
Grosse L, Carvalho LA, Wijkhuijs AJ, Bellingrath S, Ruland T, Ambree O, Alferink J, Ehring T, Drexhage HA, Arolt V (2015) Clinical characteristics of inflammation-associated depression: monocyte gene expression is age-related in major depressive disorder. Brain Behav Immun 44:48–56
Drexhage RC, Knijff EM, Padmos RC, Heul-Nieuwenhuijzen L, Beumer W, Versnel MA, Drexhage HA (2010) The mononuclear phagocyte system and its cytokine inflammatory networks in schizophrenia and bipolar disorder. Expert Rev Neurother 10:59–76
Hodes GE, Kana V, Menard C, Merad M, Russo SJ (2015) Neuroimmune mechanisms of depression. Nat Neurosci 18:1386–1393
Dantzer R, O’Connor JC, Freund GG, Johnson RW, Kelley KW (2008) From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci 9:46–56
Krishnan R, Cella D, Leonardi C, Papp K, Gottlieb AB, Dunn M, Chiou CF, Patel V, Jahreis A (2007) Effects of etanercept therapy on fatigue and symptoms of depression in subjects treated for moderate to severe plaque psoriasis for up to 96 weeks. Br J Dermatol 157:1275–1277
Simen BB, Duman CH, Simen AA, Duman RS (2006) TNFalpha signaling in depression and anxiety: behavioral consequences of individual receptor targeting. Biol Psychiatry 59:775–785
Yamada K, Iida R, Miyamoto Y, Saito K, Sekikawa K, Seishima M, Nabeshima T (2000) Neurobehavioral alterations in mice with a targeted deletion of the tumor necrosis factor-alpha gene: implications for emotional behavior. J Neuroimmunol 111:131–138
Karson A, Demirtas T, Bayramgurler D, Balci F, Utkan T (2013) Chronic administration of infliximab (TNF-alpha inhibitor) decreases depression and anxiety-like behaviour in rat model of chronic mild stress. Basic Clin Pharmacol Toxicol 112:335–340
Krugel U, Fischer J, Radicke S, Sack U, Himmerich H (2013) Antidepressant effects of TNF-alpha blockade in an animal model of depression. J Psychiatric Res 47:611–616
Raison CL, Rutherford RE, Woolwine BJ, Shuo C, Schettler P, Drake DF, Haroon E, Miller AH (2013) A randomized controlled trial of the tumor necrosis factor antagonist infliximab for treatment-resistant depression: the role of baseline inflammatory biomarkers. JAMA Psychiatry 70:31–41
Maes M, Anderson G, Kubera M, Berk M (2014) Targeting classical IL-6 signalling or IL-6 trans-signalling in depression? Expert Opin Ther Targets 18:495–512
Bay-Richter C, Linderholm KR, Lim CK, Samuelsson M, Traskman-Bendz L, Guillemin GJ, Erhardt S, Brundin L (2015) A role for inflammatory metabolites as modulators of the glutamate N-methyl-D-aspartate receptor in depression and suicidality. Brain Behav Immun 43:110–117
Anisman H, Ravindran AV, Griffiths J, Merali Z (1999) Interleukin-1 beta production in dysthymia before and after pharmacotherapy. Biol Psychiatry 46:1649–1655
Corwin EJ, Johnston N, Pugh L (2008) Symptoms of postpartum depression associated with elevated levels of interleukin-1 beta during the first month postpartum. Biol Res Nurs 10:128–133
Diniz BS, Teixeira AL, Talib L, Gattaz WF, Forlenza OV (2010) Interleukin-1beta serum levels is increased in antidepressant-free elderly depressed patients. Am J Geriatr Psychiatry 18:172–176
Koo JW, Duman RS (2009) Evidence for IL-1 receptor blockade as a therapeutic strategy for the treatment of depression. Curr Opin Investig Drugs 10:664–671
Maes M, Song C, Yirmiya R (2012) Targeting IL-1 in depression. Expert Opin Ther Targets 16:1097–1112
Koo JW, Duman RS (2008) IL-1beta is an essential mediator of the antineurogenic and anhedonic effects of stress. Proc Natl Acad Sci U S A 105:751–756
Zhang Y, Liu L, Liu YZ, Shen XL, Wu TY, Zhang T, Wang W, Wang YX, Jiang CL (2015) NLRP3 inflammasome mediates chronic mild stress-induced depression in mice via neuroinflammation. Int J Neuropsychopharmacol 18(8):pyv006
Song C, Phillips AG, Leonard B (2003) Interleukin 1 beta enhances conditioned fear memory in rats: possible involvement of glucocorticoids. Eur J Neurosci 18:1739–1743
Iwata M, Ota KT, Li XY, Sakaue F, Li N, Dutheil S, Banasr M, Duric V, Yamanashi T, Kaneko K, Rasmussen K, Glasebrook A, Koester A, Song D, Jones KA, Zorn S, Smagin G, Duman RS (2016) Psychological stress activates the inflammasome via release of adenosine triphosphate and stimulation of the purinergic type 2X7 receptor. Biol Psychiatry 80:12–22
Csolle C, Baranyi M, Zsilla G, Kittel A, Goloncser F, Illes P, Papp E, Vizi ES, Sperlagh B (2013) Neurochemical changes in the mouse hippocampus underlying the antidepressant effect of genetic deletion of P2X7 receptors. PLoS One 8:e66547
Solle M, Labasi J, Perregaux DG, Stam E, Petrushova N, Koller BH, Griffiths RJ, Gabel CA (2001) Altered cytokine production in mice lacking P2X(7) receptors. J Biol Chem 276:125–132
Caseley EA, Muench SP, Roger S, Mao HJ, Baldwin SA, Jiang LH (2014) Non-synonymous single nucleotide polymorphisms in the P2X receptor genes: association with diseases, impact on receptor functions and potential use as diagnosis biomarkers. Int J Mol Sci 15:13344–13371
Jiang LH, Baldwin JM, Roger S, Baldwin SA (2013) Insights into the molecular mechanisms underlying mammalian P2X7 receptor functions and contributions in diseases, revealed by structural modeling and single nucleotide polymorphisms. Front Pharmacol 4:55
Stokes L, Fuller SJ, Sluyter R, Skarratt KK, Gu BJ, Wiley JS (2010) Two haplotypes of the P2X(7) receptor containing the Ala-348 to Thr polymorphism exhibit a gain-of-function effect and enhanced interleukin-1beta secretion. FASEB J 24:2916–2927
Feng WP, Zhang B, Li W, Liu J (2014) Lack of association of P2RX7 gene rs2230912 polymorphism with mood disorders: a meta-analysis. PLoS One 9:e88575
Goshen I, Kreisel T, Ben-Menachem-Zidon O, Licht T, Weidenfeld J, Ben-Hur T, Yirmiya R (2008) Brain interleukin-1 mediates chronic stress-induced depression in mice via adrenocortical activation and hippocampal neurogenesis suppression. Mol Psychiatry 13:717–728
Rao JS, Harry GJ, Rapoport SI, Kim HW (2010) Increased excitotoxicity and neuroinflammatory markers in postmortem frontal cortex from bipolar disorder patients. Mol Psychiatry 15:384–392
Gubert C, Fries GR, Pfaffenseller B, Ferrari P, Coutinho-Silva R, Morrone FB, Kapczinski F, Battastini AM (2016) Role of P2X7 receptor in an animal model of mania induced by D-amphetamine. Mol Neurobiol 53(1):611–620
Bhattacharya A, Wang Q, Ao H, Shoblock JR, Lord B, Aluisio L, Fraser I, Nepomuceno D, Neff RA, Welty N, Lovenberg TW, Bonaventure P, Wickenden AD, Letavic MA (2013) Pharmacological characterization of a novel centrally permeable P2X7 receptor antagonist: JNJ-47965567. Br J Pharmacol 170:624–640
Lord B, Aluisio L, Shoblock JR, Neff RA, Varlinskaya EI, Ceusters M, Lovenberg TW, Carruthers N, Bonaventure P, Letavic MA, Deak T, Drinkenburg W, Bhattacharya A (2014) Pharmacology of a novel central nervous system-penetrant P2X7 antagonist JNJ-42253432. J Pharmacol Exp Ther 351:628–641
Financial Disclosures
The authors report potential conflicts of interest: A.B. and W.C.D. are employees of Janssen Research & Development, LLC, of Johnson & Johnson, and are stock-holders in Johnson & Johnson, Inc.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Bhattacharya, A., Drevets, W.C. (2016). Role of Neuro-Immunological Factors in the Pathophysiology of Mood Disorders: Implications for Novel Therapeutics for Treatment Resistant Depression. In: Dantzer, R., Capuron, L. (eds) Inflammation-Associated Depression: Evidence, Mechanisms and Implications. Current Topics in Behavioral Neurosciences, vol 31. Springer, Cham. https://doi.org/10.1007/7854_2016_43
Download citation
DOI: https://doi.org/10.1007/7854_2016_43
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-51151-1
Online ISBN: 978-3-319-51152-8
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)