
Abstract
ZEPATIER® (MK-5172A; elbasvir and grazoprevir, Merck & Co., Inc.) is a fixed-dose combination treatment for individuals with chronic hepatitis C virus (HCV) infection. This novel direct-acting antiviral (DAA) regimen combines elbasvir, a selective inhibitor of the HCV nonstructural protein 5A, and grazoprevir, a reversible competitive inhibitor of the HCV nonstructural protein 3/4A protease. After extensive preclinical testing and evaluation of safety and pharmacokinetics (PK) in healthy volunteers, the efficacy of these agents was evaluated in a systematic and comprehensive clinical development program culminating in phase 3 clinical trials in a broad population of participants with HCV infection, including treatment-naive and treatment-experienced participants, those with chronic kidney disease or inherited blood disorders, and those receiving opioid agonist therapy. These studies led to the approval of the elbasvir/grazoprevir combination therapy for the treatment of people with HCV genotype 1 or genotype 4 infection in the United States, Europe, Canada, and many other countries worldwide.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Overview
ZEPATIER® (MK-5172A; elbasvir and grazoprevir, Merck & Co., Inc.) is a fixed-dose combination of two novel direct-acting antivirals (DAAs) directed at well-validated targets within the hepatitis C virus (HCV): elbasvir (also known as MK-8742), a selective inhibitor of the HCV nonstructural protein 5A (NS5A) [1], and grazoprevir (also known as MK-5172), a novel, orally administered, reversible competitive inhibitor of the HCV nonstructural protein 3/4A (NS3/4A) protease [2]. Both of these novel agents were developed at Merck Research Laboratories through a concerted research effort focused on improving potency across a broad spectrum of HCV genotypes and maintaining potency against many of the viral variants with mutations that confer resistance to earlier-generation agents from these drug classes. After extensive preclinical testing [3,4,5,6,7] and evaluation of safety and pharmacokinetics in healthy volunteers, the efficacy of elbasvir and grazoprevir, both separately and as a fixed-dose combination therapy, was evaluated in a systematic and comprehensive clinical development program.
The objective of the ZEPATIER clinical development program was to develop a well-tolerated, convenient, and simple regimen that would be highly effective in clearing HCV infection and thereby reduce the burden of HCV-related morbidity and mortality across the spectrum of this disease. To meet this objective, the clinical development program evaluated “standard” segments of the HCV-infected population (such as treatment-naive, noncirrhotic individuals) as well as populations with a high unmet medical need (e.g., HCV-infected people with chronic kidney disease [CKD] grades 4/5 on hemodialysis; HCV-infected individuals receiving opioid agonist therapy). Figure 1 presents the entire spectrum of people with HCV infection and displays the diversity of study participants in whom the efficacy of elbasvir/grazoprevir, alone or in combination with other agents (ribavirin, sofosbuvir), was evaluated.

Populations in which the efficacy of elbasvir/grazoprevir, alone or with other agents, has been evaluated. CBP child-bearing potential, CKD chronic kidney disease, DAA direct-acting antiviral agents, HBV hepatitis B virus, HIV human immunodeficiency, IBLD inherited blood disorders, IVDU intravenous drug user, NAFLD non-alcoholic fatty liver disease, NIDDM non-insulin-dependent diabetes mellitus, PEG-IFN pegylated interferon, RBV ribavirin
The following sections describe the clinical trials that supported the licensure of ZEPATIER. Concepts common to all studies are briefly addressed here. In the phase 2 and 3 clinical trials, the primary efficacy end point was sustained virologic response (HCV RNA levels below the assay lower limit of quantitation [LLoQ]) 12 weeks after completion of study medication (SVR12). Plasma HCV RNA levels were measured using the COBAS® AmpliPrep/COBAS® TaqMan® HCV test (version 2.0, Roche Molecular Diagnostics, Branchburg, NJ, USA) with a LLoQ of 25 IU/mL in the phase 2 studies and 15 IU/mL in the phase 3 studies. Determination of HCV genotyping was primarily conducted using VERSANT® HCV genotype assay (line probe assay [LiPA] 2.0) reverse hybridization technology (Innogenetics, Ghent, Belgium) or the Abbott RealTime HCV Genotype II polymerase chain reaction assay (Abbott Park, IL, USA). In all studies, relapse was defined as HCV RNA levels above the LLoQ after having previously achieved HCV RNA below the LLoQ at the end of therapy. Virologic breakthrough was defined as the presence of confirmed on-treatment detectable HCV RNA after a previous HCV RNA level below the LLoQ while on treatment. Elbasvir and grazoprevir were administered as separate, single-entity tablets in the phase 2 and early phase 3 studies and as a fixed-dose combination tablet in the later phase 3 studies. Ribavirin, when used, was administered using weight-based administration at doses of 800–1,400 mg/day. The participant populations varied across the different studies, but in general all participants had chronic HCV infection with a baseline viral load of greater than 10,000 IU/mL. Participants with cirrhosis were enrolled in several studies; however, in all studies addressed in this chapter, these participants had well-compensated liver disease, usually defined as one of the following: liver biopsy consistent with a METAVIR fibrosis score of F4 at any time prior to entry into the study, FibroScan® (Echosens, Waltham, MA) greater than 12.5 kPa within 12 months of study entry, or an aspartate aminotransferase (AST)-to-platelet ratio greater than 2.0 and FibroTest greater than 0.75 within 12 months of study entry. All studies excluded individuals with hepatitis B virus coinfection, evidence of decompensated liver disease (such as the presence or history of ascites, esophageal or gastric variceal bleeding, hepatic encephalopathy, or other signs of advanced liver disease), or evidence of hepatocellular carcinoma. Because people with HIV infection constitute a key segment of the HCV-infected population, some studies enrolled participants coinfected with human immunodeficiency virus (HIV). A summary of virologic outcomes from phase 2 studies is presented in Table 1 [8,9,10,11,12,13,14,15], and a summary of virologic outcomes from phase 3 studies is presented in Table 2 [16,17,18,19,20,21,22,23,24,25,26].
2 Phase 1 Trials
The elbasvir/grazoprevir clinical development program consisted of 58 phase 1 studies in a total of 1,234 healthy male and female volunteers, 139 participants infected with HCV, and 66 non-HCV-infected people with liver or kidney dysfunction who received elbasvir, grazoprevir, or both compounds simultaneously. Key intrinsic and extrinsic factors were evaluated in these populations, and thorough QTc studies were also conducted to rigorously assess the effect of elbasvir and grazoprevir on the QTc interval.
2.1 Elbasvir Monotherapy Proof of Concept Study
Elbasvir was administered as monotherapy to individuals with HCV genotype (GT) 1 and GT3 infection at doses ranging from 5 to 100 mg once daily for 5 days [27]. Participants administered elbasvir had dose-dependent reductions in HCV RNA at all doses compared with those who received placebo. Observed mean maximal viral load reductions on day 5 of dosing exceeded 3 log10 IU/mL at doses of 5 mg or higher in participants with HCV GT1 infection and exceeded 2 log10 IU/mL at doses of 50 mg or higher in those with HCV GT3 infection.
2.2 Grazoprevir Monotherapy Proof of Concept Study
Grazoprevir was administered as a monotherapy to participants with HCV GT1 and GT3 infection at doses ranging from 30 to 800 mg once daily for 7 days [27]. Observed mean maximum viral load reductions exceeding 3 log10 were achieved by day 7 at all doses in participants with HCV GT1 infection and at doses of 400 mg or higher in participants with GT3 infection. In participants with HCV GT1 infection, viral load reduction appeared to plateau at doses between 50 and 800 mg, whereas in those with GT3 infection, a dose-dependent reduction in viral load was observed at doses between 100 and 600 mg.
3 Phase 2 Trials
3.1 Phase 2 Dose-Ranging Trials of Grazoprevir in Combination with Pegylated Interferon and Ribavirin
MK-5172 Protocol 003 was a phase 2 randomized, double-blind, active-controlled, dose-ranging study (NCT01353911) in which treatment-naive participants with HCV GT1 infection were randomized to receive once-daily grazoprevir at doses of 100, 200, 400, or 800 mg or boceprevir (800 mg three times daily), each in combination with pegylated interferon (PEG-IFN) and ribavirin [8]. A high proportion (89–93%) of participants achieved SVR12 at all grazoprevir doses evaluated, with no clear dose-response relationship. However, elevations of alanine aminotransferase (ALT) and/or AST levels were observed late in the course of therapy (i.e., after treatment week 4) among a proportion of participants who received grazoprevir at doses of 200, 400, or 800 mg. Late ALT/AST increases above 2× upper limit of normal (ULN) were observed in up to 23% of participants, and increases above 5× ULN were observed in up to 9% of participants at grazoprevir doses of 400 mg or higher. Given that these events were not observed in participants receiving the 100-mg dose and that doses higher than 100 mg did not increase the proportion who achieved SVR12, the 100-mg dose was selected for further evaluation in subsequent studies of participants with HCV infection. Further PK/safety analyses of the data from this study confirmed that the 100-mg dose had an adequate safety margin, with a population geometric mean (GM) area under the plasma concentration-time curve from time 0–24 h (AUC0–24) that is greater than 14× below that associated with a predicted population rate of late transaminase elevations of 5%.
Because high SVR rates were observed at all doses evaluated in Protocol 003, a second phase 2 dose-ranging study, Protocol 038 (MK-5127 Protocol 038; NCT01710501), was conducted to further define the lower end of the grazoprevir dose–SVR relationship [9]. Protocol 038 was a phase 2, double-blind, dose-ranging study that randomized treatment-naive participants with HCV GT1 infection to receive grazoprevir doses of 25, 50, or 100 mg in combination with PEG-IFN/ribavirin for 12 weeks. This study confirmed the efficacy of the 100-mg dose and demonstrated a dose-response trend at lower doses: the proportion of participants who achieved SVR12 was numerically higher in the group receiving 100 mg (87% [95% confidence interval [CI], 69.3–96.2%]) compared with the group receiving 50 mg (75% [95% CI, 55.1–89.3%]), and efficacy was substantially lower in the group receiving 25 mg (48% [95% CI, 29.4–67.5]).
These results supported the selection of the 100-mg dose of grazoprevir for further evaluation. The choice of the 100-mg dose also offered an advantage in that factors that might result in a decrease in grazoprevir levels, such as drug–demographic, drug–disease, and drug–drug interactions, would be less likely to result in lower efficacy.
3.2 C-WORTHY: Elbasvir and Grazoprevir Among a Broad Population of HCV GT1–Infected Participants
C-WORTHY (MK-5172 Protocol 035; NCT1717326) was a phase 2 multicenter, randomized, parallel-group trial that evaluated grazoprevir plus elbasvir with or without ribavirin in patients with HCV GT1 and GT3 infection [10, 11]. The study was conducted in multiple parts. Part A evaluated elbasvir plus grazoprevir with or without ribavirin administered for 12 weeks in 65 treatment-naive, noncirrhotic participants with GT1 infection. A total of 52 participants were randomized in a 1:1 ratio to two treatment arms (A1 and A2) in which open-label grazoprevir at a dose of 100 mg once daily was administered concomitantly with double-blind elbasvir doses of either 20 or 50 mg once daily, plus twice-daily ribavirin [11]. A third arm (A3) including 13 participants with HCV GT1b infection received a regimen of 100 mg of grazoprevir once daily and 50 mg of elbasvir once daily (without ribavirin). All regimens were administered for 12 weeks, and all participants were followed for an additional 24 weeks after the end of treatment. SVR12 was achieved in more than 95% of participants receiving both the 20- and 50-mg doses of elbasvir, with no apparent dose-response relationship. Because the safety profile was also similar at both dose levels and in vitro studies suggested that the elbasvir exposures associated with the 50-mg dose are more likely to suppress HCV variants containing common NS5A resistance-associated substitutions (RASs) than the 20-mg dose [4, 6], the 50-mg dose was selected for subsequent evaluation. The choice of the 50-mg dose of elbasvir also offered the advantage that factors that might result in decreases in elbasvir levels, such as drug–drug interactions, would be less likely to result in lower efficacy.
Although SVR12 was achieved by more than 95% of treatment-naive, noncirrhotic participants with HCV GT1 infection receiving elbasvir and grazoprevir plus ribavirin for 12 weeks in Part A, it is also well-recognized that the optimal duration of therapy may differ in the presence of disease factors associated with an unfavorable response (e.g., cirrhosis, prior treatment failure). Various alternative treatment durations were therefore explored in Parts B and C of C-WORTHY [10, 11]. The study populations evaluated in these latter parts of the study were divided into two broad categories encompassing easier-to-cure (n = 279) and harder-to-cure patient populations (n = 253).
Easier-to-cure patients included those with favorable disease factors, such as those who were treatment-naive and noncirrhotic [11]. In parts A and B of the C-WORTHY study, participants with HCV GT1a and GT1b infection and disease-favorable characteristics received elbasvir plus grazoprevir, with or without ribavirin, for 8 or 12 weeks. Participants with HIV coinfection were also enrolled in Part B of the study. In treatment-naive, noncirrhotic participants with HCV monoinfection, a 12-week regimen of 50 mg of elbasvir plus 100 mg of grazoprevir administered once daily without ribavirin resulted in SVR12 rates of 98% (43/44) in those with GT1a or GT1b infection. The addition of ribavirin did not increase the proportion of participants who achieved SVR12. In a similar patient population but including those with HIV coinfection, the same 12-week treatment regimens achieved SVR12 rates of 97% (28/29) and 87% (26/30) in participants receiving elbasvir and grazoprevir with or without ribavirin, respectively. The lower SVR12 rate in the ribavirin-free arm was owing to the fact that two patients were lost to follow-up or withdrawn from the trial who had had undetectable HCV RNA at their last visit. An SVR12 rate of 80% (24/30) was achieved in patients with HCV GT1a infection receiving an 8-week regimen of 100 mg of grazoprevir and 50 mg of elbasvir plus ribavirin. This suboptimal response rate was the result of a higher frequency of virologic relapse in the 8-week compared with the 12-week regimen. Conversely, an 8-week regimen of 100 mg of grazoprevir administered with 50 mg of elbasvir, with or without ribavirin, resulted in SVR12 rates of 93 and 94% in patients with HCV GT1b infection in Part C of C-WORTHY. The higher SVR12 rate among patients with HCV GT1b infection compared with those with GT1a infection is consistent with the greater decrease in HCV RNA levels seen in patients with HCV GT1b infection compared with those with GT1a infection following administration of elbasvir as monotherapy [27]. These results are also consistent with in vitro data demonstrating that elbasvir has greater potency against GT1b replicons and that several common mutations that confer resistance to NS5A inhibitors in a GT1a backbone do not cause comparable half-maximal response (EC50) shifts in replicons with a GT1b backbone [6].
Harder-to-cure patients enrolled in Part B of C-WORTHY included those with either cirrhosis, prior PEG-IFN/ribavirin null response, or both [10]. This part of the study included a 2 × 2 factorial evaluation separately for cirrhotic patients and prior PEG-IFN/ribavirin–null responders, which included ribavirin (yes, no) and treatment duration (12 weeks, 18 weeks) as variables. SVR12 was achieved by 97% (28/29) of treatment-naive participants with cirrhosis receiving elbasvir plus grazoprevir for 12 weeks, with no improvement in response when treatment duration was extended from 12 weeks to 18 weeks or by the inclusion of ribavirin. In prior PEG-IFN/ribavirin–null responders with or without cirrhosis, administration of elbasvir plus grazoprevir with or without ribavirin for 12 or 18 weeks resulted in SVR12 in more than 90% of participants. The highest efficacy (SVR12 of 100%; 33/33) was achieved in PEG-IFN/ribavirin–null responders receiving elbasvir plus grazoprevir with ribavirin for 18 weeks, although confidence intervals overlapped across the treatment arms, making it difficult to definitively ascertain the accuracy of the observed differences. To further refine these observations, the efficacy of 12- and 16-week regimens with or without ribavirin was evaluated among PEG-IFN/ribavirin treatment-experienced patients in the phase 3 Protocol 068 C-EDGE Treatment-Experienced study [17] discussed in more detail later in this chapter.
3.3 C-SCAPE: Elbasvir plus Grazoprevir, with or Without Ribavirin, Among Participants with HCV GT2, GT4, GT5, or GT6 Infection
The C-SCAPE study evaluated the efficacy and safety of elbasvir and grazoprevir, with or without ribavirin, in participants with HCV GT2, GT4, GT5, or GT6 infection (MK-5172 protocol 047; NCT 01932762) [12]. This part-randomized, open-label, parallel-group study of treatment-naive, noncirrhotic participants was conducted in two parts. In Part A, 30 treatment-naive, noncirrhotic participants with GT2 infection received elbasvir plus grazoprevir with ribavirin for 12 weeks. In Part B, a further 30 treatment-naive, noncirrhotic participants with GT2 infection received grazoprevir with ribavirin for 12 weeks; and participants with GT4, GT5, or GT6 infection were randomized to receive elbasvir plus grazoprevir with or without ribavirin for 12 weeks.
Among participants with GT2 infection, SVR12 rates were slightly higher in those receiving elbasvir plus grazoprevir with ribavirin compared with participants receiving grazoprevir plus ribavirin (80% [24/30] vs 73% [19/26]). GT2 virions contain naturally occurring variants that encode for either methionine or lysine residues at amino acid 31 of the NS5A protein. Among participants receiving elbasvir and grazoprevir plus ribavirin, SVR12 rates were higher in those with the 31L subtype compared with the 31M subtype (93% [13/14] vs 67% [10/15]), but SVR12 rates were similarly low in participants with 31L and 31M subtypes receiving grazoprevir plus ribavirin (73% [8/11] vs 75% [9/12]). Thus, among participants with the 31M polymorphism, SVR rates were 67% (10/15) when elbasvir/grazoprevir plus ribavirin was administered and 75% (9/12) when grazoprevir plus ribavirin was administered, indicating that elbasvir offers little or no contribution to efficacy in these patients. This is consistent with in vitro studies showing that the potency of elbasvir is reduced by approximately 1,000-fold in replicons containing the 31M compared with the 31L substitution [7].
Treatment with elbasvir and grazoprevir for 12 weeks was highly effective in participants with HCV GT4 infection. Nine of ten participants achieved SVR12, no virologic failures occurred, and only one participant, who discontinued treatment for reasons unrelated to study medication, failed to achieve SVR12.
In participants with GT5 infection, elbasvir plus grazoprevir with ribavirin was more effective than the same regimen without ribavirin. Three of four participants with GT5 infection receiving elbasvir with grazoprevir had virologic failure (two relapsed and one had virologic breakthrough) compared with one of four participants receiving elbasvir plus grazoprevir with ribavirin (SVR12 was 25% vs 75%, respectively). Based on this small number of noncirrhotic participants with GT5 infection, the addition of ribavirin appears important in attaining high rates of SVR.
In contrast, elbasvir with grazoprevir alone was effective in treating HCV infection in noncirrhotic, treatment-naive participants with HCV GT6 infection. Of the four participants treated, three achieved SVR, and one had virologic breakthrough.
Data from this study were used to inform participant selection for the phase 3 clinical development program of elbasvir/grazoprevir. The data supported the inclusion of participants with HCV GT4 or GT6 infection in these studies, but elbasvir/grazoprevir with or without ribavirin was unsatisfactory for participants with HCV GT2 or GT5 infection. Although treatment with elbasvir plus grazoprevir showed efficacy in participants with the GT2 31L variant, it was decided that inclusion of participants with GT2 infection would not be pursued in the phase 3 program because of the subsequent requirement for baseline sequencing to select out those with the 31M variant. Similarly, the low rates of SVR seen in participants with GT5 infection receiving elbasvir with grazoprevir precluded their further inclusion in the phase 3 clinical program.
3.4 C-SALVAGE: Elbasvir/Grazoprevir with Ribavirin Among GT1–Infected Participants Who Failed Prior Treatment with Boceprevir, Telaprevir, or Simeprevir
C-SALVAGE (MK-5172 Protocol 048; NCT2105454) was an open-label, single-arm study of elbasvir/grazoprevir with ribavirin in participants with HCV GT1 infection who had failed a prior regimen of boceprevir, telaprevir, or simeprevir taken concomitantly with PEG-IFN/ribavirin [13, 14]. Of the 79 participants who received study drug, 66 (84%) had a history of virologic failure on a regimen containing a first-generation NS3/4A protease inhibitor; and of the remaining 13 participants, 12 had discontinued prior treatment because of an adverse experience. At baseline, 34 (43.6%) participants harbored NS3 RASs and 8 harbored NS5A RASs. SVR12 was achieved by 76 of 79 (96.2%) participants overall, including 28 of 30 (93.3%) with HCV GT1a infection, 63 of 66 (95.5%) with prior virologic failure, and 32 of 34 (94.1%) of those with cirrhosis. With regard to the impact of baseline RASs, SVR12 was achieved by 43 of 43 (100%) without baseline RASs, 31 of 34 (91.2%) with baseline NS3 RASs, 6 of 8 (75.0%) with baseline NS5A RASs, and 4 of 6 (66.7%) with both baseline NS3 and NS5A RASs.
3.5 C-SWIFT: Short-Duration Treatment with Elbasvir plus Grazoprevir and Sofosbuvir Among GT1– or GT3–Infected Treatment-Naive Participants with or Without Cirrhosis
The objective of the C-SWIFT study (MK-5172 protocol 074; NCT02133131) was to identify the minimum effective treatment duration across multiple genotypes [15]. C-SWIFT was an open-label, single-center trial in treatment-naive participants with chronic HCV GT1 or GT3 infection. All participants received 50 mg of elbasvir and 100 mg of grazoprevir plus sofosbuvir 400 mg for 4–12 weeks; those with GT1 infection who failed therapy were eligible for re-treatment with elbasvir plus grazoprevir with sofosbuvir and ribavirin for 12 weeks.
Rates of SVR12 were 32% (10 of 31) and 87% (26 of 30) in noncirrhotic participants with HCV GT1 infection treated for 4 and 6 weeks, respectively, and 80% (16 of 20) and 81% (17 of 21) in cirrhotic participants with GT1 infection treated for 6 and 8 weeks, respectively. Genotyping of plasma samples taken at the time of virologic failure indicated that in one of the cirrhotic participants with HCV GT1 infection treated for 8 weeks, GT2 infection was detected at the time of virologic failure, and thus, this participant was reclassified as having a reinfection. Twenty-three HCV GT1–infected participants who experienced relapse following initial treatment with elbasvir plus grazoprevir with sofosbuvir were re-treated with elbasvir/grazoprevir plus sofosbuvir and ribavirin for 12 weeks; all achieved SVR12.
Among participants with GT3 infection, SVR12 rates were 93% (14 of 15) and 100% (14 of 14) with 8- and 12-week treatment regimens. The SVR12 rate in cirrhotic participants with GT3 infection was 83% (10 of 12) after 12 weeks of treatment.
4 Phase 3 Trials
4.1 C-EDGE Treatment-Naive: Elbasvir/Grazoprevir in Treatment-Naive Participants with HCV Infection, with or Without Cirrhosis
The C-EDGE Treatment-Naive study (MK-5172 protocol 060; NCT02105467) was a randomized, double-blind, placebo-controlled, parallel-group trial of elbasvir/grazoprevir in treatment-naive cirrhotic and noncirrhotic participants with chronic HCV GT1, GT4, or GT6 infections [16]. To assess safety, participants were randomized 3:1 in a double-blinded fashion to receive either elbasvir (50 mg)/grazoprevir (100 mg) (immediate-treatment group) or a matched placebo for 12 weeks; after completing 12 weeks of randomized treatment and an additional 4-week follow-up period, placebo recipients received open-label elbasvir (50 mg)/grazoprevir (100 mg) (deferred-treatment group) so that all randomized participants would receive active therapy during the study, regardless of their initial treatment group.
Of the 316 participants in the immediate-treatment group, 299 (95%) achieved SVR12. SVR12 rates were 92% (144 of 157) in those with HCV GT1a infection, 99% (129 of 131) in those with GT1b infection, 100% (18 of 18) in those with GT4 infection, and 80% (8 of 10) in those with GT6 infection. SVR12 was achieved in 97% (68 of 70) of cirrhotic and 94% (231 of 246) of noncirrhotic participants. Subgroup analyses did not identify meaningful effects of age, sex, race, ethnicity, or IL28B genotype on treatment outcome. SVR12 was achieved in 100% of participants with baseline HCV RNA levels of 800,000 IU/mL or less compared with 92% of patients with baseline HCV RNA levels of greater than 800,000 IU/mL.
Elbasvir/grazoprevir was generally well tolerated in this study. The safety profile was similar in the elbasvir/grazoprevir and placebo treatment groups and in cirrhotic and noncirrhotic participants receiving elbasvir/grazoprevir. During the immediate-treatment period, drug-related adverse events occurred in 114 (36.1%) and 41 (39.0%) participants in the active elbasvir/grazoprevir and placebo groups, respectively. Serious adverse events during treatment and the first 14 follow-up days were reported in nine (2.8%) and three (2.9%) patients in the active and placebo groups, respectively; none were considered drug-related.
During the immediate-treatment period, treatment was discontinued because of adverse events in three (0.9%) elbasvir/grazoprevir recipients (two participants with elevated aminotransferase levels and one with palpitations and anxiety on treatment day 4) and one (0.9%) placebo recipient (rash on treatment day 2). One cirrhotic and three noncirrhotic elbasvir/grazoprevir recipients (1.3%) developed late elevations of aminotransferase level more than 5× ULN, without an associated increase in bilirubin. Two of these four participants discontinued treatment because of these late aminotransferase elevations at treatment week 8 (one cirrhotic patient) and week 10 (one noncirrhotic patient), as stipulated by protocol. In both patients, aminotransferase elevations resolved rapidly after cessation of study therapy and SVR12 was achieved.
4.2 C-EDGE Treatment-Experienced: Elbasvir/Grazoprevir in Participants with HCV Infection Who Experienced Virologic Failure After Prior Treatment with Pegylated Interferon Alfa and Ribavirin
C-EDGE Treatment-Experienced (MK-5172 protocol 068; NCT02105701) was a randomized, parallel-group, multisite, open-label trial of elbasvir/grazoprevir administered once daily with or without ribavirin for 12 or 16 weeks in participants with HCV GT1, GT4, or GT6 infection who had experienced virologic failure after prior treatment with PR [17]. Participants coinfected with HIV were also eligible for enrollment. In total, 420 participants were randomized in a 1:1:1:1 ratio to treatment with elbasvir (50 mg)/grazoprevir (100 mg) once daily for 12 weeks with or without ribavirin or for 16 weeks with or without ribavirin. Randomization was stratified by the presence or absence of cirrhosis and by prior PEG-IFN/ribavirin treatment response (relapse, partial response, or null response). The investigators and participants were blinded to the assigned treatment duration during the period from randomization through treatment week 12.
SVR12 rates were 92.4% (97/105) in the 12-week elbasvir/grazoprevir arm, 94.2% (98/104) in the 12-week elbasvir/grazoprevir plus ribavirin arm, 92.4% (97/105) in the 16-week elbasvir/grazoprevir arm, and 98.1% (104/106) in the 16-week elbasvir/grazoprevir plus ribavirin arm. Pooling across treatment durations, the difference in SVR12 between the participants who received ribavirin and those who did not was 3.8%. Pooling arms with and without ribavirin, the difference in SVR12 rates between participants who received 16 weeks of treatment and those who received 12 weeks of treatment was 2.0%.
A per-protocol analysis, which focused on virologic failures, was conducted to evaluate the efficacy of elbasvir/grazoprevir among participant subgroups. Across arms, 207 of 218 (95.0%), 143 of 145 (98.6%), 32 of 36 (88.9%), and 5 of 6 (83.3%) participants with GT1a, GT1b, GT14, and GT16 infection, respectively, achieved SVR12. Overall, the SVR12 rates were 93.8% (135 of 144) in participants with cirrhosis and 96.6% (255 of 264) in those without cirrhosis. Across all treatment arms, SVR12 was achieved by 98% (202 of 207) of participants with a baseline viral load of 2,000,000 IU/mL or less and by 94% (188 of 201) of those with a baseline viral load greater than 2,000,000 IU/mL. Among those who received the 12-week regimen, SVR12 rates were highest in participants with HCV GT1b infection (34 of 34 [100%]), those with prior relapse after treatment with PEG-IFN/ribavirin (35 of 35 [100%]), or those with partial response (17 of 18 [94.4%]). Efficacy among those with GT1a infection (55 of 59 [93.2%]) and those with prior null response (45 of 49 [91.8%]) was lower. SVR12 rates were 100% for all participants who received elbasvir/grazoprevir with ribavirin for 16 weeks, including those with HCV GT1a infection and prior null response (20 of 20), participants with baseline NS3 RASs (37 of 37), and those with NS5A baseline RASs (6 of 6).
Across all treatment arms, drug-related adverse events were reported in 56% (235/420) of participants with higher rates in the ribavirin-containing compared with no ribavirin arms (64–76% vs 39–44%). Serious adverse events occurred in 3.3% of patients, with similar frequencies across the four treatment arms. Discontinuations due to adverse events occurred in 1.7% of patients, most often in the treatment arm that received 16 weeks of treatment with elbasvir/grazoprevir plus ribavirin (n = 5). However, none of the discontinuations were attributed to the study drugs. Hemoglobin levels of 9.9 g/dL or less were reported in 31 of 210 (14.8%) participants in the ribavirin-containing arms and no participants (0 of 210) in the ribavirin-free treatment arms. Decreases in hemoglobin levels were managed by dose reductions of ribavirin, and no treatment discontinuations owing to anemia occurred. Four participants (1.0%) had late elevations of ALT/AST above 5× ULN, but these elevations were transient and did not require interruption or discontinuation of treatment with elbasvir/grazoprevir. All ALT elevations returned to baseline after study medication was discontinued, and all participants with an ALT elevation above 5× ULN achieved SVR.
4.3 C-SURFER: Elbasvir/Grazoprevir in HCV GT1–Infected Participants with Advanced Chronic Kidney Disease
C-SURFER was a randomized, parallel-group, multisite, placebo-controlled trial of elbasvir/grazoprevir, administered for 12 weeks without ribavirin in HCV GT1–infected participants with advanced chronic kidney disease (CKD) stages 4 and 5, including those receiving hemodialysis (MK-5172 protocol 052; NCT02092350) [18, 19]. Ribavirin was not included in the regimen, since it is contraindicated in people with advanced CKD. Cirrhotic, noncirrhotic, treatment-naive, and treatment-experienced adults were eligible for enrollment. CKD stage 4 was defined as an estimated glomerular filtration rate (eGFR) of 15–29 mL/min/1.73 m2 and CKD stage 5 as an eGFR less than 15 mL/min/1.73 m2, including dialysis dependence.
Overall, 224 participants were randomized in a 1:1 ratio to receive immediate or deferred treatment with elbasvir (50 mg)/grazoprevir (100 mg). In total, 179 (76.2%) participants were receiving maintenance hemodialysis (including those awaiting renal transplant or with a previous failed kidney transplant who were no longer on immunosuppressant therapy). A total of 111 participants were enrolled in the immediate-treatment group and received elbasvir/grazoprevir for 12 weeks. An additional 113 participants who were enrolled in the deferred-treatment group received placebo for 12 weeks, followed by a 4-week unblinding period, open-label elbasvir/grazoprevir for 12 weeks, and then an additional 24 weeks of follow-up after treatment with study medication was completed. Eleven participants (six on hemodialysis and five not on hemodialysis) were also enrolled in an open-label intensive PK arm and received elbasvir/grazoprevir for 12 weeks while undergoing intensive PK sampling. The deferred-treatment group was used to provide a comparator for safety data collected in the immediate-treatment group, given the substantial comorbidities seen in patients with stage 4–5 CKD. Randomization in this study was stratified by the presence of diabetes (a predictor for serious cardiovascular adverse events that occur at an increased frequency among participants with CKD stages 4–5) and by dialysis dependence.
The primary analysis population was the modified full analysis set (mFAS) population, which excluded participants who failed to complete treatment due to death or early discontinuation for reasons unrelated to their response to the HCV treatment. This population was selected as the primary analysis population in C-SURFER because people with CKD stages 4–5 have a high incidence of major cardiovascular events that may lead to study discontinuation. Any bias that may have been incurred through considering non–drug and non–HCV-related discontinuations as treatment failures is therefore removed.
Of the 122 patients in the immediate-treatment group and intensive PK arms, 6 were excluded from the mFAS population for reasons other than virologic failure (death, lost to follow-up, noncompliance, participant withdrawal, and withdrawal by physician owing to violent behavior). All six participants had an HCV RNA level of less than 15 IU/mL at the time of discontinuation. Of the 116 remaining participants, 115 (99%) achieved SVR12. Relapse occurred in one noncirrhotic participant with HCV GT1b infection and CKD stage 5. High response rates were observed in all subgroups, including hemodialysis and nonhemodialysis, and participants with characteristics historically associated with poor response to HCV therapy. In particular, SVR12 was achieved in 100% (51/51) of African-American participants, 99% (86/87) of participants with the IL28B non-CC genotype, 98% of (40/41) participants with diabetes, and all 6 participants with cirrhosis.
Drug-related adverse events were reported in 38 (34.2%) participants in the immediate-treatment group and 39 (34.5%) of those in the deferred-treatment group during the placebo phase. Serious adverse events were also reported at similar frequencies in both treatment arms (14% vs 17%, respectively), most of which were consistent with the underlying comorbidities and complications within this population. Serious adverse events that occurred in more than one participant receiving elbasvir/grazoprevir in the immediate-treatment group were hypertension and pneumonia (n = 2 each), and none were considered to be drug-related. Treatment discontinuations due to an adverse event occurred in five patients in the deferred-treatment group and none in the immediate-treatment group. Increases in liver transaminase levels during treatment were more common in participants receiving deferred treatment than in those receiving immediate treatment. Increases in ALT and AST levels more than 2.5× baseline in the deferred-treatment group were reported in six (5.3%) and four (4.6%) participants, respectively, compared with one (0.8%) and zero participants in the immediate-treatment group.
Adverse events related to the renal system also occurred at similar frequencies in both treatment groups. During treatment, maintenance dialysis was initiated by two participants in the immediate-treatment group, and renal function in six participants (four in the immediate-treatment group, two in the deferred-treatment group) changed from 15 to 29 mL/min/1.73 m2 at baseline to less than 15 mL/min/1.73 m2 during the study.
4.4 C-EDGE CO-INFECTION: Elbasvir/Grazoprevir in Treatment-Naive, HCV-/HIV-Coinfected Participants with or Without Cirrhosis
The C-EDGE CO-INFECTION study (MK-5172 protocol 061; NCT02105662) was an open-label, multicenter study that evaluated the safety, tolerability, and efficacy of elbasvir (50 mg)/grazoprevir (100 mg) in treatment-naive, HIV-coinfected, and HCV GT1–, GT4–, and GT6–infected participants with or without cirrhosis [20]. A total of 218 participants were enrolled: all were coinfected with HIV-1 and were either naive to antiretroviral therapy or on stable antiretroviral therapy with tenofovir or abacavir and either emtricitabine or lamivudine plus raltegravir, dolutegravir, or rilpivirine for at least 8 weeks before enrollment. Antiretroviral therapy-naive patients had CD4 T-cell counts greater than 500 cells/μL and an HIV RNA viral load of less than 50,000 copies/mL; participants on stable antiretroviral therapy had CD4 T-cell counts greater than 200 cells/μL and undetectable HIV RNA (less than 20 copies/mL). Because of the potential for drug–drug interactions, boosted HIV-1 protease inhibitors or efavirenz are not recommended for use in combination with elbasvir/grazoprevir.
Overall, 210 of the 218 enrolled participants (96%) achieved SVR12. Five participants relapsed: all were noncirrhotic and included four with HCV GT1 infection and one with GT4 infection. Among this small number of relapsed participants, no clear association was observed between any individual patient characteristic and the propensity for relapse. Two additional participants who did not achieve SVR12 were infected with a different HCV genotype during follow-up (one with HCV GT1a and one with GT1b infection at enrollment and both with GT3 infection at follow-up week 12). In the primary analysis, these participants were classified as having relapsed, but sequencing data are consistent with reinfection after treatment. One participant did not achieve SVR12 for a nonvirologic reason.
Two participants who were receiving antiretroviral therapy had transient HIV viremia during the treatment period. Both participants subsequently achieved undetectable HIV RNA with additional compliance education and without a change in antiretroviral regimen. Throughout the trial, there were no notable changes in the CD4 T-cell count or percentage at treatment week 12 or follow-up week 12.
A total of 75 (34%) participants experienced drug-related adverse events, the most common of which were fatigue (13%), headache (12%), and nausea (9%). Six participants experienced serious adverse events, of which four occurred after dosing was complete (pneumonia and generalized seizure during treatment and erysipelas, acute psychosis, ulnar fracture, and spontaneous bacterial peritonitis during follow-up). None of the serious adverse events required discontinuation of study drug, and none were considered to be related to treatment. Two participants had late ALT/AST increases above 5× ULN (one at treatment week 6 and the other at treatment week 10) and both normalized without discontinuation of treatment.
4.5 C-EDGE CO-STAR: Elbasvir and Grazoprevir in HCV-Infected Participants Receiving Opioid Agonist Therapy
The aim of the CO-STAR (Hepatitis C Patients on Opioid Substitution Therapy Antiviral Response) study (MK-5172 protocol 062; NCT02105688) was to assess the efficacy and safety of elbasvir (50 mg)/grazoprevir (100 mg) administered for 12 weeks in persons who inject drugs (PWID) who had HCV GT1, GT4, or GT6 infection and who were receiving opiate agonist therapy [21].
CO-STAR was a randomized, placebo-controlled, double-blind trial. Similar to the C-SURFER and C-EDGE Treatment-Naive studies, C-EDGE CO-STAR had an immediate-treatment arm in which participants received elbasvir/grazoprevir for 12 weeks and a deferred-treatment arm in which participants received placebo for 12 weeks followed by deferred active therapy with elbasvir/grazoprevir for 12 weeks. As in C-SURFER, the deferred-treatment group served as a comparator for safety data collected in the immediate-treatment group, given the substantial comorbidities seen in PWID. Following completion of treatment and a 24-week follow-up period, participants were eligible to enroll in a 3-year observational study to assess the durability of SVR, incidence of HCV reinfection, and drug use behaviors.
SVR12 was achieved by 91.5% (184/201) of participants in the immediate-treatment group and 89.5% (85/95) of those receiving deferred treatment with elbasvir/grazoprevir. Although SVR12 rates were similar in participants with HCV GT1a, GT1b, and GT4 infection in the immediate-treatment group (93.5% [144/154], 93.3% [28/30], and 91.7% [11/12], respectively), it was lower in the few participants with GT6 infection (20% [1/5]). Of the 17 patients who failed to achieve SVR12, 12 had viral recurrence and 5 had nonvirologic failure (discontinuation due to an adverse event [n = 1], an administrative reason [n = 1], or loss to follow-up [n = 3]). Seven of the 12 patients with viral recurrence had findings consistent with relapse (based on GT assessment, sequencing, and phylogenetic analysis), and 5 had signs consistent with probable reinfection. The SVR12 rate in the immediate-treatment group was 94.0% (189 of 201), when participants with probable reinfection were considered to have initially cleared the virus prior to reinfection.
Ongoing drug use during the study did not appear to impact adherence to study medication. Urine drug screen (UDS) was positive for at least one potential drug of abuse at each clinic visit (excluding methadone and buprenorphine) in more than 50% of participants in both the immediate-treatment and deferred-treatment groups, remaining relatively stable throughout treatment. During the same period, 96.5% of participants (192/199) in the immediate-treatment group and 100% of those in the deferred-treatment group during the placebo phase (97/97) were more than 95% adherent.
At follow-up week 24, recurrent viremia was reported in 18 participants (immediate-treatment group, n = 14; deferred-treatment group [elbasvir/grazoprevir], n = 4). Five participants in the immediate-treatment group were considered to have probable reinfection, all with recurrent viremia at follow-up week 8, and one participant in the deferred-treatment group (during the active treatment phase) was considered to have probable reinfection with recurrent viremia at follow-up week 24. In four of six probable reinfections, the HCV GT detected at the time of recurrence differed from that present at baseline, and in all six participants, the virus present at recurrence was from a distinct lineage compared with the virus detected at baseline. Ultradeep sequencing of plasma samples taken at baseline failed to amplify when GT-dependent primers based on the virus present at recurrence were used, indicating that the virus present at recurrence was not present at baseline, that these participants acquired a new virus, and that they did not have a mixed infection at baseline. Of note, in three of the six cases, recurrent viremia was transient, with subsequent samples taken after recurrence having undetectable HCV RNA. Four of the participants with probable reinfection tested positive for opioids other than opiate agonist therapy. From the end of treatment through follow-up week 24, the incidence of reinfection was 4.6 reinfections (CI, 1.7–10.0) per 100 person-years (130.6 person-years of follow-up).
Drug-related adverse events were reported in 41.3% (83/201) of participants in the immediate-treatment group and in 34.0% (34/100) and 26.3% (25/95) of those in the deferred-treatment group during the placebo phase and active treatment phase, respectively. The frequency of serious adverse events (3.5% in the immediate-treatment group and 4% in the deferred-treatment group) and discontinuations due to adverse events (less than 1% in the immediate-treatment group and 1% in the deferred-treatment group) were also low in both arms. One serious adverse event in each treatment group was considered to be drug-related, and one participant in each treatment arm discontinued treatment owing to an adverse event.
Despite the general perception that PWID would not be able to adhere to HCV therapy, this study demonstrated high efficacy and safety coupled with excellent treatment adherence in PWIDs receiving stable opiate agonist therapy despite ongoing drug use among most participants. In particular, the potential impact of HCV reinfection following successful treatment is of considerable clinical and public health interest. High levels of HCV reinfection might undermine any benefit associated with initially successful treatment, from both individual and public health perspectives. Data from the CO-STAR study indicate that HCV reinfection in the early posttreatment period (to 24 weeks) does occur in PWIDs, with six cases of probable HCV reinfection in this study population. The observation that four of six participants with reinfections had positive results on opioid testing during posttreatment follow-up suggests that injection drug use was the probable source of reinfection.
4.6 C-EDGE IBLD: Elbasvir/Grazoprevir in Participants with HCV Infection and Inherited Blood Disorders
Before the introduction of screening of blood donors and blood-derived clotting factors, HCV infection was common among people with inherited blood disorders (IBLDs), including those with hemoglobinopathies such as sickle cell disease and β-thalassemia or clotting factor deficiencies such as hemophilia and von Willebrand disease. With improved medical care, patients with IBLDs are living longer but remain at risk for the significant morbidity and mortality associated with HCV infection.
The C-EDGE IBLD study (MK-5172 protocol 065; NCT02252016) was a randomized, double-blind, phase 3 study of elbasvir (50 mg)/grazoprevir (100 mg) in participants with HCV GT1, GT4, or GT6 infection and an IBLD [22]. The study design incorporated randomization to immediate treatment or deferred treatment, similar to the study design of the previously described phase 3 studies. Participants in the immediate-treatment group received elbasvir/grazoprevir for 12 weeks, and those in the deferred-treatment group received placebo for 12 weeks, followed by a 4-week follow-up period and then elbasvir/grazoprevir for 12 weeks. As in the C-SURFER and C-EDGE CO-STAR studies, the deferred-treatment group was used to provide a comparator for safety data collected in the immediate-treatment group, given the substantial comorbidities seen in patients with IBLDs.
In the immediate-treatment group, 100 of 107 participants (93.5%) achieved SVR12. Among those participants who failed to achieve SVR12, six experienced relapse and one was lost to follow-up. SVR12 rates were 91.5% (43/47), 95.7% (44/46), and 91.7% (11/12) in participants with HCV GT1a, GT1b, or GT4 infection and 100.0% (26 of 26) and 91.4% (74 of 81) in those with and without cirrhosis, respectively. High rates of SVR12 were also achieved regardless of IBLD comorbidities in participants with sickle cell disease (94.7%, 18/19), β-thalassemia (97.6%, 40/41), and hemophilia/von Willebrand disease (89.4%, 42/47).
The safety profile was similar in participants receiving elbasvir/grazoprevir in the immediate-treatment group compared with those receiving placebo in the deferred-treatment group. Drug-related adverse events were reported in 36 (33.6%) participants who received elbasvir/grazoprevir in the immediate-treatment group and 16 (30.8%) of those in the deferred-treatment group during the placebo phase. Three participants in the immediate-treatment group reported serious adverse events: one participant with β-thalassemia and erosive gastritis and hypophosphatemia, one with sickle cell disease with crisis, and one with hemophilia A and rectal hemorrhage (only the serious adverse event of erosive gastritis with hypophosphatemia was considered to be drug-related). Five serious adverse events were related to the underlying blood disorder: sickle cell disease with crisis and rectal hemorrhage in two participants receiving elbasvir/grazoprevir in the immediate-treatment group and sickle cell disease with crisis (n = 2) and anemia in three participants in the deferred-treatment group during the placebo phase. No discontinuations due to adverse events occurred in the immediate-treatment group, and one participant in the deferred-treatment group had increased ALT/AST levels that met the protocol-specified criteria for treatment discontinuation.
4.7 C-EDGE Head-2-Head: Elbasvir/Grazoprevir Versus Sofosbuvir plus Pegylated Interferon and Ribavirin in Participants with HCV Infection
C-EDGE Head-2-Head (MK-5172 protocol 077; NCT02358044) was a randomized, open-label, phase 3 trial comparing the safety and efficacy of elbasvir/grazoprevir with sofosbuvir plus PEG-IFN/ribavirin in treatment-naive and treatment-experienced participants with HCV infection [23]. Two hundred fifty-seven participants with HCV GT1 or GT4 infection were randomized to receive 12 weeks of treatment with elbasvir (50 mg)/grazoprevir (100 mg) (n = 129) or sofosbuvir (400 mg) plus PEG-IFN/ribavirin (n = 128). The primary efficacy objective was SVR12, and the primary safety objective was the proportion of patients experiencing a tier 1 safety event (serious drug-related adverse event, any drug-related adverse event leading to treatment discontinuation, neutrophil count less than 0.75 × 109/L, hemoglobin level of less than 10 g/dL, or any safety event meeting the hepatic transaminase stopping criteria).
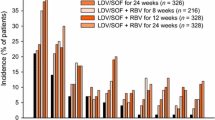
The majority of patients were noncirrhotic (83.1%), were treatment-naive (74.9%), and had HCV GT1b infection (82.0%). SVR12 rates were 99.2% (128/129) and 90.5% (114/126) in the elbasvir/grazoprevir and sofosbuvir plus PEG-IFN/ribavirin groups, respectively. The estimated adjusted difference in SVR12 was 8.8% (95% CI, 3.6–15.3%). Because the lower bound of the one-sided one-sample exact test was greater than −10% and greater than zero, both noninferiority and superiority of elbasvir/grazoprevir compared with sofosbuvir plus PEG-IFN/ribavirin were established. In subgroup analyses, all participants with HCV GT1a infection in both treatment arms achieved SVR12. However, SVR12 rates were higher in participants receiving elbasvir/grazoprevir compared with those receiving sofosbuvir plus PEG-IFN/ribavirin across multiple subgroup populations, including those with HCV GT1b infection (99% [104/105] vs 90% [94/104]), GT4 infection (100% [6/6] vs 60% [3/5]), and cirrhosis (100% [22/22] vs 76% [16/21]), and in prior PEG-IFN/ribavirin-null responders (100% [11/11] vs 50% [7/14]) and partial responders (100% [6/6] vs 88% [7/8]).
Overall, the frequency of tier 1 safety events was lower among patients receiving elbasvir/grazoprevir than those receiving sofosbuvir plus PEG-IFN/ribavirin (0.8% vs 27.8%, between-group difference, 27.0% [95% CI, −35.5% to −19.6%; P < 0.001]). Drug-related adverse events were reported in 90.5% (114/126) of participants receiving sofosbuvir plus PEG-IFN/ribavirin and 24.8% (32/129) of those receiving elbasvir/grazoprevir. Three serious drug-related adverse events occurred in participants receiving sofosbuvir plus PEG-IFN/ribavirin (perirectal abscess, anemia, and heroin abuse); however, the only serious adverse event among participants receiving elbasvir/grazoprevir was a participant with periodontal abscess, which was not considered drug-related. One participant receiving sofosbuvir plus PEG-IFN/ribavirin discontinued treatment at treatment week 1 because of the drug-related adverse events of headache, nausea, myalgia, and decreased appetite. No late ALT/AST events were reported, and no participants discontinued study medication as a result of protocol-specified hepatic laboratory abnormalities.
4.8 C-CORAL: Elbasvir/Grazoprevir in HCV GT1–, GT4–, or GT6–Infected People from the Asia-Pacific Region and Russia
C-CORAL (MK-5172 protocol 067; NCT02251990) was a phase 3, randomized, placebo-controlled, double-blind study conducted in China, Australia, South Korea, Taiwan, Thailand, Vietnam, and Russia [24]. Similar to other phase 3 studies, the study design was again based on randomization of participants to immediate- and deferred-treatment arms, permitting a placebo-controlled comparison of safety events. Treatment-naive, HIV-negative, cirrhotic, and noncirrhotic participants with chronic HCV GT1, GT4, or GT6 infection were randomized 3:1 to receive elbasvir (50 mg)/grazoprevir (100 mg) for 12 weeks (immediate-treatment group) or placebo for 12 weeks followed by deferred treatment with elbasvir/grazoprevir for 12 weeks (deferred-treatment group).
In the immediate-treatment group, 94.2% (344/365) of participants receiving elbasvir/grazoprevir achieved SVR12, and, when combined with participants who received deferred active treatment, the overall SVR12 rate for the total study population was 94.4% (459/486). SVR12 rates were 98.2% (382/389) in participants with HCV GT1b infection and 91.9% (34/37) in those with GT1a infection but were lower at 66.7% (34/51) in those with GT6 infection. The reduced efficacy of elbasvir/grazoprevir in participants with GT6 infection was the main contributing factor to the lower response rates in countries that enrolled a high proportion of people with HCV GT6 infections, such as Vietnam (81.8% [27/33]) and Thailand (57.1% [12/21]). Notably, the population from Thailand included six participants with HCV GT6f infection, of whom only one achieved SVR12 (16.7%). Subgroup analyses revealed that SVR12 rates for the combined immediate-treatment and deferred-treatment populations were consistently high across most major participant subgroups. SVR12 was achieved by 93.3% (84/90) of participants with cirrhosis, 91.1% (205/225) of those with baseline viral load of greater than 2,000,000 IU/mL, and 92.9% (39/42) of those aged ≥65 years. Efficacy was also high in Russian (99.2% [117/118]), Taiwanese (97.6% [83/85]), Chinese (96.7% [146/151]), and South Korean (96.0% [48/50]) participants.
The incidence of drug-related adverse events was similar in the immediate-treatment group and during the placebo phase of deferred treatment (21.4% [78/365] vs 21.1% [26/123]), and drug-related adverse events were also reported by 10.7% (13/121) of participants receiving deferred active treatment. One participant in the immediate-treatment group and the placebo phase of deferred treatment discontinued treatment because of an adverse event. Serious adverse events were reported by five participants in the immediate-treatment group (suicide, contusion, Evans syndrome, lymphoma, and enteritis), by two participants in deferred-treatment group during the placebo phase (influenza and foot fracture), and three participants in the deferred-treatment group during the active treatment phase (ankle fracture, atrial fibrillation, and uterine hemorrhage). Only the serious adverse event of atrial fibrillation was considered drug-related. Late on-treatment ALT/AST elevations of more than 2.0 to 5.0× ULN were reported in 1.4% (5/363) of participants in the immediate-treatment group and in 2.5% (3/122) and 1.7% (2/121) of those in the deferred-treatment group during the placebo phase and active treatment phase, respectively. Late ALT/AST elevations above 5× ULN occurred in four participants (1.1%) in the immediate-treatment group and in three participants receiving elbasvir/grazoprevir in the deferred-treatment group (2.5%). Two of these participants discontinued therapy, and the remainder continued therapy and experienced a gradual reduction in ALT/AST while on treatment, with eventual normalization.
4.9 Japanese Phase 2/3 Study: Elbasvir/Grazoprevir in Japanese Participants with HCV GT1 Infection
Protocol 058 (NCT02203149) was a phase 2/3 trial of the safety and efficacy of elbasvir and grazoprevir in Japanese participants with HCV GT1 infection [25]. The study was conducted in two parts. In Part 1, noncirrhotic participants were randomized 1:1 to receive elbasvir (50 mg) in combination with grazoprevir (50 or 100 mg) once daily for 12 weeks. Participants randomized to receive 100 mg of grazoprevir received two 50-mg tablets once daily, and those randomized to receive 50 mg of grazoprevir received one 50-mg tablet once daily plus a matching placebo tablet. The objective of Part 1 of the study was to confirm that the 100-mg dose (the dose used in other regions) was the appropriate dose for Japanese patients.
The rates of virologic response in Part 1 were similar with the 50- and 100-mg doses of grazoprevir between treatment arms. In all patients, HCV RNA was undetectable by the end of treatment, and at follow-up week 4, all participants in both treatment arms had undetectable HCV RNA. One participant in the grazoprevir 100-mg arm relapsed at follow-up week 12, resulting in SVR12 rates of 100% (31/31) in the grazoprevir 50-mg arm and 96.8% (30/31) in the grazoprevir 100-mg arm. Overall tolerability was similar between the groups, and therefore based on these results, a dose of 100 mg of grazoprevir was selected for use in combination with EBR in Part 2 of the study.
In Part 2, noncirrhotic patients were randomized 3:1 to receive immediate or deferred treatment with elbasvir (50 mg) and grazoprevir (100 mg, as determined in Part 1) for 12 weeks; cirrhotic patients received open-label immediate treatment. SVR12 was achieved by 96.5% (219/227) of participants receiving elbasvir/grazoprevir in the immediate-treatment group. Eight participants failed to achieve SVR12: three discontinued because of nonvirologic failure (adverse event, n = 2; administrative reasons, n = 1) and five relapsed. In a supportive analysis that included treatment-naive participants (and excluded those who discontinued treatment for reasons unrelated to study medication), SVR12 was achieved by 98.6% (142/144) of participants. Subgroup analyses of participants who received elbasvir/grazoprevir in the immediate-treatment group indicated high efficacy across the most important populations. SVR12 was achieved in 99% (122/123) of participants aged younger than 65 years and 93% (70/75) of those aged 65 years and older (7/8 participants who failed to achieve SVR12 were aged ≥65 years). All 5 patients with HCV GT1a infection and 34/35 cirrhotic participants (97.1%) achieved SVR.
In the randomized phase 3 part of the study (Part 2), drug-related adverse events were reported by 58 participants in the immediate-treatment group (25.6%) and 14 (18.9%) in the deferred-treatment group during the placebo phase; serious adverse events were reported by 11 (4.8%) and 1 (1.4%) participants, respectively. In the immediate-treatment group, cataract was the only serious adverse event reported by more than one participant (n = 2), and two drug-related serious adverse events of cerebral infarction and increased ALT/AST occurred. Three participants (13%) in the immediate-treatment group discontinued treatment because of an adverse event (cardiac sarcoidosis, cerebral infarction, and increased ALT/AST level) compared with one participant (1.4%) in the deferred immediate-treatment group who discontinued owing to hepatocellular carcinoma. Four of 227 participants in the immediate-treatment group had late ALT/AST elevations above 5× ULN between treatment weeks 8 and 12. Late ALT/AST level elevations above 5× ULN also occurred in 2 of 34 participants with cirrhosis (5.9%), and in both cases transaminase elevations were accompanied by slight increases in levels of bilirubin and eosinophils but no change in international normalized ratio.
4.10 C-ISLE: Elbasvir/Grazoprevir and Sofosbuvir in Participants with HCV GT3 Infection and Cirrhosis
C-ISLE was an open-label study in participants with HCV GT3 infection and compensated cirrhosis (Protocol MK-5172-083; NCT02601573) [26]. The study population included treatment-naive and treatment-experienced participants and both monoinfected and HCV-/HIV-coinfected individuals. All participants received elbasvir (50 mg)/grazoprevir (100 mg) plus sofosbuvir (400 mg) once daily. Treatment-naive participants were randomized to receive treatment for 8 weeks with ribavirin (8,000–1,400 mg) or 12 weeks without ribavirin; and treatment-experienced participants were randomized to receive elbasvir/grazoprevir plus sofosbuvir with or without ribavirin for 12 weeks or elbasvir/grazoprevir plus sofosbuvir (without ribavirin) for 16 weeks.
One hundred predominantly white (69%) and male (68%) participants were enrolled. Among the treatment-naive participants, SVR12 was achieved by 91% (21/23) treated for 8 weeks with ribavirin and 96% (23/24) of those treated for 12 weeks without ribavirin. Two participants in the 8-week arm relapsed, and one participant in the 12-week arm was lost to follow-up. Among treatment-experienced individuals treated with elbasvir/grazoprevir for 12 weeks, SVR12 was achieved by 94% (17/18) and 100% (17/17) of those treated with and without ribavirin, respectively. The participant who did not achieve SVR in the 12-week elbasvir/grazoprevir plus ribavirin arm withdrew consent after 7 days of therapy. In the 16-week arm, SVR12 was 94% (17/18), with one participant discontinuing treatment because of adverse events of vomiting and cellulitis. Thus, overall, the only two participants with virologic failure in this study were treatment-naive individuals randomized to the 8-week treatment arm.
Adverse events tended to be more common among participants receiving ribavirin compared with those receiving elbasvir/grazoprevir plus sofosbuvir alone, with fatigue (56% [10/18] vs 34% [14/41]), nausea (33% [6/18] vs 15% [6/41]), and headache (61% [11/18] vs 29% [12/41]) all increased in participants who received ribavirin, when considering only participants treated for 12 weeks (ribavirin vs no ribavirin). Drug-related adverse events were reported by 60.9% (14/23) and 83.3% (15/18) of participants receiving a ribavirin-containing regimen for 8 or 12 weeks compared with 43.9% (18/41), and 61.1% (11/18) of those receiving ribavirin-free treatment for 12 or 16 weeks. Five participants reported serious adverse events: three were receiving ribavirin (pneumonia, chest pain, opiate overdose) and two were receiving elbasvir/grazoprevir plus sofosbuvir alone (cellulitis and decreased creatinine, with both considered to be drug-related). Three participants had on-treatment hemoglobin levels of less than 10 g/dL (two were receiving ribavirin and required ribavirin dose reduction), and no ALT/AST elevations above 5× ULN were reported.
5 Integrated Analyses
5.1 Patients with Compensated Cirrhosis
An integrated safety and efficacy analysis was performed that included 402 participants with compensated cirrhosis who received elbasvir/grazoprevir with or without ribavirin for 12, 16, or 18 weeks [28]. Most participants in this retrospective analysis were originally treated within the C-WORTHY, C-SALVAGE, C-EDGE Treatment-Naive, C-EDGE Treatment-Experienced, and C-EDGE CO-INFECTION studies. To be included in this analysis, participants had Child–Pugh class A compensated cirrhosis defined as: liver biopsy consistent with a METAVIR fibrosis score of F4 at any time prior to entry into the study; FibroScan greater than 12.5 kPa within 12 months of starting treatment; or an AST-to-platelet ratio greater than 2.0 and FibroTest greater than 0.75 within 12 months of starting treatment.
Overall, 42% (169/402) of participants in this analysis were treatment-naive and 58% (233/402) were treatment-experienced. The treatment-experienced participants included 34 participants from the C-SALVAGE study who had failed previous treatment with PEG-IFN/ribavirin plus a first-generation protease inhibitor. Overall, 54% had HCV GT1a infection and 39% had HCV GT1b infection. Sixty-four percent of participants had cirrhosis diagnosed through FibroScan, of whom 36% had values greater than 25.0 kPa. In total, 6% of participants had albumin levels of less than 3.5 g/dL and 25% had platelet counts lower than 100,000 cells/μL.
Among the treatment-naive population, SVR12 rates were 97.8% (135/138) in those treated with elbasvir (50 mg)/grazoprevir (100 mg) for 12 weeks and 90.3% (28/31) in those treated with elbasvir/grazoprevir with ribavirin for 16 or 18 weeks. In the treatment-experienced population receiving elbasvir/grazoprevir for 12 weeks, SVR12 rates were 88.9% (48/54), while among treatment-experienced participants treated for 16 or 18 weeks, SVR12 was achieved by 100% (49/49) and 93.9% (46/49) of those receiving elbasvir/grazoprevir with or without ribavirin, respectively. Subgroup analyses showed uniformly high rates of SVR12 across a broad spectrum of participants. SVR12 rates were high regardless of severity of cirrhosis, as indicated by the generally high response rates in patients with albumin levels less than 3.5 g/dL (96%, 24/25), platelets less than 100 × 103 cells/μL (90%, 91/101), and FibroScan values greater than 25.0 kPa (89%, 83/93). All 69 participants with HCV GT1b infection and 10 of 12 (83%) participants with GT4 infection who received elbasvir/grazoprevir for 12 weeks achieved SVR. In treatment-naive and treatment-experienced cirrhotic participants with HCV GT1a infection, SVR rates were 96.1% (73/76) and 88.6% (31/35), respectively.
5.2 HCV GT1a-Infected Patients
In the clinical trials of elbasvir/grazoprevir, rates of virologic failure tended to be higher among participants with HCV GT1a infection compared with those with GT1b infection when treatment with elbasvir (50 mg)/grazoprevir (100 mg) for 12 weeks was administered; however, comparable efficacy was observed across both genotypes in those receiving elbasvir (50 mg)/grazoprevir (100 mg) plus ribavirin for 16 weeks. In an analysis performed by the US Food and Drugs Administration, the presence of baseline NS5A RASs was identified as a predictor of lower efficacy in patients with HCV GT1a infection but not in those with GT1b or GT4 infection receiving elbasvir/grazoprevir for 12 weeks [29]. This analysis revealed that SVR12 rates were ~25% lower in treatment-naive participants with HCV GT1a infection and baseline NS5A RASs compared with those with wild-type virus at baseline. However, all participants with HCV GT1a infection who received elbasvir/grazoprevir with ribavirin for 16 weeks achieved SVR12, regardless of the presence of baseline NS5A RASs. As a result, in the United States testing for variants associated with resistance to EBR is routinely performed prior to the initiation of treatment with elbasvir/grazoprevir in people with HCV GT1a infection. People with HCV GT1a wild-type virus at baseline receive elbasvir/grazoprevir for 12 weeks, and those with RASs at the NS5A positions 28, 30, 31, or 93 receive elbasvir/grazoprevir with ribavirin for 16 weeks [30]. Stratification according to the presence of baseline NS5A RASs assigns approximately 11% of patients with HCV GT1a infection to the extended 16-week elbasvir/grazoprevir plus ribavirin treatment regimen [31].
In the European Union, testing for RASs at baseline is not adopted as standard practice in the treatment of HCV infection, and therefore an alternative approach using baseline viral load is employed to identify people with HCV GT1a infection who would benefit from an extended treatment regimen. European guidelines recommend that patients with HCV GT1a infection and a baseline viral load of 800,000 IU/mL or less receive treatment with elbasvir/grazoprevir for 12 weeks and those with a baseline viral load of more than 800,000 IU/mL receive elbasvir/grazoprevir plus ribavirin for 16 weeks [32]. This recommendation is based on an analysis of 506 participants with HCV GT1a infection who received elbasvir/grazoprevir for 12 weeks in five elbasvir/grazoprevir clinical trials. This analysis showed numerically lower SVR12 rates with increasing viral load strata, and no virologic failures among those who received elbasvir/grazoprevir with ribavirin for 16 weeks [33]. Overall, this approach has a high positive predictive value (98.9% of those with low baseline viral load achieve SVR12) but a very low negative predictive value (only 7.3% of those with high baseline viral load failed to achieve SVR12), resulting in a relatively weak overall accuracy for this approach of 38.9%. In this analysis, 331 of 506 participants were categorized as having high baseline viral load, of whom 307 (93%) achieved SVR12 when treated with elbasvir/grazoprevir for 12 weeks. If those with high baseline viral load had been stratified to receive elbasvir/grazoprevir with ribavirin for 16 weeks based solely on their viral load, 61% (307 of 506) of the population would have been over-treated.
5.3 HCV GT1b-Infected Patients
A retrospective analysis of data from participants with chronic HCV GT1b infection enrolled in 11 phase 2/3 clinical trials was performed [34]. One thousand and seventy participants who received elbasvir (50 mg)/grazoprevir (100 mg) once daily for 12 weeks without ribavirin in 11 phase 2/3 clinical trials were included in this analysis. A high proportion (43%) of those enrolled were from Asian countries, including Japan, Taiwan, and South Korea, 16% were from the United States, and 8% were from Russia. Most (80%) participants were treatment-naive. Comorbidities among the enrolled population included compensated cirrhosis (18%), HIV coinfection (5%), CKD stage 4–5 (10%), and inherited blood disorders (4%). Overall, the SVR12 rate was 97.2% (1,040/1,070). Of the 30 participants who failed to attain SVR12, 15 experienced relapse and 15 had nonvirologic failure. Among participant subgroups, SVR12 rates were high in those with compensated cirrhosis (188/189, 99.5%), HIV coinfection (51/54, 94.4%), and baseline viral load of more than 800,000 IU/mL (705/728, 96.8%). Resistance-associated substitutions at NS5A positions 28, 30, 31, or 93 were present in 21.6% of participants at baseline. SVR12 rates were 99.6% (820/823) in participants without baseline NS5A RASs and 94.7% (215/227) in those with baseline NS5A RASs. A total of 104 participants in this analysis had variants at the Y93 position (primarily Y93H), of whom 99 (95.2%) achieved SVR12. This integrated analysis demonstrates that elbasvir/grazoprevir for 12 weeks represents an effective treatment option for people with HCV GT1b infection, regardless of baseline viral load or the presence of baseline NS5A RASs. Pretreatment resistance testing in individuals with HCV GT1b infection is not required prior to initiation of treatment with elbasvir/grazoprevir for 12 weeks.
5.4 HCV GT4-Infected Patients
One hundred and fifty-five participants with HCV GT4 infection were enrolled in eight international clinical trials across the elbasvir/grazoprevir phase 2/3 clinical program [35]. Most participants in this analysis had HCV GT4a (47%) or 4d (41%) infection, and this was a primarily white (85%) and male (68%) population. Approximately 21% of the population had cirrhosis and 22% had HCV/HIV coinfection. In total, 111/117 (95.0%) of treatment-naive and treatment-experienced participants with GT4 infection achieved SVR12. Of the six participants who failed to achieve SVR12, three experienced relapse, two were lost to follow-up and one participant died. SVR12 rates were comparable in cirrhotic and noncirrhotic participants (91% vs 96%), those with baseline viral load of 800,000 or less and greater than 800,000 IU/mL (94% vs 95%), those with HCV monoinfection and HCV/HIV coinfection (94% vs 97%), and those with HCV GT4a, GT4d, or GT4-other infection (96% vs 94% vs 93%). NS5A RASs at positions 24, 28, 30, 31, 32, 38, 58, 92, or 93 were present in 42 of 114 (37%) participants who received elbasvir/grazoprevir for 12 weeks. SVR12 rate was 97.2% (41/42) in those with baseline NS5A RASs and 97.6% (70/72) in those with no baseline NS5A RASs. In the United States, elbasvir/grazoprevir for 12 weeks is a recommended treatment regimen for people with HCV GT4 infection, regardless of the presence of NS5A RASs or other baseline demographic characteristics.
5.5 Integrated Safety Analysis
A comprehensive integrated analysis of 1,690 participants who received elbasvir/grazoprevir with or without ribavirin in five phase 2 and three phase 3 clinical trials has also been reported (Table 3) [36]. This analysis included 1,033 participants who received elbasvir/grazoprevir alone and 657 who received elbasvir/grazoprevir plus ribavirin. A further 105 participants who received placebo for 12 weeks prior to deferred therapy with elbasvir/grazoprevir in the C-EDGE Treatment-Naive study were also included to provide a direct comparison of safety events. The analysis population included participants with compensated cirrhosis, HIV coinfection, prior treatment failure, and infection with HCV GT1–6.
In participants receiving elbasvir/grazoprevir, the most frequent adverse events were fatigue (71.4%), headache (16.2%), nausea (18.0%), and insomnia (4.1%). Drug-related adverse events were reported in 40.1% of participants, and five participants discontinued treatment because of an adverse event (in three of these cases, the adverse event was considered related to study medication [ALT level increase, n = 2; anxiety, n = 1]). In this integrated population, three deaths occurred among participants receiving elbasvir/grazoprevir (post-appendectomy complication, n = 1; coronary artery disease, n = 1) or elbasvir/grazoprevir plus ribavirin (motor vehicle accident, n = 1). The overall safety profile of elbasvir/grazoprevir for 12 weeks was generally similar to that of placebo, with similar frequencies of drug-related adverse events (40.1% vs 39.0%), serious adverse events (2.4% vs 2.9%), and discontinuations due to adverse events (0.5% vs 1.0%).
During the phase 2/3 trials of elbasvir/grazoprevir, 13 of 1,690 (0.8%) participants experienced elevation of ALT levels above 5× ULN. These events occurred generally at or after treatment week 8 (mean onset, 10 weeks; range, 6–12 weeks) and were typically asymptomatic. Most late ALT elevations resolved with ongoing therapy or after completion of therapy; however, in three participants (0.2%), treatment was discontinued early. The incidence of late ALT elevations was not affected by treatment duration, and the presence of compensated cirrhosis was not a risk factor for late ALT elevations. Clinically significant elevations of bilirubin or changes in liver function were also not observed.
6 Summary
The objective of the elbasvir/grazoprevir clinical development program was to develop a well-tolerated, convenient, and simple regimen highly effective in clearing HCV infection. Following an extensive program of clinical trials encompassing a broad spectrum of participants with HCV infection, elbasvir/grazoprevir was approved for the treatment of people with HCV GT1 and GT4 infection. These studies showed consistently high rates of SVR of more than 90% in participants with HCV GT1 and GT4 infection, together with an acceptable safety profile. In addition, this clinical development program also provided unique insights into the management of several important HCV populations, including those with stage 4–5 CKD in the C-SURFER study and those receiving opioid agonist therapy in the CO-STAR study. Overall, all-oral DAA regimens have revolutionized the treatment of HCV infection, offering the hope of virologic cure to the vast majority of affected individuals. Elbasvir/grazoprevir represents an important all-oral DAA treatment option for many people with HCV infection, combining high rates of sustained virologic response with a well-established safety profile across a broad patient population.
References
Coburn C (2018) Discovery of elbasvir. Top Med Chem. https://doi.org/10.1007/7355_2018_44
McCauley J, Rudd M (2018) The invention of grazoprevir: an HCV NS3/4a protease inhibitor. Top Med Chem. https://doi.org/10.1007/7355_2018_41
Lahser FC, Bystol K, Curry S, McMonagle P, Xia E, Ingravallo P et al (2016) The combination of grazoprevir, a hepatitis C virus (HCV) NS3/4A protease inhibitor, and elbasvir, an HCV NS5A inhibitor, demonstrates a high genetic barrier to resistance in HCV genotype 1a replicons. Antimicrob Agents Chemother 60(5):2954–2964
Harper S, McCauley JA, Rudd MT, Ferrara M, DiFilippo M, Crescenzi B et al (2012) Discovery of MK-5172, a macrocyclic hepatitis C virus NS3/4a protease inhibitor. ACS Med Chem Lett 3(4):332–336
Summa V, Ludmerer SW, McCauley JA, Fandozzi C, Burlein C, Claudio G et al (2012) MK-5172, a selective inhibitor of hepatitis C virus NS3/4a protease with broad activity across genotypes and resistant variants. Antimicrob Agents Chemother 56(8):4161–4167
Liu R, Curry S, McMonagle P, Yeh WW, Ludmerer SW, Jumes PA et al (2015) Susceptibilities of genotype 1a, 1b, and 3 hepatitis C virus variants to the NS5A inhibitor elbasvir. Antimicrob Agents Chemother 59(11):6922–6929
Coburn CA, Meinke PT, Chang W, Fandozzi CM, Graham DJ, Hu B et al (2013) Discovery of MK-8742: an HCV NS5A inhibitor with broad genotype activity. ChemMedChem 8(12):1930–1940
Manns MP, Vierling JM, Bacon BR, Bruno S, Shibolet O, Baruch Y et al (2014) The combination of MK-5172, peginterferon, and ribavirin is effective in treatment-naive patients with hepatitis C virus genotype 1 infection without cirrhosis. Gastroenterology 147(2):366–376
Lagging M, Brown A, Mantry PS, Ramji A, Weilert F, Vierling JM et al (2016) Grazoprevir plus peginterferon and ribavirin in treatment-naive patients with hepatitis C virus genotype 1 infection: a randomized trial. J Viral Hepat 23(2):80–88
Lawitz E, Gane E, Pearlman B, Tam E, Ghesquiere W, Guyader D et al (2015) Efficacy and safety of 12 weeks versus 18 weeks of treatment with grazoprevir (MK-5172) and elbasvir (MK-8742) with or without ribavirin for hepatitis C virus genotype 1 infection in previously untreated patients with cirrhosis and patients with previous null response with or without cirrhosis (C-WORTHY): a randomised, open-label phase 2 trial. Lancet 385:1075–1086
Sulkowski M, Hezode C, Gerstoft J, Vierling JM, Mallolas J, Pol S et al (2015) Efficacy and safety of 8 weeks versus 12 weeks of treatment with grazoprevir (MK-5172) and elbasvir (MK-8742) with or without ribavirin in patients with hepatitis C virus genotype 1 mono-infection and HIV/hepatitis C virus co-infection (C-WORTHY): a randomised, open-label phase 2 trial. Lancet 385(9973):1087–1097
Brown A, Hezode C, Zuckerman E, Foster GR, Zekry A, Roberts SK et al (2018) Efficacy and safety of 12 weeks of elbasvir +/− grazoprevir +/− ribavirin in participants with HCV genotype 2, 4, 5, or 6 infection: the C-SCAPE study. J Viral Hepat 25(5):457–464
Forns X, Gordon SC, Zuckerman E, Lawitz E, Calleja JL, Hofer H et al (2015) Grazoprevir and elbasvir plus ribavirin for chronic HCV genotype-1 infection after failure of combination therapy containing a direct-acting antiviral agent. J Hepatol 63(3):564–572
Buti M, Gordon SC, Zuckerman E, Lawitz E, Calleja JL, Hofer H et al (2016) Grazoprevir, elbasvir, and ribavirin for chronic hepatitis C virus genotype 1 infection after failure of pegylated interferon and ribavirin with an earlier-generation protease inhibitor: final 24-week results from C-SALVAGE. Clin Infect Dis 62(1):32–36
Lawitz E, Poordad F, Gutierrez JA, Wells JT, Landaverde CE, Evans B et al (2017) Short-duration treatment with elbasvir/grazoprevir and sofosbuvir for hepatitis C: a randomized trial. Hepatology 65(2):439–450
Zeuzem S, Ghalib R, Reddy KR, Pockros PJ, Ben AZ, Zhao Y et al (2015) Grazoprevir-elbasvir combination therapy for treatment-naive cirrhotic and noncirrhotic patients with chronic HCV genotype 1, 4, or 6 infection: a randomized trial. Ann Intern Med 163(1):1–13
Kwo P, Gane E, Peng CY, Pearlman B, Vierling JM, Serfaty L et al (2017) Effectiveness of elbasvir and grazoprevir combination, with or without ribavirin, for treatment-experienced patients with chronic hepatitis C infection. Gastroenterology 152(1):164–175
Roth D, Nelson DR, Bruchfeld A, Liapakis A, Silva M, Monsour Jr H et al (2015) Grazoprevir plus elbasvir in treatment-naive and treatment-experienced patients with hepatitis C virus genotype 1 infection and stage 4-5 chronic kidney disease (the C-SURFER study): a combination phase 3 study. Lancet 386(10003):1537–1545
Bruchfeld A, Roth D, Martin P, Nelson DR, Pol S, Londono MC et al (2017) Elbasvir plus grazoprevir in patients with hepatitis C virus infection and stage 4-5 chronic kidney disease: clinical, virological, and health-related quality-of-life outcomes from a phase 3, multicentre, randomised, double-blind, placebo-controlled trial. Lancet Gastroenterol Hepatol 2(8):585–594
Rockstroh JK, Nelson M, Katlama C, Lalezari J, Mallolas J, Bloch M et al (2015) Efficacy and safety of grazoprevir (MK-5172) and elbasvir (MK-8742) in patients with hepatitis C virus and HIV co-infection (C-EDGE CO-INFECTION): a non-randomised, open-label trial. Lancet HIV 2(8):e319–e327
Dore GJ, Altice F, Litwin AH, Dalgard O, Gane EJ, Shibolet O et al (2016) Elbasvir-grazoprevir to treat hepatitis C virus infection in persons receiving opioid agonist therapy: a randomized trial. Ann Intern Med 165(9):625–634
Hezode C, Colombo M, Bourliere M, Spengler U, Ben-Ari Z, Strasser SI et al (2017) Elbasvir/grazoprevir for patients with hepatitis C virus infection and inherited blood disorders: a phase III study. Hepatology 66(3):736–745
Sperl J, Horvath G, Halota W, Ruiz-Tapiador JA, Streinu-Cercel A, Jancoriene L et al (2016) Efficacy and safety of elbasvir/grazoprevir and sofosbuvir/pegylated interferon/ribavirin: a phase III randomized controlled trial. J Hepatol 65(6):1112–1119
George J, Burnevich E, Sheen IS et al (2018) Elbasvir/grazoprevir in Asia/Pacific/Russian participants with chronic hepatitis C virus genotype 1, 4, or 6 infection. Hepatol Comm 2:595–606. https://doi.org/10.1002/hep4.1177/full
Kumada H, Suzuki Y, Karino Y, Chayama K, Kawada N, Okanoue T et al (2017) The combination of elbasvir and grazoprevir for the treatment of chronic HCV infection in Japanese patients: a randomized phase II/III study. J Gastroenterol 52(4):520–533
Foster GR, Agarwal K, Cramp ME, Moreea S, Barclay S, Collier J et al (2018) Elbasvir/grazoprevir and sofosbuvir for HCV genotype 3 infection with compensated cirrhosis: a randomized trial. Hepatology 67:2113. https://doi.org/10.1002/hep.29852
Yeh WW, Fraser IP, Jumes P et al (2018) Antiviral activity, safety, and tolerability of multiple ascending doses of elbasvir or grazoprevir in participants with hepatitis C virus genotype-1 or -3. Clin Ther 40:704–718. https://doi.org/10.1016/j.clinthera.2018.03.002
Jacobson IM, Lawitz E, Kwo PY, Hezode C, Peng CY, Howe AY et al (2017) Safety and efficacy of elbasvir/grazoprevir in patients with hepatitis C virus infection and compensated cirrhosis: an integrated analysis. Gastroenterology 152(6):1372–1382
Boyd SD, Tracy L, Komatsu TE, Harrington PR, Viswanathan P, Murray J et al (2017) US FDA perspective on elbasvir/grazoprevir treatment for patients with chronic hepatitis c virus genotype 1 or 4 infection. Clin Drug Investig 37(4):317–326
Zepatier [package insert] (2018) Merck, Kenilworth
Komatsu TE, Boyd S, Sherwat A, Tracy L, Naeger LK, O’Rear JJ et al (2017) Regulatory analysis of effects of hepatitis C virus NS5A polymorphisms on efficacy of elbasvir and grazoprevir. Gastroenterology 152(3):586–597
European Association for the Study of the Liver (2017) EASL recommendations on treatment of hepatitis C 2016. J Hepatol 66(1):153–194
European Medicines Agency (2016) Zepatier EMA CHMP assessment report. Contract No.: EMA/419807/2016, European Medicines Agency, London. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/004126/WC500211237.pdf
Zeuzem S, Serfaty L, Vierling J, Cheng W, George J, Sperl J et al (2018) The safety and efficacy of elbasvir and grazoprevir in participants with hepatitis C virus genotype 1b infection. J Gastroenterol 53:679. https://doi.org/10.1007/s00535-018-1429-3
Asselah T, Reesink H, Gerstoft J, de Ledinghen V, Pockros PJ, Robertson M et al (2018) Efficacy of elbasvir and grazoprevir in participants with hepatitis C virus genotype 4 infection: a pooled analysis. Liver Int. https://doi.org/10.1111/liv.13727
Dusheiko GM, Manns MP, Vierling JM, Reddy KR, Sulkowski MS, Kwo PY (2015) Safety and tolerability of grazoprevir/elbasvir in patients with chronic hepatitis C: integrated analysis of phase 2–3 trials [Abstract 712]. Hepatology 62(Suppl S1):562A
Acknowledgments
We extend our gratitude to the participants, their families, investigators, and site personnel who participated in the elbasvir/grazoprevir clinical trials. Medical writing and editorial assistance were provided by Tim Ibbotson, PhD, of ApotheCom, Yardley, PA, and funded by Merck & Co., Inc., Kenilworth, NJ, USA.
Compliance with Ethical Standards
Funding Funding for the studies described in this book chapter was provided by Merck & Co., Inc., Kenilworth, NJ, USA.
Conflict of Interest Drs Robertson and Barr are employees of, and hold stock in, Merck & Co., Inc., Kenilworth, NJ, USA.
Ethical Approval
All studies were carried out in accordance with the Declaration of Helsinki, current guidelines on Good Clinical Practices and local ethical and legal requirements. For each study, independent institutional review boards or ethics committees reviewed and approved the protocol and applicable amendments.
Informed Consent
In all studies, all participants gave written informed consent.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Robertson, M.N., Barr, E. (2019). Development of ZEPATIER® . In: Sofia, M. (eds) HCV: The Journey from Discovery to a Cure. Topics in Medicinal Chemistry, vol 32. Springer, Cham. https://doi.org/10.1007/7355_2018_54
Download citation
DOI: https://doi.org/10.1007/7355_2018_54
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-28399-5
Online ISBN: 978-3-030-28400-8
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)