
Abstract
The single-tablet regimen of sofosbuvir (SOF), an HCV nucleotide analog NS5B polymerase inhibitor; velpatasvir (VEL), an HCV NS5A inhibitor; and voxilaprevir (VOX), an NS3/4 protease inhibitor, provides a highly efficacious, safe, and salvage regimen for patients with genotype 1 to 6 HCV infection with and without compensated cirrhosis who were previously unsuccessfully treated with direct-acting antivirals (DAAs). The clinical development program for SOF/VEL/VOX focused on generating safety and efficacy data in DAA-experienced patients without retreatment options as well as assessing the possibility of shortening treatment duration for DAA-naive patients. The Phase 3 studies enrolled and treated over 1,000 genotype 1–6 HCV-infected patients with SOF/VEL/VOX. In DAA-experienced patients treated with 12 weeks of SOF/VEL/VOX, the overall SVR rate was 97%, and high SVR rates were observed across all genotypes irrespective of prior DAA regimen, cirrhosis status, or the presence of baseline resistance-associated substitutions (RASs), supporting its use as a retreatment regimen. In DAA-naive patients treated with 8 weeks of SOF/VEL/VOX, the overall SVR rate was 95% making it an alternative treatment option for regions in which a shorter duration is of particular interest.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
1 Introduction
In 2014, at the time initiation of the clinical development program for VOX, there was much activity in the development of DAA combinations for HCV treatment. However, these combinations were limited to NS5A inhibitors with sofosbuvir or with protease inhibitors. From the outset of the clinical program, the plan for VOX was to develop it with SOF and VEL in a pangenotypic three-DAA combination, with the goal of providing a salvage therapy for the most difficult-to-cure patients who failed prior highly effective DAA therapies and also, potentially, to shorten treatment duration for the relatively easy-to-cure DAA-naive patients. At the time of VOX Phase 1 initiation, the marketed protease inhibitors were only approved for the treatment of HCV genotype 1 and associated with the potential to cause liver injury. Voxilaprevir was specifically designed as a next-generation NS3/4A protease inhibitor with potent antiviral activity against HCV genotypes 1–6, and an improved resistance profile when compared with earlier approved protease inhibitors. In addition, VOX was specifically designed to minimize the potential for drug-induced liver injury (see [1]).
2 Phase 1 Studies
Voxilaprevir was initially characterized as an individual agent in healthy patients in 11 Phase 1 studies. The information from 27 other Phase 1 studies conducted during the initial development of SOF and VEL were supportive of the clinical pharmacology of SOF/VEL/VOX (see [2]). Further, seven Phase 1 studies were performed with SOF/VEL/VOX in a fixed-dose combination, specifically in scenarios in which SOF, VEL, or VOX was a perpetrator of a potential drug-drug interaction or in which using the three drugs in combination was particularly clinically important.
The median peak plasma concentration of VOX was observed 4 h postdose. Voxilaprevir is >99% bound to human plasma proteins and is primarily a substrate of CYP3A4 with slow turnover. Following a single dose of labeled VOX, the majority (approximately 91%) of radioactivity in plasma was parent drug. Biliary excretion of parent drug was determined as the major route of elimination for VOX. The median terminal half-life of VOX following administration of SOF/VEL/VOX was approximately 33 h.
No clinically relevant differences in VOX pharmacokinetics were observed between healthy patients and patients with severe renal impairment. No dose adjustment of SOF/VEL/VOX is warranted for patients with mild or moderate renal impairment. This recommendation for use is guided by its most restrictive component, SOF, and the increased exposure of its major metabolite GS-331007 in renal impairment. Relative to patients with normal hepatic function, the VOX AUCinf values were 299 and 500% higher in patients with moderate and severe hepatic impairment, respectively. Population pharmacokinetic analysis in patients with HCV infection indicated that patients with compensated cirrhosis had 73% higher VOX exposure than those without cirrhosis. No dose adjustment of SOF/VEL/VOX is therefore required for patients with compensated cirrhosis; SOF/VEL/VOX has not been evaluated in patients with decompensated cirrhosis and is not recommended in this population.
Sofosbuvir, VEL, and VOX are substrates of drug transporters P-glycoprotein (P-gp) and breast cancer resistance protein (BCRP), while GS-331007 is not (Table 1). Velpatasvir is poorly transported by OATP1B1 and OATP1B3. Voxilaprevir is a substrate of OATP1B1 and OATP1B3. Drugs that are potent inducers of P-gp and/or moderate to potent inducers of CYP2B6, CYP2C8, or CYP3A4 will reduce plasma concentrations of SOF, VEL, and/or VOX, and coadministration of these agents with SOF/VEL/VOX, similar to SOF/VEL, is not recommended. As a decrease in plasma concentration of efavirenz is a result of an interaction with VEL, coadministration of efavirenz is not recommended with SOF/VEL/VOX, again similar to SOF/VEL. However, the addition of VOX to the fixed-dose combination results in a more restrictive drug-drug interaction profile of SOF/VEL/VOX compared to SOF/VEL. With SOF/VEL/VOX, statin exposure is expected to be higher than with SOF/VEL resulting in more restrictions (rosuvastatin is not recommended, and pravastatin dose is not to exceed 40 mg). As the plasma concentration of VOX is increased by OATP inhibitors, coadministration of potent OATP inhibitors, such as cyclosporine A, atazanavir, and lopinavir, with SOF/VEL/VOX is not recommended. Acid-reducing agents do not impact the absorption of VOX, and the dosing recommendations for SOF/VEL/VOX with acid-reducing agents are dependent on the effect on the pH-dependent absorption of VEL. Because the VEL concentration is higher with SOF/VEL/VOX than with SOF/VEL and because SOF/VEL/VOX is always dosed with food, the concomitant use of SOF/VEL/VOX with proton pump inhibitors is less restrictive.
When SOF/VEL/VOX or its components taken together were administered with food, SOF AUCinf was 64–144% higher; VEL AUCinf was 40–166% higher; and VOX AUCinf was 112–435% higher compared with the exposure under fasted conditions. The increase in VOX exposure when administered with food is the net effect of multiple factors, including increased solubility of VOX and mitigation of the drug-drug interaction in which VOX exposure is decreased by VEL (likely via inhibition of the intestinal uptake transporter OATP2B1). Based on these data, SOF/VEL/VOX was administered with food in the Phase 1b, Phase 2, and Phase 3 studies; collective safety, efficacy, and pharmacokinetic data support administration of SOF/VEL/VOX with food.
3 Phase 1b Study
The pharmacokinetic/pharmacodynamic relationship for antiviral activity was evaluated in a Phase 1b study, GS-US-338-1121, of VOX in patients with HCV infection [3]. The study was double-blind, multicenter, randomized, and placebo-controlled and had an adaptive design to allow testing in a fasted or fed state. In the first completed five cohorts, patients with HCV genotype 1a, HCV genotype 2, and HCV genotype 3 received double-blinded VOX (50, 100, or 300 mg for patients with HCV genotype 1a and 3 and 100 mg for patients with HCV genotype 2) or placebo once daily under fasting conditions for 3 days; VOX 100 mg was administered once daily for 3 days under fasting conditions to patients with HCV genotype 1b and HCV genotype 4.
Of the 67 patients who received treatment, 65 patients completed 10 days of follow-up. Of the two patients who discontinued prior to day 10 of the study, one with genotype 3 infection withdrew consent following treatment with 1 dose of VOX 100 mg, and a second patient with genotype 1a infection treated with VOX 300 mg was lost to follow-up after completion of study treatment. The mean age of study participants was 49 years, and most patients included in this study were male (70%) and white (69%). The viral RNA load was comparable across treatment groups, and the mean viral RNA load at baseline was 6.2 log10 IU/mL. Overall, 11 patients (16%) experienced adverse events, 9 of whom were dosed with VOX (9 of 59; 15%) and 2 of whom received placebo (2 of 8; 25%). No serious adverse events, adverse events leading to study drug discontinuation, or deaths occurred during the study. All adverse events were mild or moderate in severity. The most common adverse events were diarrhea, occurring in 5% (3 of 59) of patients receiving VOX and in 13% (1 of 8) of patients receiving placebo, and headache, occurring in 2% (1 of 59) of patients receiving VOX and in 25% (2 of 8) of patients treated with placebo. The incidence of adverse events was not correlated with the dose of the study drug. There were no clinically significant changes in laboratory abnormalities, vital signs, physical exam findings, or ECGs. Voxilaprevir exhibited linear pharmacokinetics and was associated with a median half-life of 29–42 h, supporting once-daily dosing.
Administration of VOX daily for 3 days resulted in a rapid decline in HCV RNA from pretreatment levels at all doses and across all genotypes, except among patients with genotype 3a infection who received VOX 50 mg (Fig. 1). Following treatment with 100 mg of VOX, median maximum decline in all groups was >3 log10 IU/mL: the median maximum HCV RNA reduction was 4.5 log10 IU/mL for patients with HCV genotype 1a, 3.9 log10 IU/mL for patients with HCV genotype 1b infection, 3.6 log10 IU/mL for patients with HCV genotype 2, 3.6 log10 IU/mL for patients with HCV genotype 3a, and 4.1 log10 IU/mL for patients with HCV genotype 4. Patients with HCV genotype 3 receiving VOX 50 mg or 100 mg for 3 days had more rapid virologic rebound after treatment than patients with other HCV genotypes. The presence of NS3 RASs at baseline had no impact on response to 3 days of monotherapy with VOX [4].

Median change from baseline HCV RNA over time in patients with genotypes 1–4 HCV infection following administration of VOX (GS-9857) at 0 (day 1), 24 (day 2), and 48 (day 3) hours. (a) Genotype 1a. (b) Genotype 1b. (c) Genotype 2. (d) Genotype 3. (e) Genotype 4 [3]
The exposure-response relationship with VOX dose in patients with genotype 3 infection could be adequately described using a simple maximal anti-HCV activity (Emax) model that used AUC0–24 on day 3 of treatment. Using PK and antiviral response data following VOX monotherapy and the known increase in VOX exposure with food, Emax modeling predicted VOX exposures at a 100 mg dose when administered as SOF/VEL/VOX with food would achieve near maximal (≥90%) antiviral effect.
Based on the safety, pharmacokinetic, and antiviral activity, the 100 mg dose of VOX with food was selected to move forward in combination with SOF 400 mg and VEL 100 mg in Phase 2 trials with HCV-infected patients.
4 Phase 2 Studies
Based on the potent pangenotypic antiviral activity, improved coverage of clinically important NS3 RASs, and high barrier to resistance (see [1]), VOX was an excellent candidate to pair with SOF and VEL. At the time of Phase 2 initiation of SOF/VEL/VOX clinical development, Phase 3 studies had already demonstrated that the combination of SOF 400 mg and VEL 100 mg administered for 12 weeks was well tolerated and resulted in high SVR rates across all HCV genotypes (see [2]). The goal of adding the third DAA to a fixed-dose combination was to address the two largest remaining questions in the field: How should patients who fail first-line DAA treatment be retreated? What is the shortest duration possible for DAA-based initial HCV treatment?
The primary goal for the SOF/VEL/VOX Phase 2 program was to determine the appropriate duration of treatment based on patient characteristics. The efficacy and safety of SOF/VEL plus VOX or SOF/VEL/VOX for 4–12 weeks of treatment were evaluated in four Phase 2 studies in DAA-experienced and DAA-naive patients with HCV infection with or without cirrhosis, described separately below. Across the SOF/VEL/VOX clinical development program, cirrhosis was determined similarly to other SOF-containing protocols (liver biopsy, FibroTest score >0.75 and AST-platelet ratio index >2, or transient elastography result >12.5 kPa), and key exclusion criteria included hepatic decompensation and coinfection with HBV or HIV.
4.1 Study GS-US-337-1468 (LEPTON)
Study GS-US-337-1468 (LEPTON) was a Phase 2, open-label study conducted at two sites in New Zealand [5]. It enrolled 161 treatment-naive and previously treated patients with genotype 1 and 3 HCV infection between September 2014 and March 2015. Patients were enrolled into one of ten groups, and the duration of therapy with SOF/VEL plus VOX was determined by baseline patient characteristics: 4 or 6 weeks for treatment-naive patients without cirrhosis, 6 weeks for treatment-naive patients with cirrhosis, and 6 or 8 weeks for treatment-experienced patients with or without cirrhosis. Table 2 shows the demographic, disease, and baseline characteristics by treatment group. The DAA-experienced patients with genotype 1 HCV infection had failed prior treatment with DAAs from two classes (protease inhibitor plus non-nucleoside NS5B polymerase inhibitor for six patients, NS5A inhibitor plus NS5B nucleotide polymerase inhibitor for four patients). All patients completed the assigned treatment.
The virologic outcomes are shown in Table 3. No patients experienced virologic breakthrough, and one patient with genotype 3 HCV infection and cirrhosis was lost to follow-up. Among treatment-naive patients with genotype 1 infection without cirrhosis, SVR12 was achieved in 4 of 15 (27%) receiving SOF/VEL plus VOX for 4 weeks and in 14 of 15 (93%) receiving SOF/VEL plus VOX for 6 weeks. Of the 15 treatment-naive patients with genotype 1 HCV and cirrhosis receiving 6 weeks of treatment, 13 (87%) achieved SVR12. Six weeks of treatment led to SVR12 in 20 of 30 (67%) patients with and without cirrhosis who failed previous treatment that contained two DAAs. Eight weeks of SOF/VEL plus VOX led to SVR12 in 17 of 17 (100%) patients with cirrhosis and with genotype 1 HCV who had previously been treated with pegylated interferon plus ribavirin and in 25 of 28 (89%) patients with or without cirrhosis and with genotype 1 HCV who failed a previous protease inhibitor-containing regimen. Among treatment-naive patients with genotype 3 HCV and cirrhosis, SVR12 was achieved by 15 of 18 (83%) receiving 6 weeks of treatment. Eight weeks of SOF/VEL plus VOX led to SVR12 in 19 of 19 (100%) patients with cirrhosis and with genotype 3 HCV who had previously been treated with pegylated interferon plus ribavirin and in 4 of 4 (100%) patients with or without cirrhosis and with genotype 3 who failed a previous DAA-containing regimen.
Overall, RASs forming at least 15% of the viral population in at least one of the three target genes – NS3, NS5A, and NS5B – were detected at baseline in 38% of patients. The SVR12 rate in patients with RASs was 85%, which was similar to the SVR12 rate of 84% in patients without RASs suggesting that baseline resistance did not impact treatment outcome. No treatment-emergent NS3, NS5A, or NS5B RASs were detected at the 15% assay cutoff in the 28 patients who relapsed, consistent with a high barrier to resistance of the regimen.
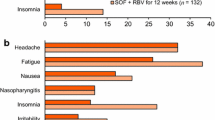
Adverse events were reported by 80% (128 of 161) of patients overall, and the most common adverse events were headache (23%), nausea (21%), fatigue (17%), and diarrhea (12%). All adverse events were mild or moderate. There were three serious adverse events, none of which were reported by more than one patient and none of which were related to study drug. There were no clinically meaningful changes in ALT or total bilirubin values during treatment.
In LEPTON, the three drug combination of SOF/VEL plus VOX demonstrated similar efficacy in patients with genotype 1 or genotype 3 and was well tolerated. Although the 6-week treatment duration achieved SVR rates >90% in treatment-naive genotype 1 patients without cirrhosis, results in more difficult-to-treat patients with cirrhosis and DAA-experienced patients were suboptimal. These data supported the further exploration of the 3-DAA regimen for 6 and 8 weeks of DAA-naive patients and 12 weeks in DAA-experienced patients in other larger Phase 2 studies.
4.2 Studies GS-US-367-1168 and GS-US-367-1169
Study GS-US-367-1168 was a Phase 2, multicenter, open-label study to assess the safety, tolerability, and efficacy of SOF/VEL plus VOX in treatment-naive and DAA-experienced patients with genotype 1 HCV infection [6]. The study enrolled subjects between March and September 2015 at 34 sites in the United States and New Zealand. Treatment-naive patients without cirrhosis received SOF/VEL plus VOX for 6 or 8 weeks, treatment-naive patients with cirrhosis received SOF/VEL plus VOX with or with ribavirin for 8 weeks, and DAA-experienced patients who previously failed an NS5A inhibitor or at least two classes of DAA with or without cirrhosis received SOF/VEL plus VOX for 12 weeks. Study GS-US-342-1169 was a Phase 2, multicenter, open-label study that assessed the safety, tolerability, and efficacy of SOF/VEL plus VOX treatment-naive and treatment-experienced patients with genotype 2, 3, 4, 5, or 6 HCV infection (i.e., non-genotype 1; [7]). Treatment-naive patients without cirrhosis received SOF/VEL plus VOX for 6 weeks, treatment-naive patients with cirrhosis received SOF/VEL plus VOX for 8 weeks, and treatment-experienced patients who previously failed a DAA-based or interferon-based regimen with or without cirrhosis received SOF/VEL plus VOX for 12 weeks. Study GS-US-342-1168 and study GS-US-342-1169 enrolled subjects between March and September 2015 at the same 34 sites in the United States and New Zealand. Based on the similar objectives and design of these two studies, the data will be presented together by prior treatment experience.
The studies enrolled 128 treatment-experienced patients, all but one of whom completed 12 weeks of SOF/VEL plus VOX for 12 weeks. There were 63 patients infected with HCV genotype 1 in GS-US-367-1168 and 65 patients infected with HCV genotype 2, 3, 4, or 6 in GS-US-367-1169, of whom 48% had cirrhosis (Table 4). Among the DAA-experienced patients with genotype 1 infection, 46% previously received a NS5A inhibitor (ledipasvir, 7 patients; daclatasvir, 11 patients; other, 11 patients), and 54% previously received a protease inhibitor and a NS5B polymerase inhibitor (simeprevir with sofosbuvir, 25 patients; other, 9 patients). Among the treatment-experienced patients with non-genotype 1 infection, 42% previously received an interferon-based treatment (27 patients), and 58% had been previously treated with a DAA (38 patients), most of whom with SOF. At baseline, 59% had RASs in NS3, NS5A, and/or NS5B using a 15% assay cutoff. Overall, 99% of treatment-experienced patients (127 of 128) who were treated with SOF/VEL plus VOX in the two studies achieved SVR12 (Fig. 2). The only treatment-experienced patient who relapsed had genotype 3 HCV infection and cirrhosis and had previously been treated with SOF and ribavirin. This patient had the NS5A RAS Y93H at baseline and at relapse and had treatment-emergent Q80R which does not confer in vitro resistance to VOX.

SVR12 rates by treatment and duration in study GS-US-367-1168 and study GS-US-367-1169
The studies enrolled 197 treatment-naive patients; 3 patients discontinued treatment due to adverse events. There were 135 patients infected with genotype 1 HCV and 62 patients infected with non-genotype 1 HCV, and 48% had cirrhosis (Table 4). At baseline, 50% had RASs in NS3, NS5A, and/or NS5B using a 15% assay cutoff. For the treatment-naive patients, the SVR12 rate among those without cirrhosis who received 6 weeks of SOF/VEL plus VOX was 79% (53 of 67 with 14 relapsers; Fig. 2). Overall, 96% (95 of 99) of patients who received 8 weeks of SOF/VEL plus VOX achieved SVR12, 100% (36 of 36 patients) of those without cirrhosis and 94% of those with cirrhosis (59 of 63 patients). Of the four patients who relapsed in this group, all had cirrhosis, two had genotype 1, one had genotype 3, and one had genotype 4. In the group of patients with genotype 1 infection and cirrhosis who received SOF/VEL plus VOX plus RBV for 8 weeks, the SVR12 rate was 81% (25 of 31patients with 6 relapsers). Of the 24 treatment-naive patients who relapsed, none had treatment-emergent RASs at the time of virologic failure using a 15% assay cutoff.
SOF/VEL plus VOX, with and without RBV, was generally safe and well tolerated. Across treatment groups, most patients had at least one adverse event (68%, 219 of 325 patients). The majority of adverse events were Grade 1 or Grade 2 in severity. The most common adverse events were headache (23%, 75 patients), nausea (18%, 58 patients), fatigue (18%, 58 patients), and diarrhea (15%, 50 patients). Patients in the ribavirin-containing group had higher rates of fatigue (32%, 10 of 31 patients), anemia (23%, 7 of 31 patients), and decreased hemoglobin (13%, 3 of 31 patients) than patients in the other groups.
These two 12-week Phase 2 multicenter studies of the three-DAA regimen containing SOF, VEL, and VOX led to high SVR12 rates across HCV genotypes in treatment-experienced patients with and without cirrhosis, including those with DAA experience with NS5A and/or NS5B inhibitors. Among patients who were treatment naive, 8 weeks of SOF/VEL plus VOX was highly effective in patients with HCV genotypes 1–6 with and without cirrhosis. Together, these data supported the 12- and 8-week durations of SOF/VEL/VOX treatment in the DAA-experienced and DAA-naive populations, respectively, in the Phase 3 program.
4.3 Study GS-US-367-1871 (TRILOGY-3)
Study GS-US-367-1871 (TRILOGY-3) was a Phase 2, open-label study conducted at one site in Texas [8]. This study enrolled patients with chronic HCV genotype 1 infection who were previously treated with a DAA and randomized them to receive SOF/VEL/VOX with or without ribavirin stratified by the presence of cirrhosis and prior exposure to an NS5A inhibitor. This study enrolled patients between August and October 2015 beginning after the multicenter Phase 2 studies GS-US-367-1168 and GS-US-367-1169. In advance of the start of this study, the results from the Phase 1 bioavailability study were available which demonstrated that the pharmacokinetics of the SOF/VEL/VOX fixed-dose combination (400/100/100 mg) tablets was similar to that of the coadministered SOF/VEL (400/100 mg) and VOX single-agent (100-mg) tablets, and GS-US-367-1871 was the first clinical study to use the SOF/VEL/VOX fixed-dose combination for treatment of patients infected with chronic HCV.
Of the 49 patients enrolled, 24 received SOF/VEL/VOX without ribavirin, and 25 received SOF/VEL/VOX with ribavirin. All patients completed SOF/VEL/VOX; ribavirin dosing was discontinued by three patients and interrupted or modified by three patients. Table 5 shows the demographic and baseline characteristics by study treatment. Overall, 51% of patients had cirrhosis, 41% had failed prior treatment with an NS5A inhibitor, and 73% had RASs in NS3, NS5A, and/or NS5B. Of the patients who received SOF/VEL/VOX without and with ribavirin, 100% and 96% achieved SVR12, respectively (Table 6). The one patient who relapsed in the SOF/VEL/VOX plus ribavirin treatment group had genotype 1a infection, cirrhosis, previously failed LDV/SOF treatment and treatment-emergent NS3 (V36M, Q41R, D168G) and NS5A (M28T, Q30R), and RASs detected at relapse using a 15% assay cutoff.
Treatment-emergent adverse events were reported by 46% of patients receiving SOF/VEL/VOX and 60% of patients receiving SOF/VEL/VOX plus ribavirin. The most common adverse events were diarrhea (13%) in patients receiving SOF/VEL/VOX. Patients receiving SOF/VEL/VOX plus ribavirin had more ribavirin-associated toxicities: the most common adverse events were fatigue (36%) and anemia (16%), and the most common laboratory abnormality was decreased hemoglobin (16%).
In TRILOGY-3, a small, single-site Phase 2 study, 12 weeks of treatment with a fixed-dose combination of SOF/VEL/VOX was effective and well tolerated among patients with genotype 1 infection who had previously failed a DAA-based regimen. The addition of ribavirin did not improve efficacy but did contribute to the safety profile. Based on these results, ribavirin was not further assessed in the SOF/VEL/VOX Phase 3 program.
5 Phase 3 Studies
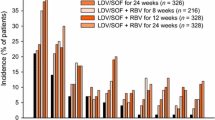
The efficacy of SOF/VEL/VOX was evaluated in four Phase 3 registrational studies in patients with HCV infection without cirrhosis or with compensated cirrhosis; two studies investigated 12 weeks of treatment in DAA-experienced patients, and two studies investigated 8 weeks of treatment in DAA-naive patients (Fig. 3). These studies enrolled patients between November 2015 and April 2016 at 117 sites in the United States, Canada, the United Kingdom, France, Germany, Australia, and New Zealand.

Design of SOF/VEL/VOX Phase 3 studies (GS-US-367-1171 [POLARIS-1], GS-US-367-1172 [POLARIS-2], GS-US-367-1173 [POLARIS-3], and GS-US-367-1170 [POLARIS-4])
5.1 Efficacy in DAA-Experienced Patients
The primary goal of the clinical development program was to demonstrate that SOF/VEL/VOX was a highly effective and safe salvage therapy for the most difficult-to-cure patients. The Phase 3 program was conducted to specifically assess the efficacy of SOF/VEL/VOX in DAA-experienced patients for a treatment duration of 12 weeks as supported by the Phase 2 studies, GS-US-367-1168, GS-US-367-1169, and TRILOGY-3. Initially, a single study was proposed which would enroll all DAA-experienced patients to receive SOF/VEL/VOX or blinded placebo for 12 weeks. In response to a request from the US Food and Drug Administration, the DAA-experienced patient population was separated into two separate studies, one with the original placebo-controlled design for NS5A inhibitor-experienced patients (POLARIS-1) and another with open-label SOF/VEL for 12 weeks as an active comparator for DAA-experienced patients who have not received an NS5A inhibitor (POLARIS-4).
5.1.1 GS-US-367-1171 (POLARIS-1)
Study GS-US-367-1171 (POLARIS-1) was a Phase 3, randomized, double-blind, placebo-controlled study which assessed the antiviral efficacy, safety, and tolerability of SOF/VEL/VOX compared with placebo for 12 weeks in NS5A inhibitor-experienced patients with chronic HCV infection [9]. The placebo control was chosen for this study as there was no available approved treatment for NS5A inhibitor-experienced patients. Additionally, this design allowed for an assessment of the safety of SOF/VEL/VOX as compared with the patients who received placebo. Patients with genotype 1 HCV infection were randomized 1:1 to receive SOF/VEL/VOX or placebo for 12 weeks, stratified according to cirrhosis status. Patients with other HCV genotypes were not randomized and were enrolled to receive SOF/VEL/VOX for 12 weeks. The primary efficacy end point of the study was the SVR12 rate in the SOF/VEL/VOX 12-week group was compared with a pre-specified SVR performance goal of 85% using a two-sided exact one-sample binomial test at the 0.05 significance level.
Overall, 415 patients were enrolled and began treatment with SOF/VEL/VOX (263 patients) or placebo (152 patients). Five patients prematurely discontinued study treatment, two in the SOF/VEL/VOX group (one due to an adverse event and one was lost to follow-up) and three patients in the placebo group due to AEs. The demographics and baseline characteristics of the patients treated with SOF/VEL/VOX for 12 weeks are shown in Table 7. The majority of the patients had genotype 1 HCV infection (57%, 150 patients) or genotype 3 HCV infection (30%, 78 patients), and 46% (121 patients) had cirrhosis. Most patients (93%) previously received an NS5A inhibitor in combination with another DAA. The most common NS5A inhibitors used in previous unsuccessful treatments were ledipasvir (55%), daclatasvir (23%), and ombitasvir (13%). Among the 248 patients with available baseline sequencing, 205 (83%) had NS5A and/or NS3 RASs.
In POLARIS-1, 96% (253 of 263) of patients achieved SVR12, and this rate met the primary efficacy end point of being significantly superior to the pre-specified performance goal of 85% ( p < 0.001). As shown in Table 8, treatment with SOF/VEL/VOX for 12 weeks in NS5A inhibitor-experienced patients demonstrated consistently high SVR12 rates across genotypes and regardless of cirrhosis status. Seven patients had virologic failure: six patients relapsed, and one patient had on-treatment virologic failure with PK data consistent with nonadherence. All six patients who relapsed had cirrhosis: one patient had genotype 1a HCV infection, four patients had genotype 3 HCV infection, and one patient had genotype 4 HCV infection. Baseline RASs did not impact treatment outcome: the SVR12 rate in patients with baseline RASs was 97% and in patients without baseline RASs was 98%. Of the six patients who relapsed, only one patient had treatment-emergent resistance (the patient with genotype 4 infection developed Y93H).
Of the 152 initially randomized to receive placebo in the primary study of POLARIS-1, 147 enrolled in a subsequent substudy to receive 12 weeks of open-label SOF/VEL/VOX [10]. All 147 patients completed treatment, and 97% (143 of 147 patients) achieved SVR12. Four patients experienced virologic relapse; all had HCV genotype 1a, one had cirrhosis, and two had treatment-emergent RASs.
5.1.2 GS-US-367-1170 (POLARIS-4)
Study GS-US-367-1170 (POLARIS-4) was a Phase 3, randomized, open-label study which assessed the antiviral efficacy, safety, and tolerability of 12 weeks of SOF/VEL/VOX and 12 weeks of SOF/VEL in DAA-experienced patients with chronic HCV infection with or without cirrhosis who had not previously received an inhibitor of the HCV NS5A protein, (with the exception that those who had received only a PI with pegylated interferon and ribavirin were not included, since these patients had approved retreatment options) [9]. The use of SOF/VEL/VOX and SOF/VEL within the same study allowed an assessment of the contribution of VOX to efficacy as well as to the safety of the treatment regimen. Patients with genotype 1, 2, or 3 HCV infection were randomized 1:1 to receive SOF/VEL/VOX or SOF/VEL once daily for 12 weeks, stratified according to HCV genotype and cirrhosis status. Patients infected with other HCV genotypes with or without cirrhosis were enrolled in the SOF/VEL/VOX 12-week group. In the primary efficacy analysis, the SVR12 rate in the SOF/VEL/VOX 12-week and SOF/VEL 12-week groups was compared with a pre-specified SVR performance goal of 85% using a two-sided exact one-sample binomial test at the 0.025 significance level.
A total of 333 patients were enrolled in POLARIS-4, 182 in the SOF/VEL/VOX group and 151 in the SOF/VEL group. Two patients did not complete SOF/VEL treatment, one due to an adverse event and another due to a lack of efficacy. The demographics and baseline characteristics of the patients treated with SOF/VEL/VOX for 12 weeks are shown in Table 7. Similar to POLARIS-1, the majority of the patients enrolled in POLARIS-4 had genotype 1 HCV infection (43%, 144 patients) or genotype 3 HCV infection (32%, 106 patients), and 46% (153 patients) had cirrhosis. Most patients (72%) previously received an NS5A inhibitor without another DAA, and 85% of patients had received sofosbuvir as a part of previous unsuccessful treatment. At baseline sequencing, 49% of patients had NS5A and/or NS3 RASs.
In POLARIS-4, treatment with SOF/VEL/VOX for 12 weeks resulted in an SVR12 rate of 98%, which was statistically superior to the performance goal of 85% at the pre-specified 0.025 significance level ( p < 0.001), meeting the primary efficacy end point. Treatment with SOF/VEL for 12 weeks resulted in an SVR12 rate of 90%, which was not statistically superior to the performance goal of 85% at the pre-specified 0.025 significance level ( p = 0.092). Although not pre-specified in the POLARIS-4 statistical analysis plan, an ad hoc evaluation of the superiority of SOF/VEL/VOX treatment for 12 weeks compared with SOF/VEL treatment for 12 weeks was performed in patients with genotypes 1, 2, or 3 HCV infection. Treatment with SOF/VEL/VOX for 12 weeks was statistically superior to treatment with SOF/VEL for 12 weeks ( p = 0.005).
As shown in Table 8, patient subgroups by genotype and by cirrhosis status, SOF/VEL/VOX for 12 weeks led to higher SVR12 rates compared with SOF/VEL for 12 weeks, demonstrating the contribution of VOX to the regimen. There was only one patient with virologic failure in the SOF/VEL/VOX group who had genotype 1a HCV infection and cirrhosis and was previously treated with SOF plus simeprevir who had no RASs at the time of relapse. There were 15 patients experienced virologic failure in the SOF/VEL group. There was one patient with genotype 2 HCV infection without cirrhosis who experienced virologic breakthrough with treatment-emergent resistance with the infrequently seen SOF signature mutation S282T in addition to Y93H. Of the 14 patients who relapsed following SOF/VEL treatment for 12 weeks, 8 patients had genotype 3 HCV infection, and 7 of these patients also had cirrhosis. Six patients who relapsed had genotype 1 HCV infection: three patients with genotype 1a with cirrhosis, two patients with genotype 1a without cirrhosis, and one patient with genotype 1b without cirrhosis who completed only 56 days of study treatment (discontinued treatment due to headache). Ten of these 14 patients with relapse had treatment-emergent RASs, most of which were in the NS5A gene at amino acid position 93. In both treatment groups, the presence of baseline RASs did not impact treatment outcome: in the SOF/VEL/VOX group, the SVR12 rates in patients with and without baseline RASs were 100 and 99%, respectively; in the SOF/VEL group, the SVR12 rates in patients with and without baseline RASs were 90 and 89%, respectively.
5.2 Efficacy in DAA-Naive Patients
The second goal of the SOF/VEL/VOX clinical development program was to assess whether the addition of a third potent DAA to the regimen would allow shortening treatment duration for the relatively easy-to-cure DAA-naive patients. To this end, the Phase 3 program included an evaluation in DAA-naive patients of the efficacy of SOF/VEL/VOX for 8 weeks, as supported by the Phase 2 studies, LEPTON, GS-US-367-1168, and GS-US-367-1169, compared to SOF/VEL for 12 weeks. Initially, a single study was initially proposed which would enroll all DAA-naive patients to receive SOF/VEL/VOX for 8 weeks or SOF/VEL for 12 weeks (POLARIS-2). In response to a request from the US Food and Drug Administration, a separate study with a similar design was conducted in patients with genotype 3 HCV infection and cirrhosis (POLARIS-2), and this population was removed from POLARIS-3. For both POLARIS-2 and POLARIS-3, SOF/VEL for 12 weeks was chosen as the comparator for this study based on the data from the Phase 3 studies ASTRAL-1, ASTRAL-2, and ASTRAL-3 (see [2]) and because it was anticipated to be a standard of care regimen for DAA-naive patients with genotype 1–6 HCV infection during the development of SOF/VEL/VOX, including those without cirrhosis or with compensated cirrhosis. In addition, the choice of comparator allowed for the assessment of the safety profile of VOX in the SOF/VEL/VOX regimen.
5.2.1 GS-US-367-1172 (POLARIS-2)
Study GS-US-367-1172 (POLARIS-2) was a Phase 3, randomized, open-label study which assessed the antiviral efficacy, safety, and tolerability of 8 weeks of SOF/VEL/VOX compared with 12 weeks of SOF/VEL in DAA-naive patients with chronic HCV infection [11]. Patients with genotype 1, 2, or 4 HCV infection with or without cirrhosis or genotype 3 HCV infection without cirrhosis were randomized 1:1 to receive SOF/VEL/VOX for 8 weeks or SOF/VEL for 12 weeks (patients with genotype 3 HCV infection and cirrhosis were enrolled in POLARIS-3). Patients with other genotypes with or without cirrhosis were enrolled into the SOF/VEL/VOX 8-week group. In POLARIS-2, the primary efficacy analysis assessed the noninferiority of the rate of SVR among patients receiving SOF/VEL/VOX to the rate among patients receiving SOF/VEL using a noninferiority margin of 5%. A two-sided 95% confidence interval was constructed for the difference in the rates of SVR between the two treatment groups using stratum-adjusted Mantel–Haenszel proportions. Noninferiority was established if the lower bound was greater than −5%.
A total of 941 patients were enrolled and treated in POLARIS-2, 501 in the SOF/VEL/VOX group and 440 in the SOF/VEL group. One patient did not complete SOF/VEL/VOX treatment due to pregnancy, and three patients did not complete SOF/VEL treatment, two due to adverse events and one was lost to follow-up. The demographics and baseline characteristics of the patients enrolled in POLARIS-2 are shown in Table 9. The majority of patients had genotype 1 (49%) or genotype 3 (19%) HCV infection; 12% of patients had genotype 2, 13% had genotype 4, 2% had genotype 5, and 4% had genotype 6. Overall, 19% of patients had cirrhosis. A total of 218 patients (23%) had prior treatment with an interferon-based regimen; the majority of these patients (80%; 174 of 218 patients) had failed prior treatment with pegylated interferon plus ribavirin. At baseline sequencing, 50% of patients had NS5A and/or NS3 RASs.
In the POLARIS-2 trial, the SVR12 rate was 95% (95% CI 93–97%) among patients receiving 8 weeks of SOF/VEL/VOX and 98% (95% CI 96–99%) among those receiving 12 weeks of SOF/VEL (Table 10). The SVR12 rate for the SOF/VEL/VOX 8-week group did not demonstrate noninferiority to the SVR12 rate for the SOF/VEL 12-week group. The strata-adjusted difference (95% CI) in the proportions was −3% (−6% to −<1%), the lower bound of which is not greater than the pre-specified noninferiority margin of −5%.
The lower SVR12 rate observed in the SOF/VEL/VOX 8-week group compared with the SOF/VEL 12-week group was primarily due to a lower SVR rate among patients with genotype 1a HCV infection, particularly among those enrolled at US sites. Overall, the SVR12 rate for patients with genotype 1a infection who were treated with SOF/VEL/VOX for 8 weeks was 92% (155 of 169); among those in the United States, the SVR12 rate was 89% (95 of 107), and among those outside the United States, the SVR12 rate was 97% (60 of 62). Of the 21 patients in the SOF/VEL/VOX 8-week group who relapsed, 14 had genotype 1a HCV infection. The other seven patients with virologic relapse included two patients with genotype 1b, one of whom had cirrhosis; two patients with genotype 2 HCV infection without cirrhosis; two patients with genotype 4 infection, one of whom had cirrhosis; and one patient with genotype 5 infection without cirrhosis. Although the SVR12 rate was lower among patients receiving SOF/VEL/VOX for 8 weeks with cirrhosis (91%) compared with those without cirrhosis (96%), most of the patients with cirrhosis who relapsed had genotype 1a HCV infection (5 of 7 patients). The SVR12 rate was 94% for patients with baseline NS5A and/or NS3 RASs and 98% for patients without baseline NS5A and/or NS3 RASs. For patients with HCV genotype 1a, the rates of SVR in patients with and without baseline RASs were 89% and 95%, respectively. The Q80K RAS was the most commonly observed NS3 variant; although it confers no change to VOX susceptibility in vitro, the SVR12 rate was lower for genotype 1a patients with baseline Q80K compared to those without (88 and 94%, respectively). Of the 21 patients with virologic relapse at posttreatment week 12, one patient had treatment-emergent resistance (NS5A RASs Q30R and L31M).
In the SOF/VEL 12-week group, three patients had virologic relapse, one patient with genotype 1a with cirrhosis, one patient with genotype 1b without cirrhosis, and one patient with genotype 4a HCV infection without cirrhosis. The patient with genotype 1a who relapsed developed treatment-emergent Y93N. The presence of baseline RASs did not impact the SVR12 rate for the SOF/VEL 12-week group; 100% (217 of 218 patients) of the patients with RASs and 99% (206 of 208 patients) of the patients without RASs achieved SVR12.
5.2.2 GS-US-367-1173 (POLARIS-3)
Study GS-US-367-1173 (POLARIS-3) was a Phase 3, randomized, open-label study which assessed the antiviral efficacy, safety, and tolerability of 8 weeks of SOF/VEL/VOX compared with 12 weeks of SOF/VEL in DAA-naive, cirrhotic patients with chronic genotype 3 HCV infection [11]. Patients were randomized 1:1 to receive SOF/VEL/VOX once daily for 8 weeks or SOF/VEL once daily for 12 weeks, stratified by treatment experience. In POLARIS-3, the primary efficacy analysis assessed first the SVR12 rate among patients in the SOF/VEL/VOX group against a performance goal of 83% using a two-sided exact one-sample binomial test at the 0.05 significance level. If this group met this criterion, the SVR12 rate in the SOF/VEL group also would be assessed against the performance goal of 83% at the 0.05 significance level. The performance goal of 83% was based on the prior results of SOF/VEL for 12 weeks in this patient population in the ASTRAL-3 trial (SVR12 rate 91%; 95% CI 83–96%, see [2]).
A total of 219 patients were enrolled and treated in POLARIS-3, 110 in the SOF/VEL/VOX group and 109 in the SOF/VEL group. Two patients did not complete SOF/VEL treatment, one due to an adverse event and one due to a lack of efficacy. The demographics and baseline characteristics of the patients enrolled in POLARIS-3 are shown in Table 9. Per protocol, all patients had genotype 3 HCV infection and cirrhosis. Overall, 31% (67 of 219 patients) of patients had prior treatment with an interferon-based regimen, and the majority of these patients (91% 61 of 67 patients) had failed prior treatment with pegylated interferon plus ribavirin. Baseline NS5A and/or NS4 RASs were observed in 46 patients (21%).
In POLARIS-3, the SVR12 rate was 96% of patients (106 of 110) in the SOF/VEL/VOX 8-week group and 96% of patients (105 of 109) in the SOF/VEL 12-week group (Table 10). The SVR12 rate for each treatment group was statistically superior to the pre-specified performance goal of 83% ( p < 0.001 for both groups). Two patients in the SOF/VEL/VOX group had a virologic relapse, both of whom were treatment experienced and had drug concentrations that were low for at least one study visit, suggesting that the patient was not fully adherent to study dosing: One patient who did not achieve on-treatment HCV RNA suppression had a virologic rebound by week 8 of treatment, and one patient had virologic relapse. Baseline RASs had no impact on virologic outcome in the SOF/VEL/VOX 8-week or SOF/VEL 12-week group; all patients with baseline NS3 and/or NS5A RASs achieved SVR12. The two patients who relapsed following treatment with SOF/VEL/VOX for 8 weeks did not have treatment-emergent RASs, in contrast to the two virologic failures in the SOF/VEL 12-week group, both of whom developed treatment-emergent Y93H.
5.3 Safety of SOF/VEL/VOX
The Integrated Phase 3 Safety Population provided the largest dataset to support the safety profile of SOF/VEL/VOX. It was comprised of 1908 patients enrolled in the four Phase 3 clinical studies, including 445 DAA-experienced patients who received SOF/VEL/VOX for 12 weeks in POLARIS-1 and POLARIS-4; 611 DAA-naive patients who received SOF/VEL/VOX for 8 weeks regimen in POLARIS-2 and POLARIS-3; 700 patients who received SOF/VEL for 12 weeks in POLARIS-2, POLARIS-3, and POLARIS-4; and 152 patients who received placebo for 12 weeks in POLARIS-1.
In general, the adverse event profile was similar between patients receiving SOF/VEL/VOX for 12 or 8 weeks, SOF/VEL, and placebo, and rates were low for patients with Grade 3 or 4 adverse events, serious adverse events, and adverse events leading to discontinuation with no trends observed across the treatment groups (Table 11). The comparable incidence of most adverse events among the SOF/VEL/VOX groups versus the placebo 12-week group suggests relatively high background rates of these adverse events in patients with HCV infection.
All treatment groups had the same frequently occurring adverse events (>10% of patients), headache, fatigue, diarrhea, and nausea. Headache and fatigue were reported by 25 and 22% of patients overall, respectively, with similar frequencies reported in each treatment group. Consistent with the known effects of some protease inhibitors, patients receiving SOF/VEL/VOX had a higher incidence of gastrointestinal adverse events compared with patients receiving SOF/VEL. The diarrhea and nausea reported in patients receiving SOF/VEL/VOX were mostly mild and not treatment limiting with no patient discontinuing or interrupting treatment due to diarrhea or nausea. The duration of SOF/VEL/VOX treatment did not significantly impact the rates.
Most adverse events across all treatment groups were Grade 1 or 2 in severity. The incidence of Grade 3 or above adverse events in the SOF/VEL/VOX groups was low (2%) and similar to the incidence in the SOF/VEL group (2%) and placebo group (3%). The incidence was similar whether SOF/VEL/VOX treatment occurred for 8 (2%) or 12 (2%) weeks.
A total of two deaths were reported in the Integrated Phase 3 Safety Population. There was one treatment-emergent death in the SOF/VEL/VOX 12-week group (patient died 2 days after completion of study treatment from an illicit drug overdose) and one nontreatment-emergent death in the SOF/VEL/VOX 8-week group (patient had a medical history of hypertension and died 78 days after completion of study treatment from “hypertension” per the coroner’s report). Both deaths were considered unrelated to study drug by the investigator.
Few patients (2%, 47 of 1908) in the Integrated Phase 3 Safety Population had serious adverse events. The highest rate of serious adverse events was reported in the placebo 12-week group (5%). There were no serious adverse events in any treatment group that were considered related to study drug.
The incidence of adverse events leading to discontinuation of study drugs was low across all treatment groups (<1%). Only patient among those who received SOF/VEL/VOX for 8 or 12 weeks prematurely discontinued treatment due to an adverse event (a Grade 3 adverse event of angioedema considered unrelated to study drug and attributed by the investigator to ramipril initiated the day prior to the event).
In the Integrated Phase 3 Safety Population, graded laboratory abnormalities were observed more often in the placebo 12-week group (76%) and SOF/VEL/VOX 12-week group (69%) compared with the SOF/VEL/VOX 8-week group (58%) and SOF/VEL 12-week group (59%), most likely reflecting the higher percentage of patients with cirrhosis in the placebo 12-week and SOF/VEL/VOX 12-week groups. The higher rates of laboratory abnormalities in the patients receiving placebo were largely due to the higher rates of alanine aminotransferase (ALT) and aspartate aminotransferase abnormalities consistent with untreated HCV infection.
The higher incidence of graded laboratory abnormalities in the SOF/VEL/VOX 12-week group compared with the 8-week group was mostly due to more patients in the 12-week group with Grade 1 or 2 laboratory abnormalities (62%), notably in decreased platelets and increased total bilirubin, consistent with more patients with cirrhosis being enrolled in the 12-week duration regimen in the studies for DAA-experienced patients. For patients receiving SOF/VEL/VOX, there was a higher rate of Grade 1 hyperbilirubinemia compared with patients receiving SOF/VEL or placebo (Table 11). Similar to other protease inhibitors, VOX is an inhibitor of OATP1B1 and OATP1B3, which resulted in an increase in Grade 1 total bilirubin in patients receiving SOF/VEL/VOX. This increase is observed more often in patients with cirrhosis (10%) than in patients without cirrhosis (4%). There were no adverse events of jaundice. There was no pattern of VOX associated with ALT elevation (Table 12). Grade 1 or 2 ALT elevations occurred early in SOF/VEL/VOX treatment and were consistent with expected fluctuations prior to viral suppression. Of the 1,056 patients receiving SOF/VEL/VOX in the Integrated Phase 3 Safety Population, 1 patient had a Grade 3 elevation in ALT (<1%), and none had a Grade 4 elevation in ALT.
The rates of Grade 3 or 4 chemistry laboratory abnormalities in the SOF/VEL/VOX groups were similar to the SOF/VEL group and lower than the placebo 12-week group. Increased glucose, lipase, and creatine kinase were the most common Grade 3 or 4 chemistry laboratory abnormalities with SOF/VEL/VOX. Glucose elevations were observed primarily among patients with a history of diabetes or those with high glucose prior to study drug initiation. Elevations in lipase were generally isolated or intermittent; all were asymptomatic, and there were no adverse events of pancreatitis. Similarly, creatine kinase elevations were mostly transient and asymptomatic.
5.4 Summary of Phase 3 Data Supporting the Initial Registration of SOF/VEL/VOX
In POLARIS-1 and POLARIS-4, 445 DAA-experienced patients with and without compensated cirrhosis were treated with 12 weeks of SOF/VEL/VOX, and the overall SVR12 rate was 97%. Treatment was highly efficacious across HCV genotypes and regardless of prior DAA regimen, cirrhosis status, or the presence of baseline RASs. In POLARIS-2 and POLARIS-3, 611 DAA-naive patients with and without compensated cirrhosis were treated with 8 weeks of SOF/VEL/VOX, and the overall SVR12 rate was 95%. The SVR12 rate was lower in patients with genotype 1a HCV infection than in patients with other HCV genotypes. Treatment-emergent resistance following treatment with SOF/VEL/VOX was uncommon in both the DAA-experienced and DAA-naive populations consistent with the regimen having a high barrier to resistance. The regimen was well-tolerated with similar frequently occurring adverse events compared with SOF/VEL and placebo, with higher rates of mild diarrhea and nausea compared with SOF/VEL. There were no clinically meaningful laboratory abnormalities. Slight increases in total bilirubin were observed, consistent with VOX inhibition of OATP1B1 and OATP1B3.
These data supported the approval of SOF/VEL/VOX (Vosevi®) in the United States on July 18, 2017, as the first retreatment option for patients who have failed prior treatment with NS5A inhibitor and/or sofosbuvir [12]. Vosevi was also approved in the European Union shortly afterwards on July 28, 2017, where it is recommended for all HCV genotypes for 12 weeks in DAA-experienced patients with and without cirrhosis and DAA-naive patients with compensated cirrhosis and for 8 weeks in DAA-naive patients without cirrhosis [13].
6 Conclusion
The development of DAAs led to a transformation in the treatment of HCV. However, even with an anticipated 95% cure rate, there is a need for a treatment to address the small percentage of patients who are not cured with first-line therapy. The once-daily, single-tablet regimen of SOF/VEL/VOX for 12 weeks is a highly effective and safe treatment for DAA-experienced patients with chronic HCV infection and will provide this growing population with an option for retreatment and a high likelihood for cure, regardless of genotype, the presence of cirrhosis, RASs, or prior treatment regimen. For regions in which a shorter duration of initial HCV treatment is of particular interest, SOF/VEL/VOX for 8 weeks is also a safe and effective option. Vosevi was the fourth HCV treatment developed by Gilead to be approved in 4 years and completed the HCV portfolio providing safe and effective treatment options for nearly all patient populations.
References
Taylor JG (2019) Discovery of voxilaprevir (GS-9857): the pan-genotypic hepatitis C virus NS3/4A protease inhibitor utilized as a component of Vosevi®. Top Med Chem. https://doi.org/10.1007/7355_2018_61
Brainard DM, McHutchison JG (2019) The clinical development of sofosbuvir/velpatasvir (SOF/VEL, Epclusa®). Top Med Chem. https://doi.org/10.1007/7355_2018_43
Rodriguez-Torres M, Glass S, Hill J et al (2016) GS-9857 in patients with chronic hepatitis C virus genotype 1-4 infection: a randomized, double-blind, dose-ranging phase 1 study. J Viral Hepat 23:614–622
Lawitz E, Yang JC, Stamm LM et al (2017) Characterization of HCV resistance from a 3-day monotherapy study of voxilaprevir, a novel pangenotypic NS3/4A protease inhibitor. Antivir Ther. https://doi.org/10.3851/IMP3202
Gane EJ, Schwabe C, Hyland RH et al (2016) Efficacy of the combination of sofosbuvir, velpatasvir, and the NS3/4A protease inhibitor GS-9857 in treatment-naive or previously treated patients with hepatitis C virus genotype 1 or 3 infections. Gastroenterology 151:448–456
Gane EJ, Kowdley KV, Pound D et al (2016) Efficacy of sofosbuvir, velpatasvir, and GS-9857 in patients with hepatitis C virus genotype 2, 3, 4, or 6 infections in an open-label, phase 2 trial. Gastroenterology 151:902–909
Lawitz E, Reau N, Hinestrosa F et al (2016) Efficacy of sofosbuvir, velpatasvir, and GS-9857 in patients with genotype 1 hepatitis C virus infection in an open-label, Phase 2 trial. Gastroenterology 151:893–901
Lawitz E, Poordad F, Wells J et al (2017) Sofosbuvir-velpatasvir-voxilaprevir with or without ribavirin in direct-acting antiviral-experienced patients with genotype 1 hepatitis C virus. Hepatology 65:1803–1809
Bourliere M, Gordon SC, Flamm SL et al (2017) Sofosbuvir, velpatasvir, and voxilaprevir for previously treated HCV infection. N Engl J Med 376:2134–2146
Bourliere M, Gordon SC, Shiff ER et al (2018) Deferred treatment with sofosbuvir-velpatasvir-voxilaprevir for patients with chronic hepatitis C virus who were previously treated with an NS5A inhibitor: an open-label substudy of POLARIS-1. Lancet Gastroenterol Hepatol. May 30 (epub ahead of print)
Jacobson IM, Lawitz E, Gane EJ et al (2017) Efficacy of 8 weeks of sofosbuvir, velpatasvir, and voxilaprevir in patients with chronic HCV infection: 2 phase 3 randomized trials. Gastroenterology 153:113–122
Vosevi US prescribing information
Vosevi EU summary of product characteristics
Acknowledgments
The authors would like to thank the patients and their families as well as study site staff who participated in the clinical trials of Vosevi.
Compliance with Ethical Standards
Conflict of Interest Luisa M. Stamm and John G. McHutchison are employees of Gilead Sciences, Inc.
Ethical Approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Informed Consent
Informed consent was obtained from all individual participants included in the study.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer International Publishing AG, part of Springer Nature
About this chapter

Cite this chapter
Stamm, L.M., McHutchison, J.G. (2019). The Clinical Development of Sofosbuvir/Velpatasvir/Voxilaprevir (SOF/VEL/VOX, Vosevi®). In: Sofia, M. (eds) HCV: The Journey from Discovery to a Cure. Topics in Medicinal Chemistry, vol 32. Springer, Cham. https://doi.org/10.1007/7355_2018_49
Download citation
DOI: https://doi.org/10.1007/7355_2018_49
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-28399-5
Online ISBN: 978-3-030-28400-8
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)