
Abstract
The cellular plasma membrane, a seemingly chaotic structure composed of thousands of lipids and proteins, empowers cellular life and represents a versatile hub for signalling. Transient interactions of membrane components integrate to large-scale membrane properties such as packing, tension, curvature, or viscosity and reorganise the membrane in response to physiological needs. How do nanoscale interactions and heterogeneities determine cellular plasma membrane properties? This question is not trivial to address with technologies limited by the diffraction of light. Here, we discuss Stimulated Emission Depletion (STED) nanoscopy and its combination with fluorescence correlation spectroscopy (FCS, STED-FCS) as a technique to shed light onto the relevant molecular scales. While imaging with STED allows for tremendous insights into the spatial domain of organisation, the combination of STED with FCS provides increased temporal resolution and yields insights into the nature of the molecular motion on the relevant scales, allowing us to study nanoscale diffusion modes. We provide a comprehensive overview of the field, introduction to the technique and discuss common pitfalls with its implementation as well as alternative methodologies to measure the diffusion modes on the nanoscale.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
Every mammalian cell is enclosed by a plasma membrane (PM) which serves as a semi-permeable boundary to the surrounding medium and as a reaction platform composed of thousands of different types of lipids and proteins [1, 2]. The PM is known to be heterogeneously organised over a wide range of temporal and spatial scales ranging from nanometres to micrometres and nanoseconds to tens of seconds, respectively [3,4,5]. Fascinatingly, short-lived, nanoscale interactions and the heterogeneous membrane organisation contribute to its functions, to the point that the membrane should be pictured as much more than a disorganised fluid [6]. Deciphering the details of membrane organisation allows us to better understand signalling mechanisms, biophysical principles of cellular life, dysregulations of physiological processes and ultimately a variety of diseases. However, the dynamic nature of membrane organisation is challenging to access directly. Imaging methodologies can only provide snapshots or time-averaged insights into the principles of organisation [7]. Short-lived interactions integrating to the overall collective behaviour escape standard imaging technologies. Thus, in membrane research, the use of dynamic techniques such as fluorescence recovery after photobleaching (FRAP), single particle tracking (SPT), or fluorescence correlation spectroscopy (FCS) is very common [4]. While these methodologies provide insights into the behaviour of molecular motion and interactions, they struggle to probe interactions on the relevant scales: standard confocal, total internal reflection (TIR) or wide field setups cannot resolve two objects when they are closer than ~200 nm to each other [8]. Yet, lipids, proteins, and even protein clusters are <10 nm in size and their interactions may only last several milliseconds. [3] Even considering larger scales, for example, the formation of functional signalling platforms, lipid domains, or lipid rafts are well beyond what a standard microscope can elucidate. Consequently, the biology dictates that we need to employ technologies that allow us to probe interactions below the diffraction limit of light – on the nanoscale.
2 A Brief Introduction to Super-Resolution Microscopy
Several super-resolution methodologies have emerged over the past two decades and the development of photoactivated localisation microscopy (PALM), stochastic optical reconstruction microscopy (STORM) and stimulated emission depletion (STED) microscopy has been awarded with the Nobel Prize in Chemistry in 2014 [9, 10]. While it’s often referred to as “breaking the diffraction limit”, these techniques rather use smart tricks to circumvent it [8]. In single molecule localisation microscopy (SMLM) methods (such as PALM and STORM) the exact position of a fixed, isolated molecule in space can be approximated with sub-diffraction accuracy by fitting the emission profile of a single fluorophore to a model (for example, to a Gaussian in 2D). Of course, standard labelled samples contain more than thousands of single molecules very close to each other (closer than the diffraction limit and the microscope’s resolution). Thus, the key is to switch them on and off to only allow isolated emitters at a time (i.e., per camera frame) which can be achieved chemically by tuning buffer conditions or photo-physically with a switching laser. A similar concept was later employed in the so-called points accumulation for imaging in nanoscale topography (PAINT) technologies, where emitters are only fluorescent when inserted into the membrane or in DNA-PAINT only transiently bound to a probe with a complimentary DNA-oligo [11, 12]. This acquisition strategies come with the necessity for long acquisition times and many frames (with sparse emitters) to reconstruct the position of all molecules within the sample which makes it challenging to combine with live-cell imaging and especially for studying PM dynamics. STED nanoscopy also employs the trick of switching molecules on and off but uses a spatial pattern (central zero) of a depletion beam to achieve this (Fig. 1a) [13, 14]. Like in confocal imaging this beam is then scanned to construct an image with resolution dictated by the depletion beam efficiency and laser power (Fig. 1b). By itself, also STED microscopy suffers from rather slow acquisition times per frame as it is based on laser scanning acquisitions (Fig. 1c). However, it can be straightforwardly combined with fluorescence correlation spectroscopy in a point measurement fashion which makes it an instant super-resolution technique with very high temporal resolution [15].

STED microscopy. (a) Principle of STED microscopy. The excitation beam (blue) is superimposed with a doughnut-shaped STED beam (magenta). Fluorescence is depleted, cancelled, by the STED beam in the periphery of the excitation spot but the central zero of the STED beam allows to observe emission from the centre. The resulting, effective, emission or observation spot (green) is smaller than the diffraction limited excitation spot (blue). (b) The effective observation spot size, i.e., the resolution of the STED acquisition can be tuned by increasing the STED laser power. (c) Simplified implementation of a STED microscope (left) and mechanism of action (right). The excitation beam (blue) is combined with the doughnut-shaped STED beam (magenta) and scanned across the sample. Fluorescence is recorded by a point detector. Fluorescence excitation acts on the ground state S0 which causes red-shifted emission after vibrational relaxations. The STED beam acts on the excited state S1 and forces the molecule back to the ground state S0
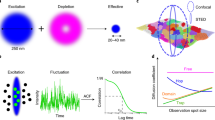
FCS only requires intensity data over time to report on the diffusion dynamics in the observation volume or in membranes in the observation spot (Fig. 2a). By parking the beam in the membrane and recording and analysing the intensity fluctuations, the underlying dynamics can be revealed. The STED beam constrains the observable fluorescence and the effective observation spot/volume to a sub-diffraction size (Figs. 1 and 2a). Consequently, the resulting intensity fluctuations used for the FCS analysis report directly on the nanoscale diffusion dynamics [16].

Fluorescence correlation spectroscopy combined with STED offers to measure nanoscale diffusion modes. (a) Principle of FCS and STED-FCS. Fluorophores (grey stars) can be excited by the confocal excitation beam (blue, emitting molecules are depicted as green stars). When the confocal excitation beam is overlaid with a STED beam, the resulting observation spot is smaller. The recorded intensity profiles in confocal (green) and STED (magenta) illustrate lower counts for STED illumination as less molecules are excited than in the confocal case. Autocorrelation of the intensity traces results in the correlation curves which reveal the underlying dynamics. The transit time τD which is the average time a molecule needs to cross the observation spot is larger for the big confocal observation spot and shorter for the smaller STED observation spot (scaling with STED laser power). (b) STED-FCS diffusion modes. Molecules can undergo a variety of diffusion behaviours: They can simply diffuse freely (left), undergo trapped diffusion with transient halts in their diffusion path (binding to an interaction partner, middle), or their diffusion can be confined by a meshwork structure such as the cortical actin cytoskeleton (hop diffusion right). The diffusion law plot reveals this diffusion modes by plotting the apparent diffusion coefficient D against the observation spot diameter d (full width at half max of the observation spot). The size of the observation area can be tuned by the STED laser power
3 STED-FCS as a Window to Nanoscale Dynamics
STED-FCS allows for directly measuring the diffusion dynamics with a given resolution tuneable by the STED beam laser power which enables to measure diffusion dynamics on different length scales [7, 16]. This concept has been exploited before in spot-variation FCS (svFCS) where the back aperture of the objective is underfilled and the observation area enlarged [17, 18]. Thus, in svFCS the dynamics from confocal to longer length-scales (up to μm) can be revealed with high temporal resolution. Plotting the diffusion time or diffusion coefficient against observation length-scale introduces the so-called diffusion law plot [18, 19]. The diffusion law gives insights how diffusion scales with space. It is very powerful in discriminating free diffusion from constrained diffusion or trapped diffusion and can help estimating sizes of domains or constraint length-scales. With this, the diffusion law plot is also similar to plotting the mean-squared-displacement (MSD) against time in SPT, which can also reveal hindrances in molecular diffusion [20].
STED-FCS takes the principles of the diffusion law to the nanoscale. By increasing the STED laser power, the observation spot sizes shrinks and the molecular transit time deceases (FCS diffusion time, τD, through the focus is shorter as the volume is smaller, Fig. 2a) [15, 16]. By calibration of the resolution, we can calculate the apparent diffusion coefficient D, \( D=\frac{d^2}{8\cdotp \ln (2)\cdotp {\tau}_D} \), and plot it against observation spot diameter d (defined as the full width at half max of the point spread function) [21]. Molecules undergoing free, Brownian, diffusion will show no change in diffusion coefficient with observation spot diameter d. However, any sort of hindrances will result in a deviation from this behaviour: Trapped diffusion, meaning nanoscale interactions with short halts in the diffusion path, will result in a reduction of apparent diffusion coefficient D with decreasing observation spot diameter as the trapping events become more pronounced in smaller observation areas (Fig. 2b). In contrast, hop diffusion, also termed compartmentalised diffusion, where molecules are constrained within a semi-permeable mesh, will cause an increase in D with smaller observation spot diameter as its length-scale approaches mesh size and starts probing free diffusion within compartments [22]. The presence of nanoscale domains appears as a decrease in apparent diffusion coefficient as the observation diameter decreases (similar to trapping) but stays constant or increases at very short length-scales [7] (Fig. 2b).
STED-FCS has extensively been used in the membrane field to investigate presence, distribution and impact of nanoscale hindrances, the influence of the actin cytoskeleton on membrane organisation, and ultimately cellular processes such as signalling [16, 23, 24]. The first experimental verification of the presence of nanoscale hindrances with STED-FCS were the measurements from Eggeling et al. in 2009 [16]. By comparing the diffusion mode of a fluorescently tagged (Atto647N) sphingomyelin (SM) and phospholipid (PE) completely different behaviours were observed and attributed to SM’s ability to bind to other molecules in the PM [16]. The origin of this trapping behaviour was linked to the cortical actin cytoskeleton and cholesterol [16, 21, 23]. As a matter of fact, cortical actin cytoskeleton generally plays a crucial role in trapping and hopping of molecules; however, not every anomalous diffusion mode is dependent on it. For instance, ganglioside GM1 exhibits domain-like diffusion in cells, and this diffusion mode is preserved in cytoskeleton-free cell-derived vesicles (Fig. 3) [23]. Trapped diffusion has also been observed for Ras-kinases in tumour cells or for molecules in the context of HIV infection [24, 25]. To elucidate the origin of hindered diffusion, extensive studies on model membrane systems have been performed to, for instance, investigate the influence of phase separation, phase preference or presence of pore-forming proteins on the diffusion law [26,27,28].

Nanoscale diffusion modes of lipids in cells and cell-derived vesicles devoid of the cortical actin cytoskeleton. (a) PtK2 cells labelled with Cell Mask as PM stain and Lifeact-GFP (top) show cortical actin cytoskeleton underneath the PM while giant plasma membrane vesicles (GPMVs) derived from these cells (bottom) do not show the organized actin cytoskeleton. Diffusion mode of (b) Atto647N-labelled DPPE, (c) Atto647N-labelled DOPE, (d) Atto647N-labelled SM and (e) Atto647N-labelled GM1. Atto647N-DOPE shows both hop (pool 1, P1) and free diffusion (pool 2, P2) in live PtK2 cells, but only free diffusion in GPMVs. Atto647N-DPPE shows free diffusion both in cells and GPMVs. Atto647N-SM shows confined diffusion in cells and free diffusion in GPMVs. Atto647N-GM1 shows domain-like diffusion both in cells and in GPMVs. The diffusion behaviour can quickly be assessed by plotting the diffusion coefficient in STED (DSTED) divided by the diffusion coefficient in confocal (DConf). Scale bars 10 μm. Indicated STED laser powers in b were measured at the back focal plane. Figure adapted from ref. [23]
In addition to obtaining the diffusion law in the membrane, STED-FCS can be used to reveal the diffusion in small entities (i.e., smaller than the diffraction limit), such as viruses, or virus budding sites [25, 29, 30]. Besides on 2D systems such as membranes, STED-FCS in solution is a rapidly developing field. With the use of new depletion patterns and adaptive optical elements such as spatial light modulators the diffusion law can also be measured in the three dimensional space with applications in measuring cytosolic trapping or interactions in liquid–liquid phase separated systems [31,32,33,34].
4 Practical Considerations and Implementations of STED-FCS
Performing STED-FCS experiments relies in the first instance only on having a STED microscope which is basically an extended laser scanning confocal microscope (Fig. 1b). However, many practical considerations have to be taken into account and we will briefly address the most important ones here and refer the reader for more details to [7].
-
1.
Fluctuation data acquisition
Fluctuation data can be acquired and processed in many different ways. The data can be acquired as simple point measurement over time (t-scan) and then post-processed [35]. Alternatively, the detector can be connected to a hardware correlator which outputs the auto-correlation curves ready to be fitted. While this is convenient, it does not allow for any photon filtering or post processing. It is also important to consider the data type saved and exported. Photon counting data allow for additional flexibility like lifetime gating in post-processing but also increase the size of the files as well the time to correlate and obtain the raw correlation curves [7, 36, 37]. It is useful to have a robust display of the correlation curves while acquiring the data.
-
2.
Calibration
For accurate and interpretable diffusion law measurements it is crucial to calibrate the observation spot size in confocal and at different STED laser powers. This can be achieved by imaging sub-diffraction fluorescent beads or by using a lipid bilayer as a freely diffusing system. Calibration should be performed daily and should not vary. Changes in apparent spot-sizes indicate drifts or misalignment of the optical system.
-
3.
Experimental requirements for STED-FCS
Most important requirement for a successful STED-FCS experiment is a good fluorescent label. The dye should have high molecular brightness and should be easily “depletable” (i.e., work well with STED). The molecular brightness is important as it determines the signal to noise ratio (SNR) in FCS [38, 39]. The brighter the dye, the better the signal, which is especially important when considering the short transit times under high STED laser beam intensity when only a few photons can be collected [39]. Ideally, the resolution should be calibrated using the same dye as in the experiment (see above for Calibration). In general, high-quality optics corrected for optical aberrations are required in STED microscopy to achieve low background levels. In STED-FCS higher background levels cause uncorrelated signal which dampens the autocorrelation function’s amplitude complicating data analysis. Choice of depletion wavelength can have a great influence on the achievable resolution. Usually a wavelength far red-shifted from the emission maximum is chosen (Fig. 1c). The optimal wavelength is also a property of the fluorescent dye and needs to be determined empirically.
-
4.
Data analysis and processing
As mentioned above, the raw data type depends on the acquisition mode. If the data are acquired as photon streams they need to be correlated, if the data are acquired using a hardware correlator, they are ready to be analysed. The autocorrelation curves are fitted to theoretical models derived for various physical situations such as 2D or 3D diffusion, blinking, binding, etc. Fitting the data needs to be performed with care and requires some training and expertise. As a general rule: Always fit the data to the simplest model (fewest fitting parameters) possible. From the fits the transit time τD as well as the average number of molecules N and the molecular brightness can be extracted. The transit time as average time of molecules to transverse the focus relates inversely to the apparent diffusion coefficient D (Fig. 2b). The average number of molecules can be used to measure concentration of molecules, for example, as particle per μm2 in membranes, yet, this is not reliable at higher STED powers since noise can dominate the amplitude. The molecular brightness can be used to determine degree of oligomerisation of the investigated fluorescent particle.
To see examples of good and bad curves as well as good and bad fitting, please see ref. [7]
5 Variations of STED-FCS
As the basis of STED-FCS is a STED laser scanning microscope, many other fluorescence fluctuation techniques can be combined with STED to shed light on the nanoscale which is a unique feature of the technology.
As already mentioned before, the fluctuation data can be acquired not only from a single point but also from multiple points in space: by scanning the confocal (or STED) focus quickly along one line or circle, fluctuation data for multiple points in space (pixels) are obtained. This approach is termed scanning STED-FCS and has helped to elucidate the spatial heterogeneity in nanoscale diffusion hindrances [40,41,42]. It allows mapping diffusion times or coefficient over space. By interleaving confocal and STED excitation, line-interleaved excitation scanning STED-FCS (LIESS-FCS) maps the diffusion modes across space [43]. Instead of only acquiring a line over time, a series of frames can be acquired and analysed by raster image correlation spectroscopy (RICS) in conjunction with STED [44, 45]. As a bigger area is integrated, the nanoscale dynamics are a result of the robust average. The combination with pair-correlation analysis allows the identification nanoscale obstacles in the diffusion path and reveals interconnectivity [46].
With the increased availability of commercial STED microscopes, we anticipate that the combination with various fluctuation analyses will become of more wide-spread use [7, 39]. In addition, the increased availability of functional probes, reporting on biophysical properties such as viscosity or lipid order has become more common and offers complementary insights using STED imaging as well as spectroscopy [37, 47,48,49].
6 Alternative Ways to Investigate Nanoscale
The investigation of nanoscale dynamics by STED microscopy and STED-FCS has proven as a powerful tool to elucidate membrane heterogeneities. While ease of use and ease of implementation as well as the extension of the fluctuation techniques are convenient, employing the high-power STED beam for long-term measurements can have negative effects on sensitive samples [50]. Additionally, STED microscopy relies on dyes that have the right photo-physical properties and requires specialised equipment for STED beam shaping [7].
The gold standard for nanoscale diffusion dynamics (first elucidated by STED-FCS) has been the free diffusion of phosphatidylethanolamine (PE) and the trapped diffusion of sphingomyelin (SM) in the PM of PtK2 cells [16, 21, 23, 43]. This very different nanoscale behaviour is not apparent by imaging (homogeneous PM stain in both cases) [7]. This PE/SM system has now been extensively used to demonstrate the power of alternative techniques to investigate nanoscale diffusion and hindrances.
To trick the diffraction limit, fluorescence in the periphery of the observation area needs to be rejected. In STED, this is achieved photo-physically by fluorescence depletion. In another clever attempt to do this physically, nano-apertures have been combined with FCS. In near-field scanning optical microscopy (NSOM) FCS (NSOM-FCS), the fluorescence is excited through a nanometric aperture [51]. By using different sizes of the aperture, FCS data on different scales can be obtained. Consistently, with the results from STED-FCS, very different behaviours were observed for PE and SM in the PM.
Building on the NSOM approach, nanoantennas have been employed to observe diffusion dynamics in sub-diffraction areas [52, 53]. By fabricating arrays of nanoantennas to confine illumination as well as to enhance fluorescence in the electromagnetic near-field, intensity fluctuations over time can be observed and the nanoscale diffusion dynamics probed [54]. Changing the size or distance of the antennas allows to probe dynamics at different length-scales. This method was applied to measure the diffusion of PE and SM in the PM and the results are in agreement with the previous measurements from NSOM-FCS and STED-FCS.
The above techniques are reporting on true nanoscale interactions by (photo-)physically restricting the area of collected light. NSOM-FCS and the nanoantenna based approaches, however, are near-field approaches necessitating the direct contact of sample and optics. As an alternative to STED-FCS and the near-field optics, confocal-based sFCS in combination with statistical analysis can be used to probe hindrances in diffusion [55]. Large sets of measurements can pick up on small fractions of different sub-diffusive behaviours and can be used to differentiate Brownian from trapped diffusion which has been experimentally shown using the PE/SM system. An additional alternative approach could be the image mean-squared-displacement (iMSD) methodology which accordingly takes the fluctuations from a larger area of pixels (and their statistics) into account [56, 57]. However, to the best of our knowledge, this has not been used systematically to study the PE/SM system in Ptk2 cells.
SPT methods could of course be used to study the nanoscale diffusion dynamics [20]. However, to obtain sufficient SNR and localisation precision, large structures such as fluorescent beads, quantum dots or gold particles need to be employed potentially disturbing the system under study [58]. Very fast frame-rates are necessary and great care needs to be taken during data analysis in terms of model selection and correction for camera blur [20, 59,60,61]. Yet, especially the combination of SPT with scattering techniques such as interferometric scattering (iSCAT) microscopy is attractive as the sample does not bleach and bleaching is arguably one of the biggest causes for artefacts in fluorescence and fluorescence fluctuation analysis [62].
7 Future Developments
The wealth of technology to study nanoscale dynamics is astonishing and developing further every day. The mapping of diffusion modes in LIESS-FCS (STED-FCS based) opened the door to spatially resolved diffusion maps [43, 63]. Pushing the spatial component further to image correlation methods will undoubtedly allow to resolve diffusion maps across entire cells at the nanoscale. Yet, one crucial component missing is the direct identification of interactions in a two-colour fashion. Due to the high spectral cross-talk and technical requirements for gating and pulse-interleaving schemes for excitation and depletion, STED fluorescence cross correlation spectroscopy (STED-FCCS) has not been realised yet. Though, we anticipate that such a tool will be invaluable and enable us to directly proof and characterise nanoscale interactions and heterogeneities.
Another very promising tool to study the nanoscale is MINFLUX which can be seen as a particle tracking approach with very high temporal and spatial resolution [64]. It avoids or minimises the typical issues of SPT such as photobleaching or the need for large labels and can easily achieve high frame-rates and large track length [65, 66]. Yet, many particles need to be sampled and the stability of the biological sample assured. We anticipate MINFLUX to evolve as a new gold standard for super-resolved tracking methodologies.
We are living in the exciting times of super-resolution microscopy enabling us to look beyond the curtain of diffraction. The discussed methods and their applications to nanoscale diffusion phenomena represent an extremely exciting field of study. However, all these technologies have been mostly applied on model membrane systems and immortalised cell lines. Future work will need to be undertaken to bring nanoscale diffusion measurements into tissues and whole organisms. Labelling the molecules in living cells and tissues still remains a bottleneck for STED and STED-FCS. STED-capable fluorescent proteins that can easily be implemented in living tissues will without doubt increase the applicability of STED and STED-FCS in the future. In addition, the combination of omics technologies with the biophysical characterisation of PM properties will bring us in a new age of research.
References
Sezgin E, Levental I, Mayor S, Eggeling C (2017) The mystery of membrane organization: composition, regulation and roles of lipid rafts. Nat Rev Mol Cell Biol 18:361–374
Semrau S, Schmidt T (2009) Membrane heterogeneity – from lipid domains to curvature effects. Soft Matter 5:3174
Kadir SR et al (2021) Nanoscape, a data-driven 3D real-time interactive virtual cell environment. eLife 10:1–36
Jacobson K, Liu P, Lagerholm BC (2019) The lateral organization and mobility of plasma membrane components. Cell 177:806–819
Schneider F, Colin-York H, Fritzsche M (2021) Quantitative bio-imaging tools to dissect the interplay of membrane and cytoskeletal actin dynamics in immune cells. Front Immunol 11:1–13
Nicolson GL (2014) The fluid—mosaic model of membrane structure: still relevant to understanding the structure, function and dynamics of biological membranes after more than 40 years. Biochim Biophys Acta Biomembr 1838:1451–1466
Sezgin E et al (2019) Measuring nanoscale diffusion dynamics in cellular membranes with super-resolution STED–FCS. Nat Protoc 14:1054–1083
Sezgin E et al (2017) Super-resolution optical microscopy for studying membrane structure and dynamics. J Phys Condens Matter 273001:273001
Hell SW et al (2015) The 2015 super-resolution microscopy roadmap. J Phys D Appl Phys 48:443001
Vangindertael J et al (2018) Methods and applications in fluorescence - an introduction to optical super-resolution microscopy for the adventurous biologist. Methods Appl Fluoresc 6:55
Sharonov A, Hochstrasser RM (2006) Wide-field subdiffraction imaging by accumulated binding of diffusing probes. Proc Natl Acad Sci 103:18911–18916
Jungmann R et al (2014) Multiplexed 3D cellular super-resolution imaging with DNA-PAINT and exchange-PAINT. Nat Methods 11:313–318
Hell SW, Wichmann J (1994) Breaking the diffraction resolution limit by stimulated emission: stimulated-emission-depletion fluorescence microscopy. Opt Lett 19:780
Klar TA, Hell SW (1999) Subdiffraction resolution in far-field fluorescence microscopy. Opt Lett 24:954–956
Kastrup L, Blom H, Eggeling C, Hell SW (2005) Fluorescence fluctuation spectroscopy in subdiffraction focal volumes. Phys Rev Lett 94:178104
Eggeling C et al (2009) Direct observation of the nanoscale dynamics of membrane lipids in a living cell. Nature 457:1159–1162
Lenne P-F et al (2006) Dynamic molecular confinement in the plasma membrane by microdomains and the cytoskeleton meshwork. EMBO J 25:3245–3256
Wawrezinieck L, Rigneault H, Marguet D, Lenne P-F (2005) Fluorescence correlation spectroscopy diffusion laws to probe the submicron cell membrane organization. Biophys J 89:4029–4042
Šachl R, Bergstrand J, Widengren J, Hof M (2016) Fluorescence correlation spectroscopy diffusion laws in the presence of moving nanodomains. J Phys D Appl Phys 49:114002
Lagerholm BC, Andrade DM, Clausen MP, Eggeling C (2017) Convergence of lateral dynamic measurements in the plasma membrane of live cells from single particle tracking and STED-FCS. J Phys D Appl Phys 50:063001
Mueller V et al (2011) STED nanoscopy reveals molecular details of cholesterol- and cytoskeleton-modulated lipid interactions in living cells. Biophys J 101:1651–1660
Andrade DM et al (2015) Cortical actin networks induce spatio-temporal confinement of phospholipids in the plasma membrane – a minimally invasive investigation by STED-FCS. Sci Rep 5:11454
Schneider F et al (2017) Diffusion of lipids and GPI-anchored proteins in actin-free plasma membrane vesicles measured by STED-FCS. Mol Biol Cell 28:1507–1518
Guzmán C et al (2014) The efficacy of raf kinase recruitment to the GTPase H-ras depends on H-ras membrane conformer-specific nanoclustering. J Biol Chem 289:9519–9533
Chojnacki J et al (2017) Envelope glycoprotein mobility on HIV-1 particles depends on the virus maturation state. Nat Commun 8:545
Sarangi NK, Roobala C, Basu JK (2017) Unraveling complex nanoscale lipid dynamics in simple model biomembranes: insights from fluorescence correlation spectroscopy in super-resolution stimulated emission depletion mode. Methods 140–141:198–211
Honigmann A, Mueller V, Hell SW, Eggeling C (2013) STED microscopy detects and quantifies liquid phase separation in lipid membranes using a new far-red emitting fluorescent phosphoglycerolipid analogue. Faraday Discuss 161:77–89
Sarangi NK et al (2016) Super-resolution stimulated emission depletion-fluorescence correlation spectroscopy reveals nanoscale membrane reorganization induced by pore-forming proteins. Langmuir 32:9649–9657
Favard C et al (2019) HIV-1 Gag specifically restricts PI(4,5)P2 and cholesterol mobility in living cells creating a nanodomain platform for virus assembly. Sci Adv 5(10):eaaw8651
Urbančič I et al (2018) Lipid composition but not curvature is the determinant factor for the low molecular mobility observed on the membrane of virus-like vesicles. Viruses 10:415
Barbotin A, Galiani S, Urbančič I, Eggeling C, Booth MJ (2019) Adaptive optics allows STED-FCS measurements in the cytoplasm of living cells. Opt Express 27:23378
Barbotin A, Urbančič I, Galiani S, Eggeling C, Booth M (2020) Background reduction in STED-FCS using a bi-vortex phase mask. ACS Photon. https://doi.org/10.1021/acsphotonics.0c00388
Shakya A, King JT (2018) Non-Fickian molecular transport in protein-DNA droplets. ACS Macro Lett 7:1220–1225
Galiani S et al (2021) Diffusion and interaction dynamics of the cytosolic peroxisomal import receptor PEX5. Biophys Rep 2(2):100055. https://doi.org/10.1016/j.bpr.2022.100055
Waithe D et al (2017) Optimized processing and analysis of conventional confocal microscopy generated scanning FCS data. Methods 140–141:62–73
Clausen MP et al (2015) A straightforward approach for gated STED-FCS to investigate lipid membrane dynamics. Methods 88:67–75
Lanzanò L et al (2017) Measurement of nanoscale three-dimensional diffusion in the interior of living cells by STED-FCS. Nat Commun 8:65
Wohland T, Rigler R, Vogel H (2001) The standard deviation in fluorescence correlation spectroscopy. Biophys J 80:2987–2999
Schneider F et al (2020) High photon count rates improve the quality of super-resolution fluorescence fluctuation spectroscopy. J Phys D Appl Phys 53:164003
Honigmann A et al (2014) Scanning STED-FCS reveals spatiotemporal heterogeneity of lipid interaction in the plasma membrane of living cells. Nat Commun 5:5412
Benda A, Ma Y, Gaus K (2015) Self-calibrated line-scan STED-FCS to quantify lipid dynamics in model and cell membranes. Biophys J 108:596–609
Maraspini R, Beutel O, Honigmann A (2017) Circle scanning STED fluorescence correlation spectroscopy to quantify membrane dynamics and compartmentalization. Methods 140–141:188–197
Schneider F et al (2018) Nanoscale spatiotemporal diffusion modes measured by simultaneous confocal and stimulated emission depletion nanoscopy imaging. Nano Lett 18:4233–4240
Hedde PN et al (2013) Stimulated emission depletion-based raster image correlation spectroscopy reveals biomolecular dynamics in live cells. Nat Commun 4:1–8
Garcia E, Bernardino de la Serna J (2018) Dissecting single-cell molecular spatiotemporal mobility and clustering at focal adhesions in polarised cells by fluorescence fluctuation spectroscopy methods. Methods 140–141:85–96
Bianchini P, Cardarelli F, Di Luca M, Diaspro A, Bizzarri R (2014) Nanoscale protein diffusion by STED-based pair correlation analysis. PLoS One 9:e99619
Sezgin E et al (2017) Polarity-sensitive probes for superresolution stimulated emission depletion microscopy. Biophys J 113:1321–1330
Carravilla P et al (2021) Long-term STED imaging of membrane packing and dynamics by exchangeable polarity-sensitive dyes. Biophys Rep 1:100023
Urbančič I et al (2021) Aggregation and mobility of membrane proteins interplay with local lipid order in the plasma membrane of T cells. FEBS Lett. https://doi.org/10.1002/1873-3468.14153
Kilian N et al (2018) Assessing photodamage in live-cell STED microscopy. Nat Methods 15:755–756
Manzo C, van Zanten TS, Garcia-Parajo MF (2011) Nanoscale fluorescence correlation spectroscopy on intact living cell membranes with NSOM probes. Biophys J 100:L8–L10
Winkler PM et al (2017) Transient nanoscopic phase separation in biological lipid membranes resolved by planar plasmonic antennas. ACS Nano. https://doi.org/10.1021/acsnano.7b03177
Regmi R et al (2017) Planar optical nanoantennas resolve cholesterol-dependent nanoscale heterogeneities in the plasma membrane of living cells. Nano Lett 17:6295–6302
Winkler PM, García-Parajo MF (2021) Correlative nanophotonic approaches to enlighten the nanoscale dynamics of living cell membranes. Biochem Soc Trans 0:1–13
Schneider F et al (2018) Statistical analysis of scanning fluorescence correlation spectroscopy data differentiates free from hindered diffusion. ACS Nano 12:8540–8546
Di Rienzo C, Gratton E, Beltram F, Cardarelli F (2013) Fast spatiotemporal correlation spectroscopy to determine protein lateral diffusion laws in live cell membranes. Proc Natl Acad Sci 110:12307–12312
Di Rienzo C, Gratton E, Beltram F, Cardarelli F (2016) Spatiotemporal fluctuation analysis: a powerful tool for the future nanoscopy of molecular processes. Biophys J 111:679–685
Clausen MP, Christoffer Lagerholm B (2011) The probe rules in single particle tracking. Curr Protein Pept Sci 12:699–713
Kusumi A et al (2005) Paradigm shift of the plasma membrane concept from the two-dimensional continuum fluid to the partitioned fluid: high-speed single-molecule tracking of membrane molecules. Annu Rev Biophys Biomol Struct 34:351–378
Reina F, Eggeling C, Lagerholm BC (2021) Lipid tracking at kilohertz sampling rates on live cell membranes through interferometric scattering microscopy. bioRxiv. https://doi.org/10.1101/2021.08.06.455401
Reina F et al (2021) TRAIT2D: a software for quantitative analysis of single particle diffusion data. bioRxiv 5390. https://doi.org/10.1101/2021.03.04.433888
Reina F (2018) Applications of interferometric scattering (iSCAT) microscopy to single particle tracking in model and cell membranes. Doctoral dissertation, University of Oxford
Bernabé-Rubio M, Bosch-Fortea M, Alonso MA, Bernardino de la Serna J (2021) Multi-dimensional and spatiotemporal correlative imaging at the plasma membrane of live cells to determine the continuum nano-to-micro scale lipid adaptation and collective motion. Methods 193:136–147
Balzarotti F et al (2017) Nanometer resolution imaging and tracking of fluorescent molecules with minimal photon fluxes. Science 355:606–612
Eilers Y, Ta H, Gwosch KC, Balzarotti F, Hell SW (2018) MINFLUX monitors rapid molecular jumps with superior spatiotemporal resolution. Proc Natl Acad Sci 115:6117–6122
Schmidt R et al (2021) MINFLUX nanometer-scale 3D imaging and microsecond-range tracking on a common fluorescence microscope. Nat Commun 12:1–12
Acknowledgements
We thank SciLifeLab Advanced Light Microscopy facility and National Microscopy Infrastructure (VR-RFI 2016-00968) for their support on imaging. ES is supported by grants from Swedish Research Council Starting Grant (2020-02682), from Karolinska Institutet and from the SciLifeLab National COVID-19 Research Program, financed by the Knut and Alice Wallenberg Foundation. FS acknowledges support by EMBO (ALTF 849-2020) and HFSP (LT000404/2021-L).
Author Contributions
ES and FS wrote the manuscript and prepared the figures. Both authors contributed equally to the chapter.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2022 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Schneider, F., Sezgin, E. (2022). Diffusion Measurements at the Nanoscale with STED-FCS. In: Šachl, R., Amaro, M. (eds) Fluorescence Spectroscopy and Microscopy in Biology. Springer Series on Fluorescence, vol 20. Springer, Cham. https://doi.org/10.1007/4243_2022_27
Download citation
DOI: https://doi.org/10.1007/4243_2022_27
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-031-30361-6
Online ISBN: 978-3-031-30362-3
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)