
Abstract
Metabolic stress is an important stimulus that promotes apoptosis-mediated tumor suppression. Metabolic stress arises in tumors from multiple factors that include insufficient nutrient supply caused by deficient angiogenesis and high metabolic demand of unrestrained cell proliferation. The high metabolic demand of tumor cells is only exacerbated by reliance on the inefficient process of glycolysis for energy production. Recently it has become clear that tumor cells survive metabolic stress through the catabolic process of autophagy. Autophagy also functions as a tumor suppression mechanism by preventing cell death and inflammation and by protecting the genome from damage and genetic instability. How autophagy protects the genome is not yet clear but may be related to its roles in sustaining metabolism or in the clearance of damaged proteins and organelles and the mitigation of oxidative stress. These findings illuminate the important role of metabolism in cancer progression and provide specific predictions for metabolic modulation in cancer therapy.
Access provided by Autonomous University of Puebla. Download conference paper PDF
Similar content being viewed by others


Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Origins of Metabolic Stress in Tumors
Metabolic stress is a common occurrence in human tumors and is caused by multiple factors. Tumors initially lack a blood supply and when growth progresses towards a mass in the range of 1 mm in diameter, passive diffusion of nutrients is insufficient to sustain those tumor cells in the center of the mass, which suffer metabolic stress (Folkman 2006). Continuation of tumor growth requires angiogenesis to supply nutrients, and recruitment of a blood supply to the tumor can ameliorate metabolic stress. Using hypoxia as a marker for metabolic stress in tumors, induction of metabolic stress during tumor formation and stress abatement following angiogenesis is readily apparent (Nelson et al. 2004). Following successful tumor angiogenesis, metabolic stress is still a factor since blood vessel formation is abnormal and subject to intermittent collapse that inflicts metabolic stress in established tumors. Thus, unlike normal tissues, tumors lack a constant, uninterrupted nutrient supply and thereby are regularly assaulted by bouts of metabolic stress.
Another contributing factor to metabolic stress in tumors is their high metabolic demand caused by unregulated, unrelenting tumor cell growth, a hallmark of cancer (Hanahan and Weinberg 2000). Normal tissues have strict controls that limit cell division to specific developmental periods and circumstances that are tightly linked to nutrient and growth factor availability. Tumor cells lack these controls and proliferate independently, by either autocrine mechanisms or despite the absence of growth factors and nutrients. This dissociation of growth control from nutrient availability is a significant contributor to metabolic stress in tumors that leads to cellular damage and cell death by metabolic catastrophe (Jin et al. 2007). Finally, inefficient energy production in tumor cells due to metabolism dominated by glycolysis has long been recognized as critical distinction between normal and tumor cells (Warburg 1956). The juxtaposition of restricted nutrient supply, high energy demand, and inefficient energy production exemplifies the fundamental importance of metabolic stress to tumor physiology.
2 Activation of Tumor Suppression by Apoptosis in Response to Cellular Stress
Cellular stress is a well-known trigger of cell death by apoptosis, which is a critical tumor suppression mechanism. Conceptually, cellular stress that results in excessive cell damage activates apoptosis as a means to eliminate dysfunctional and potentially dangerous cells that can acquire mutations and progress toward cancer. There are multiple independent pathways by which apoptosis is triggered in tumors (Adams and Cory 2007; Gelinas and White 2005). Two stress-related pathways that signal apoptosis are the DNA damage and cellular stress response pathway controlled by the p53 tumor suppressor, and the metabolic stress pathway (Fig. 1) (Jin et al. 2007; Jin and White 2007; White 2006).

Activation of the apoptotic tumor suppression response to metabolic stress. See text for explanation
p53 activates apoptosis in part through transcriptional upregulation of the pro-apoptotic BH3-only proteins Puma and Noxa (Vousden and Lane 2007), whereas metabolic stress signals through the pro-apoptotic BH3-only protein Bim (Nelson et al. 2004; Tan et al. 2005). BH3-only proteins, in turn, signal apoptosis through the multidomain proapoptotic Bax and Bak proteins, which permeabilize the outer mitochondrial membrane causing the release of factors that promote caspase activation leading to cell death (Adams and Cory 2007). How Bim is activated in response to metabolic stress to induce apoptosis is not known, but Bim is a critical epithelial tumor suppressor in the p53-independent apoptotic pathway (Degenhardt et al. 2002; Tan et al. 2005).
3 Modulation of the Apoptotic Response by Cancer Therapy
One of the advantages of a comprehensive knowledge of apoptosis regulation is the ability to use that information to rationally tailor cancer therapies aimed at activating or enhancing the apoptotic response. As such, the development of cancer treatments specifically targeting the apoptotic pathway shows great promise (Fesik 2005; Wu et al. 2007). For example, promoting p53 activation by inducing DNA damage with cytotoxic chemotherapy is one means to shift tumor cells toward apoptosis during cancer therapy (Fig. 1). Another example is the induction of Bim by the taxane class of chemotherapeutic drugs, which results in Bim-mediated apoptosis (Fig. 1) and tumor regression in preclinical models (Tan et al. 2005). These types of therapeutic strategies can shift the balance in tumor cells from survival to death to facilitate treatment response. Indeed, many of the newer noncytotoxic targeted therapies that inhibit signal transduction, relieve apoptosis suppression, thereby favoring tumor cell death.
4 Inactivation of Apoptosis in Tumor Progression
Cancer cells, however, can defeat proapoptotic tumor suppression mechanisms by inactivating apoptosis in various ways that enable tumor progression and treatment resistance (Fig. 2) (Adams and Cory 2007). For example, anti-apoptotic Bcl-2 family members are overexpressed in many tumors, bind and inhibit Bax and Bak to block apoptosis, enabling tumor cell survival, tumor progression and treatment resistance. Analogously, many viruses have acquired these same Bcl-2-like anti-apoptotic mechanisms among their repertoire of oncogenes (Cuconati and White 2002). One example is the E1B19K gene product of the DNA tumor virus adenovirus (Fig. 2), which blocks apoptosis by binding and inhibiting Bax and Bak to facilitate oncogenesis (Cuconati and White 2002; Nelson et al. 2004; White 2006). Tumors acquire mechanisms for inactivating apoptosis that can also be indirect. One example is the upregulation of the ubiquitin ligase for p53, Mdm-2, which promotes p53 degradation in the ubiquitin proteasome pathway to promote tumorigenesis (Vousden and Lane 2007). Another is the phosphorylation and proteasome-mediated degradation of Bim by the Map kinase Erk, which inactivates apoptosis, thereby promoting tumorigenesis and resistance to taxane chemotherapy (Fig. 2) (Tan et al. 2005). Defining how tumors defeat the apoptotic response as exemplified above can dictate rational approaches to anti-cancer therapies.

Inactivation of apoptosis in cancer cells. See text for explanation
5 Autophagy Mediates Tumor Cell Survival to Metabolic Stress
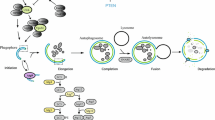
Once tumor cells evolve defects in apoptosis, they fail to die in response to metabolic stress, but this does not suffice to explain how these undead cells survive long periods of metabolic stress. By simulating in vivo metabolic stress in tumors in vitro, we discovered that metabolic stress activates the catabolic survival pathway of autophagy (Degenhardt et al. 2006). This autophagy allows tumor cells to cannibalize themselves to sustain their metabolism during extended periods of starvation (Degenhardt et al. 2006). Furthermore, autophagy in tumors in vivo localizes to regions of metabolic stress and tumor cells with defects in autophagy fail to survive in these tumor regions (Fig. 3A) (Degenhardt et al. 2006; Karantza-Wadsworth et al. 2007; Mathew et al. 2007b). These findings revealed that autophagy is an important component of tumor physiology linked to metabolic stress survival.

Dual roles for autophagy in suppressing tumorigenesis. A Cells at the center of a tumor are subjected to metabolic stress can either undergo apoptosis or, in this illustration, can activate autophagy to enable long-term survival. Eventually, with acquisition of a blood supply through angiogenesis, tumor progression can occur. B Defects in autophagy promote either necrotic or apoptotic cell death and the inflammation and cytokine production that results can promote tumorigenesis by a non-cell-autonomous mechanism. C Defects in autophagy cause DNA damage and genome instability due to the failure to mitigate metabolic stress. The accelerated mutation rate that results can facilitate tumor progression by a cell-autonomous mechanism
Starvation promotes autophagy, which through the action of a host of essential autophagy regulators such as Beclin1 and ATG proteins, double membrane vesicles form that engulf cytoplasm and organelles that traffic this cargo to lysosomes for degradation (Klionsky 2007; Levine and Kroemer 2008; Mizushima 2007). By recycling intracellular nutrients, autophagy can support cellular metabolism during nutrient and growth factor deprivation. In mammalian development, autophagy is essential to sustain metabolism to enable survival during the neonatal starvation period (Kuma et al. 2004). Autophagy also plays a critical role in maintaining protein and organelle quality control exemplified by the accumulation of protein aggregates and aberrant mitochondria associated with neuronal degeneration in mice (Hara et al. 2006; Komatsu et al. 2005, 2006, 2007). Thus, autophagy is an important component of the metabolic stress response that promotes normal cell survival by providing an alternate energy source and by degrading damaged proteins and organelles.
6 Autophagy Is a Tumor Suppression Mechanism
Although autophagy promotes tumor cell survival, paradoxically, allelic loss of the essential autophagy gene beclin1 is found with high frequency in human cancers, and beclin1 +/– mutant mice are tumor-prone (Liang et al. 1999; Qu et al. 2003; Yue et al. 2003). Furthermore, allelic loss of beclin1 and defective autophagy promotes tumorigenesis of immortal mouse kidney and mammary epithelial cells (Degenhardt et al. 2006; Karantza-Wadsworth et al. 2007; Mathew et al. 2007b). It then became a challenge to decipher the mechanism by which loss of function of a survival pathway of autophagy promotes tumor growth. Although on the surface it may appear counterintuitive that loss of a survival function such as autophagy causes tumorigenesis, this is not the first time this situation has occurred. Defects in DNA repair, for example, compromise cell survival to DNA damage, yet the accelerated mutation rate promotes tumorigenesis (Hanahan and Weinberg 2000). In this setting, the elevated mutation rate that results from DNA repair deficiency compensates for the reduced survival to DNA damage by expediting tumor evolution. As such, enhanced tumor cell survival is not always associated with promotion of tumorigenesis.
7 Autophagy Suppresses Cell Death and Inflammation to Limit Tumor Progression
In tumor cells as in normal cells, autophagy promotes survival to metabolic stress. In apoptosis-competent tumor cells, autophagy suppresses apoptosis in response to metabolic stress (Boya et al. 2005; Degenhardt et al. 2006; Karantza-Wadsworth et al. 2007; Mathew et al. 2007b). In apoptosis-defective tumor cells, autophagy enables long-term survival to metabolic stress such that restoration of nutrients permits efficient recovery (Degenhardt et al. 2006; Karantza-Wadsworth et al. 2007; Mathew et al. 2007b). This prosurvival function of autophagy prevents tumor cell death, and as a result, the inflammation, macrophage infiltration, and tumor-promoting cytokine production that result from excessive tumor cell death is prevented (Degenhardt et al. 2006; Jin and White 2007; Karantza-Wadsworth and White 2007; Mathew et al. 2007a; Mathew and White 2007). This chronic inflammation in autophagy-defective tumors is associated with enhanced tumor growth (Fig. 3B,C). In tumors where apoptosis is intact, autophagy defects can promote chronic apoptosis in response to metabolic stress, and excess apoptotic tumor cells can promote macrophage infiltration associated with poor prognosis (Fig. 3B) (Condeelis and Pollard 2006). In tumors with defects in apoptosis, deficient autophagy results in chronic necrotic cell death due to metabolic catastrophe in response to metabolic stress analogous to a corrupted would-healing response associated with poor prognosis (Fig. 3B) (Degenhardt et al. 2006; Jin et al. 2007; Jin and White 2007; Mathew et al. 2007a). While the role of inflammation in tumorigenesis is complex, many aspects of the inflammatory response are associated with tumor progression (Balkwill et al. 2005). Thus, by preserving cell health and viability, autophagy serves as an important non-cell-autonomous tumor-suppression mechanism by limiting inflammation (Degenhardt et al. 2006; Mathew et al. 2007a).
8 Autophagy Prevents Genome Damage to Suppress Tumorigenesis
While autophagy permits tumor cells to tolerate metabolic stress, it serves an important protective function by limiting the accumulation of damaged proteins and organelles (Komatsu et al. 2007; Komatsu et al. 2005), which may ultimately protect the genome and suppresses tumor progression (Mathew et al. 2007a). Tumorigenesis commonly requires stable genetic or epigenetic changes acquired through mutation (Hanahan and Weinberg 2000). Interestingly, accumulation of damaged proteins and organelles can be associated with elevated oxidative stress that may directly or indirectly promote genome damage and mutation. Indeed, autophagy-defective immortal mouse kidney and mammary epithelial cells have an elevated DNA damage response in metabolic stress and are prone to gene amplification and chromosome gains and losses (Karantza-Wadsworth et al. 2007; Mathew et al. 2007b). These findings provide evidence that autophagy protects the genome, providing the first link between metabolism and limiting DNA damage and mutation that can promote tumorigenesis by a cell-autonomous mechanism (Fig. 3C) (Jin and White 2007; Karantza-Wadsworth and White 2007; Mathew et al. 2007a; Mathew and White 2007). Determining the molecular mechanism by which autophagy protects cells from genome damage will be of great interest.
9 Modulation of Tumor Cell Metabolism in Cancer Therapy
The findings described above illustrate that the metabolic stress response is linked to both apoptosis and autophagy, and understanding these pathways is essential for developing cancer therapies. As autophagy is clearly a survival pathway utilized by tumor cells to survive metabolic stress, inhibitors of autophagy may be therapeutically useful (Jin et al. 2007; Jin and White 2007; Karantza-Wadsworth and White 2007; Mathew et al. 2007a; Mathew and White 2007). In this setting, it will be important to induce acute rather than chronic tumor cell death to avoid inflammation and genome instability associated with impairment of autophagy. Furthermore, since autophagy provides a protective tumor-suppression function, autophagy stimulators may be particularly useful in the setting of cancer prevention. Therapeutically stimulating autophagy in individuals at risk may suppress cell death, inflammation, and genome damage, thereby restricting tumor progression (Jin et al. 2007; Jin and White 2007; Karantza-Wadsworth and White 2007; Mathew et al. 2007a; Mathew and White 2007). Finally, as many tumors have defects in autophagy in conjunction with high metabolic demand that is reliant on the inefficient process of glycolysis for energy production, strategies to inflict therapeutic starvation should be considered (Jin et al. 2007). In that way, we can take advantage of the most fundamental factor that discriminates normal cells from cancer cells (Warburg 1956).
References
Adams JM, Cory S (2007) Bcl-2-regulated apoptosis: mechanism and therapeutic potential. Curr Opin Immunol 19:488–496
Balkwill F, Charles KA, Mantovani A (2005) Smoldering and polarized inflammation in the initiation and promotion of malignant disease. Cancer Cell 7:211–217
Boya P, Gonzalez-Polo RA, Casares N, Perfettini JL, Dessen P, Larochette N, Metivier D, Meley D, Souquere S, Yoshimori T et al (2005) Inhibition of macroautophagy triggers apoptosis. Mol Cell Biol 25:1025–1040
Condeelis J, Pollard JW (2006) Macrophages: obligate partners for tumor cell migration, invasion and metastasis. Cell 124:263–266
Cuconati A, White E (2002) Viral homologs of BCL-2: role of apoptosis in the regulation of virus infection. Genes Dev 16:2465–2478
Degenhardt K, Chen G, Lindsten T, White E (2002) BAX and BAK mediate p53-independent suppression of tumorigenesis. Cancer Cell 2:193–203
Degenhardt K, Mathew R, Beaudoin B, Bray K, Anderson D, Chen G, Mukherjee C, Shi Y, Gelinas C, Fan Y et al (2006) Autophagy promotes tumor cell survival and restricts necrosis, inflammation and tumorigenesis. Cancer Cell 10:51–64
Fesik SW (2005) Promoting apoptosis as a strategy for cancer drug discovery. Nat Rev 5:876–885
Folkman J (2006) Angiogenesis. Annu Rev Med 57:1–18
Gelinas C, White E (2005) BH3-only proteins in control: specificity regulates MCL-1 and BAK-mediated apoptosis. Genes Dev 19:1263–1268
Hanahan D, Weinberg RA (2000) The hallmarks of cancer. Cell 100:57–70
Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H et al (2006) Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 441:885–889
Jin S, White E (2007) Role of autophagy in cancer: management of metabolic stress. Autophagy 3:28–31
Jin S, DiPaola RS, Mathew R, White E (2007) Metabolic catastrophe as a means to cancer cell death. J Cell Sci 120:379–383
Karantza-Wadsworth V, White E (2007) Role of autophagy in breast cancer. Autophagy 3:610–613
Karantza-Wadsworth V, Patel S, Kravchuk O, Chen G, Mathew R, Jin S, White E (2007) Autophagy mitigates metabolic stress and genome damage in mammary tumorigenesis. Genes Dev 21:1621–1635
Klionsky DJ (2007) Autophagy: from phenomenology to molecular understanding in less than a decade. Nat Rev Mol Cell Biol 8:931–937
Komatsu M, Waguri S, Ueno T, Iwata J, Murata S, Tanida I, Ezaki J, Mizushima N, Ohsumi Y, Uchiyama Y et al (2005) Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol 169:425–434
Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E et al (2006) Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 441:880–884
Komatsu M, Waguri S, Koike M, Sou YS, Ueno T, Hara T, Mizushima N, Iwata JI, Ezaki J, Murata S et al (2007) Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell 131:1149–1163
Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, Ohsumi Y, Tokuhisa T, Mizushima N (2004) The role of autophagy during the early neonatal starvation period. Nature 432:1032–1036
Levine B, Kroemer G (2008) Autophagy in the pathogenesis of disease. Cell 132:27–42
Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H, Levine B (1999) Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 402:672–676
Mathew R, White E (2007) Why sick cells produce tumors: the protective role of autophagy. Autophagy 3:502–505
Mathew R, Karantza-Wadsworth V, White E (2007a) Role of autophagy in cancer. Nat Rev Cancer 7:961–967
Mathew R, Kongara S, Beaudoin B, Karp CM, Bray K, Degenhardt K, Chen G, Jin S, White E (2007b) Autophagy suppresses tumor progression by limiting chromosomal instability. Genes Dev 21:1367–1381
Mizushima N (2007) Autophagy: process and function. Genes Dev 21:2861–2873
Nelson DA, Tan TT, Rabson AB, Anderson D, Degenhardt K, White E (2004) Hypoxia and defective apoptosis drive genomic instability and tumorigenesis. Genes Dev 18:2095–2107
Qu X, Yu J, Bhagat G, Furuya N, Hibshoosh H, Troxel A, Rosen J, Eskelinen EL, Mizushima N, Ohsumi Y et al (2003) Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest 112:1809–1820
Tan TT, Degenhardt K, Nelson DA, Beaudoin B, Nieves-Neira W, Bouillet P, Villunger A, Adams JM, White E (2005) Key roles of BIM-driven apoptosis in epithelial tumors and rational chemotherapy. Cancer Cell 7:227–238
Vousden KH, Lane DP (2007) p53 in health and disease. Nat Rev Mol Cell Biol 8:275–283
Warburg O (1956) On respiratory impairment in cancer cells. Science 124:269–270
White E (2006) Mechanisms of apoptosis regulation by viral oncogenes in infection and tumorigenesis. Cell Death Differ 13:1371–1377
Wu H, Tschopp J, Lin SC (2007) Smac mimetics and TNFalpha: a dangerous liaison? Cell 131:655–658
Yue Z, Jin S, Yang C, Levine AJ, Heintz N (2003) Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc Natl Acad Sci U S A 100:15077–15082
Author information
Authors and Affiliations
Corresponding author
Editor information
Rights and permissions
Copyright information
© 2008 Springer-Verlag Berlin Heidelberg
About this paper
Cite this paper
White, E. (2008). Role of the Metabolic Stress Responses of Apoptosis and Autophagy in Tumor Suppression. In: Kroemer, G., Mumberg, D., Keun, H., Riefke, B., Steger-Hartmann, ., Petersen, K. (eds) Oncogenes Meet Metabolism. Ernst Schering Foundation Symposium Proceedings, vol 2007/4. Springer, Berlin, Heidelberg. https://doi.org/10.1007/2789_2008_087
Download citation
DOI: https://doi.org/10.1007/2789_2008_087
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-540-79477-6
Online ISBN: 978-3-540-79478-3
eBook Packages: MedicineMedicine (R0)