
Abstract
For the past two decades several scholarly reviews have appeared on the inwardly rectifying potassium (Kir) channels. We would like to highlight two efforts in particular, which have provided comprehensive reviews of the literature up to 2010 (Hibino et al., Physiol Rev 90(1):291–366, 2010; Stanfield et al., Rev Physiol Biochem Pharmacol 145:47–179, 2002). In the past decade, great insights into the 3-D atomic resolution structures of Kir channels have begun to provide the molecular basis for their functional properties. More recently, computational studies are beginning to close the time domain gap between in silico dynamic and patch-clamp functional studies. The pharmacology of these channels has also been expanding and the dynamic structural studies provide hope that we are heading toward successful structure-based drug design for this family of K+ channels. In the present review we focus on placing the physiology and pharmacology of this K+ channel family in the context of atomic resolution structures and in providing a glimpse of the promising future of therapeutic opportunities.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Cytosolic G-loop gate
- GIRK
- Helix bundle crossing gate
- K+ transport channel
- KATP
- Phosphoinositides
- Potassium inward rectifiers
- Resting potential
1 Historical Perspective: The Pre-structure Era
Inward Rectification and Dependence of Conductance on [K+]o: Kir currents were first described in skeletal muscle fibers, where Bernard Katz observed (Katz 1949a, b) that when these cells were immersed in solutions containing high potassium the membrane conductance was larger at hyperpolarizing potentials. This behavior was the opposite from what Cole and Curtis had reported in squid giant axon (Cole and Curtis 1941), where the depolarization-induced non-linear conductance had been described as rectification, borrowing from electrical engineering terminology used for diodes. The fact that the first example of outward rectification, demonstrated by the delayed rectifier Kv current, was also in Katz’s laboratory with Hodgkin and Huxley (Hodgkin et al. 1952; Katz 1949a, b), led Katz to describe the skeletal muscle conductance in high K+ as “anomalous rectification” (“propriétés détectrices anormales,” from the French description). This anomalous rectification was later (Adrian et al. 1970) renamed “inward rectification,” the term most used at the present. Another difference between the Kir and the outwardly rectifying Kv currents was their respective voltage dependence. Kir currents were voltage-independent but instead their activation depended on the driving force (Vm-EK, i.e. the distance of the membrane potential Vm from the equilibrium potential EK for K+ ions). Activation was shown to depend on (Vm-EK) if [K+]o, but not [K+]i, was changed (Hagiwara and Yoshii 1979; Leech and Stanfield 1981; Stanfield et al. 2002). The dependence of Kir current rectification and conductance on [K+]o is shown in Fig. 1a (Hagiwara et al. 1976). Early on, the inward rectification was attributed to block of the channel by particles from the cytosol (Armstrong 1969; Hille and Schwarz 1978; Standen and Stanfield 1978), and later the intracellular blocking substances were identified to be the millimolar levels of Mg2+ as well as polyamines that exist in sub-millimolar concentrations inside of cells (Lopatin et al. 1994; Matsuda et al. 1987). Upon hyperpolarization, the inward Kir current showed a time-independent increase due to fast Mg2+ unblock, followed by a time-dependent increase due to slow polyamine unblock (Lopatin et al. 1995).

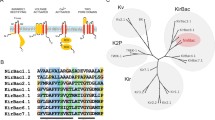
Kir channel inward rectification and phylogenetic/functional classification. (a) I-V relationship of the starfish Kir at four different [K+]o. Continuous and broken lines indicate instantaneous and steady-state currents, respectively (from Hagiwara et al. 1976) (b) Phylogenetic analysis of the 16 known subunits of human Kir channels (for identity/similarity see Fig. 10). These subunits have been classified into four functional groups
1.1 Tissue Distribution of Kir Family Members
Kir channels have been found in a wide variety of cells, including: cardiac myocytes (Beeler and Reuter 1970; Kurachi 1985; McAllister and Noble 1966; Rougier et al. 1967), neurons (Brown and Carpentier 1990; Gahwiler and Brown 1985; Lacey et al. 1988; North et al. 1987; Takahashi 1990; Williams et al. 1988), blood cells (Lewis et al. 1991; McKinney and Gallin 1988), osteoclasts (Sims and Dixon 1989), endothelial cells (Silver and DeCoursey 1990), glial cells (Kuffler and Nicholls 1966; Newman 1984), epithelial cells (Greger et al. 1990; Hebert et al. 2005; Lorenz et al. 2002; Lu et al. 2002), and oocytes (Hagiwara et al. 1978; Hagiwara et al. 1976; Hagiwara and Takahashi 1974). Figure 2 shows the tissue/organ expression of each Kir subfamily throughout the body. The Kir tissue expression has been comprehensively reviewed (de Boer et al. 2010; Hibino et al. 2010). The role of Kir channels lies in contributing to the cellular resting potential and keeping Vm near EK. This serves to reduce action potential firing in excitable cells, to control K+ transport in non-excitable cells or to transduce extracellular (by external stimuli) to intracellular (by internal metabolites) communication and vice versa. In 1993 the first 3 of the 16 members of the Kir channel family were cloned (Dascal et al. 1993; Ho et al. 1993; Kubo et al. 1993a; Kubo et al. 1993b). Based on sequence alignment (see Fig. 9) and phylogenetic analysis, the 16 human isoforms have been classified into seven subfamilies (Kir1-7) and four functional groups (a. the K+ transport, b. the classical, c. the ATP-sensitive, and d. the G protein-gated K+ channels) (Fig. 1b). The strong sequence similarity among family members (30–99%) (Fig. 10) allows for formation of heteromeric assemblies along with homomeric ones, in one case across subfamilies (Kir4.1 and Kir5.1) and in most cases within subfamilies (e.g., Kir3.1/3.2 in neuronal tissues and Kir3.1/3.4 in atrial cells of the heart). The resulting heteromers display unique functional properties compared to the homomers that will be discussed individually for each subfamily.

Expression in indicated organs and tissues of Kir channel members of each of the seven subfamilies. Blank indicates no, striations intermediate, and black prominent levels of expression. Adapted (permission obtained) from de Boer et al. (2010)
1.2 Kir Channel Gating
The signaling phospholipid, phosphatidylinositol 4,5-bisphosphate [PI(4,5)P2 or PIP2] found in the inner leaflet of the plasma membrane plays an essential role in supporting Kir channel gating (Hilgemann and Ball 1996; Huang et al. 1998; Logothetis et al. 2015; Petit-Jacques et al. 1999; Sui et al. 1998). Excision into inside-out patches in ATP-free solutions showed a typical gradual decline in channel activity, referred to as “run-down” (Hilgemann and Ball 1996). Run-down activity could be restored by hydrolyzable forms of ATP or by PIP2 (Hilgemann and Ball 1996; Huang et al. 1998; Petit-Jacques et al. 1999; Sui et al. 1998). The seven Kir subfamily members could be classified into four groups depending on the degree of stereospecificity and affinity to PI(4,5)P2 (over PI(3,4,5)P3 and PI(3,4)P2) (Rohacs et al. 2003): Highest or Group 1 (Kir2.1, Kir2.4, Kir4.1), Moderate or Group 2 (Kir1.1, Kir2.2, Kir2.3, Kir4.2, Kir7.1), Weak or Group 3 (Kir3.4, Kir3.1/3.4), Lowest or Group 4 (Kir6.2, Kir6.2/SURs). Mutagenesis studies identified channel regions critical for the effects of PIP2 and linked basic and non-basic residues to sensitivity to PIP2 and Kir channelopathies (Lopes et al. 2002).
1.3 Kir Channel Trafficking
Kir channel mutations that lead to channelopathies, especially those that affect trafficking, have been recently reviewed (Zangerl-Plessl et al. 2019). As other transmembrane proteins, Kir channels are translated in the endoplasmic reticulum (ER) and transported through the Golgi apparatus and the trans-Golgi network to the plasma membrane through the process known as forward trafficking. Upon removal from the plasma membrane, channel proteins enter the degradation pathway through trafficking to the early endosome and the multivesicular body to the lysosome, through a process known as backward trafficking. Kir channels could instead of becoming degraded be recycled back to the plasma membrane from the early endosome to the trans-Golgi network. Kir channel mutations resulting in loss (LOF) or gain (GOF) of function are associated with a variety of human diseases, including Bartter syndrome type II (Kir1.1 LOF), Andersen-Tawil syndrome (Kir2.1 LOF and GOF), thyrotoxic hypokalemic periodic paralysis (Kir2.6 LOF), Keppen-Lubinsky syndrome (Kir3.2 LOF), familial hyperaldosteronism type III and long QT syndrome 12 (Kir3.4 LOF), EAST/SeSAME syndrome (Kir4.1 LOF), Cantú syndrome (Kir6.1 or SUR2 GOF) and hyperinsulinism and hypoglycemia (Kir6.2 LOF) and diabetes (Kir6.2 GOF), Leber congenital amaurosis type 16 and Snowflake vitreoretinal degeneration (Kir7.1 LOF). In many of the diseases mentioned above LOF has been associated with defective forward trafficking, while GOF mutations or loss of specificity mutations (e.g., some Kir3.4 mutations) are not likely to be related to trafficking abnormalities. The likely causes of trafficking defects are either (1) defects in trafficking motifs compromising interactions with the proteins involved in the trafficking machinery, or (2) protein structure defects leading to channel misfolding, destabilization and ER-associated degradation (Zangerl-Plessl et al. 2019).
1.4 The Structural Era
1.4.1 Kir Structures by 2020
The first Kir channel high-resolution crystal structure to be solved in 2002 was the cytoplasmic domain (CTD) of Kir3.1 (Nishida and MacKinnon 2002). In this crystal structure, the transmembrane domain of the channel was removed, and the N-terminus was fused to the CTD and expressed in bacteria (PDBID: 1N9P). Soon after this structure, the CTDs of Kir2.1 (PDBID: 1U4F, 2005), (Pegan et al. 2005) pointing to the potential of a cytosolic constriction, coined as the G-loop gate, and Kir3.2 (PDBID: 2E4F, 2007) (Inanobe et al. 2007) were solved. Subsequently, in 2007, a crystal structure of a Kir3.1 prokaryotic Kir channel chimera was solved (PDBID: 2QKS) (Table 1). In this structure, two thirds of the transmembrane domains (TMDs) were replaced with the corresponding region of a homologous prokaryotic Kir channel, revealing the continuity of the permeation pathway from the membrane (2/3 prokaryotic, 1/3 mammalian) to the cytosolic (mammalian) domains of the Kir3.1 channel. Two distinct conformations of the G-loop gate were captured, in one structure the apex of the CTD was open (or dilated), and in the other it was closed (or constricted) (Nishida et al. 2007). The transmembrane gate, referred to as the helix bundle crossing (HBC) gate was captured in the closed conformation in both structures. Even though Nishida and colleagues were not able to show that the chimera they constructed was functional (Nishida et al. 2007), a subsequent paper showed that the purified chimera once reconstituted in planar lipid bilayers with PIP2 was indeed functional (Leal-Pinto et al. 2010). In 2009, a crystal structure of the chicken Kir2.2, 90% identical to the human Kir2.2, was solved (PDBID:3JYC), where large structured turrets and an unusual selectivity filter entryway were seen, which may explain the relative insensitivity of eukaryotic Kir channels to toxins (Tao et al. 2009). In 2011, the crystal structure of Kir2.2 channel in complex with a short-chain derivative of PIP2 was solved [PDBID:3SPI] (see Fig. 3b). This structure showed that PIP2 binds at an interface between the TMD and CTD of the Kir2.2 channel. Upon PIP2 binding, a flexible linker between the TMD and CTD transforms to a helical structure, which causes a 6 Å translation of the CTD toward the TMD, as the HBC gate begins to open (Hansen et al. 2011). In the same year (2011), the crystal structure of Kir3.2 in complex with sodium and PIP2 was solved (PDBID:3SYA). The structures suggested that the presence of PIP2 couples the G loop and HBC gates to open in a coordinated manner. The intracellular Na+ binding site was also confirmed in this structure (Whorton and MacKinnon 2011). In 2013, the crystal structure of Kir3.2-βγ G-protein (Gβγ) complex was solved (PDBID:4KFM). In this structure, the Gβγ subunit bound to the interfaces between four pairs of adjacent Kir3.2 channel CTD subunits that were also bound to sodium and a PIP2 analog. The structure was thought to represent a “pre-open” state, an intermediate between the closed and open but still a non-conducting conformation of the channel (Whorton and MacKinnon 2013) (shown later in Fig. 7b). In 2017, a hetero-octameric pancreatic KATP channel in complex with the non-competitive inhibitor glibenclamide was solved by cryo-electron microscopy (EM) at a 5.6-Å resolution [PBDID:5WUA]. This structure showed four SUR1 regulatory subunits located in the periphery of a centrally located Kir6.2 channel tetramer (Li et al. 2019). In 2017, Wu and colleagues solved complexes of the KATP channel (KIR6.2/SUR1) with ATP, Mg-ADP, and ATP/glibenclamide, respectively, by using cryo-EM (PDBID: 5YW8, 5YWC, 5YKE). These structures depict the binding site of glibenclamide, ATP, and suggested a mechanism of how Mg-ADP binding on nucleotide-binding domains drives a conformational change of the SUR1 subunit (Wu et al. 2018). Martin and colleagues solved a cryo-EM structure of SUR1/Kir6.2 channel bound to glibenclamide and ATP at 3.63 Å resolution (PDBID: 6BAA). The structure showed that the drug bonded to the transmembrane bundle of SUR1, and mutation of the interacting residues in the binding site reduced the channel sensitivity to the drug (Martin et al. 2017). Lee and colleagues solved two cryo-EM structures of the human KATP channel (Kir6.2/SUR1) in complex with Mg2+ and nucleotides, referred to as the quatrefoil (PDBID: 6C3O) and propeller (PDBID: 6C3P) forms. In both forms, ATP binds to the inhibitory site in Kir6.2. Mg2+-ATP and Mg2+-ADP bind to the degenerate and consensus sites, respectively, in the nucleotide-binding domains of SUR1. A lasso extension interface between Kir6.2 and SUR1 formed in the propeller form but was disrupted in the quatrefoil form (Lee et al. 2017). In 2019, a higher resolution (3.3 Å) cryo-EM structure of the same pancreatic KATP channel was achieved in complex with the short-acting insulin secretagogue repaglinide and adenosine-5′-(γ-thio)-triphosphate (ATPγS) (PDBID: 6JB1) (Ding et al. 2019) (shown later in Fig. 8). Table 1 summarizes crystal and cryo-EM Kir channel structures for which functional expression has been demonstrated.

(a) Crystal structure of Kir2.2 (PDBID: 3SPI) in complex with PIP2. (b) Selectivity Filter. (c) Helix bundle crossing gate. (d) G-loop gate. (e) PIP2 binding site
1.4.2 Structural Features of Kir Channels
The Kir channel structures have provided new insights into the mechanism of channel function, such as channel gating, selectivity, rectification, and modulation by PIP2, sodium ions, alcohol, G proteins, ATP, etc. Figure 3 shows the classical Kir2.2 crystal structure with which we will illustrate some key features of this channel family. Kir channels are either homotetramers or heterotetramers formed by four subunits. Each subunit contains a transmembrane region, the TM1 (or outer) and TM2 (or inner) helices (see Fig. 3b), and a cytoplasmic domain (Fig. 3d).
1.4.2.1 Selectivity Filter (SF)
The selectivity filter (SF) of the Kir channel is similar to that of other potassium channels (Fig. 3b), but instead of the canonical filter sequence TXGYGDX (X: aliphatic amino acid), the corresponding sequence in Kir channels is TIGYGXR (X: Y/F/H, V/L, T, G), with very few exceptions (Kir2.4-S147, Kir6.1/6.2-F143/133, Kir7.1-M125) (see Fig. 9). The conserved Kir2.2(R149) forms an ionized hydrogen bond with E139 in the pore helix. Kir channels also contain a highly conserved disulfide bond flanking the pore region (C123-C155) (see Figs. 3b and 9). Between the outer helix and pore region “turret,” there is the conserved 3–10 helical HGDL signature sequence for Kir channels (Tao et al. 2009), with very few exceptions: Kir3.1/3.3/3.4 (RGDL), Kir3.2 (RGDM), Kir1.1 (HKDL), and Kir7.1 (NGDL) (see Figs. 3b and 9).
1.4.2.2 Gates
The residues I177 and M181 on the TM2 helix in Kir2.2 form two hydrophobic seals that close the path from the pore to the CTD, which is referred to as the helix bundle crossing (HBC) gate (Fig. 3c). In other K+ channels, such as KcsA and Kv channels, a small or polar amino acid exists at the position corresponding to I177. Alternate residues can be utilized in other Kirs at these positions [at I177: V/L/T/C and at M181: Kir2 (M), Kir3/6 (F), Kir1/4/5 (L), Kir7.1 (V)] (see Figs. 3c and 9). In addition to the HBC gate, Kir channels have a second unique cytoplasmic gate (G loop) at the apex of the cytoplasmic domain (see Figs. 3d and 9) (Tao et al. 2009).
1.4.2.3 PIP2 Binding Site
PIP2 binds to the interface between TMD and CTD and is thought to cause a 6 Å translation of the CTD toward the TMD (Fig. 3a, e). The negatively charged phosphate groups interact with the Kir channel through highly conserved salt bridge interactions. The 1′ phosphate interacts with the R78 and R80 of the RWR highly conserved sequence motif with a few exceptions (Kir2.2 numbers, unless otherwise specified): at R78 – Kir2/7 (R), Kir1/3.1/3.2/3.4/4.2/5/6 (K), Kir3.3/4 (Q) and at R80 – Kir6.2(P69) (see Figs. 3e and 9) at the N-terminus of the outer helix. The 4′ and 5′ phosphates interact with the TM2 residue K183 (absolutely conserved) and the highly conserved residues of the B-loop region sequence motif – 186-RPKKR-190 – with the following exceptions: R186 (Kir3/6 (Q), Kir5.1(T174)); K188 (Kir6 (H), Kir3.3 (N)); and K189 (Kir6 (R); Kir7.1(N165)) (see Figs. 3e and 9). These basic amino acids are critical for PIP2 activation of Kir channels (Hansen et al. 2011; Tao et al. 2009).
1.4.2.4 Cholesterol Regulation
Cholesterol enrichment or depletion was shown to decrease or increase, respectively, Kir2.1 currents in endothelial cells (Romanenko et al. 2002). Through mutagenesis structural determinants could be identified, suggesting strongly that specific interactions of cholesterol with the Kir proteins were responsible for the observed effects (Epshtein et al. 2009; Rosenhouse-Dantsker 2019). Cholesterol enrichment caused significant inhibition to several active Kir channels (Kir1.1, Kir2.1, Kir3.1(F137S), Kir6.2Δ36) and activation (rather than inhibition) to others (Kir3.2, Kir3.4(S143T)). Modeling studies and site-directed mutagenesis suggested that the interaction sites were located between α-helices of two adjacent channel subunits and involved hydrophobic and aromatic residues (Rosenhouse-Dantsker 2019). Even though some key determinant residues for cholesterol sensitivity overlapped with those for PIP2 sensitivity, others did not, making the interrelationship between cholesterol and PIP2 influences on Kir channels unclear. A recent cryo-EM structure of Kir3.2 with a cholesterol analog (in the presence and absence of PIP2) suggests that cholesterol binds near PIP2 potentiating its effects (Mathiharan et al. 2020).
1.4.2.5 Rectification Determinants
Kir channels can be classified as strong, intermediate, and weak rectifiers based on their rectification properties (Table 2; Yellow – Kir2, Blue – Kir3, Green – Transport, Orange – Kir6). The rectification features of Kir channel are generally believed to occur through blocking of the channels by Mg2+ and polyamines (spermine, spermidine, and putrescine) (Nichols and Lee 2018). Key acidic residues involved in the rectification of Kir channels have been identified through mutagenesis. A TM2 aspartate residue (corresponding to D173 in the Kir2.2 channel) was the first residue identified as critical for inward rectification. Mutation of this residue affected both polyamine-blocking affinity and voltage dependence (Fakler et al. 1996; Lopatin et al. 1994; Wible et al. 1994; Yang et al. 1995). The corresponding residue in intermediate/weak rectifiers such as Kir1.1 and Kir6.2 is an asparagine. Mutation of this asparagine to an aspartate residue converted these channels to strong rectifier channels (Lopatin et al. 1994; Lu and MacKinnon 1994; Shyng et al. 1997). Thus, this residue has been termed as the “rectification controller.” Several other negatively charged residues in the pore-lining region of the CTD in Kir channels are important for rectification. For example, mutation of E224, E229, D259, and E299 of Kir2.1 (numbers are one less than Kir2.2) to a neutral residue reduced the intensity of inward rectification (Fujiwara and Kubo 2006; Guo and Lu 2003; Kubo and Murata 2001; Taglialatela et al. 1995; Xie et al. 2002; Yang et al. 1995). Figure 4 shows a comparison of these residues between the strong rectifier Kir2.2 and the weak rectifier Kir6.2. Table 2 shows that the number of acidic residues per Kir channel subunit correlates well with the functional rectification properties of each subfamily member, such that 4–5 negatively charged residues per subunit (Q/s) result in strong, 3 Q/s in intermediate and 1 Q/s in weak rectification.

Critical residues for Kir channel rectification. (a) Kir2.2 (PDBID: 3SPI); (b) Kir6.2 (PDBID: 6C3O)
Recent microsecond MD simulation studies on polyamine blocking of the mKir3.2 channel were conducted by Chen et al. (2020). Two binding sites for putrescine were identified, one located close to the E236 (hE234) in the CTD, and another located close to the T154 (hT152) from the SF. These observations were consistent with previous experimentally identified residues. By applying a cross-membrane electric field, putrescine was transferred from the CTD binding site to the SF site. In contrast to putrescine (+2 charges), spermine (+4 charges) did not transfer to the SF site, but remained bound to the CTD site. An additional force was needed to be applied to make spermine transfer from the CTD to the SF binding site (Chen et al. 2020). These results contribute toward a dynamic molecular insight of the rectification mechanism through polyamines. In future studies, additional dynamic simulations will be needed to provide further molecular insight into the dynamic rectification mechanisms of Kir channels, such as how polyamines limit potassium ion permeation.
2 Classical Kir2 Channels
2.1 Historical Perspective
The classical (also sometimes referred to as “canonical”) rectifiers display strong inward rectification, with little outward current at depolarizing potentials. As we mentioned already in Sect. 1, they were differentiated early on from voltage-gated K+ (Kv) channels (outward rectifiers) in that at membrane potentials around the K+ equilibrium potential (EK), where Kv channels are closed, they are constitutively open, albeit conducting small but physiologically relevant outward currents. They conduct very little to not at all at depolarized membrane potentials, where Kv channels are open and conduct the most. This property of strong inward rectifiers is critical for setting a background K+ conductance in excitable tissues, such as in muscle (both skeletal and cardiac, where Kir2 channels are expressed – see below), driving the resting potential near EK and enabling rapid conduction of excitation and coordinated contraction (Nichols et al. 1996). As already mentioned, the molecular mechanisms underlying Kir rectification, such as the blockade by polyamines, have been studied extensively (Fujiwara and Kubo 2006; Guo and Lu 2003; Ishihara and Ehara 2004; Kubo and Murata 2001; Nishida et al. 2007; Taglialatela et al. 1995; Xie et al. 2003; Yang et al. 1995). Yet, several aspects of the mechanisms involved remain unclear (Liu et al. 2012; Xu et al. 2009). One of these is the voltage dependence of inward rectification (i.e., polyamine block) displaying shifts that occur with changes in EK when [K+]o but not [K+]i is varied (see Fig. 1a) (Hagiwara and Takahashi 1974; Hagiwara and Yoshii 1979; Hestrin 1981; Kubo 1996; Kubo et al. 1993a; Lopatin and Nichols 1996; Nishida et al. 2007). Thus, the probability of channels being open (Popen) shifts along the voltage axis by 25 mV for an e-fold change in [K+]o and the open channel conductance (inward or outward) is proportional to [K+]o with a power of 0.2–0.6, referred to as a “square root” proportionality (Hagiwara and Takahashi 1974; Hille and Schwarz 1978; Kubo et al. 1993a; Matsuda 1988; Ohmori 1978; Sackmann et al. 1984). It has been argued that this dependence of the Kir conductance on [K+]o, particularly the outward conductance, plays a significant role in regulating the myocardial action potential duration (Matsuoka et al. 2003; Nichols et al. 1996; Shimoni et al. 1992), cardiac contractility (Bouchard et al. 2004), and arrhythmogenicity (Asakura et al. 2014; Ishihara et al. 2009; Maruyama et al. 2011). Ishihara has recently shown (2018) that in the absence of Na+ in the external solutions, the open conductance of Kir2.1 does not change over a wide [K+]o range but in its presence, Na+ competitively inhibits K+ conductance in a voltage-dependent manner. Thus, it is an impermeant physiological cation that mediates the apparent [K+]o dependence of the open Kir channel conductance (Ishihara 2018). The structural determinants of the Na+ binding site have not yet been elucidated. A guiding clue comes from Kir7.1, that differs from other Kir channels in lacking to a great extent in the property of being activated by [K+]o. We will revisit this clue of the structural determinants for the dependence of the K+ conductance on [K+]o and how Na+ may be involved when we discuss Kir7.1 in the “K+ transport Kir channels” in Sect. 5.3.
2.2 Subfamily Members and Tissue Distribution
The first subfamily member Kir2.1 (also referred to as IRK1) was cloned in 1993 from a mouse macrophage cell line (Kubo et al. 1993a). In the following 2 years three additional members were cloned, Kir2.2, Kir2.3, and Kir2.4 (Bond et al. 1994; Bredt et al. 1995; Morishige et al. 1994). Kir2.5 was cloned from fish in 2008 (Crucian carp; Carassius carassius) (Hassinen et al. 2008), while Kir2.6 was revealed as the fifth mammalian isoform in 2010 (Ryan et al. 2010).
In excitable cells, Kir2 channels are expressed in skeletal muscle (Kir2.1, Kir2.2, Kir2.6), brain (Kir2.1, Kir2.2, Kir2.3, Kir2.4), and heart (Kir2.1, Kir2.2, Kir2.3), while an intermediate level of expression is seen in smooth muscle tissue and the retina (Fig. 2) (de Boer et al. 2010; Hibino et al. 2010). Their role in non-excitable cells remains to be elucidated (e.g., in macrophages where Kir2.1 was first cloned from) (Kubo et al. 1993a). Not only these channel subunits assemble as homotetramers but also as heterotetramers, both in heterologous expression systems and in native tissues where they are expressed, endowing tissues with greater versatility in the physiological roles they play (Hibino et al. 2010). Only a small fraction of Kir2.6 manages to leave the endoplasmic reticulum to make it to the cell surface, whilst the almost identical subunit Kir2.2 (6 differences in the human clones Kir2.6-Kir2.2: L15S, A56E, V100I, H118R, L156P, G430E) is trafficked robustly. Dassau and colleagues showed that the L156P and P156L accounted for the majority of the endoplasmic reticulum rescue and retention of Kir2.6 and Kir2.2, respectively. In addition, they showed that the wild-type Kir2.6 co-assembles with Kir2.1 and Kir2.2 in vitro and in skeletal muscle acting as a dominant negative subunit, limiting Kir2.1 and Kir2.2 localization to the cell surface (Dassau et al. 2011).
2.3 Physiology/Pathophysiology
The physiology and pathophysiology of Kir2 channels has been previously reviewed (de Boer et al. 2010; Hibino et al. 2010). Here, we will highlight the major roles of this channel subfamily members in the various tissues they are expressed.
Heart: Kir2 channels are critically involved in determining the shape of the cardiac action potential by (1) setting the resting potential, (2) permitting the plateau phase (through their Mg2+ and polyamine block at depolarized potentials), and (3) inducing a rapid final stage of repolarization. Contributions of different Kir2 subunits in comprising cardiac Kir currents have revealed that Kir2.1 knockout in mice abolishes ventricular Kir2 currents, while Kir2.2 knockout reduces the currents by 50% (Zaritsky et al. 2001). This suggests that Kir2.1 is the dominant expressing subunit and Kir2.2 contributes to the current by assembling with Kir2.1. Kir2.1 dysfunction due to 21 mutations identified in 30 families have been identified to cause Andersen-Tawil syndrome (ATS) by multiple mechanisms, including allosteric decreases in channel-PIP2 interactions (Donaldson et al. 2004; Lopes et al. 2002). ATS is an autosomal-dominant disorder resulting in cardiac arrhythmias (long Q-T syndrome 7, LQT7), periodic paralysis, and dysmorphic bone structure in the face and fingers (Tawil et al. 1994). The cardiac symptoms entail a depolarized VREST and this loss of the stabilization of Vm can trigger arrhythmias. The inability to contribute to the late phase of the repolarization of the action potential results in a prolonged action potential duration, hence the LQT7 arrhythmia. Differential expression is seen within tissues of the same organ, as in the heart for example, where dominant expression in the atria is seen for Kir2.3, while in the ventricles for Kir2.1 (Anumonwo and Lopatin 2010). Besides ATS, Kir2.1 has been implicated in atrial fibrillation (AF), the most prevalent arrhythmia that ranges from 1–2% in the general population to 9–10% in the elderly population (Dobrev et al. 2005). Kir3.1 and Kir3.4 have also been implicated in AF but will be discussed in the next section devoted to Kir3 channels. Enhanced expression of the Kir2.1 channel leads to AF and this has been linked with downregulation for two microRNAs: miR-1 (Girmatsion et al. 2009; Yang et al. 2007) and miR-26 (Luo et al. 2013). Additionally, the Kir2.1(V93I) is a gain-of-function mutation that has been linked to hereditary AF (Xia et al. 2005).
Skeletal muscle: The periodic paralysis symptom of ATS is due to the effects of the IKir2.1 reduction in skeletal muscle. There, the depolarized VREST inactivates Nav channels making them unavailable for initiation and propagation of the action potential, leading to paralysis. Kir2.1 has been reported to be essential for myoblast differentiation (Konig et al. 2004) and also for fusion of mononucleated myoblasts to form multinucleated skeletal muscle fibers (Fischer-Lougheed et al. 2001). Kir2.1 underlies a 60 mV hyperpolarization in the VREST of myoblasts during development, which drives Ca2+ entry through Ca2+-permeant ion channels that promotes differentiation and fusion of myoblasts. Even though there are no gross skeletal muscle developmental defects in ATS patients, their “slender” built could be related to the mild developmental defects caused by the Kir2.1 dysfunction. Kir2.6 is also expressed in skeletal muscle and as already mentioned is nearly identical to Kir2.2 (Ryan et al. 2010). It was discovered in a screen for candidate genes responsible for thyrotoxic hypokalemic periodic paralysis, a serious complication of hyperthyroidism characterized by skeletal muscle paralysis and hypokalemia, affecting young adult male patients of Asian descent (Kung et al. 2006). The gene coding for Kir2.6 (KCNJ18) is transcriptionally regulated by thyroid hormone via a thyroid-responsive element in its promoter region. Thirty three percent of the patients afflicted by this condition bear mutations mainly localized in the C-terminus causing decreased current densities (de Boer et al. 2010). Destabilizing VREST would inactivate Nav channels yielding paralysis.
Bone: The dysmorphic facial bone structure defects point out the role of Kir2.1 channels in bone development. Indeed, it has been recently reported that Kir2.1 is important for efficient signaling by the bone morphogenic proteins in mammalian face development (Belus et al. 2018).
Blood vessels – Endothelial cells: Both the endothelial and smooth muscle cells that comprise the vasculature express Kir2 channels (Adams and Hill 2004; Nilius and Droogmans 2001). In fact, for vascular endothelial cells these are considered the most prominent channels expressed (Nilius and Droogmans 2001; Nilius et al. 1993; von Beckerath et al. 1996). By setting VREST near EK, they drive Ca2+ entry into endothelial cells (Kwan et al. 2003; Wellman and Bevan 1995) that triggers NO-mediated vasodilation. More recently, Kir2.1 channels were shown to boost the endothelial cell-dependent vasodilation generated by Ca2+-dependent activation of small and intermediate conductance Ca2+-activated K+ channels in resistance-sized arteries (Sonkusare et al. 2016). In aortic endothelial cells evidence has suggested that Kir2.2 channels are the dominant Kir2 conductance (Fang et al. 2005).
Smooth muscle cells: A mild increase in [K+]o (from 6 to 15 mM) hyperpolarizes the Vm of smooth muscle cells by 15 mV (from −45 to −60 mV) by increasing Kir conductance and vasodilating cerebral and coronary arteries (Knot et al. 1996; McCarron and Halpern 1990; Nelson et al. 1995). The hyperpolarization closes Cav channels reducing [Ca2+]i leading to vasodilation (Knot and Nelson 1998). In cerebral arteries, evidence has been presented arguing for astrocyte secretion of K+ upon neuronal stimulation, suggesting a way to couple neuronal activity to local blood flow in the brain (Filosa et al. 2006). Kir2.1, rather than Kir2.2 or Kir2.3, is thought to underlie these hyperpolarization effects in vascular smooth muscle cells (Bradley et al. 1999; Zaritsky et al. 2000). This capillary-to-arteriole coupling, whereby neuronal activity results in a small increase in extracellular K+, is sensed by the Kir2.1 channels of capillaries to increase Kir2.1 outward current and hyperpolarization that in turn vasodilates the arterioles to cause an increase in local cerebral blood flow (Longden et al. 2017). Interestingly, this is a regulated process, as signaling through Gq-protein coupled receptors (GqPCRs) that hydrolyzes PIP2 prevents activation of Kir2.1 by the [K+]o increase and uncouples this intricate sensing mechanism (Harraz et al. 2018).
Neurons: Kir2 channels are abundantly and differentially expressed in somata and dendrites of neurons in the brain: diffusely and weakly in the whole brain (Kir2.1), moderately throughout the forebrain and strongly in the cerebellum (Kir2.2), mainly in the forebrain and olfactory bulb (Kir2.3), and in the cranial nerve motor nuclei in the midbrain, pons (Kir2.4) (Hibino et al. 2010). PIP2 depletion, once again, modulates activity, this time of striatopallidal neurons, as stimulation of their dendritic spines that contain Kir2.3 channels results in enhancement of dendritic excitability (Shen et al. 2007). The microvilli of Schwann cells at the node of Ranvier express Kir2.1 and Kir2.3 which may be serving a buffering role to maintain [K+]o by absorbing excess K+ released by excited neurons as done by astroglia (Mi et al. 1996).
2.4 Pharmacology
Kir2 channel inhibitors: A number of small molecule inhibitors for Kir2 channels have been reported. Tamoxifen, an estrogen receptor antagonist for breast cancer treatment, inhibits Kir2.1, Kir2.2, and Kir2.3. The experimental results suggested the compound inhibits the channels by interfering with channel–PIP2 interactions (Ponce-Balbuena et al. 2009). Chloroquine, an important anti-malaria drug, inhibits the Kir2.1 channel and can induce lethal ventricular arrhythmias. Molecular modeling and mutagenesis suggested that chloroquine blocks the Kir2.1 channel by plugging the cytoplasmic conduction pathway, interacting with the negatively charged and aromatic residues within the central pocket (Rodriguez-Menchaca et al. 2008).
Gambogic acid (GA) was discovered as a Kir2.1 inhibitor through a library screening of 720 naturally occurring compounds. GA acts slowly at nanomolar concentrations to abolish Kir2.1 but not Kv2.1, HERG or Kir1.1 channel activity. GA could interfere with Kir2.1 channel trafficking to the cell surface (Zaks-Makhina et al. 2009). VU573 was discovered through a thallium (Tl+) flux-based high-throughput screen of a Kir1.1 inhibitor library. The compound inhibits Kir3, Kir2.3, and Kir7.1 over Kir1.1 and Kir2.1 channels (Raphemot et al. 2011). ML133 was discovered through a high-throughput screen (HTS) of a library (>300,000 small molecules). It inhibits Kir2.1 with little selectivity against other Kir2 family channels. However, ML133 has no effect on Kir1.1, and a weak activating effect on Kir4.1 and Kir7.1 channels. Using a chimera between Kir2.1 and Kir1.1, the molecular determinants were identified to be residues D172 (the “rectification controller” residue) and I176 (one of the two HBC gates, I177 in Kir2.2) of the TM2 segment in Kir2.1 (Wang et al. 2011). Chloroethylclonidine (CEC) has been reported as an agonist for α2-adrenergic receptors and an antagonist for α1x-receptors. It inhibits Kir2.1 by directly blocking the channel pore, possibly at an intracellular polyamine binding site. Mutation of E172 in Kir2.1 to asparagine abolished CEC inhibition (Barrett-Jolley et al. 1999). Celastrol has been discovered as an inhibitor for the Kir2.1 and hERG channels and causes QT prolongation. It also alters the rate of channel transport and causes a reduction of channel density at the cell surface (Sun et al. 2006a). 3-bicyclo [2.2.1] hept-2-yl-benzene-1,2-diol was discovered as an inhibitor of the Kir2.1 and Kv2.1 channels through screening of 10,000 small molecules from a combinatorial chemical library. It is a weak inhibitor of Kir2.1 compared to Kv2.1 channels (Zaks-Makhina et al. 2004).
Kir2 channel activators: Pregnenolone sulfate (PREGS) belongs to the neurosteroid family and has been shown to be a Kir2.3 activator from the extracellular side of the membrane with no effect from the intracellular side. The activation was not affected by changes in the external pH. Other Kir channels, such as, Kir1.1, Kir2.1, Kir2.2, and Kir3 channels, were insensitive to PREGS (Kobayashi et al. 2009). Tenidap was discovered as a potent activator of Kir2.3 channels using an 86Rb+ efflux assay. The action of tenidap was from the extracellular side of the membrane. It had little or no effect on Kir2.1, Kv1.5, and Nav channels. Tenidap could serve as a pharmacological tool, an opener of Kir2.3 channels (Liu et al. 2002) (Table 3).
2.5 Structural Studies
Channel activation of all Kir channels requires PIP2, but Kir2 channels have been shown to require an additional secondary non-specific phospholipid (PL-) (i.e., PA, phosphatidic acid; PG, phosphatidylglycerol; PS, phosphatidylserine; PI, phosphatidylinositol) for high PIP2 sensitivity. Lee et al. (2013) using molecular docking simulations and experimental validation identified a putative anionic phospholipid [PL(−)] binding site, which is located adjacent to the PIP2 binding site formed by two lysine residues, K64 and K219. Neutralization of either of these residues to Cys reduces channel activity, but lipid-tethering K64C with decyl-MTS (methanethiosulfonate) induces high-affinity PIP2 activation even in the absence of PL(−). These results suggest a molecular mechanism for a PL(−) synergistic effect on PIP2-dependent activation of Kir2 channels. Interestingly, the residues K64 and K219 in Kir2.1 are not highly conserved in the Kir channel family. For example, the corresponding residues in Kir 3.2 are E71 and N229 (Fig. 9). Thus, for Kir3 channels, instead of PL(−), it is the regulators Gβγ and Na+ that play a critical role in stimulating channel activity by enhancing channel-PIP2 interactions (Lee et al. 2013). Lee and colleagues further characterized the PL(−) site by introducing a Trp mutation at the K62 position in Kir2.2 channel (corresponding to the K64 in Kir2.1), which enhances PIP2 sensitivity even in absence of PL(−). High-resolution crystal structures of Kir2.2(K62W) were solved in the presence and absence of PIP2. The mutation caused a tight tethering of the CTD to the TMD of the channel regardless of the presence of PIP2 and mimicked the PL(−) binding to the second site, inducing formation of the high affinity primary PIP2 site. MD simulations have revealed a more extensive hydrogen bonding to basic residues at the PIP2 binding site in the Kir2.2 (K62W) mutant compared to the wild-type channel (Lee et al. 2016). To elucidate the open conformation and mechanism of K+ ion permeation, Zangerl-Plessl et al. (2020) introduced an additional G178D mutation in the Kir2.2 (K62W) mutant channel to generate the “forced open” mutant channel (KW/GD). Crystal structures of the KW/GD mutant channel were solved in the presence and absence of PIP2. The PIP2 bound form of the KW/GD structure showed a slight widening of the HBC gate (∼1.5 Å) compared to the PIP2 bound K62W structure. However, microsecond MD simulations for KW/GD in lipid bilayer showed opening of the HBC gate and K+ permeation. Simulation results showed K+ ion permeation through the SF via a strict ion-ion knock-on mechanism with rates comparable to the experimentally measured ion conductance (Zangerl-Plessl et al. 2020).
3 G Protein Kir3 Channels
3.1 Historical Perspective
In the mid-1950s after the advent of intracellular microelectrodes and voltage-clamp techniques Burgen and Terroux (1953) revisited the vagal induced hyperpolarization of heart muscle reported by Gaskell in the mid-1880s (Gaskell 1886; Gaskell 1887). They first confirmed that hyperpolarization is induced by ACh application (Burgen and Terroux 1953) and then by measuring the effect of external K+ concentration on resting potential in the absence and presence of ACh, they also demonstrated that an increased cell permeability to potassium may underlie hyperpolarization. Del Castillo and Katz used microelectrodes to directly show hyperpolarization of the sinus node upon vagal stimulation (Del Castillo and Katz 1955). Voltage clamp allowed Trautwein and Dudel to directly confirm changes in cell potassium permeability by measuring K+ reversal potentials (Trautwein and Dudel 1958). Noma and Trautwein studied activation kinetics of ACh-induced K+ currents and concluded that ACh binding activates a specific ion channel, KACh (Noma and Trautwein 1978). The introduction of the patch-clamp technique (Hamill et al. 1981) led to the first single-channel recordings of KACh currents (IK-ACh) (Sakmann et al. 1983), which clearly demonstrated kinetic properties distinct from other background potassium channels. In the mid-1980s five studies shed light as to how ACh activated IK-ACh. First, following the advent of the patch-clamp technique, Soejima and Noma demonstrated the membrane-delimited nature of IK-ACh activation, when in cell-attached recordings they showed that ACh could only activate the channels if it was perfused (or loaded) directly in the pipette but not in the bath, arguing that no second messenger was involved in activating IK-ACh (Soejima and Noma 1984). The following year two reports published back-to-back provided evidence that pertussis toxin- (PTX-) sensitive Gi/o proteins coupled muscarinic receptors to IK-ACh, either by showing that PTX treatment disabled ACh stimulation of channel activity (Pfaffinger et al. 1985) or showing that non-hydrolyzable GTP analogs activated IK-ACh constitutively (Breitwieser and Szabo 1985). The following year using inside-out patches from atrial cells Kurachi and colleagues showed that in the presence of adenosine (or ACh) in the pipette GTP perfusion in the bath supported activation of IK-ACh (Kurachi et al. 1986). In 1987, Logothetis and colleagues showed that purified Gβγ (but not Gα subunits) activated IK-ACh in a manner similar to non-hydrolyzable GTP analogs, identifying IK-ACh as the first direct effector of the Gβγ subunits (Fig. 5a – top). A controversy arose as Birnbaumer and Brown argued that the activated Gα (i.e., Gα-GTPγS) and not the Gβγ subunits were the activators of IK-ACh (Birnbaumer and Brown 1987). The controversy was resolved 7 years later when Reuveny and colleagues confirmed with recombinant channels in Xenopus oocytes that Gβγ subunits were the G protein activators of IK-ACh (Reuveny et al. 1994). Intracellular Na+ ions were also shown to activate Kir3.4 and Kir3.2 homo- and heterotetramers in a G protein-independent manner (Ho and Murrell-Lagnado 1999; Sui et al. 1996). Ethanol was also shown to activate Kir3 currents (Kobayashi et al. 1999; Lewohl et al. 1999). In 1998, Kir3 currents activated by Gβγ or Na+ were shown to require PIP2 in the plasma membrane for activation (Huang et al. 1998; Petit-Jacques et al. 1999; Sui et al. 1998). Although for classical Kir channels, PIP2 was sufficient to stimulate activity, Kir3 channels required a gating molecule (e.g., Na+, Gβγ, alcohol, etc.) together with PIP2 to stimulate activity (Huang et al. 1998; Sui et al. 1998). These gating molecules seemed to strengthen the interactions of Kir3 channels with PIP2, as a single point mutant (Kir3.4-I229L) removed the requirement for gating molecules, allowing PIP2 to activate on its own (Zhang et al. 1999).

The Kir3 GEMMA (Gi/opcr-Effector-Macromolecular-Membrane-Assembly) before activation (name/concept adapted from Ferré et al. 2021)
3.2 Subfamily Members and Tissue Distribution
It wasn’t until 1993 that the first member Kir3.1 of this subfamily was cloned (Dascal et al. 1993; Kubo et al. 1993b). The following year the neuronal Kir3.2 and Kir3.3 subunits were identified (Lesage et al. 1994). Finally, in 1995 the Kir3.4 subunit was cloned and when co-expressed together with Kir3.1 was shown to produce the biophysical properties of IK-ACh expressed in heterologous cells (Chan et al. 1996; Krapivinsky et al. 1995). Kir3.2 and Kir3.4 are capable of conducting currents through homotetramers, while Kir3.1 and Kir3.3 need to heteromerize with other subunits to produce functional channels. In the brain, multiple signals including ACh, adenosine, dopamine, opioids, GABA, etc. bind their respective Gi/o protein-coupled receptors (Gi/oPCRs) to activate Kir3 channels using the Gβγ subunits of PTX-sensitive G proteins (Fig. 5b – bottom). Expression of different subunits within distinct neuronal populations defines their responsiveness to neurotransmitters. The ventral tegmental area (VTA) in the reward centers of the brain illustrates well the relevance of the Kir3 subunit composition (Lujan et al. 2014). Dopaminergic neurons within the VTA do not express Kir3.1 but do express Kir3.2 and Kir3.3, producing Kir3.2 homotetramers as well as Kir3.2/3.3 heterotetramers (Cruz et al. 2004; Jelacic et al. 2000; Kotecki et al. 2015). GABAergic neurons within the VTA also express Kir3.2 and Kir3.3 along with Kir3.1 producing also the dominating Kir3.1/3.2 heterotetramers. VTA dopaminergic neurons show less sensitivity to GABA than GABAergic neurons do (Cruz et al. 2004; Labouebe et al. 2007). Genetic ablation of Kir3.3 in the dopaminergic neurons enhances their sensitivity to GABA, suggesting an inhibitory role by Kir3.3. Thus, low concentrations of agonists preferentially inhibit GABAergic neurons and thereby disinhibit dopaminergic neurons. This disinhibition might confer reinforcing properties on addictive GABAB receptor agonists (Labouebe et al. 2007).
Three major alternatively spliced isoforms of Kir3.2 are differentially expressed in the brain Kir3.2a-c. Kir3.2c is longer than Kir3.2a, which is longer than Kir3.2b and also shows differences in 8 C-terminal aa (Hibino et al. 2010). In the periphery, Kir3 channels show moderate expression in the heart and pancreas (Fig. 2) as well as in the testis and the pituitary and adrenal glands.
Heart: Kir3.1 and Kir3.4 subunits are predominantly expressed in the atria, in nodal cells and pulmonary vein myocardial sleeves (Ehrlich et al. 2004; Greener et al. 2011; Krapivinsky et al. 1995). Expression in mouse ventricles has also been reported but it is not thought to impact cardiac physiology or ventricular arrhythmogenesis (Anderson et al. 2018).
Pancreas: Four Kir3 isoforms have been reported to be expressed and play important roles in the physiology of the pancreas. Kir3.4 subunits exist in α, β, and δ cells of pancreatic islets and in the exocrine pancreas, whereas the Kir3.2c isoform is expressed in α and δ cells (Ferrer et al. 1995; Iwanir and Reuveny 2008; Vaughn et al. 2000; Yoshimoto et al. 1999).
Testis: Kir3.1 and Kir3.2d (another splice isoform, 18 aa shorter than Kir3.2c, that has been described only in the testis) have been reported to be expressed in the spermatids and in spermatogonia and spermatocytes (Inanobe et al. 1999).
Anterior pituitary lobe: Kir3.1, Kir3.2, and Kir3.4 have all been reported to be expressed in the anterior pituitary (Gregerson et al. 2001; Morishige et al. 1999).
Adrenal gland: Since 2011 it was reported that in patients with severe hereditary hypertension Kir3.4 mutations were found in adrenal aldosterone-producing adenomas to cause hyperaldosteronism. These findings brought a greater appreciation of the role of Kir3.4 in aldosterone production and disease (Choi et al. 2011; Gomez-Sanchez and Oki 2014). Other tissues including the eye, skin, and the colon have also been reported to express Kir3 channels (Huang et al. 2018a; Yamada et al. 1998).
Central nervous system (CNS): Kir3.1-Kir3.3 are expressed throughout, with highest levels seen in the olfactory bulb, neocortex, hippocampus, and the granule cell layer of the cerebellum but notably also in the amygdala, thalamus, substantia nigra, ventral tegmental area, locus coeruleus, some nuclei of the brainstem, and the spinal cord (Kobayashi and Ikeda 2006; Lujan and Aguado 2015). In contrast, Kir3.4 is expressed in relatively fewer brain regions with highest levels in deep cortical pyramidal neurons, the endopiriform nucleus and claustrum of the insular cortex, the globus pallidus, the ventromedial hypothalamic nucleus, parafascicular and paraventricular thalamic nuclei, and a few brainstem nuclei, such as the inferior olive and vestibular nuclei (Wickman et al. 2000).
3.3 Physiology/Pathophysiology
Physiology: The Kir3 channels and the adenylyl cyclase enzymes have been studied extensively as prototypical effectors coupled to G proteins and their corresponding GPCRs. There is a vast body of literature supporting the existence of pre-coupled macromolecular complexes that have been coined as GEMMAs, GPCR-Effector-Μacromolecular-Μembrane-Αssemblies (Ferré et al. 2021). Thus, the various Gi/oPCRs-Gαi/oβγ-Kir3 channel tetramers constitute unique GEMMAs (Fig. 5). The GEMMA concept is significant in that it extends the collision coupling model by which receptors, G proteins, and effectors communicate and transduce signals. In a GEMMA, agonists set a series of conformational changes in the pre-coupled elements that communicate with each other like clock gears rather than billiard balls. Clearly, the best evidence for GEMMAs is yet to come from high-resolution structures capturing them in their pre-coupled inactive and active states and elucidating their interactions at atomic resolution. With cryogenic electron microscopy (cryo-EM) having achieved technological breakthroughs in the past decade, structures of complexes are becoming a reality and it is only a matter of time before we obtain high-resolution images of GEMMAs. Computational power to perform dynamic simulations of large systems is also in the midst of technological breakthroughs, such that the workings of GEMMAs can be captured in the course of a computer simulation. This unprecedented progress in our ability to make reliable dynamic structure-based models of macromolecular membrane assemblies of proteins promises to illuminate our understanding of the molecular physiology and pharmacology of GEMMAs.
GEMMAs use a core assembly, as shown in Fig. 5 with the examples of the KACh and KMOR GEMMAs, to carry out their basic function but can accommodate in a dynamic manner the coming and going of multiple other partners to fine-tune effector function in response to external and internal signals. Each member of the core GEMMA can recruit additional regulatory partners or scaffold proteins that orchestrate a symphony of regulatory partners with the GEMMA core proteins. RGS (regulator of G protein signaling) proteins, for example, enhance the GTPase activity of G proteins and as such they accelerate receptor-induced Kir3 current deactivation kinetics and inhibit GPCR-Kir3 signaling (Doupnik 2015; Doupnik et al. 1997; Sjogren 2011). Molecular platforms to achieve subcellular localization of membrane proteins or post-translational modifications needed to serve the physiology of the GEMMA have been reviewed by Ferré and colleagues (2021).
Utilization of motifs in the C-termini of any of the core proteins of the GEMMA can recruit specific modulatory proteins. As an example, let us consider the Kir3 channels using their PDZ domains to recruit scaffold proteins. The PDZ domain is a common structural domain of 80–90 amino acids found in signaling proteins from bacteria to animals. Proteins containing PDZ domains play a key role in anchoring receptor proteins in the membrane to cytoskeletal components. Proteins with these domains help hold together and organize signaling complexes at cellular membranes. PDZ got its name by combining the first letters of the first three proteins discovered to share the domain – post synaptic density protein (PSD95), Drosophila disc large tumor suppressor (Dlg1), and zonula occludens-1 protein (zo-1). Scaffold proteins of the PSD-95 and Shank families recognize class I PDZ motifs (x-S/T-x-F: F is a hydrophobic aa and x is any aa) and can bind the C-termini of these channel subunits. Kir3.2c and Kir3.3 possess a C-terminal motif for class I PDZ domain-containing proteins (ESKV) that is absent in Kir3.2a. PDZ-scaffold proteins are considered to be one of the major determinants of postsynaptic localization of various proteins (Sheng and Sala 2001). Thus, Kir3.2c and Kir3.3 channels could be localized post-synaptically through direct or indirect interactions with PDZ-scaffold proteins. Classical Kir2 channels, for which there is no evidence that they are part of a GEMMA, also contain class I PDZ motifs. Interestingly, Kir2, but not Kir3 subunits, has been found to interact directly with PSD-95 (Nehring et al. 2000), indicating that the mere presence of the binding motif does not necessarily imply a direct binding of the scaffold protein. Instead, the scaffold protein sorting nexin 27 (SNX27) binds the Kir3.2c and Kir3.3 channels but not the Kir2 (or a Kir4 channel that also contains the motif (Balana et al. 2011; Lunn et al. 2007)). SNX27 association with the Kir3 subunits leads to a reduction of signaling at the plasma membrane, likely by promoting internalization of the channel (Lunn et al. 2007). This example points to how subunit composition (in this case Kir3.2c and Kir3.3) makes the channel and potentially its GEMMA prone to another regulatory control of channel density at the plasma membrane to control cellular function. Interactions of Kir3.1/3.2 with NCAM (neural cell adhesion molecule) in lipid raft microdomains of hippocampal neurons have been reported to decrease Kir3.1/3.2 cell surface density (Delling et al. 2002).
Finally, let us consider post-translational modifications of Kir3 channels, such as phosphorylation. Kir3 (as well as several other Kir) channels have been reported to be modified in a functionally meaningful manner by phosphorylation, e.g. by protein kinases A and C (PKA, PKC): (Keselman et al. 2007; Leaney et al. 2001; Lei et al. 2001; Lopes et al. 2007; Mao et al. 2004; Medina et al. 2000; Mullner et al. 2000; Sharon et al. 1997; Sohn et al. 2007; Stevens et al. 1999; Witkowski et al. 2008; Gada and Logothetis 2021). Another large (more than 50-member) class of scaffolding proteins called AKAPs (A kinase anchoring proteins) binds to a common motif on effectors or GPCRs to organize recruitment of specific kinases and phosphatases (e.g., AKAP5 anchors PKA, PKC, calcineurin to proteins it binds) to modify the protein(s) of interest (Ferré et al. 2021). Is phosphorylation of Kir3 channels involving the use of scaffolding partner proteins, such as AKAPs? Although direct evidence is not yet available for Kir channels, GPCRs have been studied extensively and as parts of a GEMMA they could provide the organization needed for phosphorylation/dephosphorylation of Kir channels. Pathophysiology Studies on mutant mice lacking specific Kir3 subunits have provided evidence for physiological/pathophysiological effects suggesting these channels as therapeutic targets and highlighting the usefulness of potential Kir3 activators/inhibitors (Fig. 6).

Pathophysiological Kir3 conditions and drug regulators needed as potential therapeutics (A) Activators (I) Inhibitors. CB, cannabinoids, ETOH ethanol, MOR μ-opioid receptor, PSVT paroxysmal supraventricular tachycardia
Cardiac arrhythmias: As we have discussed above, stimulation of M2R in supraventricular tissues by ACh released by the vagus nerve activates IK-ACh slowing down heart rate and endowing the heart with the adaptability needed to respond to rate adjustments (heart rate variability or HRV) and meet the demands of the body, as in exercise. Adenosine has similar effects on IK-ACh (Kurachi et al. 1986), acting through adenosine 1 receptors (A1Rs). Although Kir3.4 or Kir3.1 knockout in mice resulted at best in mild resting tachycardia, these deletions strongly decreased chronotropic response and HRV (Bettahi et al. 2002; Wickman et al. 1998). Kir3.4 loss of function mutations in people have been described resulting in Long QT (LQT) syndrome (Yang et al. 2010). Similarly, in cases of paroxysmal supraventricular tachycardia (PSVT), adenosine and other A1R receptor agonists have been used successfully for its acute treatment (Prystowsky et al. 2003). Vagal denervation (or knockout of the Kir3.4 channel) prevents the induction of atrial fibrillation (AF), the most common arrhythmia in clinical practice (Kovoor et al. 2001). On the other hand, gain of function conditions, such as vagal stimulation, predispose to AF. In AF, a constitutive increase in IKACh, in the absence of channel stimulation by acetylcholine, promotes the shortening of action potential duration (Carlsson et al. 2010; Dobrev et al. 2005; Kovoor et al. 2001). This increase in basal activity of the channel is ATP dependent and is enhanced in the presence of phosphatase inhibitors (Makary et al. 2011), suggesting kinase-mediated activity. Conventional PKC isoforms inhibit, while novel PKC isoforms stimulate IK-ACh (Makary et al. 2011). A decrease in PKCα and a concomitant increase in PKCε expression have been reported from patients with AF (Voigt et al. 2014; Gada and Logothetis 2021).
Hyperaldosteronism: As mentioned above, Kir3.4 mutations were found in adrenal aldosterone-producing adenomas causing hyperaldosteronism in patients with severe hereditary hypertension (Choi et al. 2011).
Hyperalgesia and decreased analgesia: Kir3.1 and/or Kir3.2 knockout mice exhibit hyperalgesia (Marker et al. 2004). Moreover, Gi/oPCR ligands for opioid, M2 muscarinic, α2 adrenergic, GABAB, and cannabinoid receptors show reduced analgesic effects in Kir3.2 and/or Kir3.3 knockout mice (Blednov et al. 2003; Cruz et al. 2008; Lotsch et al. 2010; Lujan et al. 2014; Luscher and Slesinger 2010; Mitrovic et al. 2003; Nishizawa et al. 2009; Smith et al. 2008; Tipps and Buck 2015). Gene polymorphisms in KCNJ6 (coding of Kir3.2) in humans have been associated with analgesia and pain sensitivity (Bruehl et al. 2013; Nishizawa et al. 2014). Thus, a plethora of studies converge on the mechanism that Kir3 channel activation induces analgesia accounting for the action of many drugs that reduce pain.
Drug addiction: As discussed earlier, Kir3 channels are thought to play an important role in the communication between the midbrain GABAergic and VTA dopaminergic neurons in the mesolimbic reward system, resulting in disinhibition of the VTA dopaminergic neurons which have been connected to reward behaviors (Kotecki et al. 2015; Marron Fernandez de Velasco et al. 2015; Tipps and Buck 2015). Kir3 channels play a role in mediating the rewarding effects of ethanol and addictive drugs that target GPCRS, such as the MOR and cannabinoid type 1 receptor (Blednov et al. 2003; Blednov et al. 2001a; Blednov et al. 2001b; Marron Fernandez de Velasco et al. 2015; Nagi and Pineyro 2014; Nassirpour et al. 2010; Rifkin et al. 2017; Tipps and Buck 2015). Kir3.2 and Kir3.3 knockout mice showed reduced self-administration of cocaine compared to wild-type mice indicating that in the absence of Kir3 channels the behavioral response to drugs of abuse is altered (Morgan et al. 2003). It has been shown in multiple cell lines that exposure to these addictive drugs reduces Kir3 currents (Marron Fernandez de Velasco et al. 2015; Rifkin et al. 2017; Tipps and Buck 2015). Recent studies have attempted to generate cell-specific Kir3 channel knockouts with cre-lox methodologies (Kotecki et al. 2015). Whether specific Kir3 subunit composition selective modulators could become useful pharmacotherapies for managing addiction remains to be tested.
Epilepsy: Unlike Kir3.4 (Wickman et al. 2000), Kir3.2 knockout mice exhibit spontaneous seizures and increased susceptibility to a convulsant agent (Signorini et al. 1997). Adenosine-induced hyperpolarization in hippocampal brain slice recordings could be blocked by a Kir3 blocker, which induced seizure activity (Hill et al. 2020). In temporal lobe epilepsy, the most common epilepsy in adults (Tellez-Zenteno and Hernandez-Ronquillo 2012) that progresses to drug-resistant seizures, N-methyl-D-aspartate receptor (NMDAR) hyperactivity triggers apoptotic mechanisms, including activation of cysteinyl aspartate-specific proteases (caspases) that characterize the severity of epileptic seizures. It has been recently shown that caspase-3 targets the Kir3.1 and Kir3.2 proteins, cleaving their C-terminal ends to compromise Gβγ activation and cell surface localization of these channels, implicating them strongly in the progression of this disease (Baculis et al. 2017).
Down syndrome: The KCNJ6 that encodes for Kir3.2 is located in human chromosome 21. In Down syndrome there is a trisomy of chromosome 21, resulting in a duplication of the KCNJ6 gene that alters synaptic transmission (Reeves et al. 1995; Sago et al. 1998). Full or partial duplication of the corresponding mouse gene that contains KCNJ6 produced hallmark characteristics of the disease (Reeves et al. 1995; Sago et al. 1998). Kir3.2 protein upregulation resulted in altered reward mechanisms, cognitive functions, and synaptic plasticity (Reeves et al. 1995; Sago et al. 1998; Siarey et al. 1999). To control for the fact that there are several gene duplications that make the pathophysiology more complex in this disorder, a single trisomy of the KCNJ6 gene was produced in mice recapitulating deficits characteristic of the syndrome, suggesting that triplication of only this gene is key in some of the abnormal neurological phenotypes seen in Down syndrome (Cooper et al. 2012).
Alzheimer’s disease (AD): AD is characterized by progressive cognitive decline accompanied by formation of amyloid-beta (Aβ) plaques, neurofibrillary tangles, and aggregates of hyperphosphorylated tau protein. The main brain regions affected by AD are the hippocampus and amygdala, where early onset AD pathogenesis is seen (Arriagada et al. 1992; Haass and Selkoe 2007; Hardy and Selkoe 2002; Huang and Mucke 2012; Swanberg et al. 2004; Zald 2003). Imbalances of excitatory/inhibitory synaptic transmission occur early in the pathogenesis of AD, leading to hippocampal hyperexcitability and causing synaptic, network, and cognitive dysfunction. Neuronal Kir3 channels control neuronal excitability contributing to the inhibitory signaling in the hippocampus. The tonic Kir3 currents related to GABAB receptors contribute critically to the balancing of membrane excitability and synaptic transmission (Lujan et al. 2009; Nava-Mesa et al. 2013; Sanchez-Rodriguez et al. 2020). Intracerebroventricular injections of Aβ impaired inhibitory signaling and disrupted synaptic plasticity (Sanchez-Rodriguez et al. 2017). Aβ25–35 significantly decreased Kir3 channel conductance in the hippocampus (Mayordomo-Cava et al. 2015). RT-qPCR revealed that in CA3-CA1 hippocampal neurons application of Aβ decreased mRNA transcripts for Kir3.2, Kir3.3, and Kir3.4 subunits (Mayordomo-Cava et al. 2015). Contradictory results have also been published suggesting that a Kir3-mediated potassium efflux triggers apoptosis in hippocampal neurons (May et al. 2017). These conflicting results will need to be resolved in future studies.
Fear: Kir3.2 knockout mice showed impairments in both hippocampal-dependent contextual fear conditioning and hippocampal-independent cue fear conditioning (Victoria et al. 2016). Mice subjected to auditory cue-induced fear extinguish the fear by activating pathways between basolateral amygdala (BLA) and the nucleus accumbens (NAc) in the ventral striatum. BLA expresses all Kir3 isoforms, except Kir3.4, and displays baclofen-induced Kir3 currents through GABAB receptors (Xu et al. 2020).
3.4 Pharmacology
Kir3 channel inhibitors: Table 4 lists a number of inhibitors of Kir3 currents that have been studied in supraventricular cardiac arrhythmias and AF in particular.
3.4.1 Inhibitors in Disease
Atrial fibrillation (AF): The discovery that potent low nanomolar inhibition of Kir3.1/3.4 could be achieved by tertiapin (TPN), the 21 aa peptide extracted from the honey bee venom that also inhibits similarly Kir1.1 channels (Jin and Lu 1998), provided a useful tool to explore the role of this channel in AF. A non-oxidizable variant of TPN(M13Q) (or TPNQ) shows a similar Ki value to TPN and is often used instead (Jin and Lu 1999). Its specificity limitation, however, encouraged exploration for small molecule inhibitors.
Three compounds in the benzopyran class emerged, NIP-142, NIP-151, and NTC-801 with outstanding potency but not high enough specificity, especially against the neuronal Kir3.1/3.2. NTC-801 in particular inhibited Kir3.1/3.4 channels with a sub-nanomolar IC50 but inhibited Kir3.1/3.2 with an IC50 of 24 nM (Machida et al. 2011).
Although NTC-801 was able to convert AF to normal sinus rhythm in canine tachypacing models (Yamamoto et al. 2014), in clinical trials it failed to reduce AF burden in 20 patients with paroxysmal AF (Podd et al. 2016). The reason for this failure was likely that due to lack of specificity between Kir3.1/3.2 and Kir3.1/3.4 it was not possible to reach concentrations required for antiarrhythmic effect without producing CNS side effects. Cui and colleagues have revealed the mechanism of action of this class of compounds by the prototypic Benzopyran-G1 (likely to be NTC-801) revealing its binding site in Kir3.1 and showing that it acts non-specifically in Kir3.1 heteromeric channels (Cui et al. 2021). Recently, XAF-1407 was reported to inhibit Kir3.1/3.4 with an IC50 of 1 nM and to decrease the AF rate in an equine model of persistent AF (Fenner et al. 2020) but whether due to specificity issues it will have a similar fate as NTC-801 remains to be seen.
Pain: TPNQ (tertiapin-Q) was shown to reduce MOR agonist activity in the immersion tail flick test (Marker et al. 2004). Additionally, intrathecal administration of TPNQ attenuated oxycodone-induced antinociceptive effects in mice (Nakamura et al. 2014).
Epilepsy: TPNQ administered intrathecally induced seizures (Mazarati et al. 2006). Several drugs used clinically as antipsychotics and antidepressants in the early 2000s (e.g., haloperidol, clozapine, desipramine, fluoxetine) were identified to inhibit Kir3 channels at higher concentrations and to induce seizures as a side effect (Kobayashi et al. 2000; Kobayashi et al. 2003; Kobayashi et al. 2004; Kobayashi et al. 2006; Luscher and Slesinger 2010; Ulens et al. 1999; Yamakura et al. 2001).
Kir3 channel activators: Table 4 also lists a number of Kir3 activators. Specifically, there is a highly potent urea scaffold class that gave rise to a completely neuronal specific Kir3.1/3.2 activator with no effect on the cardiac Kir3.1/3.4 channels (i.e., GAT1508 – see below – Xu et al. 2020).
Ethanol was the first direct Kir3 channel drug activator, as a series of N-alkyl alcohols were shown to activate Kir3.1/3.2 and Kir3.1/3.4 channels with 1-propanol being the most effective and potent activator (Kobayashi et al. 1999; Lewohl et al. 1999). In 2011 it was identified that at high micromolar concentrations the flavonoid Naringin from grapefruit could activate Kir3.1/3.2 channels (Yow et al. 2011). In 2013, data mining and development of the thallium flux fluorescent assay for Kir channels allowed for early identification of CID736191 (nF-ML297), revealing an urea core scaffold that was connecting a left side benzene ring, to the N-phenyl pyrazole on the right side (see Fig. 11, Kir3 Channel Activators, ML297) (Kaufmann et al. 2013; Weaver et al. 2004; Wen et al. 2013). Structure-activity relationship studies produced compound ML297 [1-(3,4-difluorophenyl)-3-(3-methyl-1-phenyl-1H-pyrazol-5-yl) urea]. ML297 has a Kir3.1/3.2 EC50 of 0.160 μM and a Kir3.1/3.4 EC50 of 1.8 μM and did not show any local motor impairments at efficacious doses in the rotor rod behavioral test in mice. ML297 was effective in two models of epilepsy in mice (Kaufmann et al. 2013). A slight chemical modification of the ML297 scaffold changing a phenyl group to a benzyl group produced compound VU0466551, 1-(1-benzyl-3-methyl-1H-pyrazol-5-yl)-3-(3,4-difluorophenyl) urea. VU0466551 has a Kir3.1/3.2 EC50 of 0.07 μM and Kir3.1/3.4 EC50 of 0.11 μM and was efficacious in two rodent models of pain (Abney et al. 2019; Wen et al. 2013). A second scaffold was data mined and screened for Kir3.1-containing channel activity by Weaver and colleagues, VU0259369, (2-(2-chlorophenyl)-N-(3-(N,N-dimethylsulfamoyl)-4-methylphenyl) acetamide) with an acetamide core, similar to the Urea-pyrazole class of compounds. However, this compound lacked selectivity and potency and was not pursued (Ramos-Hunter et al. 2013). The following SAR reported in 2017 was a scaffold merging of ML297’s pyrazole core and replacing the urea with the acetamide core from VU0259369 to yield the hybrid compound VU0810464, 2-(3-chloro-4-fluorophenyl)-N-(1-cyclohexyl-3-methyl-1H-pyrazol-5-yl) acetamide. VU0810464 was synthesized to improve blood brain barrier (BBB) penetration (Wieting et al. 2017). In 2018, VU0810464 was evaluated in cultured hippocampal neurons and a stress induced hyperthermia model for anxiety and was found to be effective at reducing induced hyperthermia and activated Kir3 currents in hippocampal neurons (Vo et al. 2019). However, VU0810464 was not efficacious in the elevated plus maze model of anxiety in mice, similar to its parent compound ML297 (Vo et al. 2019). Recent studies identified GAT1508, a highly selective potent and efficacious Kir3.1/3.2 activator, with an EC50 of 0.075 μM, which is devoid of cardiac Kir3.1/3.4 channel activity (Xu et al. 2020). GAT1508 was evaluated in a rat auditory cue-induced fear extinction model of post-traumatic stress disorder (PTSD) that was found to be effective at extinguishing conditioned fear (Xu et al. 2020). GAT1508 also was found to induce Kir3 currents in basolateral amygdala (BLA) brain slice recordings where at low ineffective concentrations it potentiated baclofen-induced current showing synergism (Xu et al. 2020). Additionally, in 2017 a larger macrocyclic antiparasitic drug, Ivermectin was also shown to activate Kir3.1/3.2 channels with an EC50 of 3.5 μM and Kir3.1/3.4 channel with an EC50 of 7.5 μM (Chen et al. 2017).
3.4.2 Activators in Disease
Pain: Flupirtine, a non-opioid analgesic that non-selectively activates potassium channels and indirectly antagonizes NMDA receptors, activated Kir3 channels in rat hippocampal neurons and in cultured retinal ganglion cells (Jakob and Krieglstein 1997; Sattler et al. 2008). When given in combination with morphine it potentiated antinociception in two rat models of pain, suggesting a role for Kir3 in flupirtine-induced analgesia (Devulder 2010; Jakob and Krieglstein 1997).
Epilepsy: Gi/oPCR agonists, like a selective A1 adenosine receptor ligand, could be effective in suppressing seizures in a model of pharmaco-resistant epilepsy in mice (Gouder et al. 2003). Urea-based activators (e.g., ML297) of Kir3.1/3.2 heteromers have been shown to exert antiepileptic efficacy both in maximal electric shock-induced and in chemically-induced models of epilepsy in rodents (Huang et al. 2018b; Kaufmann et al. 2013; Zhao et al. 2020).
Alzheimer’s disease (AD): Activation of Kir3 channels by ML297 was able to rescue all hippocampal deficits induced by intracerebroventricular injection of Aβ1–42, restoring proper excitability in CA3-CA1 synapses (Sanchez-Rodriguez et al. 2017). ML297 prevented increased excitability, restored long-term potentiation (LTP) hindered by Aβ, and recovered hippocampal oscillatory activity (Sanchez-Rodriguez et al. 2017). ML297 was also able to restore long-term potentiation and novel object recognition deficits (Sanchez-Rodriguez et al. 2017). These findings suggest that Kir3 channel activation may represent a novel therapeutic strategy to recover excitation imbalances conferred by Aβ in early onset AD.
3.5 Structural Studies
Numerous MD simulation studies have been performed with Kir3 channels. Pioneering MD simulations were conducted based on homology models of Kir1.1, Kir3.1, and Kir6.2 (Haider et al. 2007; Stansfeld et al. 2009). The results provided insights into the nature of Kir channel–lipid interactions, in particular the importance of the slide helix and linker of the TMD and CTD in forming interactions with the headgroups of lipids. However, the timescale of the simulations at these early studies was limited to 10 ns that is a relatively short time period for an adequate sampling of protein motions (Haider et al. 2007). Using a combination of coarse-grained (1.5 μs) and atomistic MD simulations (20 ns), the interactions of PIP2 and KirBac1.1, Kir3.1-KirBac1.3 chimera, and Kir 6.2 channels were explored. The PIP2-binding site was identified at the N-terminal end of the slide helix and interface between adjacent subunits of Kir channels (Stansfeld et al. 2009). Meng et al. (2012) provided the first dynamic molecular view of PIP2-induced channel gating by conducting MD simulations on the Kir3.1-KirBac1.3 chimera in the presence and absence of PIP2 (100 ns). Simulations of the closed state with PIP2 revealed an intermediate state between the closed and open conformations of the channel. A PIP2-driven movement of the N-terminus and C-linker led to CD-loop stabilization of the G-loop gate in the open state (Meng et al. 2012). Due to timescale limitations of the MD simulations, the opening of the HBC gate of Kir channels had not been observed in any of these early studies. A highly conserved glycine residue in the middle of TM2 plays an important pivoting role in channel gating. Replacement of the residue immediately following this glycine by a proline in Kir3 channels leads to constitutively active channels (Jin et al. 2002; Sadja et al. 2001). Meng et al. (2016) conducted MD simulations on the Kir3.1 chimera (M170P) mutant (2QKS) in the presence of PIP2 (100 ns). Interestingly, the HBC gate of the channel mutant was opened within 30 ns of the simulations. The open HBC gate reached 6 Å in diameter, which allowed partial hydrated K+ ions to pass through. During the gating process, a cooperative rotation of TM1 and TM2 in counterclockwise direction (viewed from the extracellular side) was observed. Three K+ ions passed through the HBC gate during a 100 ns simulation. Introduction of the proline mutation decreased the free energy barrier for opening the channel by 1.4 kcal/mol (Meng et al. 2016).
Lacin et al. (2017) studied the role of a conserved basic residue, Kir3.2(K200) at the tether helix region, combining functional studies with MD simulations (400 ns). Both experiments and simulations demonstrated that K200 in Kir3.2 supports a dynamic interaction with PIP2. When K200 was mutated to a Tyr, it activated the channel by enhancing the interaction with PIP2. The mutant K200Y opened the HBC gate during a 400 ns simulation (Lacin et al. 2017). Bernsteiner et al. (2019) conducted multi-microsecond–timescale MD simulations based on the crystal structures of Kir3.2 bound to PIP2. The simulations provided detailed insights into the channel’s gating dynamics, as well as the movement of K+ ions through the channel under an electric field across the membrane (Bernsteiner et al. 2019). Li et al. (2019) also conducted microsecond-scale MD simulations based on the Kir3.2 crystal structure (PDB code 4KFM) in complex with PIP2, Na+, and Gβγ to understand which gates are controlled by Na+ and Gβγ and how each regulator uses the channel domain movements to control gate transitions. The simulation results suggested that Na+ ions control the cytosolic gate of the channel through an anti-clockwise rotation, whereas Gβγ stabilizes the transmembrane gate in the open state through a rocking movement of the cytosolic domain. Both effects alter the way in which the channel interacts with PIP2 and thereby stabilize the open states of the respective gates (Fig. 7) (Li et al. 2019). Li et al. (2016a, b) predicted Tertiapin and Kir3.2 channel interactions using MD simulations and PMF (potentials of mean force) calculations. The residue K17 of TPN was predicted to protrude into the pore of the channel and form a hydrogen bond with carbonyl group of T157, and R7 of TPN to form a salt bridge with the E127 from the channel turret (Li et al. 2016b). As discussed under the “Kir3 Channel Activators” (Sect. 3.4), ML297 and particularly its derivative GAT1508 proved to be potent and specific activators of Kir3.1/3.2 channels. Molecular docking and MD simulations predicted a GAT1508 binding site validated by mutagenesis experiments, providing molecular insights into how GAT1508 interacts with the channel and allosterically modulates channel–PIP2 interactions (Xu et al. 2020).

Modulators, PIP2, Na+, and Gβγ stabilize Kir3.2 channel in open state conformation. (a) Kir3.2 (Apo) is in a close conformation. (b) Kir3.2 (Holo, with PIP2, Na+ and Gβγ) is in an open conformation. Residues F192 form HBC gate, and M319 form G loop gate. The structural coordinates were obtained from MD simulations (Li et al. 2019)
4 ATP-Sensitive Kir6 Channels
4.1 Introduction
The pharmacology of ATP-sensitive K+ channels (KATP) is in part addressed by Li and colleagues (2021, in this issue chapter). KATP channels are comprised of four pore-forming subunits (Kir6.1 or Kir6.2) and four sulfonylurea receptors subunits (SUR1, SUR2A or SUR2B) creating an octomeric structure (eight total subunits). Here we complement (Li et al. 2021) with emphasis on the Kir6 subunits of the KATP channels, as members of the Kir channel family at large. KATP channels (different combinations of SUR/Kir6 subunits) are found in cardiomyocytes, pancreatic β cells, neurons, skeletal muscle, smooth muscle, endothelial cells, and in membranes of mitochondria (Rodrigo and Standen 2005; Li et al. 2021). They couple metabolic changes (ATP to ADP ratio) to cellular excitability. They are regulated by a variety of physiological regulators including H2S, long chain CoA esters, phosphatidylinositol phosphates (PIPs), carbon monoxide (CO), and changes in intracellular pH. As the intracellular pH acidifies, the channel activates, increasing rectification (Baukrowitz et al. 1999; Li et al. 2016a; Tinker et al. 2018; Rohacs et al. 2003; Shumilina et al. 2006). Like most other ion channels, KATP is regulated by post-translational modifications, such as S-palmitoylation (Yang et al. 2020) and phosphorylation affecting open probability and functional channel cell surface expression (Beguin et al. 1999). Analogous to all other Kir channels, KATP channels are ultimately regulated by phosphatidylinositol (4,5) bisphosphate (PIP2). The sensitivity of the channel to ATP is regulated by PIP2. PIP2 decreases the ATP sensitivity resulting in a reversal of channel closure by ATP and stabilizing the open state (Shyng et al. 2000). However, these channels are the least stereospecific to regulation by PI(4,5)P2 relative to other PIPs. The electrostatic interactions, dictated by the number of negative charges in the head group of PIPs, and the stereoselective nature, length of acyl chain, is less important for KATP channels, allowing PI(4,5)P2, PI(3,4,5)P3, PI(3,4)P2, and PI(4)P to regulate the channel equally (Rohacs et al. 2003). Furthermore, long chain CoA esters activate and regulate the KATP channel by binding residues 311–332 on the C-terminus, a site distinct to both PIP2 and ATP binding sites (Branstrom et al. 2007; Shyng et al. 2000). The metabolic sensing capability and its coupling to the electrical activity of KATP channels along with its distinct tissue localization has put much of the clinical emphasis on insulin regulation, cardiac rhythm, and neurological function.
4.2 Channel Regulation and Trafficking
ATP binds directly to Kir6 while MgADP binds to SUR. As ATP levels increase, ATP binds to the Kir6 subunits causing closure of the channel gates leading to a depolarization of the membrane and thus an increase in electrical activity. Conversely, as ATP levels fall and the ADP levels increase, MgADP binds to the SUR subunits causing the channels to open leading to hyperpolarization of the membrane and reduction in electrical activity (Ashcroft 2005).
Trafficking: Both SUR and Kir have a putative ER retention signal, RKR, in the distal portion of the C-termini. The SUR and Kir subunits co-assemble and in doing so mask each other’s ER retention signals. This allows for the octameric complex to exit the ER and localize to the plasma membrane (Zerangue et al. 1999). The truncated Kir6.2 channel (Kir6.2ΔC36) is a functional channel in the absence of any SUR subunits due to its lack of ER retention motif and is an experimentally useful tool (Reimann et al. 2001; Zerangue et al. 1999). KATP channels may also be post-translationally trafficked to specific excitable membrane domains by the cytoskeletal adapter ankyrin-B (AnkB) (Kline et al. 2009). SUR and Kir6 are regulated in the ER by proteasome-mediated degradation as well as through protein recognition and translocation by Derlin-1 and folding by the chaperone protein Hsp90 (Wang et al. 2012; Yan et al. 2010).
4.3 Physiology/Pathophysiology and Tissue Distribution
We will briefly summarize major points of channel characteristics, disease implications, and tissue distribution. For a more detailed discussion of channel characteristics and disease implications, refer to (Li et al. 2021) as well as to more in-depth reviews (e.g., Tinker et al. 2018).
Pancreatic: The KATP channels of pancreatic β cells are comprised of SUR1/Kir6.2. These channels couple the metabolic state of the cell to the release of insulin. In the presence of elevated blood glucose levels, ATP levels rise, bind directly to the Kir6 subunit, and cause channel closure. In β cells this closure depolarizes the cell leading to activation of voltage-gated Ca2+ channels, thus increasing levels of intracellular Ca2+, which results in insulin vesicle fusion to the plasma membrane and secretion (Hibino et al. 2010). There are a range of polymorphisms in the SUR1 and Kir6 subunit that leads to insulin secretory disorders. Hyperinsulinemia, neonatal diabetes, and DEND syndrome have all been linked to loss of function or gain of function mutations in the Kir6.2 subunit (Ashcroft 2005).
Cardiac: Cardiac KATP channels were initially thought to be an assembly of SUR2A and Kir6.2 due to most studies being conducted with ventricular cells. However, there has been discovery of other subunit assemblies within various tissues in the heart (for a list see Table 3 in Li et al. 2021). Cardiac KATP channels play an important cardioprotective role. Depending on their location in the heart, conductance and Ki of ATP do vary. KATP channels appear to be predominantly cardiac adaptive/protective in the presence of stressors (Tinker et al. 2018). Activation of cardiac KATP channels plays a significant role in ischemic preconditioning, and by shortening the action potential duration during myocardial ischemia increases cell survival (Tamargo et al. 2004). There is also evidence of KATP channels having altered expression patterns due to exercise leading to physiological adaptation (Tinker et al. 2018; Zingman et al. 2011).
Skeletal muscle: The classical skeletal muscle KATP channel assembly is SUR2A/Kir6.2, however skeletal muscles have also been shown to contain SUR1/Kir6.2. This has been found in fast twitch muscle, where the current density is larger (Tinker et al. 2018). Like cardiac KATP channels, in the skeletal muscle these channels contribute little to the membrane potential at rest. Their roles in skeletal muscle are still being investigated but it is evident they prevent muscle fatigue and muscle damage as well as regulate glucose uptake and cellular metabolism (Tinker et al. 2018). These findings suggest potential roles in muscle disorders, weight gain, and glucose regulation.
Vasculature and smooth muscle: Vascular smooth muscle, smooth muscle, and endothelial KATP channels are all comprised of Kir6.1/SUR2B (Suzuki et al. 2001; Aziz et al. 2017). Regulation of vascular smooth muscle through KATP channels is due to the ability of channel activity to regulate membrane potential. When hyperpolarization occurs through activation, intracellular Ca2+ is reduced causing dilation. This can occur through the Gs/PKA phosphorylation pathway as well as indirectly through hypoxic conditions via the mitochondria. Contraction occurs through the Gi/PLCβ2/3/PKC pathway that inhibits channel activity, causes depolarization and therefore contraction due to increased intracellular Ca2+ (Tinker et al. 2018). Smooth muscle KATP, like cardiac and neuronal KATP, plays a protective role in hypoxia and ischemic reperfusion (Aziz et al. 2017). However, there may be variation in expression depending on the location of the smooth muscle. KATP channels have been found in smooth muscle in the GI tract, bladder, uterus, urethra, and respiratory tract.
Neuronal: There is extensive expression of KATP channels in neuronal populations including glial cells (SUR1/Kir6.1 or Kir6.2) and neurons in the cortex, hippocampus, dorsal root ganglion (Kir6.2/SUR1 or SUR2), hypothalamus (Kir6.2/SUR1), and substantia nigra pars reticulata (Kir6.2/SUR1) (Liss et al. 1999; Sun et al. 2006b). The differential expression of Kir6 and SUR in neuronal populations leads to distinct roles in metabolic and non-metabolic sensing (Sun et al. 2006b; Tinker et al. 2018). KATP channels have been implicated in pain, locomotion and behavior, nerve function, immune activity of glial cells, and most notably neuroprotection against ischemia in hypoxic conditions (Sun and Feng 2013; Tinker et al. 2018). In hypoxic conditions, KATP channels open leading to hyperpolarization which dampens neuronal firing and reduces metabolic demands. Depolarization increases Ca2+ entry which can lead to neuronal death. The neuroprotective nature of KATP appears to be linked directly to the Kir6 subunit (Sun et al. 2006b; Heron-Milhavet et al. 2004; Tinker et al. 2018).
4.4 Pharmacology
Therapeutic compounds that regulate KATP channels belong to a few main classes, the sulfonylureas and the general class of potassium channel openers (KCOs) or potassium channel blockers (KBOs). These drug classes exert their effects on KATP channels by binding to the sulfonylurea receptors (SURs) and allosterically inhibiting or activating the channel or by directly engaging with the channel pore.
Small molecule drugs may exert their regulatory properties affecting gating by allosteric modulation or direct interaction with the binding pockets of PIP2, ATP, or the channel pore. Experimental and MD studies have uncovered other specific chemical scaffolds that bind directly to the Kir6 channels to exert regulatory effects (see Fig. 11). The exact mechanism of action by these chemicals varies and, in some cases, remains elusive. Therapeutic compounds that bind directly to the Kir6 channels, which have been experimentally confirmed, include the imidazoline compounds, thiazolidinedione (“glitazone”) compounds, morpholinoguanidines, quinine, the antiarrhythmic, cibenzoline, biguanides and various anesthetics such as propofol and bupivacaine (Cui et al. 2003; Grosse-Lackmann et al. 2003; Kawahito et al. 2011; Kawano et al. 2004a; Kawano et al. 2004b; Kovalev et al. 2004; Mukai et al. 1998; Proks and Ashcroft 1997; Rui Zhang et al. 2010; Yu et al. 2012).
Computational simulations, including MD, have suggested travoprost, betaxolol, and ritodrine as other drugs that bind directly to the Kir6 subunit to exert their effects (Chen et al. 2019). These latter drugs, along with others uncovered through in silico screens and dynamic studies, will need to be experimentally validated for their binding affinity for the Kir6 subunit and the mechanistic impact on channel gating. However, they provide important insights into new therapeutic chemical scaffolds for Kir6 subunits. It remains an open question whether Kir6 targeting drugs control activity either directly by altering channel-PIP2 interactions or indirectly by altering channel-ATP interactions. With the differential expression of both SUR and Kir6 subunits, drugs that bind each with varying affinities offer substantial advantage to subtype specific targeting and reduced off target effects.
Another mechanism of KATP channel regulation is the rescue of surface expression impairments causing biogenesis or trafficking defects, through pharmacological correctors (Shyng et al. 2000). There are well-established mutations in SUR domains that cause trafficking defects of the KATP complex to the cell surface (Zhou et al. 2014). Carbamazepine is a clinically approved anticonvulsant drug, approved for the treatment of epilepsy and bipolar disorder, known to bind to NaV channels, CaV channels, and GABAA receptors (Chen et al. 2013). Carbamazepine rescued trafficking of KATP with trafficking mutations occurring in the TMD0 of SUR1 (Zhou et al. 2014). The TMD0 is part of the N-terminal region of the SUR which binds and couples to the Kir6 channel to form the KATP complex (Chan et al. 2003; Chen et al. 2013). Interestingly, carbamazepine also appears to affect MgADP binding to the NBD domains of SUR, so while it rescues trafficking and expression can inhibit the channel by abolishing the MgADP activation effect (Zhou et al. 2014). It has also been established that sulfonylurea drugs may also be able to rescue trafficking and folding in specific SUR mutants (Yan et al. 2004). Again, they also have an impact on KATP activity and conductance once folding and cell surface expression is rescued, increasing the complexity of the pharmacology of such drugs.
The following listed drugs (Table 5 and Fig. 11) were validated with the functional channels Kir6.2ΔC26 or Kir6.2ΔC36 that do not require the presence of SUR subunits, therefore suggesting they act directly on the Kir6 subunit. While they all provide valuable insights into structure-based drug discovery, they all require further computational and experimental investigation to better understand the binding sites and mechanisms of gating.
4.4.1 Drugs Acting Through the Kir6 Subunits
Imidazolines: Imidazolines, including clinically relevant clonidine and phentolamine, and others not on the clinical market, such as idazoxan, efaroxan, and RX871024, bind directly to Kir6 channels (Kawahito et al. 2011; Proks and Ashcroft 1997; Rui Zhang et al. 2010). Clonidine and phentolamine made it to the market based on their ability to bind α-adrenergic receptors but later discovered to bind endogenous imidazoline receptors (Bousquet et al. 2020). Computational studies have elucidated various putative binding sites on the Kir6 channels including the PIP2 binding site (Idazoxan) and near the slide helix (clonidine and efaroxan).
Despite such differences, they all bind close to the interface between the transmembrane domain and cytosolic domain of the Kir6 subunit (Rui Zhang et al. 2010). Functional studies of clonidine and phentolamine (Kir6.2ΔC36 channel in HEK293 cells and Xenopus oocyte macropatches, respectively) suggest they do not act through SUR (Grosse-Lackmann et al. 2003; Kawahito et al. 2011; Proks and Ashcroft 1997). This finding has been further validated (Kawahito et al. 2011). RX871024 binds in the channel pore forming two hydrogen bonds with interior pore-lining residues. This causes direct block of the channel (Rui Zhang et al. 2010). Aside from RX871024, imidazoline drugs are likely candidates to be working through direct or allosteric alteration of channel–PIP2 interactions. The ATP-insensitive mutant (Kir6.2ΔC36-K185Q) showed no reduction in current in the presence of ATP but both clonidine (1 mM) and phentolamine (1 μM) were still able to block the current as effectively as in the Kir6.2ΔC36 channel, suggesting the binding site of these two imidazolines is not directly at the ATP binding site or involving key ATP binding residues (Kawahito et al. 2011; Proks and Ashcroft 1997).
Thiazolidinediones: The thiazolidinediones (TZDs), also known as the glitazones, are a drug class whose members exert their clinical anti-diabetic effect by binding to the nuclear transcription factor, PPAR and as such they do have increased cardiovascular risks (Chen et al. 2020; Nanjan et al. 2018). Rosiglitazone (RSG) and pioglitazone are two important clinically approved anti-diabetic drugs of this class. TZDs are reported to have a direct and indirect mechanism of action in relation to diabetes. It is also well characterized that TZDs can directly bind to the Kir6 subunits of the KATP channel through functional studies (Yu et al. 2012). More interestingly, it has been shown computationally that RSG binds in close proximity to the helix bundle crossing (HBC) gate and PIP2, without complete PIP2 binding site occupation (Chen et al. 2020). The main impact of RSG on Kir6 channel current is increased mean closed times (Yu et al. 2012). Based on the putative binding site proposed by Stary-Weinzinger, RSG binds residues in or near the slide helix separating the putative binding site to be distinct from that of the ATP binding site (Chen et al. 2019; Martin et al. 2017).
Morpholinoguanidines: The most historically studied morpholinoguanidine in relation to KATP channels is PNU-37883A. It acts directly on the Kir6 subunit and has selectivity for Kir6.1 over Kir6.2 making it more selective for smooth muscle with some selectivity for the vascular KATP channels (Cui et al. 2003). The selectivity of PNU-37883A for KATP channels is largely impacted by the SUR that complexes with the Kir6.1 (Cui et al. 2003). This likely has to do with either obstruction of the drug binding pocket or an allosteric conformational change of Kir6 upon SUR binding (Humphrey 1999; Kovalev et al. 2004). The binding region for the PNU-38773A has been mapped to amino acids 200–280 on the C-terminus of Kir6 (Kovalev et al. 2004). It is still unclear if binding to the C-terminus allows the molecule to position into the channel pore or it causes a conformational rearrangement of the gating machinery to close the channel. The effect of PNU-37883A on the binding of ATP or PIP2 to the Kir6 channel has not been adequately explored. Neither the precise binding site of PNU-37883A is determined nor how its binding results in current inhibition. Could it promote ATP binding? Could it disrupt MgADP binding to the SUR, since the SUR NBD interfaces with the Kir6.2 CTD? Could it work by disrupting channel–PIP2 interactions directly or allosterically? Such questions would need to be further addressed in future studies (Lee et al. 2017). The non-KATP channel-specific nature of the drug leading to a variety of pharmacological actions does not make it a good drug candidate but its further study will provide a chemical and structural framework for developing more specific KATP channel inhibitors (Teramoto 2006).
Anesthetics (propofol, bupivacaine): Propofol is a general anesthetic that is used widely throughout the world. Its pharmacological action has been largely attributed to its action on the GABAAR (Tang and Eckenhoff 2018). Growing functional evidence has uncovered its action at KATP channels. Propofol is selective for KATP channels containing Kir6.2 over Kir6.1 and directly binds the Kir6 subunit of the KATP channel (Kawano et al. 2004b). Utilizing the ATP-insensitive mutant and the R31E mutation, inhibition was abolished indicating propofol has at least some partial overlap with the ATP binding site and with an N-terminal binding site (Kawano et al. 2004b). There is also evidence to suggest that propofol affects the expression level of Kir6.1 (Zhang et al. 2016).
Bupivacaine is a local anesthetic with a chemical structure different and more complex than that of propofol (see Fig. 11). Bupivacaine was experimentally proven to have a potency on Kir6.2ΔC36 equal to that of Kir6.1/SUR2B. It has a greater affinity for the cardiac channel (Kir6.2/SUR2A). It had higher affinity over levobupivacaine and ropivacaine due to the stereoselective nature of the Kir6 binding pocket (Kawano et al. 2004a). It likely exerts its effects by binding residues at the cytosolic end of TM2 which are important in the channel gating properties likely relinquishing any interference with ATP binding to the channel (Kawano et al. 2004a). Bupivacaine and other similar local anesthetics bind to NaV channels as well as CaV and KV channels leading to their major cardiotoxic effects. It has been proposed that local anesthetics inhibit Kir3 channels through antagonism of channel–PIP2 interactions (Zhou et al. 2001). If local anesthetics work at the same conserved site on KATP as they do on Kir3 channels, PIP2 binding residues may be important candidates to explore.
Biguanides: Metformin and phenformin are the most well-known, clinically relevant biguanides. They are used to increase insulin sensitivity in type 2 DM (Hansen 2006). Phenformin was withdrawn from the market in the 1970s. Biguanides activate AMPK with several downstream implications of the metabolic state of the cell as well as potentially increasing KATP cell surface expression with evidence that they directly affect KATP channels independently of AMPK signaling (Aziz et al. 2010). Aziz et al. (2010) were able to show that there is differential activity of phenformin based on SUR subunit composition but equivalent activity of Kir6 in the absence of SUR subunits. While they appear to bind Kir6 directly, SUR does play a role in modulating the affinity of biguanides to the channel (Aziz et al. 2010).
Drugs acting through the SURs: KATP channel openers and inhibitors acting through the SURs of the KATP channel complexes have been discussed in chapter “The pharmacology of ATP-sensitive K+ channels (KATP)” (Li et al. 2021).
Miscellaneous drugs: Quinine, chloroquine, and quinacrine are all structurally related drugs containing the quinolone bicyclic ring. These drugs have been shown to bind and alter the conductance of Kirs (Lopez-Izquierdo et al. 2011; Grosse-Lackmann et al. 2003). Quinolones are used as one of the most common antibacterial in the world and their mechanism may limit their potential use on KATP regulation for the current clinically used quinolones (Aldred et al. 2014).
The well-known antiarrhythmic agent, cibenzoline, is a diphenylmethane with inhibitory properties on the Kir6 subunit shown by functional studies. There is evidence that cibenzoline binds the K+-recognition site on KATP channels similar to how it binds the H+, K+ -ATPase (Tabuchi et al. 2001). This may prove to be a novel mechanism of binding but would decrease its selectivity for Kir6 over other Kirs given the conservation of the putative binding site. In 2018 Ramu and colleagues uncovered a 54-residue protein toxin recovered from the venom of S. polymorpha (SpTx) that inhibited Kir6 from the extracellular side (Ramu et al. 2018). SpTx binds the Kir subunit and not the SUR subunit. Further investigation is needed to determine the mechanism of channel closure but nonetheless this discovery opens a new possibility in the pharmacology of Kir6 channel gating. There is evidence to suggest that glinides or the benzylsuccinic acid derivative, mitiglinide, bind both to the SUR at a high affinity site and directly to the Kir6 subunit at a lower affinity site (Aziz et al. 2010; Reimann et al. 2001). Similarly, the repaglinide binding site is on the SUR, as resolved by cryo-EM, but it also has a partial contribution by the N-terminus of the Kir6.2 subunit. This was confirmed through binding studies in which co-expression of Kir6.2 enhanced binding affinity and deletion of the N-terminal peptide of Kir6.2 abolished the binding enhancement (Ding et al. 2019).
4.5 Structural Studies
Before atomic resolution structures of the KATP were available, in order to identify the PIP2 binding site, Haider and colleagues conducted molecular docking and MD simulations (10 ns) based on a Kir6.2 homology model. The PIP2 head group was predicted to interact with K39, N41, and R54 in the N-terminus, K67 in the transmembrane domain and R176, R177, E179, and R301 in the C-terminus. The model predictions are consistent with a large body of functional data, suggesting how PIP2 binding may lead to an increase in Kir6.2 open probability and a reduction in ATP sensitivity (Haider et al. 2007). In order to understand the mechanism by which disease mutations exert their deleterious effects, in 2017, Cooper and colleagues worked on a Cantu syndrome (CS) mutant, where the Kir6.1(V65M)/ Kir6.2(V64M) GoF mutations in the slide helix enhance the channel activity when co-expressed with the SUR1 or SUR2 regulatory subunits (Cooper et al. 2017). The Val to Leu mutation that does not cause the disease was used as a negative control. The disease mutations abolished the sensitivity of KATP to ATP and to the blocker glibenclamide, which acts on the SUR subunit. This finding suggested that sulfonylurea therapy may not be successful, at least for some CS mutations. Homology modeling and MD simulations (100 ns) were conducted on the Kir6.1(WT), Kir6.1(V65L), and Kir6.1(V65M) systems. The stabilization of the open state of the Val to Met mutations (but not to Leu) that was obtained experimentally could not be shown computationally in the 100 ns simulation time frame. The same year in 2017, near-atomic resolution structures were obtained from three independent labs (Chen in Beijing, Shyng in Oregon and MacKinnon in New York). These structures were solved for the Kir6.2/SUR1 in complex with ADP, ATP, and glibenclamide (Lee et al. 2017; Martin et al. 2017; Wu et al. 2018). In 2019, a higher resolution (3.3 Å) cryo-EM structure of Kir6.2/SUR1 in complex with ATPγS and repaglinide was solved by the Chen lab. Figure 8 shows the Kir6.2 channel surrounded by four SUR1 subunits, the two distinct binding sites for ATPγS (one in Kir6.2 and the other in SUR1), and the binding site for repaglinide in the transmembrane domain of SUR1 (Ding et al. 2019). This study also suggested through cryo-EM and binding studies that the N-terminal portion of Kir6.2 adds to the binding pocket for repaglinide. This is further supported by earlier functional studies done by Reimann et al. (2001) in which a structurally similar drug, mitiglinide, had a biphasic dose-response curve, suggesting a high affinity binding site at SUR and a low affinity binding site at Kir6.2. Chen and colleagues conducted MD simulations (at a microsecond time scale) on Kir6.1 channel to investigate the blocking mechanism by rosiglitazone (RSG). The putative RSG binding site was identified through unbiased MD simulations of Kir6.1 channel with 20 RSG molecules randomly placed in the solvent, and followed by free energy calculations. Based on the predicted RSG binding site, dynamic pharmacophore models were constructed and used for screening of hits in the DrugBank database. Three new high affinity blockers, betaxolol, ritodrine, and travoprost, were identified and subsequently tested functionally. Using the inside-out patch-clamp mode in HEK293T cells expressing Kir6.2/SUR2A, travoprost, betaxolol, and ritodrine had IC50s of 2.46 μM, 22.06 μM, and 7.09 μM, respectively (Chen et al. 2019). Given the computational identification of direct binding to Kir6, the expectation would be that these three drugs should have similar potency when tested on Kir6.2ΔC channels. Computational simulations to understand the dynamics of KATP channel structure and function are still lacking. The recent cryo-EM structures have provided us with the opportunity to characterize the channel dynamic function using long timescale MD simulations, and a molecular basis for structure-based drug discovery for these important drug targets.

Cryo-EM structure of KATP channel in complex with ATPγS and Repaglinide (PDBID:6JB1), (a) side view. (b) top view. The KATP channel and SUR1 are drawn in Newcartoon, the ATPγS and Repaglinide are drawn in VDW
5 K+ Transport Kir (1, 4, 4/5, 7) Channels
5.1 Kir1.1: Historical Perspective
The first member of the Kir family to be cloned (see below) was initially referred to as the “rat outer medullary K+” (ROMK1) channel for its prominent role in the kidney.
The task of renal epithelial cells is to maintain ionic homeostatic control of K+, Na+, and Cl− in the urine and blood and Kir1 channels play a key role in achieving this task. The channels are specifically localized in the apical rather than the basolateral membrane of these cells. To appreciate the strategic importance of Kir1 channels let us consider their role in the thick ascending loop of Henle (TAL), the segment of the nephron responsible for 25–30% of the total Na+ reabsorbed by the kidney and the site of action of loop diuretics. Here, K+ efflux through Kir1 channels also maintains the Na+-K+-2Cl− symporter active by supplying K+ to the extracellular site of the transporter and enabling the uptake of NaCl along with K+ into the TAL cells. The Na+ entering the cytoplasm from the apical (lumen or urine side) through the symporter fuels the basolateral Na+-K+ ATPase to continue transporting Na+ to the blood side. K+ channels (e.g., Kir1, Kir4, Kir7) co-expressed with the Na+-K+-ATPase in basolateral membranes supply K+ to the extracellular side of the pump to maintain its activity. This is referred to as “K+ recycling” (Hibino et al. 2010). Furthermore, the hyperpolarization caused by Kir1 channels accelerates Cl− exit from basolateral Cl− channels, establishing “the lumen positive transepithelial potential,” which serves as the main driving force for paracellular Na+, Ca2+, and Mg2+ transport from the lumen to blood side. Deletion of Kir1 channels impairs renal NaCl absorption yielding “Barter’s syndrome”-like phenotypes, as we will discuss below (Bleich et al. 1990; Greger et al. 1990). Moreover, Kir1 channels on the apical membrane of TAL epithelial cells are functionally coupled to Cl− channels (the cystic fibrosis transmembrane regulator – CFTR) in that CFTR decreases Kir1 activity. This effect can be reversed by PKA phosphorylation and may underlie the actions of the antidiuretic hormone arginine vasopressin that increases Kir1 activity, limiting K+ secretion and urinary K+ loss (Field et al. 1984).
5.1.1 Subfamily Members and Tissue Distribution
In early 1993 the first two Kir channel cDNAs were reported, Kir1.1 and Kir2.1, followed by Kir3.1 later the same year (Dascal et al. 1993; Ho et al. 1993; Kubo et al. 1993a; Kubo et al. 1993b). Six alternatively spliced isoforms of Kir1.1(a-f) have been identified but only two of these (a, c) give rise to distinct proteins from the other four isoforms (b, d-f) (Boim et al. 1995; Ho et al. 1993; Kondo et al. 1996; Shuck et al. 1994; Zhou et al. 1994). The splice isoforms differ in their N-termini with Kir1.1b being the shortest, Kir1.1a 19 aa and Kir1.1c 26 aa longer than Kir1.1b (the human isoform shown in Fig. 9 is the Kir1.1a isoform with 391 aa). No Kir1.1 heteromers with members of other Kir subfamilies have been reported. As mentioned above, Kir1.1 is expressed in the kidney (TAL, distal convoluted tubule, and cortical collecting duct) and, as shown by in situ hybridization, in cortical and hippocampal neurons (Kenna et al. 1994).
5.1.2 Physiology/Pathophysiology
Kir1.1 function is regulated by internal pH, phosphorylation by a number of different kinases, miRNAs, and ubiquitination. Acidification closes Kir1.1 channels with a pKa of ~6.5 (Choe et al. 1997; Doi et al. 1996; Fakler et al. 1996; McNicholas et al. 1998; Tsai et al. 1995). Multiple studies have implicated the involvement of several residues in pH sensitivity but structural insights are still lacking (Hibino et al. 2010). Kir1.1 has ER retention signals (368R-X/A-R370 and K370-R371) (Ma et al. 2001; Yoo et al. 2005). It is regulated by phosphorylation of S44 (by both PKA, SGK – Ser/Thr protein kinases), increasing trafficking to the plasma membrane (McNicholas et al. 1994; Xu et al. 1996; Yoo et al. 2005). Additionally, PKA-mediated phosphorylation of S219 and S313 increases the channel open probability, likely by enhancing channel–PIP2 interactions (Liou et al. 1999; MacGregor et al. 1998). Another class of Ser/Thr kinases, WNKs (With-no-K – K = Lys) decreases Kir1.1 cell surface expression by unique mechanisms (Hibino et al. 2010). A number of microRNAs have been shown to be upregulated in high-K+ diet and, in turn, they regulate Kir1.1: miR-802 increased Kir1.1 surface expression and channel activity by reducing caveolin-1 that limits Kir1.1 expression at the plasma membrane (Lin et al. 2014); miR-194 also enhanced Kir1.1 surface expression by counteracting the opposite regulation by WNK and intersectin 1 (He et al. 2007). Finally, monoubiquitination of K22 decreased cell surface Kir1.1 serving as a signal to process the channel for lysosomal degradation (Lin et al. 2005). The Kir1.1 knockout mouse resulted in mice afflicted with Barter’s syndrome (BS) (Lorenz et al. 2002; Lu et al. 2002). BS is an autosomal recessive renal tubulopathy characterized by hypokalemic metabolic alkalosis, renal salt wasting, hyperreninemia, and hyperaldosteronism (Asteria 1997; Guay-Woodford 1995; Hebert 2003; Karolyi et al. 1998; Peters et al. 2002; Rodriguez-Soriano 1998). Loss-of-function mutations of Kir1.1 (type II BS) affect many aspects of the functional integrity of the channel and its regulation, including interactions with PIP2 (Hibino et al. 2010; Lopes et al. 2002). BS patients and Kir1.1 mice are characterized by hypokalemia and excess K+ in the urine, which is contrary to the role of Kir1.1 as providing the main secretory pathway of K+ in renal tubules. Bailey and colleagues addressed the conundrum showing that in the absence of Kir1.1 in the TAL cells K+ secretion is sustained in the late distal tubule by maxi-K+ channels (Bailey et al. 2006).
5.2 Kir4/Kir5: Historical Perspective and Tissue Distribution
Kir4 are expressed as homotetramers as well as heterotetramers with Kir5.1, while when Kir5.1 is expressed alone in a heterologous expression system it shows no function.
These channels are the molecular identities of neuro- and retinal glial cell K+ channels. Their cloning provided a boost to their long-recognized importance (Kuffler and Nicholls 1966; Newman 1984). Kir4channel cDNAs were first identified in the mid-90s and Kir4.1 has been referred to under a variety of names: BIR10 (Bond et al. 1994), KAB-2 (Takumi et al. 1995), BIRK-1 (Bredt et al. 1995), and Kir1.2 (Shuck et al. 1997). In situ hybridization showed Kir4.1 predominantly expressed in glial cells in the CNS (brain and spinal cord) and retina (Müller cells) (Hibino et al. 2010; Takumi et al. 1995). Kir4.1 has also been reported to be expressed in the kidney (reviewed in Manis et al. 2020), stomach (Fujita et al. 2002), and the cochlea of the inner ear (Hibino and Kurachi 2006). Kir4.2 was first isolated from a kidney library, where it is highly expressed, along with liver, embryonic fibrocytes, and microvascular endothelial cells (Lachheb et al. 2008; Pearson et al. 1999; Shuck et al. 1997). A single member of the Kir5 subfamily, Kir5.1, was identified in 1994 (Bond et al. 1994). It is highly expressed in kidney, spleen, adrenal glands, liver, in several brain regions and in spermatozoa and spermatogenic tissue (Bond et al. 1994; Salvatore et al. 1999). Kir5.1 is only functional as a heteromer with Kir4.1 and Kir4.2. The nephron serves as a great example of where expression and heteromerization with one, the other, or both of the Kir4 isoforms best serves overall renal salt handling (see below in Sect. 5.2.1). Its distribution and co-expression with the Kir4 isoforms vary along the nephron to fulfill specific functional characteristics of each tubule segment (full validation of the expression and activity of Kir4.2 is still pending). Thus, in the proximal convoluted tubule Kir5.1 is co-expressed with Kir4.2, in the cortical collecting duct and also in the connecting tubule (connecting the distal convoluted tubule to the cortical collecting duct) it is co-expressed with Kir4.1, while in the thick ascending limb of Henle it is not expressed at all (only Kir4.1 is expressed), and in the distal convoluted tubule it is co-expressed with both Kir4 isoforms (Manis et al. 2020).
5.2.1 Physiology/Pathophysiology
Kir5.1 confers several unique functional characteristics to Kir4 isoforms: (1) greater pH sensitivity (Tucker et al. 2000) (2) maintenance of pH homeostasis (Puissant et al. 2019), (3) inhibition of the dependence of channel conductance on [K+]o (Edvinsson et al. 2011a; Edvinsson et al. 2011b) (shown only for Kir4.2 thus far), and (4) activation by [Na+]i (Rosenhouse-Dantsker et al. 2008). Kir4.1/Kir5.1 channels have been shown to act as K+ sensors in the distal nephron (Cuevas et al. 2017) regulating expression of apical Na+ transporting proteins and renal salt handling in response to dietary salt intake (Manis et al. 2020). Dysfunction of these channels has been proposed to contribute to the pathogenesis of salt-sensitive hypertension (Palygin et al. 2017; Palygin et al. 2018). Knockouts of either Kir4.1 (Cuevas et al. 2017) or Kir5.1 (Wu et al. 2019) compromise [K+]o sensing and K+ excretion. A unifying link for action of various Kir4/5 modulators could be the allosteric changes in channel–PIP2 interactions. The modulators proposed to work in this way include [Na+]i (entering through the apical side; Rosenhouse-Dantsker et al. 2008)), [K+]o at the basolateral side (Harraz et al. 2018), changes in intracellular pH or phosphorylation by kinases, such as WNK-4, (Du et al. 2004). Differences in Kir4.1 versus Kir4.2 and their relative heteromers with Kir5.1, along with their characteristic expression may underlie their distinct physiological roles. Thus, Kir4.2 is more pH sensitive than Kir4.1. Heteromerization of Kir4.2 with Kir5.1, unlike Kir4.1, converts Kir4.2 from a strong to a weak rectifier, rendering it more sensitive to intracellular pH. Knockout mice have shown that Kir4.2 plays a distinct role in acid-base homeostasis compared to Kir4.1, resulting in metabolic acidosis with intracellular alkanization and membrane depolarization in the proximal convoluted tubules of the kidney (Manis et al. 2020). In the CNS, the Kir4 isoforms and their regulatory Kir5.1 subunits exhibit very interesting physiology by allowing for control of [K+]o in nearby neurons to regulate neuronal excitability (Hibino et al. 2010).
5.3 Kir7.1: Historical Perspective
Kir7.1 is the only member identified in this subfamily and the most distant relative of all other Kir channels, showing the highest homology/identity to the Kir4 isoforms (~50% similarity and 40% identity; Fig. 10). Three groups reported cloning of Kir7.1 from CNS neurons in 1998 (Doring et al. 1998; Krapivinsky et al. 1998; Partiseti et al. 1998). A splice variant in native human retinal pigment epithelium has been described to be missing a region from exon 2 (amino acids 244–479), yielding a non-functional truncated Kir7.1 that is expressed at fourfold lower levels than the wild-type protein and does not interfere with the activity of the full-length protein (Yang et al. 2007). Another two splice variants of Kir7.1 were described recently in several mouse tissues. These lacked most of the C-terminal domain, Kir7.1-R166X (in the B-loop, Fig. 9) and Kir7.1-Q219X (in βE, Fig. 9). These truncation variants would be comparable to mutations associated with Leber congenital amaurosis, a rare recessive hereditary retinal disease that results in vision loss at early age. Simultaneous expression with the full-length Kir7.1, however, led to a reduction in activity of the wild-type channel (possibly due to partial proteasome degradation of WT-mutant channel heteromers) (Vera et al. 2019).
5.3.1 Tissue Distribution
The Kir7.1 protein has been reported to be expressed along with the Na+/K+-ATPase for “K+ recycling” either in the apical membranes of epithelial cells of the choroid plexus (Doring et al. 1998; Hasselblatt et al. 2006; Nakamura et al. 1999) and retinal pigment epithelium (Kusaka et al. 2001; Shimura et al. 2001) or in basolateral membranes of thyroid follicular cells (Nakamura et al. 1999), in the distal convoluted tubule, proximal tubule, and collecting duct of renal epithelia (Derst et al. 2001; Ookata et al. 2000) as well as in the small intestine and stomach (Partiseti et al. 1998). Kir7.1 has also been reported to be expressed in hypothalamic arcuate neurons involved in the appetite control circuit, where they are coupled to the MC4R G Protein Coupled Receptor in a G protein-independent manner (Ghamari-Langroudi et al. 2015) as well as in the myometrial smooth muscle, where it controls uterine excitability throughout pregnancy (McCloskey et al. 2014).
5.3.2 Physiology/Pathophysiology
A hallmark of Kir channels is a dependence of their conductance on [K+]o (Fig. 1a). Kir7.1 is also characterized by very weak rectification and a very low unitary conductance (Krapivinsky et al. 1998). The crucial difference appears to lie in the structure of the outer mouth of the selectivity filter, where Kir7.1 has a Met residue (M125), instead of Arg that is found in every other Kir channel (Fig. 9). M125R point mutation in Kir7.1 not only increased its unitary conductance by nearly 20-fold, strengthened its inward rectification and sensitivity to Ba2+ but also reinstated strong dependence on [K+]o (Doring et al. 1998; Krapivinsky et al. 1998). The corresponding residue R148 in Kir2.1 had been shown to have the opposite effect, namely the Kir2.1(R148Y) mutant showed a much reduced [K+]o dependence (Kubo 1996) while the R148H mutation increased sensitivity to pHo (but not pHi) (Shieh et al. 1999). Taken together, this evidence suggests that a positively charged residue is needed at the outer mouth of the selectivity filter to confer the dependence on (V-EK) shown by Kir channels. Several questions remain for a better understanding of this property. How do extracellular Na+ ions serve as blocking particles to regulate this dependence (Ishihara 2018; Soe et al. 2009) and how such interactions couple allosterically to control channel–PIP2 interactions (Harraz et al. 2018)? Dynamic computational studies will be instrumental in answering these questions. We will next focus discussion of the role of Kir7.1 channels in health and disease in the best studied system, the retinal pigment epithelium (RPE).
Retinal pigment epithelium: The RPE is a hexagonally packed, tight-junction connected monolayer of post-mitotic, pigmented cells between the neuroretina and the choroids. The apical membrane of the polarized RPE cells faces the photoreceptor outer segment (POS), while the basolateral membrane faces the fenestrated endothelium of the choriocapillaris. Interactions between the RPE and the POS are essential for visual function. The RPE’s primary functions are: a) to transport nutrients, ions, and water between the blood supply and the POS, b) to absorb light and protect against photo-oxidation of proteins and phospholipids of the POS, c) the isomerization of retinal which serves as the agonist of the rhodopsin GPCR to signal to the phosphodiesterase to cleave cGMP to 5’-GMP, d) phagocytosis of shed photoreceptor membranes, and e) secretion of essential elements for the morphological integrity of the retina (Kumar and Pattnaik 2014). In the dark, the cGMP levels are maintained to signal to the cGMP-gated (CNG) cation channels in the POS and allow the influx of Na+ and Ca2+ to the photoreceptor cell (rod or cone). This is countered by a net K+ efflux in the photoreceptor inner segment (PIS) mediated by various K+ channels (e.g., KCNQ, and 8 Kir currents, with Kir7.1 being the predominant one) giving rise to the so-called dark current. The efflux at the PIS pulls the resting Vm toward EK but the cationic influx at the POS counters the K+ efflux, maintaining a rather depolarized level in the dark. Upon light illumination, the resulting 5’-GMP no longer keeps the CNG channels open, causing the K+ efflux to dominate and an ensuing hyperpolarization of the photoreceptor cell. This signal will decrease glutamate release that once integrated to the retinal ganglion cells, it will be communicated to the brain through the optic nerve and be interpreted as “light.” The new balance of Na+ and K+ ions decreases the PIS Na+/K+ ATPase activity and the [K+]o falls from 5 mM in the dark to 2 mM in the light, making EK more negative increasing Kir currents toward re-establishing [K+]o to the 5 mM levels. The predominant Kir7.1 resting conductance, unlike other Kir currents, does not decrease its conductance with the drop of [K+]o as we discussed above, carrying the weight of re-establishing the “normal” higher [K+]o levels. This suggests another case of K+ recycling in conjunction with the Na+/K+ ATPase (Kumar and Pattnaik 2014). Loss of function mutations in Kir7.1 result in the autosomal-dominant disease called snowflake vitreoretinal degeneration, which progressively causes fibrillar degeneration of the vitreous humor, early-onset cataract, minute crystalline deposits, and retinal detachment. The R162W mutation, characteristic of this disease has been suggested to affect channel–PIP2 interactions (Pattnaik et al. 2013; Zhang et al. 2013).
5.4 Transport Channel Pharmacology
Kir1, Kir4, and Kir7 channel inhibitors: Dozens of small molecule inhibitors for Kir1, Kir4, and Kir7 channels have been reported (Table 6). VU590 was discovered through HTS of 126,009 small molecules as Kir1.1 modulators. It inhibits Kir1.1 with submicromolar affinity, but has no effect on Kir2.1 or Kir4.1. It also inhibits Kir7.1 at low micromolar concentrations. Electrophysiological studies indicated VU590 is an intracellular pore blocker (Lewis et al. 2009). In an effort to achieve more selective inhibitors for Kir1.1, VU591 was developed based on VU590. VU591 is a potent inhibitor like VU590 for Kir1.1, but it is selective for Kir1.1 over Kir7.1. It blocks the intracellular pore of the channel through interactions with V168 and N171 (Bhave et al. 2011; Swale et al. 2015). VU573 was discovered through a thallium (Tl+) flux-based high-throughput screen of a Kir1.1 inhibitor library for modulators of Kir3. It inhibits selectively Kir2.3, Kir3.X, and Kir7.1 with similar potency over Kir1.1 and Kir2.1 (Raphemot et al. 2011). ML418 was discovered based on a lead compound VU714, which was identified by a HTS screen as a novel Kir7.1 channel inhibitor. It selectively inhibits Kir7.1 and Kir6.2/SUR1 over Kir1.1, Kir2.1, Kir2.3, Kir3.1/Kir3.2, and Kir4.1 channels (Swale et al. 2016).
VU0134992 was discovered through HTS of 76,575 compounds from a Vanderbilt University library for small molecule modulators of Kir4.1. It inhibits selectively Kir4.1 and Kir3.1/3.2, Kir3.1/3.4 and Kir4.2 with similar potency over (30-fold selective) Kir1.1, Kir2.1, and Kir2.2. It also weakly inhibits Kir2.3, Kir6.2/SUR1, and Kir7.1 (Kharade et al. 2018). Tricyclic antidepressants (TCA), nortriptyline, amitriptyline, desipramine, and imipramine were found to act as inhibitors for Kir4.1 channel expressed in HEK293T cells, using the whole-cell patch-clamp technique. These compounds inhibited Kir4.1 currents in a voltage-dependent fashion, and marginally affected neuronal Kir2.1 currents (Su et al. 2007). Antidepressant, selective serotonin reuptake inhibitor (SSRI), fluoxetine, was found to inhibit the Kir4.1 channel expressed in HEK293T cells using whole-cell patch clamp. It inhibited the Kir4.1 channel in a concentration-dependent and voltage-independent manner. Fluoxetine had little or no effect on Kir1.1 and Kir2.1 channels. Other SSRIs, sertraline and fluvoxamine also inhibited the Kir4.1 channel (Ohno et al. 2007). Aréchiga-Figueroa and colleagues discovered pentamidine, a drug for treatment of protozoan infections, inhibited Kir4.1 channels. This drug potently inhibited the Kir4.1 channel from the cytoplasmic side in the inside-out patch-clamp configuration. The inhibition was voltage dependent. Molecular modeling predicted the binding of pentamidine to the transmembrane pore region interacting with residues T127, T128, and E158 (Arechiga-Figueroa et al. 2017). Quinacrine, an old antimalarial drug, was discovered as an inhibitor of the Kir4.1 channel expressed on HEK293 cells, using patch clamp. It inhibited Kir4.1 channel with an IC50 of 1.8 μM and in a voltage-dependent manner. Molecular modeling and mutagenesis studies suggested quinacrine blocks Kir4.1 through the central cavity interacting with residues E158 and T128 (Marmolejo-Murillo et al. 2017a). Similarly, chloroquine, an aminoquinoline derivative anti-malarial drug, was discovered as an inhibitor for Kir4.1 channel (Marmolejo-Murillo et al. 2017b). More recently, Morán-Zendejas and colleagues discovered that aminoglycoside antibiotics (AGAs), such as gentamicin, neomycin, kanamycin, inhibited the Kir4.1 channel. Using patch-clamp, mutagenesis, and molecular modeling, the AGAs were characterized as pore blockers plugging the central cavity of the channel. The residues T128 and E158 were identified as critical determinants for AGA inhibition activity (Moran-Zendejas et al. 2020). From these studies, we can conclude that residues E158 and T128 of the central pore in Kir4.1 are critical for the activity of several channel blockers. Insight can be gained by comparing conservation of the key residue determinants of intracellular pore blockers. The residue E158 (Kir4.1) is conserved in Kir4.2(E157) and in Kir7.1(E149), but not in Kir1.1(N171) (Fig. 9). Kir1.1-specific intracellular pore blockers of Kir1.1, e.g. VU590 or VU591, utilize position 171 to exert their specific Kir1.1 effect. Indeed, VU591 interacts with Kir1.1 through this residue as a hydrogen bond donor with the nitro group acceptor of the compound. While the negatively charged residues, such as Glu and Asp, form unfavorable, repulsive interactions with the nitro group of the compound (see Fig. 11). A double mutation, Kir1.1(N171E/K80M), abolished VU591 activity (Swale et al. 2015). This explains that VU591 selectively inhibits Kir1.1 over Kir7.1, and UV590 has no effect on Kir4.1 and Kir2.1 (D172 is the corresponding residue). On the other hand, antibiotics, chloroquine, quinacrine, pentamidine utilize their amine groups to interact with Kir4.1 through E158 as a hydrogen bond acceptor to block the channel. Although discovery of selective channel blockers is challenging, the structural information of determinant residues could be useful for future structure-based drug design for selective channel inhibitors. In addition, identification of new allosteric binding sites could be valuable for discovery of novel selective modulators.
5.4.1 Transport Channel Structural Determinants
5.4.1.1 Kir1, 4, and 7 Channel Structure and Dynamics
Linder et al. (2015) conducted MD simulations (200 ns) on wild-type KirBac1.1 and a stimulatory mutant (G143E) to investigate activation gating of KirBac channels. The simulations revealed that a Glu mutation at G143 position caused significant widening at the HBC gate, enabling water flux in the cavity. Both global and local rearrangements were observed. Opening of the HBC gate could trigger twisting of the CTD, which was mediated by electrostatic interactions between the TMD and CTD. In addition, the channel’s slide-helix and C-linker interactions with lipids were strengthened during gating the channel open (Linder et al. 2015). Hu and colleagues predicted the TPNQ (Tertiapin-Q) and Kir1.1 channel interactions using homology modeling, protein docking, and MD simulations. The results suggested TPNQ toxin interacts with Kir1.1 through its helical domain as its interacting surface along with residue H12 as a pore-blocking residue. Residues F146 and F148 in Kir1.1 formed dominant nonpolar interaction with the toxin (Hu et al. 2013). To understand the mechanism for the antidepressant–Kir4.1 channel interaction, Furutani and colleagues conducted homology modeling and molecular docking simulations on Kir4.1 channel with fluoxetine and nortriptyline. Chimeric and site-directed mutagenesis studies suggested the residues T128 and E158 on the TM2 were critical for the drug inhibitory activity. In addition, a 3D quantitative structure-activity relationship (3D-QSAR) model of antidepressants was generated, which suggested common features of a hydrogen bond acceptor and positively charged moiety from the drugs interacting with the channel (Furutani et al. 2009). Using similar homology modeling and molecular docking approaches, in combination with mutagenesis and electrophysiology experiments, several Kir4.1 channel inhibitors were characterized: Quinacrine (Marmolejo-Murillo et al. 2017a), Pentamidine (Arechiga-Figueroa et al. 2017), Chloroquine (Marmolejo-Murillo et al. 2017b), and aminoglycoside antibiotics (Moran-Zendejas et al. 2020; Arechiga-Figueroa et al. 2017; Marmolejo-Murillo et al. 2017a, b; Moran-Zendejas et al. 2020; Swale et al. 2016).
Swale et al. (2016) discovered a novel Kir7.1 channel inhibitor, VU714, through fluorescence-based high-throughput screening. Homology model and docking predicted the binding site of VU714 in Kir7.1 central pore cavity. Site-directed mutagenesis suggested that residues E149 and A150 are essential determinants of VU714 activity. Lead optimization based on VU14 generated ML418, which exhibited high potency (IC50 = 310 nM) and superior selectivity over other channels (Kir1.1, Kir2.1, Kir2.2, Kir2.3, Kir3.1/3.2, and Kir4.1) except for Kir6.2/SUR1 (Swale et al. 2016).
6 Conclusions
Kir channels are mostly active at negative potentials influencing cells to rest near EK. They are either constitutively active or opened by external signals (e.g., neurotransmitters and G protein signaling) or closed by internal signals (e.g., ATP/ADP ratio). They are fundamental to the proper functioning of many cells and systems in our body and their malfunction yields a multitude of diseases, making them desirable targets for therapeutic drug discovery and development. Great insights from 3-D atomic resolution structures of these K+ channels have begun to provide insights for the molecular basis of their fundamental functional properties, such as inward rectification, dependence on [K+]o, and interactions with PIP2 to control gating. More specialized functional attributes are also being deciphered, such as Gβγ binding and allosteric control of gating or ATP binding in relation to the gates and the interplay with PIP2 gating. Computational studies have begun to offer dynamic views of ion permeation and gating by physiological regulators. The pharmacology of these channels has also been expanding providing insights as to how to best synthesize specific and effective drugs. Dynamic structural studies are paving the way toward successful structure-based drug design for this family of K+ channels. Thus, the physiology and pharmacology of Kir channels in the context of dynamic atomic resolution structures have provided the groundwork for numerous therapeutic opportunities. The next decade promises great advances in Kir pharmacology with therapeutic potential (Figs. 9, 10, and 11).
References
Abney KK, Bubser M, Du Y et al (2019) Correction to analgesic effects of the GIRK activator, VU0466551, alone and in combination with morphine in acute and persistent pain models. ACS Chem Nerosci 10(5):2621. https://doi.org/10.1021/acschemneuro.9b00165
Adams DJ, Hill MA (2004) Potassium channels and membrane potential in the modulation of intracellular calcium in vascular endothelial cells. J Cardiovasc Electrophysiol 15(5):598–610. https://doi.org/10.1046/j.1540-8167.2004.03277.x
Adrian RH, Chandler WK, Hodgkin AL (1970) Slow changes in potassium permeability in skeletal muscle. J Physiol 208(3):645–668. https://doi.org/10.1113/jphysiol.1970.sp009140
Aldred KJ, Kerns RJ, Osheroff N (2014) Mechanism of quinolone action and resistance. Biochemistry 53(10):1565–1574. https://doi.org/10.1021/bi5000564
Anderson A, Kulkarni K, Marron Fernandez de Velasco E et al (2018) Expression and relevance of the G protein-gated K(+) channel in the mouse ventricle. Sci Rep 8(1):1192. https://doi.org/10.1038/s41598-018-19719-x
Anumonwo JM, Lopatin AN (2010) Cardiac strong inward rectifier potassium channels. J Mol Cell Cardiol 48(1):45–54. https://doi.org/10.1016/j.yjmcc.2009.08.013
Arechiga-Figueroa IA, Marmolejo-Murillo LG, Cui M et al (2017) High-potency block of Kir4.1 channels by pentamidine: molecular basis. Eur J Pharmacol 815:56–63. https://doi.org/10.1016/j.ejphar.2017.10.009
Armstrong WM (1969) Effect of external potassium and ouabain on sodium efflux from frog sartorius muscle. Proc Soc Exp Biol Med 130(4):1264–1270. https://doi.org/10.3181/00379727-130-33769
Arriagada PV, Growdon JH, Hedley-Whyte ET, Hyman BT (1992) Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer's disease. Neurology 42(3 Pt 1):631–639. https://doi.org/10.1212/wnl.42.3.631
Asakura K, Cha CY, Yamaoka H et al (2014) EAD and DAD mechanisms analyzed by developing a new human ventricular cell model. Prog Biophys Mol Biol 116(1):11–24. https://doi.org/10.1016/j.pbiomolbio.2014.08.008
Ashcroft FM (2005) ATP-sensitive potassium channelopathies: focus on insulin secretion. J Clin Invest 115(8):2047–2058. https://doi.org/10.1172/JCI25495
Asteria C (1997) Molecular basis of Bartter's syndrome: new insights into the correlation between genotype and phenotype. Eur J Endocrinol 137(6):613–615. https://doi.org/10.1530/eje.0.1370613
Aziz Q, Thomas A, Khambra T, Tinker A (2010) Phenformin has a direct inhibitory effect on the ATP-sensitive potassium channel. Eur J Pharmacol 634(1–3):26–32. https://doi.org/10.1016/j.ejphar.2010.02.023
Aziz Q, Li Y, Anderson N, Ojake L, Tsisanova E, Tinker A (2017) Molecular and functional characterization of the endothelial ATP-sensitive potassium channel. J Biol Chem 292(43):17587–17597. https://doi.org/10.1074/jbc.M117.810325
Baculis BC, Weiss AC, Pang W et al (2017) Prolonged seizure activity causes caspase dependent cleavage and dysfunction of G-protein activated inwardly rectifying potassium channels. Sci Rep 7(1):12313. https://doi.org/10.1038/s41598-017-12508-y
Bailey MA, Cantone A, Yan Q et al (2006) Maxi-K channels contribute to urinary potassium excretion in the ROMK-deficient mouse model of type II Bartter's syndrome and in adaptation to a high-K diet. Kidney Int 70(1):51–59. https://doi.org/10.1038/sj.ki.5000388
Balana B, Maslennikov I, Kwiatkowski W et al (2011) Mechanism underlying selective regulation of G protein-gated inwardly rectifying potassium channels by the psychostimulant-sensitive sorting nexin 27. Proc Natl Acad Sci U S A 108(14):5831–5836. https://doi.org/10.1073/pnas.1018645108
Barrett-Jolley R, Dart C, Standen NB (1999) Direct block of native and cloned (Kir2.1) inward rectifier K+ channels by chloroethylclonidine. Br J Pharmacol 128(3):760–766. https://doi.org/10.1038/sj.bjp.0702819
Baukrowitz T, Tucker SJ, Schulte U, Benndorf K, Ruppersberg JP, Fakler B (1999) Inward rectification in KATP channels: a pH switch in the pore. EMBO J 18(4):847–853. https://doi.org/10.1093/emboj/18.4.847
Beeler GW Jr, Reuter H (1970) The relation between membrane potential, membrane currents and activation of contraction in ventricular myocardial fibres. J Physiol 207(1):211–229. https://doi.org/10.1113/jphysiol.1970.sp009057
Beguin P, Nagashima K, Nishimura M, Gonoi T, Seino S (1999) PKA-mediated phosphorylation of the human K(ATP) channel: separate roles of Kir6.2 and SUR1 subunit phosphorylation. EMBO J 18(17):4722–4732. https://doi.org/10.1093/emboj/18.17.4722
Belus MT, Rogers MA, Elzubeir A et al (2018) Kir2.1 is important for efficient BMP signaling in mammalian face development. Dev Biol 444(Suppl 1):S297–S307. https://doi.org/10.1016/j.ydbio.2018.02.012
Bernsteiner H, Zangerl-Plessl EM, Chen X, Stary-Weinzinger A (2019) Conduction through a narrow inward-rectifier K(+) channel pore. J Gen Physiol 151(10):1231–1246. https://doi.org/10.1085/jgp.201912359
Bettahi I, Marker CL, Roman MI, Wickman K (2002) Contribution of the Kir3.1 subunit to the muscarinic-gated atrial potassium channel IKACh. J Biol Chem 277(50):48282–48288. https://doi.org/10.1074/jbc.M209599200
Bhave G, Chauder BA, Liu W et al (2011) Development of a selective small-molecule inhibitor of Kir1.1, the renal outer medullary potassium channel. Mol Pharmacol 79(1):42–50. https://doi.org/10.1124/mol.110.066928
Birnbaumer L, Brown AM (1987) G protein opening of K+ channels. Nature 327(6117):21–22. https://doi.org/10.1038/327021a0
Blednov YA, Stoffel M, Chang SR, Harris RA (2001a) GIRK2 deficient mice. Evidence for hyperactivity and reduced anxiety. Physiol Behav 74(1–2):109–117. https://doi.org/10.1016/s0031-9384(01)00555-8
Blednov YA, Stoffel M, Chang SR, Harris RA (2001b) Potassium channels as targets for ethanol: studies of G-protein-coupled inwardly rectifying potassium channel 2 (GIRK2) null mutant mice. J Pharmacol Exp Ther 298(2):521–530
Blednov YA, Stoffel M, Alva H, Harris RA (2003) A pervasive mechanism for analgesia: activation of GIRK2 channels. Proc Natl Acad Sci U S A 100(1):277–282. https://doi.org/10.1073/pnas.012682399
Bleich M, Schlatter E, Greger R (1990) The luminal K+ channel of the thick ascending limb of Henle's loop. Pflugers Arch 415(4):449–460. https://doi.org/10.1007/BF00373623
Boim MA, Ho K, Shuck ME et al (1995) ROMK inwardly rectifying ATP-sensitive K+ channel. II. Cloning and distribution of alternative forms. Am J Physiol 268(6 Pt 2):F1132–F1140. https://doi.org/10.1152/ajprenal.1995.268.6.F1132
Bond CT, Pessia M, Xia XM, Lagrutta A, Kavanaugh MP, Adelman JP (1994) Cloning and expression of a family of inward rectifier potassium channels. Recept Channels 2(3):183–191
Bouchard R, Clark RB, Juhasz AE, Giles WR (2004) Changes in extracellular K+ concentration modulate contractility of rat and rabbit cardiac myocytes via the inward rectifier K+ current IK1. J Physiol 556(Pt 3):773–790. https://doi.org/10.1113/jphysiol.2003.058248
Bousquet P, Hudson A, Garcia-Sevilla JA, Li JX (2020) Imidazoline receptor system: the past, the present, and the future. Pharmacol Rev 72(1):50–79. https://doi.org/10.1124/pr.118.016311
Bradley KK, Jaggar JH, Bonev AD et al (1999) Kir2.1 encodes the inward rectifier potassium channel in rat arterial smooth muscle cells. J Physiol 515(Pt 3):639–651. https://doi.org/10.1111/j.1469-7793.1999.639ab.x
Branstrom R, Leibiger IB, Leibiger B et al (2007) Single residue (K332A) substitution in Kir6.2 abolishes the stimulatory effect of long-chain acyl-CoA esters: indications for a long-chain acyl-CoA ester binding motif. Diabetologia 50(8):1670–1677. https://doi.org/10.1007/s00125-007-0697-x
Bredt DS, Wang TL, Cohen NA, Guggino WB, Snyder SH (1995) Cloning and expression of two brain-specific inwardly rectifying potassium channels. Proc Natl Acad Sci U S A 92(15):6753–6757. https://doi.org/10.1073/pnas.92.15.6753
Breitwieser GE, Szabo G (1985) Uncoupling of cardiac muscarinic and beta-adrenergic receptors from ion channels by a guanine nucleotide analogue. Nature 317(6037):538–540. https://doi.org/10.1038/317538a0
Brown RA, Carpentier RG (1990) Effects of acetaldehyde on membrane potentials of sinus node pacemaker fibers. Alcohol 7(1):33–36. https://doi.org/10.1016/0741-8329(90)90057-j
Bruehl S, Denton JS, Lonergan D et al (2013) Associations between KCNJ6 (GIRK2) gene polymorphisms and pain-related phenotypes. Pain 154(12):2853–2859. https://doi.org/10.1016/j.pain.2013.08.026
Burgen AS, Terroux KG (1953) On the negative inotropic effect in the cat’s auricle. J Physiol 120(4):449–464. https://doi.org/10.1113/jphysiol.1953.sp004910
Carlsson L, Duker G, Jacobson I (2010) New pharmacological targets and treatments for atrial fibrillation. Trends Pharmacol Sci 31(8):364–371. https://doi.org/10.1016/j.tips.2010.05.001
Chan KW, Langan MN, Sui JL et al (1996) A recombinant inwardly rectifying potassium channel coupled to GTP-binding proteins. J Gen Physiol 107(3):381–397. https://doi.org/10.1085/jgp.107.3.381
Chan KW, Zhang H, Logothetis DE (2003) N-terminal transmembrane domain of the SUR controls trafficking and gating of Kir6 channel subunits. EMBO J 22(15):3833–3843. https://doi.org/10.1093/emboj/cdg376
Chen PC, Olson EM, Zhou Q et al (2013) Carbamazepine as a novel small molecule corrector of trafficking-impaired ATP-sensitive potassium channels identified in congenital hyperinsulinism. J Biol Chem 288(29):20942–20954. https://doi.org/10.1074/jbc.M113.470948
Chen IS, Tateyama M, Fukata Y, Uesugi M, Kubo Y (2017) Ivermectin activates GIRK channels in a PIP2 -dependent, Gbetagamma -independent manner and an amino acid residue at the slide helix governs the activation. J Physiol 595(17):5895–5912. https://doi.org/10.1113/JP274871
Chen X, Garon A, Wieder M et al (2019) Computational identification of novel Kir6 channel inhibitors. Front Pharmacol 10:549. https://doi.org/10.3389/fphar.2019.00549
Chen X, Brundl M, Friesacher T, Stary-Weinzinger A (2020) Computational insights into voltage dependence of polyamine block in a strong inwardly rectifying K(+) channel. Front Pharmacol 11:721. https://doi.org/10.3389/fphar.2020.00721
Choe H, Zhou H, Palmer LG, Sackin H (1997) A conserved cytoplasmic region of ROMK modulates pH sensitivity, conductance, and gating. Am J Physiol 273(4):F516–F529. https://doi.org/10.1152/ajprenal.1997.273.4.F516
Choi M, Scholl UI, Yue P et al (2011) K+ channel mutations in adrenal aldosterone-producing adenomas and hereditary hypertension. Science 331(6018):768–772. https://doi.org/10.1126/science.1198785
Cole KS, Curtis HJ (1941) Membrane potential of the squid giant axon during current flow. J Gen Physiol 24(4):551–563. https://doi.org/10.1085/jgp.24.4.551
Cooper A, Grigoryan G, Guy-David L, Tsoory MM, Chen A, Reuveny E (2012) Trisomy of the G protein-coupled K+ channel gene, Kcnj6, affects reward mechanisms, cognitive functions, and synaptic plasticity in mice. Proc Natl Acad Sci U S A 109(7):2642–2647. https://doi.org/10.1073/pnas.1109099109
Cooper PE, McClenaghan C, Chen X, Stary-Weinzinger A, Nichols CG (2017) Conserved functional consequences of disease-associated mutations in the slide helix of Kir6.1 and Kir6.2 subunits of the ATP-sensitive potassium channel. J Biol Chem 292(42):17387–17398. https://doi.org/10.1074/jbc.M117.804971
Cruz HG, Ivanova T, Lunn ML, Stoffel M, Slesinger PA, Luscher C (2004) Bi-directional effects of GABA(B) receptor agonists on the mesolimbic dopamine system. Nat Neurosci 7(2):153–159. https://doi.org/10.1038/nn1181
Cruz HG, Berton F, Sollini M et al (2008) Absence and rescue of morphine withdrawal in GIRK/Kir3 knock-out mice. J Neurosci 28(15):4069–4077. https://doi.org/10.1523/JNEUROSCI.0267-08.2008
Cuevas CA, Su XT, Wang MX et al (2017) Potassium sensing by renal distal tubules requires Kir4.1. J Am Soc Nephrol 28(6):1814–1825. https://doi.org/10.1681/ASN.2016090935
Cui Y, Tinker A, Clapp LH (2003) Different molecular sites of action for the KATP channel inhibitors, PNU-99963 and PNU-37883A. Br J Pharmacol 139(1):122–128. https://doi.org/10.1038/sj.bjp.0705228
Cui M, Alhamshari Y, Cantwell L et al (2021) A benzopyran with anti-arrhythmic activity is an inhibitor of Kir3.1-containing potassium channels. J Biol Chem 296:100535. https://doi.org/10.1016/j.jbc.2021.100535
Dascal N, Schreibmayer W, Lim NF et al (1993) Atrial G protein-activated K+ channel: expression cloning and molecular properties. Proc Natl Acad Sci U S A 90(21):10235–10239. https://doi.org/10.1073/pnas.90.21.10235
Dassau L, Conti LR, Radeke CM, Ptacek LJ, Vandenberg CA (2011) Kir2.6 regulates the surface expression of Kir2.x inward rectifier potassium channels. J Biol Chem 286(11):9526–9541. https://doi.org/10.1074/jbc.M110.170597
de Boer TP, Houtman MJ, Compier M, van der Heyden MA (2010) The mammalian K(IR)2.x inward rectifier ion channel family: expression pattern and pathophysiology. Acta Physiol (Oxf) 199(3):243–256. https://doi.org/10.1111/j.1748-1716.2010.02108.x
Del Castillo J, Katz B (1955) Production of membrane potential changes in the frog’s heart by inhibitory nerve impulses. Nature 175(4467):1035. https://doi.org/10.1038/1751035a0
Delling M, Wischmeyer E, Dityatev A et al (2002) The neural cell adhesion molecule regulates cell-surface delivery of G-protein-activated inwardly rectifying potassium channels via lipid rafts. J Neurosci 22(16):7154–7164. https://doi.org/10.1523/JNEUROSCI.22-16-07154.2002
Derst C, Hirsch JR, Preisig-Muller R et al (2001) Cellular localization of the potassium channel Kir7.1 in guinea pig and human kidney. Kidney Int 59(6):2197–2205. https://doi.org/10.1046/j.1523-1755.2001.00735.x
Devulder J (2010) Flupirtine in pain management: pharmacological properties and clinical use. CNS Drugs 24(10):867–881. https://doi.org/10.2165/11536230-000000000-00000
Ding D, Wang M, Wu JX, Kang Y, Chen L (2019) The structural basis for the binding of repaglinide to the pancreatic KATP Channel. Cell Rep 27(6):1848–1857.e4. https://doi.org/10.1016/j.celrep.2019.04.050
Dobrev D, Friedrich A, Voigt N et al (2005) The G protein-gated potassium current I(K,ACh) is constitutively active in patients with chronic atrial fibrillation. Circulation 112(24):3697–3706. https://doi.org/10.1161/CIRCULATIONAHA.105.575332
Doi T, Fakler B, Schultz JH et al (1996) Extracellular K+ and intracellular pH allosterically regulate renal Kir1.1 channels. J Biol Chem 271(29):17261–17266. https://doi.org/10.1074/jbc.271.29.17261
Donaldson MR, Yoon G, Fu YH, Ptacek LJ (2004) Andersen-Tawil syndrome: a model of clinical variability, pleiotropy, and genetic heterogeneity. Ann Med 36(Suppl 1):92–97. https://doi.org/10.1080/17431380410032490
Doring F, Derst C, Wischmeyer E et al (1998) The epithelial inward rectifier channel Kir7.1 displays unusual K+ permeation properties. J Neurosci 18(21):8625–8636
Doupnik CA (2015) RGS redundancy and implications in GPCR-GIRK signaling. Int Rev Neurobiol 123:87–116. https://doi.org/10.1016/bs.irn.2015.05.010
Doupnik CA, Davidson N, Lester HA, Kofuji P (1997) RGS proteins reconstitute the rapid gating kinetics of gbetagamma-activated inwardly rectifying K+ channels. Proc Natl Acad Sci U S A 94(19):10461–10466. https://doi.org/10.1073/pnas.94.19.10461
Du X, Zhang H, Lopes C, Mirshahi T, Rohacs T, Logothetis DE (2004) Characteristic interactions with phosphatidylinositol 4,5-bisphosphate determine regulation of Kir channels by diverse modulators. J Biol Chem 279(36):37271–81
Edvinsson JM, Shah AJ, Palmer LG (2011a) Kir4.1 K+ channels are regulated by external cations. Channels (Austin) 5(3):269–279. https://doi.org/10.4161/chan.5.3.15827
Edvinsson JM, Shah AJ, Palmer LG (2011b) Potassium-dependent activation of Kir4.2 K(+) channels. J Physiol 589(Pt 24):5949–5963. https://doi.org/10.1113/jphysiol.2011.220731
Ehrlich JR, Cha TJ, Zhang L et al (2004) Characterization of a hyperpolarization-activated time-dependent potassium current in canine cardiomyocytes from pulmonary vein myocardial sleeves and left atrium. J Physiol 557(Pt 2):583–597. https://doi.org/10.1113/jphysiol.2004.061119
Epshtein Y, Chopra AP, Rosenhouse-Dantsker A, Kowalsky GB, Logothetis DE, Levitan I (2009) Identification of a C-terminus domain critical for the sensitivity of Kir2.1 to cholesterol. Proc Natl Acad Sci U S A 106(19):8055–8060. https://doi.org/10.1073/pnas.0809847106
Fakler B, Schultz JH, Yang J et al (1996) Identification of a titratable lysine residue that determines sensitivity of kidney potassium channels (ROMK) to intracellular pH. EMBO J 15(16):4093–4099
Fang Y, Schram G, Romanenko VG et al (2005) Functional expression of Kir2.x in human aortic endothelial cells: the dominant role of Kir2.2. Am J Physiol Cell Physiol 289(5):C1134–C1144. https://doi.org/10.1152/ajpcell.00077.2005
Fenner MF, Carstensen H, Dalgas Nissen S et al (2020) Effect of selective IK,ACh inhibition by XAF-1407 in an equine model of tachypacing-induced persistent atrial fibrillation. Br J Pharmacol 177(16):3778–3794. https://doi.org/10.1111/bph.15100
Ferré SCF, Dessauer CW, González-Maeso J, Hébert TE, Jockers R, Logothetis DE, Pardo L (2021) G protein-coupled receptor-effector macromolecular membrane assemblies (GEMMA). Pharmacol Ther. Under revision
Ferrer J, Nichols CG, Makhina EN et al (1995) Pancreatic islet cells express a family of inwardly rectifying K+ channel subunits which interact to form G-protein-activated channels. J Biol Chem 270(44):26086–26091. https://doi.org/10.1074/jbc.270.44.26086
Field MJ, Stanton BA, Giebisch GH (1984) Influence of ADH on renal potassium handling: a micropuncture and microperfusion study. Kidney Int 25(3):502–511. https://doi.org/10.1038/ki.1984.46
Filosa JA, Bonev AD, Straub SV et al (2006) Local potassium signaling couples neuronal activity to vasodilation in the brain. Nat Neurosci 9(11):1397–1403. https://doi.org/10.1038/nn1779
Fischer-Lougheed J, Liu JH, Espinos E et al (2001) Human myoblast fusion requires expression of functional inward rectifier Kir2.1 channels. J Cell Biol 153(4):677–686. https://doi.org/10.1083/jcb.153.4.677
Fujita A, Horio Y, Higashi K et al (2002) Specific localization of an inwardly rectifying K(+) channel, Kir4.1, at the apical membrane of rat gastric parietal cells; its possible involvement in K(+) recycling for the H(+)-K(+)-pump. J Physiol 540(Pt 1):85–92. https://doi.org/10.1113/jphysiol.2001.013439
Fujiwara Y, Kubo Y (2006) Functional roles of charged amino acid residues on the wall of the cytoplasmic pore of Kir2.1. J Gen Physiol 127(4):401–419. https://doi.org/10.1085/jgp.200509434
Furutani K, Ohno Y, Inanobe A, Hibino H, Kurachi Y (2009) Mutational and in silico analyses for antidepressant block of astroglial inward-rectifier Kir4.1 channel. Mol Pharmacol 75(6):1287–1295. https://doi.org/10.1124/mol.108.052936
Gada K, Logothetis DE (2021) Regulation of ion channels by protein kinase C isoforms. Commissioned Review, JBC. In Revision
Gahwiler BH, Brown DA (1985) GABAB-receptor-activated K+ current in voltage-clamped CA3 pyramidal cells in hippocampal cultures. Proc Natl Acad Sci U S A 82(5):1558–1562. https://doi.org/10.1073/pnas.82.5.1558
Gaskell WH (1886) The electrical changes in the quiescent cardiac muscle which accompany stimulation of the Vagus nerve. J Physiol 7(5–6):451–452. https://doi.org/10.1113/jphysiol.1886.sp000235
Gaskell WH (1887) On the action of Muscarin upon the heart, and on the electrical changes in the non-beating cardiac muscle brought about by stimulation of the inhibitory and Augmentor nerves. J Physiol 8(6):404–4i8. https://doi.org/10.1113/jphysiol.1887.sp000269
Ghamari-Langroudi M, Digby GJ, Sebag JA et al (2015) G-protein-independent coupling of MC4R to Kir7.1 in hypothalamic neurons. Nature 520(7545):94–98. https://doi.org/10.1038/nature14051
Girmatsion Z, Biliczki P, Bonauer A et al (2009) Changes in microRNA-1 expression and IK1 up-regulation in human atrial fibrillation. Heart Rhythm 6(12):1802–1809. https://doi.org/10.1016/j.hrthm.2009.08.035
Gomez-Sanchez CE, Oki K (2014) Minireview: potassium channels and aldosterone dysregulation: is primary aldosteronism a potassium channelopathy? Endocrinology 155(1):47–55. https://doi.org/10.1210/en.2013-1733
Gouder N, Fritschy JM, Boison D (2003) Seizure suppression by adenosine A1 receptor activation in a mouse model of pharmacoresistant epilepsy. Epilepsia 44(7):877–885. https://doi.org/10.1046/j.1528-1157.2003.03603.x
Greener ID, Monfredi O, Inada S et al (2011) Molecular architecture of the human specialised atrioventricular conduction axis. J Mol Cell Cardiol 50(4):642–651. https://doi.org/10.1016/j.yjmcc.2010.12.017
Greger R, Bleich M, Schlatter E (1990) Ion channels in the thick ascending limb of Henle's loop. Ren Physiol Biochem 13(1–2):37–50. https://doi.org/10.1159/000173346
Gregerson KA, Flagg TP, O'Neill TJ et al (2001) Identification of G protein-coupled, inward rectifier potassium channel gene products from the rat anterior pituitary gland. Endocrinology 142(7):2820–2832. https://doi.org/10.1210/endo.142.7.8236
Grosse-Lackmann T, Zunkler BJ, Rustenbeck I (2003) Specificity of nonadrenergic imidazoline binding sites in insulin-secreting cells and relation to the block of ATP-sensitive K(+) channels. Ann N Y Acad Sci 1009:371–377. https://doi.org/10.1196/annals.1304.050
Guay-Woodford LM (1995) Molecular insights into the pathogenesis of inherited renal tubular disorders. Curr Opin Nephrol Hypertens 4(2):121–129. https://doi.org/10.1097/00041552-199503000-00004
Guo D, Lu Z (2003) Interaction mechanisms between polyamines and IRK1 inward rectifier K+ channels. J Gen Physiol 122(5):485–500. https://doi.org/10.1085/jgp.200308890
Haass C, Selkoe DJ (2007) Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer's amyloid beta-peptide. Nat Rev Mol Cell Biol 8(2):101–112. https://doi.org/10.1038/nrm2101
Hagiwara S, Takahashi K (1974) The anomalous rectification and cation selectivity of the membrane of a starfish egg cell. J Membr Biol 18(1):61–80. https://doi.org/10.1007/BF01870103
Hagiwara S, Yoshii M (1979) Effects of internal potassium and sodium on the anomalous rectification of the starfish egg as examined by internal perfusion. J Physiol 292:251–265. https://doi.org/10.1113/jphysiol.1979.sp012849
Hagiwara S, Miyazaki S, Rosenthal NP (1976) Potassium current and the effect of cesium on this current during anomalous rectification of the egg cell membrane of a starfish. J Gen Physiol 67(6):621–638. https://doi.org/10.1085/jgp.67.6.621
Hagiwara S, Miyazaki S, Moody W, Patlak J (1978) Blocking effects of barium and hydrogen ions on the potassium current during anomalous rectification in the starfish egg. J Physiol 279:167–185. https://doi.org/10.1113/jphysiol.1978.sp012338
Haider S, Khalid S, Tucker SJ, Ashcroft FM, Sansom MS (2007) Molecular dynamics simulations of inwardly rectifying (Kir) potassium channels: a comparative study. Biochemistry 46(12):3643–3652. https://doi.org/10.1021/bi062210f
Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ (1981) Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflugers Arch 391(2):85–100. https://doi.org/10.1007/BF00656997
Hansen JB (2006) Towards selective Kir6.2/SUR1 potassium channel openers, medicinal chemistry and therapeutic perspectives. Curr Med Chem 13(4):361–376. https://doi.org/10.2174/092986706775527947
Hansen SB, Tao X, MacKinnon R (2011) Structural basis of PIP2 activation of the classical inward rectifier K+ channel Kir2.2. Nature 477(7365):495–498. https://doi.org/10.1038/nature10370
Hardy J, Selkoe DJ (2002) The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 297(5580):353–356. https://doi.org/10.1126/science.1072994
Harraz OF, Longden TA, Dabertrand F, Hill-Eubanks D, Nelson MT (2018) Endothelial GqPCR activity controls capillary electrical signaling and brain blood flow through PIP2 depletion. Proc Natl Acad Sci U S A 115(15):E3569–E3577. https://doi.org/10.1073/pnas.1800201115
Hashimoto N, Yamashita T, Tsuruzoe N (2008) Characterization of in vivo and in vitro electrophysiological and antiarrhythmic effects of a novel IKACh blocker, NIP-151: a comparison with an IKr-blocker dofetilide. J Cardiovasc Pharmacol 51(2):162–169. https://doi.org/10.1097/FJC.0b013e31815e854c
Hasselblatt M, Bohm C, Tatenhorst L et al (2006) Identification of novel diagnostic markers for choroid plexus tumors: a microarray-based approach. Am J Surg Pathol 30(1):66–74. https://doi.org/10.1097/01.pas.0000176430.88702.e0
Hassinen M, Paajanen V, Vornanen M (2008) A novel inwardly rectifying K+ channel, Kir2.5, is upregulated under chronic cold stress in fish cardiac myocytes. J Exp Biol 211(Pt 13):2162–2171. https://doi.org/10.1242/jeb.016121
He G, Wang HR, Huang SK, Huang CL (2007) Intersectin links WNK kinases to endocytosis of ROMK1. J Clin Invest 117(4):1078–1087. https://doi.org/10.1172/JCI30087
Hebert SC (2003) Bartter syndrome. Curr Opin Nephrol Hypertens 12(5):527–532. https://doi.org/10.1097/00041552-200309000-00008
Hebert SC, Desir G, Giebisch G, Wang W (2005) Molecular diversity and regulation of renal potassium channels. Physiol Rev 85(1):319–371. https://doi.org/10.1152/physrev.00051.2003
Heron-Milhavet L, Xue-Jun Y, Vannucci SJ et al (2004) Protection against hypoxic-ischemic injury in transgenic mice overexpressing Kir6.2 channel pore in forebrain. Mol Cell Neurosci 25(4):585–593. https://doi.org/10.1016/j.mcn.2003.10.012
Hestrin S (1981) The interaction of potassium with the activation of anomalous rectification in frog muscle membrane. J Physiol 317:497–508. https://doi.org/10.1113/jphysiol.1981.sp013839
Hibino H, Kurachi Y (2006) Molecular and physiological bases of the K+ circulation in the mammalian inner ear. Physiology (Bethesda) 21:336–345. https://doi.org/10.1152/physiol.00023.2006
Hibino H, Inanobe A, Furutani K, Murakami S, Findlay I, Kurachi Y (2010) Inwardly rectifying potassium channels: their structure, function, and physiological roles. Physiol Rev 90(1):291–366. https://doi.org/10.1152/physrev.00021.2009
Hilgemann DW, Ball R (1996) Regulation of cardiac Na+,Ca2+ exchange and KATP potassium channels by PIP2. Science 273(5277):956–959. https://doi.org/10.1126/science.273.5277.956
Hill E, Hickman C, Diez R, Wall M (2020) Role of A1 receptor-activated GIRK channels in the suppression of hippocampal seizure activity. Neuropharmacology 164:107904. https://doi.org/10.1016/j.neuropharm.2019.107904
Hille B, Schwarz W (1978) Potassium channels as multi-ion single-file pores. J Gen Physiol 72(4):409–442. https://doi.org/10.1085/jgp.72.4.409
Ho IH, Murrell-Lagnado RD (1999) Molecular determinants for sodium-dependent activation of G protein-gated K+ channels. J Biol Chem 274(13):8639–8648. https://doi.org/10.1074/jbc.274.13.8639
Ho K, Nichols CG, Lederer WJ et al (1993) Cloning and expression of an inwardly rectifying ATP-regulated potassium channel. Nature 362(6415):31–38. https://doi.org/10.1038/362031a0
Hodgkin AL, Huxley AF, Katz B (1952) Measurement of current-voltage relations in the membrane of the giant axon of Loligo. J Physiol 116(4):424–448. https://doi.org/10.1113/jphysiol.1952.sp004716
Hu J, Qiu S, Yang F, Cao Z, Li W, Wu Y (2013) Unique mechanism of the interaction between honey bee toxin TPNQ and rKir1.1 potassium channel explored by computational simulations: insights into the relative insensitivity of channel towards animal toxins. PLoS One 8(7):e67213. https://doi.org/10.1371/journal.pone.0067213
Huang Y, Mucke L (2012) Alzheimer mechanisms and therapeutic strategies. Cell 148(6):1204–1222. https://doi.org/10.1016/j.cell.2012.02.040
Huang CL, Feng S, Hilgemann DW (1998) Direct activation of inward rectifier potassium channels by PIP2 and its stabilization by Gbetagamma. Nature 391(6669):803–806. https://doi.org/10.1038/35882
Huang X, Lee SH, Lu H, Sanders KM, Koh SD (2018a) Molecular and functional characterization of inwardly rectifying K(+) currents in murine proximal colon. J Physiol 596(3):379–391. https://doi.org/10.1113/JP275234
Huang Y, Zhang Y, Kong S et al (2018b) GIRK1-mediated inwardly rectifying potassium current suppresses the epileptiform burst activities and the potential antiepileptic effect of ML297. Biomed Pharmacother 101:362–370. https://doi.org/10.1016/j.biopha.2018.02.114
Humphrey SJ (1999) Pharmacology of the K-ATP channel blocking morpholinoguanidine PNU-37883A. Cardiovasc Drug Rev 17(4):295–328
Inanobe A, Horio Y, Fujita A et al (1999) Molecular cloning and characterization of a novel splicing variant of the Kir3.2 subunit predominantly expressed in mouse testis. J Physiol 521(Pt 1):19–30. https://doi.org/10.1111/j.1469-7793.1999.00019.x
Inanobe A, Matsuura T, Nakagawa A, Kurachi Y (2007) Structural diversity in the cytoplasmic region of G protein-gated inward rectifier K+ channels. Channels (Austin) 1(1):39–45
Ishihara K (2018) External K(+) dependence of strong inward rectifier K(+) channel conductance is caused not by K(+) but by competitive pore blockade by external Na(.). J Gen Physiol 150(7):977–989. https://doi.org/10.1085/jgp.201711936
Ishihara K, Ehara T (2004) Two modes of polyamine block regulating the cardiac inward rectifier K+ current IK1 as revealed by a study of the Kir2.1 channel expressed in a human cell line. J Physiol 556(Pt 1):61–78. https://doi.org/10.1113/jphysiol.2003.055434
Ishihara K, Sarai N, Asakura K, Noma A, Matsuoka S (2009) Role of mg(2+) block of the inward rectifier K(+) current in cardiac repolarization reserve: a quantitative simulation. J Mol Cell Cardiol 47(1):76–84. https://doi.org/10.1016/j.yjmcc.2009.03.008
Iwanir S, Reuveny E (2008) Adrenaline-induced hyperpolarization of mouse pancreatic islet cells is mediated by G protein-gated inwardly rectifying potassium (GIRK) channels. Pflugers Arch 456(6):1097–1108. https://doi.org/10.1007/s00424-008-0479-4
Jakob R, Krieglstein J (1997) Influence of flupirtine on a G-protein coupled inwardly rectifying potassium current in hippocampal neurones. Br J Pharmacol 122(7):1333–1338. https://doi.org/10.1038/sj.bjp.0701519
Jelacic TM, Kennedy ME, Wickman K, Clapham DE (2000) Functional and biochemical evidence for G-protein-gated inwardly rectifying K+ (GIRK) channels composed of GIRK2 and GIRK3. J Biol Chem 275(46):36211–36216. https://doi.org/10.1074/jbc.M007087200
Jin W, Lu Z (1998) A novel high-affinity inhibitor for inward-rectifier K+ channels. Biochemistry 37(38):13291–13299. https://doi.org/10.1021/bi981178p
Jin W, Lu Z (1999) Synthesis of a stable form of tertiapin: a high-affinity inhibitor for inward-rectifier K+ channels. Biochemistry 38(43):14286–14293. https://doi.org/10.1021/bi991205r
Jin T, Peng L, Mirshahi T et al (2002) The (beta)gamma subunits of G proteins gate a K(+) channel by pivoted bending of a transmembrane segment. Mol Cell 10(3):469–481. https://doi.org/10.1016/s1097-2765(02)00659-7
Karolyi L, Koch MC, Grzeschik KH, Seyberth HW (1998) The molecular genetic approach to “Bartter’s syndrome”. J Mol Med (Berl) 76(5):317–325. https://doi.org/10.1007/s001090050223
Katz B (1949a) Les constants electriques de la membrane du muscle. Arch Sci Physiol 3:285–299
Katz B (1949b) The efferent regulation of the muscle spindle in the frog. J Exp Biol 26(2):201–217
Kaufmann K, Romaine I, Days E et al (2013) ML297 (VU0456810), the first potent and selective activator of the GIRK potassium channel, displays antiepileptic properties in mice. ACS Chem Nerosci 4(9):1278–1286. https://doi.org/10.1021/cn400062a
Kawahito S, Kawano T, Kitahata H et al (2011) Molecular mechanisms of the inhibitory effects of clonidine on vascular adenosine triphosphate-sensitive potassium channels. Anesth Analg 113(6):1374–1380. https://doi.org/10.1213/ANE.0b013e3182321142
Kawano T, Oshita S, Takahashi A et al (2004a) Molecular mechanisms of the inhibitory effects of bupivacaine, levobupivacaine, and ropivacaine on sarcolemmal adenosine triphosphate-sensitive potassium channels in the cardiovascular system. Anesthesiology 101(2):390–398. https://doi.org/10.1097/00000542-200408000-00020
Kawano T, Oshita S, Takahashi A et al (2004b) Molecular mechanisms of the inhibitory effects of propofol and thiamylal on sarcolemmal adenosine triphosphate-sensitive potassium channels. Anesthesiology 100(2):338–346. https://doi.org/10.1097/00000542-200402000-00024
Kenna S, Roper J, Ho K, Hebert S, Ashcroft SJ, Ashcroft FM (1994) Differential expression of the inwardly-rectifying K-channel ROMK1 in rat brain. Brain Res Mol Brain Res 24(1–4):353–356. https://doi.org/10.1016/0169-328x(94)90150-3
Keselman I, Fribourg M, Felsenfeld DP, Logothetis DE (2007) Mechanism of PLC-mediated Kir3 current inhibition. Channels (Austin) 1(2):113–123. https://doi.org/10.4161/chan.4321
Kharade SV, Kurata H, Bender AM et al (2018) Discovery, characterization, and effects on renal fluid and electrolyte excretion of the Kir4.1 potassium channel pore blocker, VU0134992. Mol Pharmacol 94(2):926–937. https://doi.org/10.1124/mol.118.112359
Kline CF, Kurata HT, Hund TJ et al (2009) Dual role of K ATP channel C-terminal motif in membrane targeting and metabolic regulation. Proc Natl Acad Sci U S A 106(39):16669–16674. https://doi.org/10.1073/pnas.0907138106
Knot HJ, Nelson MT (1998) Regulation of arterial diameter and wall [Ca2+] in cerebral arteries of rat by membrane potential and intravascular pressure. J Physiol 508(Pt 1):199–209. https://doi.org/10.1111/j.1469-7793.1998.199br.x
Knot HJ, Zimmermann PA, Nelson MT (1996) Extracellular K(+)-induced hyperpolarizations and dilatations of rat coronary and cerebral arteries involve inward rectifier K(+) channels. J Physiol 492(Pt 2):419–430. https://doi.org/10.1113/jphysiol.1996.sp021318
Kobayashi T, Ikeda K (2006) G protein-activated inwardly rectifying potassium channels as potential therapeutic targets. Curr Pharm Des 12(34):4513–4523. https://doi.org/10.2174/138161206779010468
Kobayashi T, Ikeda K, Kojima H et al (1999) Ethanol opens G-protein-activated inwardly rectifying K+ channels. Nat Neurosci 2(12):1091–1097. https://doi.org/10.1038/16019
Kobayashi T, Ikeda K, Kumanishi T (2000) Inhibition by various antipsychotic drugs of the G-protein-activated inwardly rectifying K(+) (GIRK) channels expressed in xenopus oocytes. Br J Pharmacol 129(8):1716–1722. https://doi.org/10.1038/sj.bjp.0703224
Kobayashi T, Washiyama K, Ikeda K (2003) Inhibition of G protein-activated inwardly rectifying K+ channels by fluoxetine (Prozac). Br J Pharmacol 138(6):1119–1128. https://doi.org/10.1038/sj.bjp.0705172
Kobayashi T, Washiyama K, Ikeda K (2004) Inhibition of G protein-activated inwardly rectifying K+ channels by various antidepressant drugs. Neuropsychopharmacology 29(10):1841–1851. https://doi.org/10.1038/sj.npp.1300484
Kobayashi T, Washiyama K, Ikeda K (2006) Inhibition of G protein-activated inwardly rectifying K+ channels by ifenprodil. Neuropsychopharmacology 31(3):516–524. https://doi.org/10.1038/sj.npp.1300844
Kobayashi T, Washiyama K, Ikeda K (2009) Pregnenolone sulfate potentiates the inwardly rectifying K channel Kir2.3. PLoS One 4(7):e6311. https://doi.org/10.1371/journal.pone.0006311
Kondo C, Isomoto S, Matsumoto S et al (1996) Cloning and functional expression of a novel isoform of ROMK inwardly rectifying ATP-dependent K+ channel, ROMK6 (Kir1.1f). FEBS Lett 399(1–2):122–126. https://doi.org/10.1016/s0014-5793(96)01302-6
Konig S, Hinard V, Arnaudeau S et al (2004) Membrane hyperpolarization triggers myogenin and myocyte enhancer factor-2 expression during human myoblast differentiation. J Biol Chem 279(27):28187–28196. https://doi.org/10.1074/jbc.M313932200
Kotecki L, Hearing M, McCall NM et al (2015) GIRK channels modulate opioid-induced motor activity in a cell type- and subunit-dependent manner. J Neurosci 35(18):7131–7142. https://doi.org/10.1523/JNEUROSCI.5051-14.2015
Kovalev H, Quayle JM, Kamishima T, Lodwick D (2004) Molecular analysis of the subtype-selective inhibition of cloned KATP channels by PNU-37883A. Br J Pharmacol 141(5):867–873. https://doi.org/10.1038/sj.bjp.0705670
Kovoor P, Wickman K, Maguire CT et al (2001) Evaluation of the role of I(KACh) in atrial fibrillation using a mouse knockout model. J Am Coll Cardiol 37(8):2136–2143. https://doi.org/10.1016/s0735-1097(01)01304-3
Kozek KA, Du Y, Sharma S et al (2019) Discovery and characterization of VU0529331, a synthetic small-molecule activator of Homomeric G protein-gated, inwardly rectifying, potassium (GIRK) channels. ACS Chem Nerosci 10(1):358–370. https://doi.org/10.1021/acschemneuro.8b00287
Krapivinsky G, Gordon EA, Wickman K, Velimirovic B, Krapivinsky L, Clapham DE (1995) The G-protein-gated atrial K+ channel IKACh is a heteromultimer of two inwardly rectifying K(+)-channel proteins. Nature 374(6518):135–141. https://doi.org/10.1038/374135a0
Krapivinsky G, Medina I, Eng L, Krapivinsky L, Yang Y, Clapham DE (1998) A novel inward rectifier K+ channel with unique pore properties. Neuron 20(5):995–1005. https://doi.org/10.1016/s0896-6273(00)80480-8
Kubo Y (1996) Effects of extracellular cations and mutations in the pore region on the inward rectifier K+ channel IRK1. Recept Channels 4(2):73–83
Kubo Y, Murata Y (2001) Control of rectification and permeation by two distinct sites after the second transmembrane region in Kir2.1 K+ channel. J Physiol 531(Pt 3):645–660. https://doi.org/10.1111/j.1469-7793.2001.0645h.x
Kubo Y, Baldwin TJ, Jan YN, Jan LY (1993a) Primary structure and functional expression of a mouse inward rectifier potassium channel. Nature 362(6416):127–133. https://doi.org/10.1038/362127a0
Kubo Y, Reuveny E, Slesinger PA, Jan YN, Jan LY (1993b) Primary structure and functional expression of a rat G-protein-coupled muscarinic potassium channel. Nature 364(6440):802–806. https://doi.org/10.1038/364802a0
Kuffler SW, Nicholls JG (1966) The physiology of neuroglial cells. Ergeb Physiol 57:1–90
Kumar M, Pattnaik BR (2014) Focus on Kir7.1: physiology and channelopathy. Channels (Austin) 8(6):488–495. https://doi.org/10.4161/19336950.2014.959809
Kung AW, Lau KS, Cheung WM, Chan V (2006) Thyrotoxic periodic paralysis and polymorphisms of sodium-potassium ATPase genes. Clin Endocrinol (Oxf) 64(2):158–161. https://doi.org/10.1111/j.1365-2265.2005.02442.x
Kurachi Y (1985) Voltage-dependent activation of the inward-rectifier potassium channel in the ventricular cell membrane of guinea-pig heart. J Physiol 366:365–385. https://doi.org/10.1113/jphysiol.1985.sp015803
Kurachi Y, Nakajima T, Sugimoto T (1986) On the mechanism of activation of muscarinic K+ channels by adenosine in isolated atrial cells: involvement of GTP-binding proteins. Pflugers Arch 407(3):264–274. https://doi.org/10.1007/BF00585301
Kusaka S, Inanobe A, Fujita A et al (2001) Functional Kir7.1 channels localized at the root of apical processes in rat retinal pigment epithelium. J Physiol 531(Pt 1):27–36. https://doi.org/10.1111/j.1469-7793.2001.0027j.x
Kuzhikandathil EV, Oxford GS (2002) Classic D1 dopamine receptor antagonist R-(+)-7-chloro-8-hydroxy-3-methyl-1-phenyl-2,3,4,5-tetrahydro-1H-3-benzazepine hydrochloride (SCH23390) directly inhibits G protein-coupled inwardly rectifying potassium channels. Mol Pharmacol 62(1):119–126. https://doi.org/10.1124/mol.62.1.119
Kwan HY, Leung PC, Huang Y, Yao X (2003) Depletion of intracellular Ca2+ stores sensitizes the flow-induced Ca2+ influx in rat endothelial cells. Circ Res 92(3):286–292. https://doi.org/10.1161/01.res.0000054625.24468.08
Labouebe G, Lomazzi M, Cruz HG et al (2007) RGS2 modulates coupling between GABAB receptors and GIRK channels in dopamine neurons of the ventral tegmental area. Nat Neurosci 10(12):1559–1568. https://doi.org/10.1038/nn2006
Lacey MG, Mercuri NB, North RA (1988) On the potassium conductance increase activated by GABAB and dopamine D2 receptors in rat substantia nigra neurones. J Physiol 401:437–453. https://doi.org/10.1113/jphysiol.1988.sp017171
Lachheb S, Cluzeaud F, Bens M et al (2008) Kir4.1/Kir5.1 channel forms the major K+ channel in the basolateral membrane of mouse renal collecting duct principal cells. Am J Physiol Renal Physiol 294(6):F1398–F1407. https://doi.org/10.1152/ajprenal.00288.2007
Lacin E, Aryal P, Glaaser IW et al (2017) Dynamic role of the tether helix in PIP2-dependent gating of a G protein-gated potassium channel. J Gen Physiol 149(8):799–811. https://doi.org/10.1085/jgp.201711801
Leal-Pinto E, Gomez-Llorente Y, Sundaram S et al (2010) Gating of a G protein-sensitive mammalian Kir3.1 prokaryotic Kir channel chimera in planar lipid bilayers. J Biol Chem 285(51):39790–39800. https://doi.org/10.1074/jbc.M110.151373
Leaney JL, Dekker LV, Tinker A (2001) Regulation of a G protein-gated inwardly rectifying K+ channel by a Ca(2+)-independent protein kinase C. J Physiol 534(Pt. 2):367–379. https://doi.org/10.1111/j.1469-7793.2001.00367.x
Lee SJ, Wang S, Borschel W, Heyman S, Gyore J, Nichols CG (2013) Secondary anionic phospholipid binding site and gating mechanism in Kir2.1 inward rectifier channels. Nat Commun 4:2786. https://doi.org/10.1038/ncomms3786
Lee SJ, Ren F, Zangerl-Plessl EM et al (2016) Structural basis of control of inward rectifier Kir2 channel gating by bulk anionic phospholipids. J Gen Physiol 148(3):227–237. https://doi.org/10.1085/jgp.201611616
Lee KPK, Chen J, MacKinnon R (2017) Molecular structure of human KATP in complex with ATP and ADP. eLife 6. https://doi.org/10.7554/eLife.32481
Leech CA, Stanfield PR (1981) Inward rectification in frog skeletal muscle fibres and its dependence on membrane potential and external potassium. J Physiol 319:295–309. https://doi.org/10.1113/jphysiol.1981.sp013909
Lei Q, Talley EM, Bayliss DA (2001) Receptor-mediated inhibition of G protein-coupled inwardly rectifying potassium channels involves G(alpha)q family subunits, phospholipase C, and a readily diffusible messenger. J Biol Chem 276(20):16720–16730. https://doi.org/10.1074/jbc.M100207200
Lesage F, Duprat F, Fink M et al (1994) Cloning provides evidence for a family of inward rectifier and G-protein coupled K+ channels in the brain. FEBS Lett 353(1):37–42. https://doi.org/10.1016/0014-5793(94)01007-2
Lewis DL, Ikeda SR, Aryee D, Joho RH (1991) Expression of an inwardly rectifying K+ channel from rat basophilic leukemia cell mRNA in Xenopus oocytes. FEBS Lett 290(1–2):17–21. https://doi.org/10.1016/0014-5793(91)81215-t
Lewis LM, Bhave G, Chauder BA et al (2009) High-throughput screening reveals a small-molecule inhibitor of the renal outer medullary potassium channel and Kir7.1. Mol Pharmacol 76(5):1094–1103. https://doi.org/10.1124/mol.109.059840
Lewohl JM, Wilson WR, Mayfield RD, Brozowski SJ, Morrisett RA, Harris RA (1999) G-protein-coupled inwardly rectifying potassium channels are targets of alcohol action. Nat Neurosci 2(12):1084–1090. https://doi.org/10.1038/16012
Li CG, Cui WY, Wang H (2016a) Sensitivity of KATP channels to cellular metabolic disorders and the underlying structural basis. Acta Pharmacol Sin 37(1):134–142. https://doi.org/10.1038/aps.2015.134
Li D, Chen R, Chung SH (2016b) Molecular dynamics of the honey bee toxin tertiapin binding to Kir3.2. Biophys Chem 219:43–48. https://doi.org/10.1016/j.bpc.2016.09.010
Li N, Wu JX, Ding D, Cheng J, Gao N, Chen L (2017) Structure of a pancreatic ATP-sensitive potassium channel. Cell 168(1–2):101–110.e10. https://doi.org/10.1016/j.cell.2016.12.028
Li D, Jin T, Gazgalis D, Cui M, Logothetis DE (2019) On the mechanism of GIRK2 channel gating by phosphatidylinositol bisphosphate, sodium, and the Gbetagamma dimer. J Biol Chem 294(49):18934–18948. https://doi.org/10.1074/jbc.RA119.010047
Li Y, Aziz Q, Tinker A (2021) The pharmacology of ATP-sensitive K+ channels (KATP). In: Handbook of experimental pharmacology. Springer, Cham
Lin DH, Sterling H, Wang Z et al (2005) ROMK1 channel activity is regulated by monoubiquitination. Proc Natl Acad Sci U S A 102(12):4306–4311. https://doi.org/10.1073/pnas.0409767102
Lin DH, Yue P, Zhang C, Wang WH (2014) MicroRNA-194 (miR-194) regulates ROMK channel activity by targeting intersectin 1. Am J Physiol Renal Physiol 306(1):F53–F60. https://doi.org/10.1152/ajprenal.00349.2013
Linder T, Wang S, Zangerl-Plessl EM, Nichols CG, Stary-Weinzinger A (2015) Molecular dynamics simulations of KirBac1.1 mutants reveal global gating changes of Kir channels. J Chem Inf Model 55(4):814–822. https://doi.org/10.1021/acs.jcim.5b00010
Liou HH, Zhou SS, Huang CL (1999) Regulation of ROMK1 channel by protein kinase A via a phosphatidylinositol 4,5-bisphosphate-dependent mechanism. Proc Natl Acad Sci U S A 96(10):5820–5825. https://doi.org/10.1073/pnas.96.10.5820
Liss B, Bruns R, Roeper J (1999) Alternative sulfonylurea receptor expression defines metabolic sensitivity of K-ATP channels in dopaminergic midbrain neurons. EMBO J 18(4):833–846. https://doi.org/10.1093/emboj/18.4.833
Liu Y, Liu D, Printzenhoff D, Coghlan MJ, Harris R, Krafte DS (2002) Tenidap, a novel anti-inflammatory agent, is an opener of the inwardly rectifying K+ channel hKir2.3. Eur J Pharmacol 435(2–3):153–160. https://doi.org/10.1016/s0014-2999(01)01590-4
Liu TA, Chang HK, Shieh RC (2012) Revisiting inward rectification: K ions permeate through Kir2.1 channels during high-affinity block by spermidine. J Gen Physiol 139(3):245–259. https://doi.org/10.1085/jgp.201110736
Logothetis DE, Mahajan R, Adney SK et al (2015) Unifying mechanism of controlling Kir3 channel activity by G proteins and phosphoinositides. Int Rev Neurobiol 123:1–26. https://doi.org/10.1016/bs.irn.2015.05.013
Longden TA, Dabertrand F, Koide M et al (2017) Capillary K(+)-sensing initiates retrograde hyperpolarization to increase local cerebral blood flow. Nat Neurosci 20(5):717–726. https://doi.org/10.1038/nn.4533
Lopatin AN, Nichols CG (1996) [K+] dependence of polyamine-induced rectification in inward rectifier potassium channels (IRK1, Kir2.1). J Gen Physiol 108(2):105–113. https://doi.org/10.1085/jgp.108.2.105
Lopatin AN, Makhina EN, Nichols CG (1994) Potassium channel block by cytoplasmic polyamines as the mechanism of intrinsic rectification. Nature 372(6504):366–369. https://doi.org/10.1038/372366a0
Lopatin AN, Makhina EN, Nichols CG (1995) The mechanism of inward rectification of potassium channels: “long-pore plugging” by cytoplasmic polyamines. J Gen Physiol 106(5):923–955. https://doi.org/10.1085/jgp.106.5.923
Lopes CM, Zhang H, Rohacs T, Jin T, Yang J, Logothetis DE (2002) Alterations in conserved Kir channel-PIP2 interactions underlie channelopathies. Neuron 34(6):933–944. https://doi.org/10.1016/s0896-6273(02)00725-0
Lopes CM, Remon JI, Matavel A et al (2007) Protein kinase A modulates PLC-dependent regulation and PIP2-sensitivity of K+ channels. Channels (Austin) 1(2):124–134. https://doi.org/10.4161/chan.4322
Lopez-Izquierdo A, Arechiga-Figueroa IA, Moreno-Galindo EG et al (2011) Mechanisms for Kir channel inhibition by quinacrine: acute pore block of Kir2.x channels and interference in PIP2 interaction with Kir2.x and Kir6.2 channels. Pflugers Arch 462(4):505–517. https://doi.org/10.1007/s00424-011-0995-5
Lorenz JN, Baird NR, Judd LM et al (2002) Impaired renal NaCl absorption in mice lacking the ROMK potassium channel, a model for type II Bartter's syndrome. J Biol Chem 277(40):37871–37880. https://doi.org/10.1074/jbc.M205627200
Lotsch J, Pruss H, Veh RW, Doehring A (2010) A KCNJ6 (Kir3.2, GIRK2) gene polymorphism modulates opioid effects on analgesia and addiction but not on pupil size. Pharmacogenet Genomics 20(5):291–297. https://doi.org/10.1097/FPC.0b013e3283386bda
Lu Z, MacKinnon R (1994) Electrostatic tuning of Mg2+ affinity in an inward-rectifier K+ channel. Nature 371(6494):243–246. https://doi.org/10.1038/371243a0
Lu M, Wang T, Yan Q et al (2002) Absence of small conductance K+ channel (SK) activity in apical membranes of thick ascending limb and cortical collecting duct in ROMK (Bartter's) knockout mice. J Biol Chem 277(40):37881–37887. https://doi.org/10.1074/jbc.M206644200
Lujan R, Aguado C (2015) Localization and targeting of GIRK channels in mammalian central neurons. Int Rev Neurobiol 123:161–200. https://doi.org/10.1016/bs.irn.2015.05.009
Lujan R, Maylie J, Adelman JP (2009) New sites of action for GIRK and SK channels. Nat Rev Neurosci 10(7):475–480. https://doi.org/10.1038/nrn2668
Lujan R, Marron Fernandez de Velasco E, Aguado C, Wickman K (2014) New insights into the therapeutic potential of Girk channels. Trends Neurosci 37(1):20–29. https://doi.org/10.1016/j.tins.2013.10.006
Lunn ML, Nassirpour R, Arrabit C et al (2007) A unique sorting nexin regulates trafficking of potassium channels via a PDZ domain interaction. Nat Neurosci 10(10):1249–1259. https://doi.org/10.1038/nn1953
Luo X, Pan Z, Shan H et al (2013) MicroRNA-26 governs profibrillatory inward-rectifier potassium current changes in atrial fibrillation. J Clin Invest 123(5):1939–1951. https://doi.org/10.1172/JCI62185
Luscher C, Slesinger PA (2010) Emerging roles for G protein-gated inwardly rectifying potassium (GIRK) channels in health and disease. Nat Rev Neurosci 11(5):301–315. https://doi.org/10.1038/nrn2834
Ma D, Zerangue N, Lin YF et al (2001) Role of ER export signals in controlling surface potassium channel numbers. Science 291(5502):316–319. https://doi.org/10.1126/science.291.5502.316
MacGregor GG, Xu JZ, McNicholas CM, Giebisch G, Hebert SC (1998) Partially active channels produced by PKA site mutation of the cloned renal K+ channel, ROMK2 (kir1.2). Am J Physiol 275(3):F415–F422. https://doi.org/10.1152/ajprenal.1998.275.3.F415
Machida T, Hashimoto N, Kuwahara I et al (2011) Effects of a highly selective acetylcholine-activated K+ channel blocker on experimental atrial fibrillation. Circ Arrhythm Electrophysiol 4(1):94–102. https://doi.org/10.1161/CIRCEP.110.951608
Madiera F, Park YM, Lee J, Buso N, Gur T, Madhusoodanan N, Basutka P, Tivey ARN, Potter SC, Finn RD, Lopez R (2019) The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Research. W1:W636–641. https://doi.org/10.1093/nar/gkz268
Makary S, Voigt N, Maguy A et al (2011) Differential protein kinase C isoform regulation and increased constitutive activity of acetylcholine-regulated potassium channels in atrial remodeling. Circ Res 109(9):1031–1043. https://doi.org/10.1161/CIRCRESAHA.111.253120
Manis AD, Hodges MR, Staruschenko A, Palygin O (2020) Expression, localization, and functional properties of inwardly rectifying K(+) channels in the kidney. Am J Physiol Renal Physiol 318(2):F332–F337. https://doi.org/10.1152/ajprenal.00523.2019
Mao J, Wang X, Chen F et al (2004) Molecular basis for the inhibition of G protein-coupled inward rectifier K(+) channels by protein kinase C. Proc Natl Acad Sci U S A 101(4):1087–1092. https://doi.org/10.1073/pnas.0304827101
Marker CL, Stoffel M, Wickman K (2004) Spinal G-protein-gated K+ channels formed by GIRK1 and GIRK2 subunits modulate thermal nociception and contribute to morphine analgesia. J Neurosci 24(11):2806–2812. https://doi.org/10.1523/JNEUROSCI.5251-03.2004
Marmolejo-Murillo LG, Arechiga-Figueroa IA, Cui M et al (2017a) Inhibition of Kir4.1 potassium channels by quinacrine. Brain Res 1663:87–94. https://doi.org/10.1016/j.brainres.2017.03.009
Marmolejo-Murillo LG, Arechiga-Figueroa IA, Moreno-Galindo EG et al (2017b) Chloroquine blocks the Kir4.1 channels by an open-pore blocking mechanism. Eur J Pharmacol 800:40–47. https://doi.org/10.1016/j.ejphar.2017.02.024
Marron Fernandez de Velasco E, McCall N, Wickman K (2015) GIRK channel plasticity and implications for drug addiction. Int Rev Neurobiol 123:201–238. https://doi.org/10.1016/bs.irn.2015.05.011
Martin GM, Kandasamy B, DiMaio F, Yoshioka C, Shyng SL (2017) Anti-diabetic drug binding site in a mammalian KATP channel revealed by Cryo-EM. eLife 6. https://doi.org/10.7554/eLife.31054
Maruyama M, Lin SF, Xie Y et al (2011) Genesis of phase 3 early afterdepolarizations and triggered activity in acquired long-QT syndrome. Circ Arrhythm Electrophysiol 4(1):103–111. https://doi.org/10.1161/CIRCEP.110.959064
Mathiharan YKGI, Zhao Y, Robertson MJ, Skiniotis G, Slesinger PA (2020) Structural basis of GIRK2 channel modulation by cholesterol and PIP2. bioRxiv. https://doi.org/10.1101/2020.06.04.134544
Matsuda H (1988) Open-state substructure of inwardly rectifying potassium channels revealed by magnesium block in guinea-pig heart cells. J Physiol 397:237–258. https://doi.org/10.1113/jphysiol.1988.sp016998
Matsuda H, Saigusa A, Irisawa H (1987) Ohmic conductance through the inwardly rectifying K channel and blocking by internal Mg2+. Nature 325(7000):156–159. https://doi.org/10.1038/325156a0
Matsuda T, Takeda K, Ito M et al (2005) Atria selective prolongation by NIP-142, an antiarrhythmic agent, of refractory period and action potential duration in guinea pig myocardium. J Pharmacol Sci 98(1):33–40. https://doi.org/10.1254/jphs.fpj04045x
Matsuda T, Ito M, Ishimaru S et al (2006) Blockade by NIP-142, an antiarrhythmic agent, of carbachol-induced atrial action potential shortening and GIRK1/4 channel. J Pharmacol Sci 101(4):303–310. https://doi.org/10.1254/jphs.fp0060324
Matsuoka S, Sarai N, Kuratomi S, Ono K, Noma A (2003) Role of individual ionic current systems in ventricular cells hypothesized by a model study. Jpn J Physiol 53(2):105–123. https://doi.org/10.2170/jjphysiol.53.105
May LM, Anggono V, Gooch HM et al (2017) G-protein-coupled inwardly rectifying potassium (GIRK) channel activation by the p75 Neurotrophin receptor is required for amyloid beta toxicity. Front Neurosci 11:455. https://doi.org/10.3389/fnins.2017.00455
Mayordomo-Cava J, Yajeya J, Navarro-Lopez JD, Jimenez-Diaz L (2015) Amyloid-beta(25-35) modulates the expression of GirK and KCNQ channel genes in the Hippocampus. PLoS One 10(7):e0134385. https://doi.org/10.1371/journal.pone.0134385
Mazarati A, Lundstrom L, Sollenberg U, Shin D, Langel U, Sankar R (2006) Regulation of kindling epileptogenesis by hippocampal galanin type 1 and type 2 receptors: the effects of subtype-selective agonists and the role of G-protein-mediated signaling. J Pharmacol Exp Ther 318(2):700–708. https://doi.org/10.1124/jpet.106.104703
McAllister RE, Noble D (1966) The time and voltage dependence of the slow outward current in cardiac Purkinje fibres. J Physiol 186(3):632–662. https://doi.org/10.1113/jphysiol.1966.sp008060
McCarron JG, Halpern W (1990) Potassium dilates rat cerebral arteries by two independent mechanisms. Am J Physiol 259(3 Pt 2):H902–H908. https://doi.org/10.1152/ajpheart.1990.259.3.H902
McCloskey C, Rada C, Bailey E et al (2014) The inwardly rectifying K+ channel KIR7.1 controls uterine excitability throughout pregnancy. EMBO Mol Med 6(9):1161–1174. https://doi.org/10.15252/emmm.201403944
McKinney LC, Gallin EK (1988) Inwardly rectifying whole-cell and single-channel K currents in the murine macrophage cell line J774.1. J Membr Biol 103(1):41–53. https://doi.org/10.1007/BF01871931
McNicholas CM, Wang W, Ho K, Hebert SC, Giebisch G (1994) Regulation of ROMK1 K+ channel activity involves phosphorylation processes. Proc Natl Acad Sci U S A 91(17):8077–8081. https://doi.org/10.1073/pnas.91.17.8077
McNicholas CM, MacGregor GG, Islas LD, Yang Y, Hebert SC, Giebisch G (1998) pH-dependent modulation of the cloned renal K+ channel, ROMK. Am J Physiol 275(6):F972–F981. https://doi.org/10.1152/ajprenal.1998.275.6.F972
Medina I, Krapivinsky G, Arnold S, Kovoor P, Krapivinsky L, Clapham DE (2000) A switch mechanism for G beta gamma activation of I(KACh). J Biol Chem 275(38):29709–29716. https://doi.org/10.1074/jbc.M004989200
Meng XY, Zhang HX, Logothetis DE, Cui M (2012) The molecular mechanism by which PIP(2) opens the intracellular G-loop gate of a Kir3.1 channel. Biophys J 102(9):2049–2059. https://doi.org/10.1016/j.bpj.2012.03.050
Meng XY, Liu S, Cui M, Zhou R, Logothetis DE (2016) The molecular mechanism of opening the Helix bundle crossing (HBC) gate of a Kir Channel. Sci Rep 6:29399. https://doi.org/10.1038/srep29399
Mi H, Deerinck TJ, Jones M, Ellisman MH, Schwarz TL (1996) Inwardly rectifying K+ channels that may participate in K+ buffering are localized in microvilli of Schwann cells. J Neurosci 16(8):2421–2429
Mitrovic I, Margeta-Mitrovic M, Bader S, Stoffel M, Jan LY, Basbaum AI (2003) Contribution of GIRK2-mediated postsynaptic signaling to opiate and alpha 2-adrenergic analgesia and analgesic sex differences. Proc Natl Acad Sci U S A 100(1):271–276. https://doi.org/10.1073/pnas.0136822100
Moran-Zendejas R, Delgado-Ramirez M, Xu J et al (2020) In vitro and in silico characterization of the inhibition of Kir4.1 channels by aminoglycoside antibiotics. Br J Pharmacol. https://doi.org/10.1111/bph.15214
Morgan AD, Carroll ME, Loth AK, Stoffel M, Wickman K (2003) Decreased cocaine self-administration in Kir3 potassium channel subunit knockout mice. Neuropsychopharmacology 28(5):932–938. https://doi.org/10.1038/sj.npp.1300100
Morishige K, Takahashi N, Jahangir A et al (1994) Molecular cloning and functional expression of a novel brain-specific inward rectifier potassium channel. FEBS Lett 346(2–3):251–256. https://doi.org/10.1016/0014-5793(94)00483-8
Morishige K, Inanobe A, Yoshimoto Y et al (1999) Secretagogue-induced exocytosis recruits G protein-gated K+ channels to plasma membrane in endocrine cells. J Biol Chem 274(12):7969–7974. https://doi.org/10.1074/jbc.274.12.7969
Mukai E, Ishida H, Horie M, Noma A, Seino Y, Takano M (1998) The antiarrhythmic agent cibenzoline inhibits KATP channels by binding to Kir6.2. Biochem Biophys Res Commun 251(2):477–481. https://doi.org/10.1006/bbrc.1998.9492
Mullner C, Vorobiov D, Bera AK et al (2000) Heterologous facilitation of G protein-activated K(+) channels by beta-adrenergic stimulation via cAMP-dependent protein kinase. J Gen Physiol 115(5):547–558. https://doi.org/10.1085/jgp.115.5.547
Nagi K, Pineyro G (2014) Kir3 channel signaling complexes: focus on opioid receptor signaling. Front Cell Neurosci 8:186. https://doi.org/10.3389/fncel.2014.00186
Nakamura N, Suzuki Y, Sakuta H, Ookata K, Kawahara K, Hirose S (1999) Inwardly rectifying K+ channel Kir7.1 is highly expressed in thyroid follicular cells, intestinal epithelial cells and choroid plexus epithelial cells: implication for a functional coupling with Na+,K+-ATPase. Biochem J 342(Pt 2):329–336
Nakamura A, Fujita M, Ono H et al (2014) G protein-gated inwardly rectifying potassium (KIR3) channels play a primary role in the antinociceptive effect of oxycodone, but not morphine, at supraspinal sites. Br J Pharmacol 171(1):253–264. https://doi.org/10.1111/bph.12441
Nanjan MJ, Mohammed M, Prashantha Kumar BR, Chandrasekar MJN (2018) Thiazolidinediones as antidiabetic agents: a critical review. Bioorg Chem 77:548–567. https://doi.org/10.1016/j.bioorg.2018.02.009
Nassirpour R, Bahima L, Lalive AL, Luscher C, Lujan R, Slesinger PA (2010) Morphine- and CaMKII-dependent enhancement of GIRK channel signaling in hippocampal neurons. J Neurosci 30(40):13419–13430. https://doi.org/10.1523/JNEUROSCI.2966-10.2010
Nava-Mesa MO, Jimenez-Diaz L, Yajeya J, Navarro-Lopez JD (2013) Amyloid-beta induces synaptic dysfunction through G protein-gated inwardly rectifying potassium channels in the fimbria-CA3 hippocampal synapse. Front Cell Neurosci 7:117. https://doi.org/10.3389/fncel.2013.00117
Nehring RB, Wischmeyer E, Doring F, Veh RW, Sheng M, Karschin A (2000) Neuronal inwardly rectifying K(+) channels differentially couple to PDZ proteins of the PSD-95/SAP90 family. J Neurosci 20(1):156–162
Nelson MT, Cheng H, Rubart M et al (1995) Relaxation of arterial smooth muscle by calcium sparks. Science 270(5236):633–637. https://doi.org/10.1126/science.270.5236.633
Newman EA (1984) Regional specialization of retinal glial cell membrane. Nature 309(5964):155–157. https://doi.org/10.1038/309155a0
Nichols CG, Lee SJ (2018) Polyamines and potassium channels: a 25-year romance. J Biol Chem 293(48):18779–18788. https://doi.org/10.1074/jbc.TM118.003344
Nichols CG, Makhina EN, Pearson WL, Sha Q, Lopatin AN (1996) Inward rectification and implications for cardiac excitability. Circ Res 78(1):1–7. https://doi.org/10.1161/01.res.78.1.1
Nilius B, Droogmans G (2001) Ion channels and their functional role in vascular endothelium. Physiol Rev 81(4):1415–1459. https://doi.org/10.1152/physrev.2001.81.4.1415
Nilius B, Schwarz G, Droogmans G (1993) Modulation by histamine of an inwardly rectifying potassium channel in human endothelial cells. J Physiol 472:359–371. https://doi.org/10.1113/jphysiol.1993.sp019951
Nishida M, MacKinnon R (2002) Structural basis of inward rectification: cytoplasmic pore of the G protein-gated inward rectifier GIRK1 at 1.8 a resolution. Cell 111(7):957–965. https://doi.org/10.1016/s0092-8674(02)01227-8
Nishida M, Cadene M, Chait BT, MacKinnon R (2007) Crystal structure of a Kir3.1-prokaryotic Kir channel chimera. EMBO J 26(17):4005–4015. https://doi.org/10.1038/sj.emboj.7601828
Nishizawa D, Nagashima M, Katoh R et al (2009) Association between KCNJ6 (GIRK2) gene polymorphisms and postoperative analgesic requirements after major abdominal surgery. PLoS One 4(9):e7060. https://doi.org/10.1371/journal.pone.0007060
Nishizawa D, Fukuda K, Kasai S et al (2014) Association between KCNJ6 (GIRK2) gene polymorphism rs2835859 and post-operative analgesia, pain sensitivity, and nicotine dependence. J Pharmacol Sci 126(3):253–263. https://doi.org/10.1254/jphs.14189fp
Noma A, Trautwein W (1978) Relaxation of the ACh-induced potassium current in the rabbit sinoatrial node cell. Pflugers Arch 377(3):193–200. https://doi.org/10.1007/BF00584272
North RA, Williams JT, Surprenant A, Christie MJ (1987) Mu and delta receptors belong to a family of receptors that are coupled to potassium channels. Proc Natl Acad Sci U S A 84(15):5487–5491. https://doi.org/10.1073/pnas.84.15.5487
Ohmori H (1978) Inactivation kinetics and steady-state current noise in the anomalous rectifier of tunicate egg cell membranes. J Physiol 281:77–99. https://doi.org/10.1113/jphysiol.1978.sp012410
Ohno Y, Hibino H, Lossin C, Inanobe A, Kurachi Y (2007) Inhibition of astroglial Kir4.1 channels by selective serotonin reuptake inhibitors. Brain Res 1178:44–51. https://doi.org/10.1016/j.brainres.2007.08.018
Ookata K, Tojo A, Suzuki Y et al (2000) Localization of inward rectifier potassium channel Kir7.1 in the basolateral membrane of distal nephron and collecting duct. J Am Soc Nephrol 11(11):1987–1994
Palygin O, Pochynyuk O, Staruschenko A (2017) Role and mechanisms of regulation of the basolateral Kir 4.1/Kir 5.1K(+) channels in the distal tubules. Acta Physiol (Oxf) 219(1):260–273. https://doi.org/10.1111/apha.12703
Palygin O, Pochynyuk O, Staruschenko A (2018) Distal tubule basolateral potassium channels: cellular and molecular mechanisms of regulation. Curr Opin Nephrol Hypertens 27(5):373–378. https://doi.org/10.1097/MNH.0000000000000437
Partiseti M, Collura V, Agnel M, Culouscou JM, Graham D (1998) Cloning and characterization of a novel human inwardly rectifying potassium channel predominantly expressed in small intestine. FEBS Lett 434(1–2):171–176. https://doi.org/10.1016/s0014-5793(98)00972-7
Pattnaik BR, Tokarz S, Asuma MP et al (2013) Snowflake vitreoretinal degeneration (SVD) mutation R162W provides new insights into Kir7.1 ion channel structure and function. PLoS One 8(8):e71744. https://doi.org/10.1371/journal.pone.0071744
Pearson WL, Dourado M, Schreiber M, Salkoff L, Nichols CG (1999) Expression of a functional Kir4 family inward rectifier K+ channel from a gene cloned from mouse liver. J Physiol 514(Pt 3):639–653. https://doi.org/10.1111/j.1469-7793.1999.639ad.x
Pegan S, Arrabit C, Zhou W et al (2005) Cytoplasmic domain structures of Kir2.1 and Kir3.1 show sites for modulating gating and rectification. Nat Neurosci 8(3):279–287. https://doi.org/10.1038/nn1411
Peters M, Jeck N, Reinalter S et al (2002) Clinical presentation of genetically defined patients with hypokalemic salt-losing tubulopathies. Am J Med 112(3):183–190. https://doi.org/10.1016/s0002-9343(01)01086-5
Petit-Jacques J, Sui JL, Logothetis DE (1999) Synergistic activation of G protein-gated inwardly rectifying potassium channels by the betagamma subunits of G proteins and Na(+) and Mg(2+) ions. J Gen Physiol 114(5):673–684. https://doi.org/10.1085/jgp.114.5.673
Pfaffinger PJ, Martin JM, Hunter DD, Nathanson NM, Hille B (1985) GTP-binding proteins couple cardiac muscarinic receptors to a K channel. Nature 317(6037):536–538. https://doi.org/10.1038/317536a0
Podd SJ, Freemantle N, Furniss SS, Sulke N (2016) First clinical trial of specific IKACh blocker shows no reduction in atrial fibrillation burden in patients with paroxysmal atrial fibrillation: pacemaker assessment of BMS 914392 in patients with paroxysmal atrial fibrillation. Europace 18(3):340–346. https://doi.org/10.1093/europace/euv263
Ponce-Balbuena D, Lopez-Izquierdo A, Ferrer T, Rodriguez-Menchaca AA, Arechiga-Figueroa IA, Sanchez-Chapula JA (2009) Tamoxifen inhibits inward rectifier K+ 2.x family of inward rectifier channels by interfering with phosphatidylinositol 4,5-bisphosphate-channel interactions. J Pharmacol Exp Ther 331(2):563–573. https://doi.org/10.1124/jpet.109.156075
Proks P, Ashcroft FM (1997) Phentolamine block of KATP channels is mediated by Kir6.2. Proc Natl Acad Sci U S A 94(21):11716–11720. https://doi.org/10.1073/pnas.94.21.11716
Prystowsky EN, Niazi I, Curtis AB et al (2003) Termination of paroxysmal supraventricular tachycardia by tecadenoson (CVT-510), a novel A1-adenosine receptor agonist. J Am Coll Cardiol 42(6):1098–1102. https://doi.org/10.1016/s0735-1097(03)00987-2
Puissant MM, Muere C, Levchenko V et al (2019) Genetic mutation of Kcnj16 identifies Kir5.1-containing channels as key regulators of acute and chronic pH homeostasis. FASEB J 33(4):5067–5075. https://doi.org/10.1096/fj.201802257R
Ramos-Hunter SJ, Engers DW, Kaufmann K et al (2013) Discovery and SAR of a novel series of GIRK1/2 and GIRK1/4 activators. Bioorg Med Chem Lett 23(18):5195–5198. https://doi.org/10.1016/j.bmcl.2013.07.002
Ramu Y, Xu Y, Lu Z (2018) A novel high-affinity inhibitor against the human ATP-sensitive Kir6.2 channel. J Gen Physiol 150(7):969–976. https://doi.org/10.1085/jgp.201812017
Raphemot R, Lonergan DF, Nguyen TT et al (2011) Discovery, characterization, and structure-activity relationships of an inhibitor of inward rectifier potassium (Kir) channels with preference for Kir2.3, Kir3.x, and Kir7.1. Front Pharmacol 2:75. https://doi.org/10.3389/fphar.2011.00075
Reeves RH, Irving NG, Moran TH et al (1995) A mouse model for down syndrome exhibits learning and behaviour deficits. Nat Genet 11(2):177–184. https://doi.org/10.1038/ng1095-177
Reimann F, Proks P, Ashcroft FM (2001) Effects of mitiglinide (S 21403) on Kir6.2/SUR1, Kir6.2/SUR2A and Kir6.2/SUR2B types of ATP-sensitive potassium channel. Br J Pharmacol 132(7):1542–1548. https://doi.org/10.1038/sj.bjp.0703962
Reuveny E, Slesinger PA, Inglese J et al (1994) Activation of the cloned muscarinic potassium channel by G protein beta gamma subunits. Nature 370(6485):143–146. https://doi.org/10.1038/370143a0
Rifkin RA, Moss SJ, Slesinger PA (2017) G protein-gated potassium channels: a link to drug addiction. Trends Pharmacol Sci 38(4):378–392. https://doi.org/10.1016/j.tips.2017.01.007
Rodrigo GC, Standen NB (2005) ATP-sensitive potassium channels. Curr Pharm Des 11(15):1915–1940. https://doi.org/10.2174/1381612054021015
Rodriguez-Menchaca AA, Navarro-Polanco RA, Ferrer-Villada T et al (2008) The molecular basis of chloroquine block of the inward rectifier Kir2.1 channel. Proc Natl Acad Sci U S A 105(4):1364–1368. https://doi.org/10.1073/pnas.0708153105
Rodriguez-Soriano J (1998) Bartter and related syndromes: the puzzle is almost solved. Pediatr Nephrol 12(4):315–327. https://doi.org/10.1007/s004670050461
Rohacs T, Lopes CM, Jin T, Ramdya PP, Molnar Z, Logothetis DE (2003) Specificity of activation by phosphoinositides determines lipid regulation of Kir channels. Proc Natl Acad Sci U S A 100(2):745–750. https://doi.org/10.1073/pnas.0236364100
Romanenko VG, Rothblat GH, Levitan I (2002) Modulation of endothelial inward-rectifier K+ current by optical isomers of cholesterol. Biophys J 83(6):3211–3222. https://doi.org/10.1016/S0006-3495(02)75323-X
Rosenhouse-Dantsker A (2019) Cholesterol binding sites in inwardly rectifying potassium channels. Adv Exp Med Biol 1135:119–138. https://doi.org/10.1007/978-3-030-14265-0_7
Rosenhouse-Dantsker A, Sui JL, Zhao Q et al (2008) A sodium-mediated structural switch that controls the sensitivity of Kir channels to PtdIns(4,5)P(2). Nat Chem Biol 4(10):624–631. https://doi.org/10.1038/nchembio.112
Rougier O, Vassort G, Stampfli R (1967) Voltage clamp experiments on cardiac muscle fibers with the aid of the sucrose gap technic. J Physiol Paris 59(4 Suppl):490
Rui Zhang ZW, Ling B, Liu Y, Liu C (2010) Docking and molecular dynamics studies on the interaction of four imidazoline derivatives with potassium ion channel (Kir6.2). Mol Simul 36(2):166–174. https://doi.org/10.1080/08927020903141035
Ryan DP, da Silva MR, Soong TW et al (2010) Mutations in potassium channel Kir2.6 cause susceptibility to thyrotoxic hypokalemic periodic paralysis. Cell 140(1):88–98. https://doi.org/10.1016/j.cell.2009.12.024
Sackmann E, Kotulla R, Heiszler FJ (1984) On the role of lipid-bilayer elasticity for the lipid-protein interaction and the indirect protein-protein coupling. Can J Biochem Cell Biol 62(8):778–788. https://doi.org/10.1139/o84-099
Sadja R, Smadja K, Alagem N, Reuveny E (2001) Coupling Gbetagamma-dependent activation to channel opening via pore elements in inwardly rectifying potassium channels. Neuron 29(3):669–680. https://doi.org/10.1016/s0896-6273(01)00242-2
Sago H, Carlson EJ, Smith DJ et al (1998) Ts1Cje, a partial trisomy 16 mouse model for down syndrome, exhibits learning and behavioral abnormalities. Proc Natl Acad Sci U S A 95(11):6256–6261. https://doi.org/10.1073/pnas.95.11.6256
Sakmann B, Noma A, Trautwein W (1983) Acetylcholine activation of single muscarinic K+ channels in isolated pacemaker cells of the mammalian heart. Nature 303(5914):250–253. https://doi.org/10.1038/303250a0
Salvatore L, D'Adamo MC, Polishchuk R, Salmona M, Pessia M (1999) Localization and age-dependent expression of the inward rectifier K+ channel subunit Kir 5.1 in a mammalian reproductive system. FEBS Lett 449(2–3):146–152. https://doi.org/10.1016/s0014-5793(99)00420-2
Sanchez-Rodriguez I, Temprano-Carazo S, Najera A et al (2017) Activation of G-protein-gated inwardly rectifying potassium (Kir3/GirK) channels rescues hippocampal functions in a mouse model of early amyloid-beta pathology. Sci Rep 7(1):14658. https://doi.org/10.1038/s41598-017-15306-8
Sanchez-Rodriguez I, Djebari S, Temprano-Carazo S et al (2020) Hippocampal long-term synaptic depression and memory deficits induced in early amyloidopathy are prevented by enhancing G-protein-gated inwardly rectifying potassium channel activity. J Neurochem 153(3):362–376. https://doi.org/10.1111/jnc.14946
Sattler MB, Williams SK, Neusch C et al (2008) Flupirtine as neuroprotective add-on therapy in autoimmune optic neuritis. Am J Pathol 173(5):1496–1507. https://doi.org/10.2353/ajpath.2008.080491
Sharon D, Vorobiov D, Dascal N (1997) Positive and negative coupling of the metabotropic glutamate receptors to a G protein-activated K+ channel, GIRK, in Xenopus oocytes. J Gen Physiol 109(4):477–490. https://doi.org/10.1085/jgp.109.4.477
Shen W, Tian X, Day M et al (2007) Cholinergic modulation of Kir2 channels selectively elevates dendritic excitability in striatopallidal neurons. Nat Neurosci 10(11):1458–1466. https://doi.org/10.1038/nn1972
Sheng M, Sala C (2001) PDZ domains and the organization of supramolecular complexes. Annu Rev Neurosci 24:1–29. https://doi.org/10.1146/annurev.neuro.24.1.1
Shieh RC, Chang JC, Kuo CC (1999) K+ binding sites and interactions between permeating K+ ions at the external pore mouth of an inward rectifier K+ channel (Kir2.1). J Biol Chem 274(25):17424–17430. https://doi.org/10.1074/jbc.274.25.17424
Shimoni Y, Clark RB, Giles WR (1992) Role of an inwardly rectifying potassium current in rabbit ventricular action potential. J Physiol 448:709–727. https://doi.org/10.1113/jphysiol.1992.sp019066
Shimura M, Yuan Y, Chang JT et al (2001) Expression and permeation properties of the K(+) channel Kir7.1 in the retinal pigment epithelium. J Physiol 531(Pt 2):329–346. https://doi.org/10.1111/j.1469-7793.2001.0329i.x
Shuck ME, Bock JH, Benjamin CW et al (1994) Cloning and characterization of multiple forms of the human kidney ROM-K potassium channel. J Biol Chem 269(39):24261–24270
Shuck ME, Piser TM, Bock JH, Slightom JL, Lee KS, Bienkowski MJ (1997) Cloning and characterization of two K+ inward rectifier (Kir) 1.1 potassium channel homologs from human kidney (Kir1.2 and Kir1.3). J Biol Chem 272(1):586–593. https://doi.org/10.1074/jbc.272.1.586
Shumilina E, Klocker N, Korniychuk G, Rapedius M, Lang F, Baukrowitz T (2006) Cytoplasmic accumulation of long-chain coenzyme A esters activates KATP and inhibits Kir2.1 channels. J Physiol 575(Pt 2):433–442. https://doi.org/10.1113/jphysiol.2006.111161
Shyng S, Ferrigni T, Nichols CG (1997) Control of rectification and gating of cloned KATP channels by the Kir6.2 subunit. J Gen Physiol 110(2):141–153. https://doi.org/10.1085/jgp.110.2.141
Shyng SL, Barbieri A, Gumusboga A et al (2000) Modulation of nucleotide sensitivity of ATP-sensitive potassium channels by phosphatidylinositol-4-phosphate 5-kinase. Proc Natl Acad Sci U S A 97(2):937–941. https://doi.org/10.1073/pnas.97.2.937
Siarey RJ, Carlson EJ, Epstein CJ, Balbo A, Rapoport SI, Galdzicki Z (1999) Increased synaptic depression in the Ts65Dn mouse, a model for mental retardation in down syndrome. Neuropharmacology 38(12):1917–1920. https://doi.org/10.1016/s0028-3908(99)00083-0
Signorini S, Liao YJ, Duncan SA, Jan LY, Stoffel M (1997) Normal cerebellar development but susceptibility to seizures in mice lacking G protein-coupled, inwardly rectifying K+ channel GIRK2. Proc Natl Acad Sci U S A 94(3):923–927. https://doi.org/10.1073/pnas.94.3.923
Silver MR, DeCoursey TE (1990) Intrinsic gating of inward rectifier in bovine pulmonary artery endothelial cells in the presence or absence of internal Mg2+. J Gen Physiol 96(1):109–133. https://doi.org/10.1085/jgp.96.1.109
Sims SM, Dixon SJ (1989) Inwardly rectifying K+ current in osteoclasts. Am J Physiol 256(6 Pt 1):C1277–C1282. https://doi.org/10.1152/ajpcell.1989.256.6.C1277
Sjogren B (2011) Regulator of G protein signaling proteins as drug targets: current state and future possibilities. Adv Pharmacol 62:315–347. https://doi.org/10.1016/B978-0-12-385952-5.00002-6
Smith SB, Marker CL, Perry C et al (2008) Quantitative trait locus and computational mapping identifies Kcnj9 (GIRK3) as a candidate gene affecting analgesia from multiple drug classes. Pharmacogenet Genomics 18(3):231–241. https://doi.org/10.1097/FPC.0b013e3282f55ab2
Soe R, Andreasen M, Klaerke DA (2009) Modulation of Kir4.1 and Kir4.1-Kir5.1 channels by extracellular cations. Biochim Biophys Acta 1788(9):1706–1713. https://doi.org/10.1016/j.bbamem.2009.07.002
Soejima M, Noma A (1984) Mode of regulation of the ACh-sensitive K-channel by the muscarinic receptor in rabbit atrial cells. Pflugers Arch 400(4):424–431. https://doi.org/10.1007/BF00587544
Sohn JW, Lim A, Lee SH, Ho WK (2007) Decrease in PIP(2) channel interactions is the final common mechanism involved in PKC- and arachidonic acid-mediated inhibitions of GABA(B)-activated K+ current. J Physiol 582(Pt 3):1037–1046. https://doi.org/10.1113/jphysiol.2007.137265
Sonkusare SK, Dalsgaard T, Bonev AD, Nelson MT (2016) Inward rectifier potassium (Kir2.1) channels as end-stage boosters of endothelium-dependent vasodilators. J Physiol 594(12):3271–3285. https://doi.org/10.1113/JP271652
Standen NB, Stanfield PR (1978) Inward rectification in skeletal muscle: a blocking particle model. Pflugers Arch 378(2):173–176. https://doi.org/10.1007/BF00584452
Stanfield PR, Nakajima S, Nakajima Y (2002) Constitutively active and G-protein coupled inward rectifier K+ channels: Kir2.0 and Kir3.0. Rev Physiol Biochem Pharmacol 145:47–179. https://doi.org/10.1007/BFb0116431
Stansfeld PJ, Hopkinson R, Ashcroft FM, Sansom MS (2009) PIP(2)-binding site in Kir channels: definition by multiscale biomolecular simulations. Biochemistry 48(46):10926–10933. https://doi.org/10.1021/bi9013193
Stevens EB, Shah BS, Pinnock RD, Lee K (1999) Bombesin receptors inhibit G protein-coupled inwardly rectifying K+ channels expressed in Xenopus oocytes through a protein kinase C-dependent pathway. Mol Pharmacol 55(6):1020–1027
Su S, Ohno Y, Lossin C, Hibino H, Inanobe A, Kurachi Y (2007) Inhibition of astroglial inwardly rectifying Kir4.1 channels by a tricyclic antidepressant, nortriptyline. J Pharmacol Exp Ther 320(2):573–580. https://doi.org/10.1124/jpet.106.112094
Sui JL, Chan KW, Logothetis DE (1996) Na+ activation of the muscarinic K+ channel by a G-protein-independent mechanism. J Gen Physiol 108(5):381–391. https://doi.org/10.1085/jgp.108.5.381
Sui JL, Petit-Jacques J, Logothetis DE (1998) Activation of the atrial KACh channel by the betagamma subunits of G proteins or intracellular Na+ ions depends on the presence of phosphatidylinositol phosphates. Proc Natl Acad Sci U S A 95(3):1307–1312. https://doi.org/10.1073/pnas.95.3.1307
Sun HS, Feng ZP (2013) Neuroprotective role of ATP-sensitive potassium channels in cerebral ischemia. Acta Pharmacol Sin 34(1):24–32. https://doi.org/10.1038/aps.2012.138
Sun H, Liu X, Xiong Q, Shikano S, Li M (2006a) Chronic inhibition of cardiac Kir2.1 and HERG potassium channels by celastrol with dual effects on both ion conductivity and protein trafficking. J Biol Chem 281(9):5877–5884. https://doi.org/10.1074/jbc.M600072200
Sun HS, Feng ZP, Miki T, Seino S, French RJ (2006b) Enhanced neuronal damage after ischemic insults in mice lacking Kir6.2-containing ATP-sensitive K+ channels. J Neurophysiol 95(4):2590–2601. https://doi.org/10.1152/jn.00970.2005
Suzuki M, Li RA, Miki T et al (2001) Functional roles of cardiac and vascular ATP-sensitive potassium channels clarified by Kir6.2-knockout mice. Circ Res 88(6):570–577. https://doi.org/10.1161/01.res.88.6.570
Swale DR, Sheehan JH, Banerjee S et al (2015) Computational and functional analyses of a small-molecule binding site in ROMK. Biophys J 108(5):1094–1103. https://doi.org/10.1016/j.bpj.2015.01.022
Swale DR, Kurata H, Kharade SV et al (2016) ML418: the first selective, sub-micromolar pore blocker of Kir7.1 potassium channels. ACS Chem Nerosci 7(7):1013–1023. https://doi.org/10.1021/acschemneuro.6b00111
Swanberg MM, Tractenberg RE, Mohs R, Thal LJ, Cummings JL (2004) Executive dysfunction in Alzheimer disease. Arch Neurol 61(4):556–560. https://doi.org/10.1001/archneur.61.4.556
Tabuchi Y, Yashiro H, Hoshina S, Asano S, Takeguchi N (2001) Cibenzoline, an ATP-sensitive K(+) channel blocker, binds to the K(+)-binding site from the cytoplasmic side of gastric H(+),K(+)-ATPase. Br J Pharmacol 134(8):1655–1662. https://doi.org/10.1038/sj.bjp.0704422
Taglialatela M, Ficker E, Wible BA, Brown AM (1995) C-terminus determinants for Mg2+ and polyamine block of the inward rectifier K+ channel IRK1. EMBO J 14(22):5532–5541
Takahashi T (1990) Inward rectification in neonatal rat spinal motoneurones. J Physiol 423:47–62. https://doi.org/10.1113/jphysiol.1990.sp018010
Takumi T, Ishii T, Horio Y et al (1995) A novel ATP-dependent inward rectifier potassium channel expressed predominantly in glial cells. J Biol Chem 270(27):16339–16346. https://doi.org/10.1074/jbc.270.27.16339
Tamargo J, Caballero R, Gomez R, Valenzuela C, Delpon E (2004) Pharmacology of cardiac potassium channels. Cardiovasc Res 62(1):9–33. https://doi.org/10.1016/j.cardiores.2003.12.026
Tang P, Eckenhoff R (2018) Recent progress on the molecular pharmacology of propofol. F1000Res 7:123. https://doi.org/10.12688/f1000research.12502.1
Tao X, Avalos JL, Chen J, MacKinnon R (2009) Crystal structure of the eukaryotic strong inward-rectifier K+ channel Kir2.2 at 3.1 a resolution. Science 326(5960):1668–1674. https://doi.org/10.1126/science.1180310
Tawil R, Ptacek LJ, Pavlakis SG et al (1994) Andersen’s syndrome: potassium-sensitive periodic paralysis, ventricular ectopy, and dysmorphic features. Ann Neurol 35(3):326–330. https://doi.org/10.1002/ana.410350313
Tellez-Zenteno JF, Hernandez-Ronquillo L (2012) A review of the epidemiology of temporal lobe epilepsy. Epilepsy Res Treat 2012:630853. https://doi.org/10.1155/2012/630853
Teramoto N (2006) Pharmacological profile of U-37883A, a channel blocker of smooth muscle-type ATP-sensitive K channels. Cardiovasc Drug Rev 24(1):25–32. https://doi.org/10.1111/j.1527-3466.2006.00025.x
Tinker A, Aziz Q, Li Y, Specterman M (2018) ATP-sensitive potassium channels and their physiological and pathophysiological roles. Compr Physiol 8(4):1463–1511. https://doi.org/10.1002/cphy.c170048
Tipps ME, Buck KJ (2015) GIRK channels: a potential link between learning and addiction. Int Rev Neurobiol 123:239–277. https://doi.org/10.1016/bs.irn.2015.05.012
Trautwein W, Dudel J (1958) Mechanism of membrane effect of acetylcholine on myocardial fibers. Pflugers Arch Gesamte Physiol Menschen Tiere 266(3):324–334. https://doi.org/10.1007/BF00416781
Tsai TD, Shuck ME, Thompson DP, Bienkowski MJ, Lee KS (1995) Intracellular H+ inhibits a cloned rat kidney outer medulla K+ channel expressed in Xenopus oocytes. Am J Physiol 268(5 Pt 1):C1173–C1178. https://doi.org/10.1152/ajpcell.1995.268.5.C1173
Tucker SJ, Imbrici P, Salvatore L, D'Adamo MC, Pessia M (2000) pH dependence of the inwardly rectifying potassium channel, Kir5.1, and localization in renal tubular epithelia. J Biol Chem 275(22):16404–16407. https://doi.org/10.1074/jbc.C000127200
Ulens C, Daenens P, Tytgat J (1999) The dual modulation of GIRK1/GIRK2 channels by opioid receptor ligands. Eur J Pharmacol 385(2–3):239–245. https://doi.org/10.1016/s0014-2999(99)00736-0
Vasas A, Forgo P, Orvos P et al (2016) Myrsinane, premyrsinane, and cyclomyrsinane diterpenes from Euphorbia falcata as potassium ion channel inhibitors with selective G protein-activated inwardly rectifying ion channel (GIRK) blocking effects. J Nat Prod 79(8):1990–2004. https://doi.org/10.1021/acs.jnatprod.6b00260
Vaughn J, Wolford JK, Prochazka M, Permana PA (2000) Genomic structure and expression of human KCNJ9 (Kir3.3/GIRK3). Biochem Biophys Res Commun 274(2):302–309. https://doi.org/10.1006/bbrc.2000.3136
Vera E, Cornejo I, Burgos J, Niemeyer MI, Sepulveda FV, Cid LP (2019) A novel Kir7.1 splice variant expressed in various mouse tissues shares organisational and functional properties with human Leber amaurosis-causing mutations of this K(+) channel. Biochem Biophys Res Commun 514(3):574–579. https://doi.org/10.1016/j.bbrc.2019.04.169
Victoria NC, Marron Fernandez de Velasco E, Ostrovskaya O et al (2016) G protein-gated K(+) channel ablation in forebrain pyramidal neurons selectively impairs fear learning. Biol Psychiatry 80(10):796–806. https://doi.org/10.1016/j.biopsych.2015.10.004
Vo BN, Abney KK, Anderson A et al (2019) VU0810464, a non-urea G protein-gated inwardly rectifying K(+) (Kir 3/GIRK) channel activator, exhibits enhanced selectivity for neuronal Kir 3 channels and reduces stress-induced hyperthermia in mice. Br J Pharmacol 176(13):2238–2249. https://doi.org/10.1111/bph.14671
Voigt N, Abu-Taha I, Heijman J, Dobrev D (2014) Constitutive activity of the acetylcholine-activated potassium current IK,ACh in cardiomyocytes. Adv Pharmacol 70:393–409. https://doi.org/10.1016/B978-0-12-417197-8.00013-4
von Beckerath N, Dittrich M, Klieber HG, Daut J (1996) Inwardly rectifying K+ channels in freshly dissociated coronary endothelial cells from guinea-pig heart. J Physiol 491(Pt 2):357–365. https://doi.org/10.1113/jphysiol.1996.sp021221
Wang HR, Wu M, Yu H et al (2011) Selective inhibition of the K(ir)2 family of inward rectifier potassium channels by a small molecule probe: the discovery, SAR, and pharmacological characterization of ML133. ACS Chem Biol 6(8):845–856. https://doi.org/10.1021/cb200146a
Wang F, Olson EM, Shyng SL (2012) Role of Derlin-1 protein in proteostasis regulation of ATP-sensitive potassium channels. J Biol Chem 287(13):10482–10493. https://doi.org/10.1074/jbc.M111.312223
Weaver CD, Harden D, Dworetzky SI, Robertson B, Knox RJ (2004) A thallium-sensitive, fluorescence-based assay for detecting and characterizing potassium channel modulators in mammalian cells. J Biomol Screen 9(8):671–677. https://doi.org/10.1177/1087057104268749
Wellman GC, Bevan JA (1995) Barium inhibits the endothelium-dependent component of flow but not acetylcholine-induced relaxation in isolated rabbit cerebral arteries. J Pharmacol Exp Ther 274(1):47–53
Wen W, Wu W, Romaine IM et al (2013) Discovery of ‘molecular switches’ within a GIRK activator scaffold that afford selective GIRK inhibitors. Bioorg Med Chem Lett 23(16):4562–4566. https://doi.org/10.1016/j.bmcl.2013.06.023
Whorton MR, MacKinnon R (2011) Crystal structure of the mammalian GIRK2 K+ channel and gating regulation by G proteins, PIP2, and sodium. Cell 147(1):199–208. https://doi.org/10.1016/j.cell.2011.07.046
Whorton MR, MacKinnon R (2013) X-ray structure of the mammalian GIRK2-betagamma G-protein complex. Nature 498(7453):190–197. https://doi.org/10.1038/nature12241
Wible BA, Taglialatela M, Ficker E, Brown AM (1994) Gating of inwardly rectifying K+ channels localized to a single negatively charged residue. Nature 371(6494):246–249. https://doi.org/10.1038/371246a0
Wickman K, Nemec J, Gendler SJ, Clapham DE (1998) Abnormal heart rate regulation in GIRK4 knockout mice. Neuron 20(1):103–114. https://doi.org/10.1016/s0896-6273(00)80438-9
Wickman K, Karschin C, Karschin A, Picciotto MR, Clapham DE (2000) Brain localization and behavioral impact of the G-protein-gated K+ channel subunit GIRK4. J Neurosci 20(15):5608–5615
Wieting JM, Vadukoot AK, Sharma S et al (2017) Discovery and characterization of 1H-pyrazol-5-yl-2-phenylacetamides as novel, non-urea-containing GIRK1/2 potassium channel activators. ACS Chem Nerosci 8(9):1873–1879. https://doi.org/10.1021/acschemneuro.7b00217
Williams JT, Colmers WF, Pan ZZ (1988) Voltage- and ligand-activated inwardly rectifying currents in dorsal raphe neurons in vitro. J Neurosci 8(9):3499–3506
Witkowski G, Szulczyk B, Rola R, Szulczyk P (2008) D(1) dopaminergic control of G protein-dependent inward rectifier K(+) (GIRK)-like channel current in pyramidal neurons of the medial prefrontal cortex. Neuroscience 155(1):53–63. https://doi.org/10.1016/j.neuroscience.2008.05.021
Wu JX, Ding D, Wang M, Kang Y, Zeng X, Chen L (2018) Ligand binding and conformational changes of SUR1 subunit in pancreatic ATP-sensitive potassium channels. Protein Cell 9(6):553–567. https://doi.org/10.1007/s13238-018-0530-y
Wu P, Gao ZX, Zhang DD, Su XT, Wang WH, Lin DH (2019) Deletion of Kir5.1 impairs renal ability to excrete potassium during increased dietary potassium intake. J Am Soc Nephrol 30(8):1425–1438. https://doi.org/10.1681/ASN.2019010025
Xia M, Jin Q, Bendahhou S, He Y, Larroque MM, Chen Y, Zhou Q, Yang Y, Liu Y, Liu B, Zhu Q, Zhou Y, Lin J, Liang B, Li L, Dong X, Pan Z, Wang R, Wan H, Qiu W, Xu W, Eurlings P, Barhanin J, Chen Y (2005) A Kir2.1 gain-of-function mutation underlies familial atrial fibrillation. Biochem Biophys Res Commun 332(4):1012–9
Xie LH, John SA, Weiss JN (2002) Spermine block of the strong inward rectifier potassium channel Kir2.1: dual roles of surface charge screening and pore block. J Gen Physiol 120(1):53–66. https://doi.org/10.1085/jgp.20028576
Xie LH, John SA, Weiss JN (2003) Inward rectification by polyamines in mouse Kir2.1 channels: synergy between blocking components. J Physiol 550(Pt 1):67–82. https://doi.org/10.1113/jphysiol.2003.043117
Xu ZC, Yang Y, Hebert SC (1996) Phosphorylation of the ATP-sensitive, inwardly rectifying K+ channel, ROMK, by cyclic AMP-dependent protein kinase. J Biol Chem 271(16):9313–9319. https://doi.org/10.1074/jbc.271.16.9313
Xu Y, Shin HG, Szep S, Lu Z (2009) Physical determinants of strong voltage sensitivity of K(+) channel block. Nat Struct Mol Biol 16(12):1252–1258. https://doi.org/10.1038/nsmb.1717
Xu Y, Cantwell L, Molosh AI et al (2020) The small molecule GAT1508 activates brain-specific GIRK1/2 channel heteromers and facilitates conditioned fear extinction in rodents. J Biol Chem 295(11):3614–3634. https://doi.org/10.1074/jbc.RA119.011527
Yamada M, Inanobe A, Kurachi Y (1998) G protein regulation of potassium ion channels. Pharmacol Rev 50(4):723–760
Yamakura T, Lewohl JM, Harris RA (2001) Differential effects of general anesthetics on G protein-coupled inwardly rectifying and other potassium channels. Anesthesiology 95(1):144–153. https://doi.org/10.1097/00000542-200107000-00025
Yamamoto W, Hashimoto N, Matsuura J et al (2014) Effects of the selective KACh channel blocker NTC-801 on atrial fibrillation in a canine model of atrial tachypacing: comparison with class Ic and III drugs. J Cardiovasc Pharmacol 63(5):421–427. https://doi.org/10.1097/FJC.0000000000000065
Yan F, Lin CW, Weisiger E, Cartier EA, Taschenberger G, Shyng SL (2004) Sulfonylureas correct trafficking defects of ATP-sensitive potassium channels caused by mutations in the sulfonylurea receptor. J Biol Chem 279(12):11096–11105. https://doi.org/10.1074/jbc.M312810200
Yan FF, Pratt EB, Chen PC et al (2010) Role of Hsp90 in biogenesis of the beta-cell ATP-sensitive potassium channel complex. Mol Biol Cell 21(12):1945–1954. https://doi.org/10.1091/mbc.E10-02-0116
Yang J, Jan YN, Jan LY (1995) Control of rectification and permeation by residues in two distinct domains in an inward rectifier K+ channel. Neuron 14(5):1047–1054. https://doi.org/10.1016/0896-6273(95)90343-7
Yang B, Lin H, Xiao J et al (2007) The muscle-specific microRNA miR-1 regulates cardiac arrhythmogenic potential by targeting GJA1 and KCNJ2. Nat Med 13(4):486–491. https://doi.org/10.1038/nm1569
Yang Y, Yang Y, Liang B et al (2010) Identification of a Kir3.4 mutation in congenital long QT syndrome. Am J Hum Genet 86(6):872–880. https://doi.org/10.1016/j.ajhg.2010.04.017
Yang HQ, Martinez-Ortiz W, Hwang J, Fan X, Cardozo TJ, Coetzee WA (2020) Palmitoylation of the KATP channel Kir6.2 subunit promotes channel opening by regulating PIP2 sensitivity. Proc Natl Acad Sci U S A 117(19):10593–10602. https://doi.org/10.1073/pnas.1918088117
Yoo D, Fang L, Mason A, Kim BY, Welling PA (2005) A phosphorylation-dependent export structure in ROMK (Kir 1.1) channel overrides an endoplasmic reticulum localization signal. J Biol Chem 280(42):35281–35289. https://doi.org/10.1074/jbc.M504836200
Yoshimoto Y, Fukuyama Y, Horio Y, Inanobe A, Gotoh M, Kurachi Y (1999) Somatostatin induces hyperpolarization in pancreatic islet alpha cells by activating a G protein-gated K+ channel. FEBS Lett 444(2–3):265–269. https://doi.org/10.1016/s0014-5793(99)00076-9
Yow TT, Pera E, Absalom N et al (2011) Naringin directly activates inwardly rectifying potassium channels at an overlapping binding site to tertiapin-Q. Br J Pharmacol 163(5):1017–1033. https://doi.org/10.1111/j.1476-5381.2011.01315.x
Yu L, Jin X, Cui N et al (2012) Rosiglitazone selectively inhibits K(ATP) channels by acting on the K(IR) 6 subunit. Br J Pharmacol 167(1):26–36. https://doi.org/10.1111/j.1476-5381.2012.01934.x
Zaks-Makhina E, Kim Y, Aizenman E, Levitan ES (2004) Novel neuroprotective K+ channel inhibitor identified by high-throughput screening in yeast. Mol Pharmacol 65(1):214–219. https://doi.org/10.1124/mol.65.1.214
Zaks-Makhina E, Li H, Grishin A, Salvador-Recatala V, Levitan ES (2009) Specific and slow inhibition of the kir2.1 K+ channel by gambogic acid. J Biol Chem 284(23):15432–15438. https://doi.org/10.1074/jbc.M901586200
Zald DH (2003) The human amygdala and the emotional evaluation of sensory stimuli. Brain Res Brain Res Rev 41(1):88–123. https://doi.org/10.1016/s0165-0173(02)00248-5
Zangerl-Plessl EM, Qile M, Bloothooft M, Stary-Weinzinger A, van der Heyden MAG (2019) Disease associated mutations in KIR proteins linked to aberrant inward rectifier channel trafficking. Biomol Ther 9(11). https://doi.org/10.3390/biom9110650
Zangerl-Plessl EM, Lee SJ, Maksaev G et al (2020) Atomistic basis of opening and conduction in mammalian inward rectifier potassium (Kir2.2) channels. J Gen Physiol 152(1). https://doi.org/10.1085/jgp.201912422
Zaritsky JJ, Eckman DM, Wellman GC, Nelson MT, Schwarz TL (2000) Targeted disruption of Kir2.1 and Kir2.2 genes reveals the essential role of the inwardly rectifying K(+) current in K(+)-mediated vasodilation. Circ Res 87(2):160–166. https://doi.org/10.1161/01.res.87.2.160
Zaritsky JJ, Redell JB, Tempel BL, Schwarz TL (2001) The consequences of disrupting cardiac inwardly rectifying K(+) current (I(K1)) as revealed by the targeted deletion of the murine Kir2.1 and Kir2.2 genes. J Physiol 533(Pt 3):697–710. https://doi.org/10.1111/j.1469-7793.2001.t01-1-00697.x
Zerangue N, Schwappach B, Jan YN, Jan LY (1999) A new ER trafficking signal regulates the subunit stoichiometry of plasma membrane K(ATP) channels. Neuron 22(3):537–548. https://doi.org/10.1016/s0896-6273(00)80708-4
Zhang H, He C, Yan X, Mirshahi T, Logothetis DE (1999) Activation of inwardly rectifying K+ channels by distinct PtdIns(4,5)P2 interactions. Nat Cell Biol 1(3):183–188. https://doi.org/10.1038/11103
Zhang W, Zhang X, Wang H, Sharma AK, Edwards AO, Hughes BA (2013) Characterization of the R162W Kir7.1 mutation associated with snowflake vitreoretinopathy. Am J Physiol Cell Physiol 304(5):C440–C449. https://doi.org/10.1152/ajpcell.00363.2012
Zhang J, Xia Y, Xu Z, Deng X (2016) Propofol suppressed hypoxia/reoxygenation-induced apoptosis in HBVSMC by regulation of the expression of Bcl-2, Bax, Caspase3, Kir6.1, and p-JNK. Oxid Med Cell Longev 2016:1518738. https://doi.org/10.1155/2016/1518738
Zhao Y, Ung PM, Zahoranszky-Kohalmi G et al (2020) Identification of a G-protein-independent activator of GIRK channels. Cell Rep 31(11):107770. https://doi.org/10.1016/j.celrep.2020.107770
Zhou H, Tate SS, Palmer LG (1994) Primary structure and functional properties of an epithelial K channel. Am J Physiol 266(3 Pt 1):C809–C824. https://doi.org/10.1152/ajpcell.1994.266.3.C809
Zhou W, Arrabit C, Choe S, Slesinger PA (2001) Mechanism underlying bupivacaine inhibition of G protein-gated inwardly rectifying K+ channels. Proc Natl Acad Sci U S A 98(11):6482–6487. https://doi.org/10.1073/pnas.111447798
Zhou Q, Chen PC, Devaraneni PK, Martin GM, Olson EM, Shyng SL (2014) Carbamazepine inhibits ATP-sensitive potassium channel activity by disrupting channel response to MgADP. Channels (Austin) 8(4):376–382. https://doi.org/10.4161/chan.29117
Zingman LV, Zhu Z, Sierra A et al (2011) Exercise-induced expression of cardiac ATP-sensitive potassium channels promotes action potential shortening and energy conservation. J Mol Cell Cardiol 51(1):72–81. https://doi.org/10.1016/j.yjmcc.2011.03.010
Acknowledgements
We dedicate this review to Dr. Yoshihisa Kurachi who by devoting his career to Kir channel structure, function, and pharmacology has inspired many of us to follow his example. We are grateful to Professor Leigh Plant for reading this review and providing us with critical feedback. We thank the members of the Logothetis and Plant groups for pursuing and freely sharing their mechanistic Kir channel results. Last but not least, we thank the National Institutes of Health (NIH) in the United States of America (USA) for funding our research and thus allowing us to continue working in this exciting field (supported by NIH R01-HL059949-23 to D.E.L.)
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Appendix
Appendix


Inwardly rectifying potassium channel family alignment. The current human 16 members of the Kir family were aligned using the Clustal Omega multiple sequence alignment program, which uses seeded guide trees and HMM profile–profile techniques to generate the alignments for all Kir members. Fully conserved resides are denoted with an “*”, residues with strongly similar properties are denoted with a “:”, and residues with weakly similar properties are donated with a “.”. The important structural and gating features are highlighted and labeled (Madiera et al. 2019)

Inwardly rectifying potassium channel family similarity and identity. The 16 members of the Kir family were analyzed for percent similarity and percent identity using the Sequence Identity And Similarity (SIAS) tool which uses a pairwise analyses utilizing several methods. Amino acid similarity and sequence length is used to calculate the values (% = 100*(identical| similar residues/sequence length)). Parameters were set to the standard settings relative to amino acid similarities. Sequences were input from the Clustal Omega alignment. The values are listed as percent similarity/% identity


Chemical structures of drugs targeting Kir channels
Rights and permissions
Copyright information
© 2021 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Cui, M., Cantwell, L., Zorn, A., Logothetis, D.E. (2021). Kir Channel Molecular Physiology, Pharmacology, and Therapeutic Implications. In: Gamper, N., Wang, K. (eds) Pharmacology of Potassium Channels. Handbook of Experimental Pharmacology, vol 267. Springer, Cham. https://doi.org/10.1007/164_2021_501
Download citation
DOI: https://doi.org/10.1007/164_2021_501
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-84051-8
Online ISBN: 978-3-030-84052-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)