
Abstract
Nalfurafine has been used clinically in Japan for treatment of itch in kidney dialysis patients and in patients with chronic liver diseases. A one-year post-marketing study showed nalfurafine to be safe and efficacious without producing side effects of typical KOR agonists such as anhedonia and psychotomimesis. In this chapter, we summarize in vitro characterization and in vivo preclinical studies on nalfurafine. In vitro, nalfurafine is a highly potent and moderately selective KOR full agonist; however, whether it is a biased KOR agonist is a matter of debate. In animals, nalfurafine produced anti-pruritic effects in a dose range lower than that caused side effects, including conditioned place aversion (CPA), hypolocomotion, motor incoordination, consistent with the human data. In addition, nalfurafine showed antinociceptive effects in several pain models at doses that did not cause the side effects mentioned above. It appears to be effective against inflammatory pain and mechanical pain, but less so against thermal pain, particularly high-intensity thermal pain. U50,488H and nalfurafine differentially modulated several signaling pathways in a brain region-specific manners. Notably, U50,488H, but not nalfurafine, activated the mTOR pathway, which contributed to U50,488H-induced CPA. Because of its lack of side effects associated with typical KOR agonists, nalfurafine has been investigated as a combination therapy with an MOR ligand for pain treatment and for its effects on opioid use disorder and alcohol use disorder, and results indicate potential usefulness for these indications. Thus, although in vitro data regarding uniqueness of nalfurafine in terms of signaling at the KOR are somewhat equivocal, in vivo results support the assertion that nalfurafine is an atypical KOR agonist with a significantly improved side-effect profile relative to typical KOR agonists.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
Activation of kappa opioid receptor (KOR) produce analgesic and antipruritic effects; however, clinical development of these compounds has been limited by side effects such as psychotomimesis, dysphoria, and sedation (Pande et al. 1996; Pfeiffer et al. 1986; Wadenberg 2003; Walsh et al. 2001). One exception is nalfurafine (TRK-820 in early literature). Nalfurafine (Remitch™), 17-cyclopropylmethyl-3, 14 beta-dihydroxy-4,5 alpha-epoxy-6 beta-[N-methyl-trans-3-(3-furyl) acrylamido]-morphinan, has been used in Japan for treatment of itch in patients undergoing hemodialysis and in those with chronic liver diseases (Kozono et al. 2018; Kumagai et al. 2010, 2012; Nakao and Mochizuki 2009a). Clinical reports have shown that, at therapeutic doses, nalfurafine does not produce dysphoria or psychotomimetic effects, which sets it apart from prototyipcal KOR agonists that have been tested in humans (Kozono et al. 2018; Kumagai et al. 2010, 2012). It should be noted, however, that nalfurafine did not receive approval by the European Medicines Agency because of lack of efficacy.
Nalfurafine was first synthesized by Nagase et al. (1998) and found to be a highly potent and moderately selective KOR full agonist. A unique structural feature of TRK-820 is the 4,5-epoxymorphinan structure with a tyrosine-glycine moiety of endogenous opioid peptides, which is different from other nonpeptide KOR agonists such as U50,488H and salvinorin A.
In this chapter, we summarize preclinical studies on nalfurafine, including in vitro characterization and in vivo pharmacology in laboratory animals, as well as its potential use as an adjunct for pain treatment, and as a deterrent/treatment for drug and alcohol abuse. We also review studies that examine what makes nalfurafine different from other KOR agonists, which may provide some insights into development of KOR agonists with fewer side effects.
In the earlier literature, it was named TRK-820, whereas in the more recent literature, it was called nalfurafine. In this chapter, both terms will be used to be consistent with the original literature.
2 In Vitro Pharmacological Characterization of Nalfurafine
2.1 Bioassays
Nagase et al. (1998) reported that TRK-820 inhibited electrically stimulated contraction of guinea pig ileum (GPI) with an IC50 value of 4.8 pM and KOR/MOR selectivity of 279. TRK-820 attenuated mouse vas deferens (MVD) contraction with an IC50 value of 36 pM and KOR/MOR selectivity of 104 and KOR/DOR selectivity of 135. TRK-820 was 4,000-fold more potent than morphine in both preparations and 200 and 70 times more potent than U50,488H in the GPI and MVD, respectively.
2.2 Binding Affinity and Selectivity Profile of Nalfurafine at MOR, DOR, KOR and NOR
Several groups determined affinities of nalfurafine for each receptor by competitive inhibition of radioligand binding (Table 1), yielding different results. Its Ki values for the KOR ranged from 0.075 to 3.5 nM, those for the MOR 0.43–53 nM and those for the DOR 51–1,200 nM. Its binding to the NOR was negligible. The differences in its Ki value for a given receptor are likely due to differences in the radioligand, tissues and binding conditions used (Table 1). Its KOR/MOR selectivity ratios ranged from 2.4 to 69 and those of KOR/DOR ≥ 280. Overall, these data indicate that nalfurafine is moderately selective for the KOR over the MOR and highly selective for the KOR over the DOR.
2.3 No Significant Binding to Targets Other Than Opioid Receptors
Nalfurafine at 10 μM did not bind significantly to 45 pharmacological targets (Nakao and Mochizuki 2009b), including histamine, neurokinin, bombesin, CGRP, somatostatin, ionotropic glutamate, dopamine, adrenergic, muscarinic, adenosine, IL-1β, IL-8, CCR1, CCR2, CCK, GABAA, and VIP receptors and L-type Ca++ channel. The only target for which it exhibited a moderate affinity was the M1 muscarinic receptor with a Ki value of 1.7 μM. Nalfurafine did not affect release of several inflammatory mediators (histamine, tumor necrosis factor, interleukin-1β and -6, and prostaglandins D2 and E2), nor did it influence activities of constitutive and inducible nitric oxide synthetase (Nakao and Mochizuki 2009b).
2.4 Efficacies and Potencies of Nalfurafine in Activating MOR, DOR, KOR and NOR: Inhibition of Adenylate Cyclase and Enhancement of [35S]GTPγS Binding
Seki et al. (1999) evaluated agonistic activity of TRK-820 by inhibition of forskolin (10 μM)-stimulated cAMP accumulation in CHO cells stably transfected with cloned MOR, KOR or DOR. The IC50 and Imax values for MOR, KOR and DOR are shown in Table 2. The data indicate that compared with the reference full agonists, nalfurafine is a potent full agonist at the KOR and high-efficacy partial agonist at the MOR and has a KOR/MOR selectivity factor of 55.
Nalfurafine was also examined for its potency and efficacy in enhancing [35S]GTPγS binding via the MOR, DOR, KOR and NOR. Data are shown in Table 3. Nalfurafine is a high-potency KOR full agonist with an EC50 value below 0.1 nM, and has partial agonist activities at the MOR, DOR and NOR with much lower potency. Its KOR/MOR selectivity is 32 or 128; KOR/DOR or KOR/NOR selectivity factors are much higher. Therefore, nalfurafine is considered as a moderately selective KOR full agonist.
Potency and efficacy of nalfurafine were also examined using several other functional endpoints for G protein- and β-arrestin-mediated signaling, which are discussed in Sect. 2.5.
2.5 Functional Selectivity of Nalfurafine in KOR-Mediated Activation of G Proteins and β-Arrestins
Studies on functional selectivity of nalfurafine for G proteins or β-arrestins have yielded different results, demonstrating nalfurafine to be G protein-biased (Cao et al. 2020; Kaski et al. 2019; Schattauer et al. 2017), un-biased (Liu et al. 2019) and β-arrestin-biased (Dunn et al. 2019). The cell lines, functional endpoints and reference compounds used, and the calculated agonist biases are summarized in Table 4. The discrepancy may be attributed to differences in cell line, end point, assay method for the same end point, receptor expression level and the reference balanced agonist used. Gillis et al. (2020) recently reported that responses having amplification factors and higher receptor reserves tend to enhance potency and efficacy of an agonist, thus confounding agonist bias calculation. The cAMP response has a high signal amplification, whereas [35S]GTPγS binding and β-arrestin recruitment do not, which may contribute to the findings of nalfurafine as G protein-biased agonist in the study of Cao et al. (2020); Kaski et al. (2019), but as a unbiased agonist in Liu et al. (2019).
2.6 KOR Internalization and Down-Regulation
Wang et al. (2005) reported that TRK-820 (1 nM, 30 min, 37°C) caused internalization of FLAG-hKOR stably transfected into HEK 293 cells, similar to U50,488H (1 μM). Following incubation of CHO-FLAG-hKOR cells for 4 h, TRK-820 (1 nM, 37°C) induced down-regulation of the human KOR to the same extent as U50,488H (1 μM).
3 In Vivo Pharmacological Effects of Nalfurafine
Nalfurafine has been demonstrated to have anti-pruritic effects against several pruritogens and in disease models of itch, antinociceptive effects in various pain models, and water diuretic effects. In addition, nalfurafine has been examined for many KOR-mediated side effects, such as hypolocomotion, CPA, and motor incoordination. In general, nalfurafine produces antinociceptive and anti-pruritic effects in dose ranges that are below those causing hypolocomotion, CPA and motor incoordination. This is a unique feature that separates nalfurafine from typical KOR agonists, such as U50,488H. U50,488H produces the side effects in the dose ranges that induce anti-nociceptive and anti-pruritic effects.
3.1 Antipruritic Effects
In a pioneering study, Cowan and Gmerek (1986) showed that the KOR agonists tifluadom and U50,488 attenuated bombesin-induced scratching and grooming in rats. Subsequent publications confirmed the antipruritic activities of KOR agonists, which ultimately led to the approval of nalfurafine in Japan as an antipruritic drug for treatment of itch in patients undergoing hemodialysis or with chronic liver diseases.
Togashi et al. (2002) were the first to report anti-pruritic activity of nalfurafine in rodents. They demonstrated that in male ICR mice, TRK-820 (p.o., −30 min) reduced scratching induced by substance P or histamine, with A50 values of 19.6 and 7.3 μg/kg, respectively, which was antagonized by pretreatment with norBNI (s.c., overnight), indicating a KOR-mediated effect. No obvious suppression of the spontaneous locomotor activity was observed at the doses used. TRK-820 suppressed running activity at much higher doses with an A50 of 102.8 μg/kg.
Subsequently, nalfurafine (s.c) was shown to have inhibitory effects on scratch induced by several pruritogens in a dose-dependent manner in mice, including the malaria drug chloroquine (Inan and Cowan 2004; Snyder et al. 2018), the histamine releasing agent compound 48/80 (Liu et al. 2019; Wang et al. 2005), the KOR antagonist 5’-GNTI (Inan et al. 2009a), the protease-activating receptor 2 agonist SLIGRL, histamine and serotonin (Kardon et al. 2014). The A50 values were in the range of 6.6–10 μg/kg and nalfurafine did not affect motor activity up to 30 μg/kg or produce conditioned place aversion (CPA) up to 20 μg/kg in these studies. Snyder et al. (2018) showed that the inhibitory effect of nalfurafine against chloroquine-induced scratching was absent in KOR−/− mice. In addition, Inan et al. (2009a) reported that nalfurafine was also effective as a post treatment in attenuating scratching induced by compound 48/80 and 5’-GNTI. Remarkably, mice treated once/day with 20 μg/kg nalfurafine for 10 days did not develop tolerance to the inhibitory effect of nalfurafine against scratch induced by 5’-GNTI (Inan et al. 2009a). Compound 48/80 and 5’-GNTI enhanced c-fos expression in the dorsal horn of mouse spinal cord, which was inhibited by nalfurafine (Inan et al. 2009a). Umeuchi et al. (2003) found that the anti-scratching activity of TRK-820 (s.c) was antagonized by intracerebroventricular administration of norBNI, indicating an important role of brain KOR in TRK-820 effects. Lastly, nalfurafine (0.01–0.1 mg/kg; i.p.) reduced scratching induced by intradermal injections of serotonin in male Sprague-Dawley rats (Lazenka et al. 2018). However, nalfurafine failed to block itch-induced suppression of responding for intracranial self-stimulation (ICSS) in this same report.
Nalfurafine has also been studied in disease models of itch in rodents. Umeuchi et al. (2005) demonstrated that aged MRL/lpr mice may be used as a model of pruritus related to human autoimmune diseases and found that nalfurafine (p.o.) inhibited the scratching behavior in these mice without causing gross behavioral changes. Inan and Cowan (2006) generated a rat model of pruritus associated with cholestasis by repeated injection with ethynylestradiol and showed that nalfurafine (s.c.) suppressed whole-body scratching with an A50 value of 13 μg/kg. Nalfurafine (p.o.) was effective in reducing scratching behaviors in NC/Nga mice, a model of atopic dermatitis (Nakao et al. 2008). Nalfurafine inhibited scratching related to experimentally induced dry skin in mice (Akiyama et al. 2015; Kardon et al. 2014). Topical application of nalfurafine inhibited scratching in an oxazolone-induced murine model of atopic dermatitis (Elliott et al. 2016).
3.2 Anti-nociceptive Effects
KOR agonists have long been established to have antinociceptive effects in laboratory animals (von Voigtlander et al. 1983). Nalfurafine has been found to have antinociceptive effects in rodents and nonhuman primates. Nalfurafine is more active against inflammatory pain and mechanical pain.
Mice
Endoh et al. (1999) conducted comprehensive studies in mice. They reported that in male ddY mice TRK-820 given s.c. or p.o. was 351-fold and 796-fold more potent than U50,488H in the acetic acid-induced abdominal constriction test with A50 values of 3.3 μg/kg (s.c.) and 32 μg/kg (p.o.), respectively. The effect reached a peak at 30 min and returned to baseline in 120–180 min. Its duration of the antinociceptive effect was longer than that of morphine or other KOR agonists (U50,488H, CI-977, ICI199,441 and PD117302). TRK-820 was also more readily absorbed following oral adminstration than these KOR agonists. The antinociceptive effects produced by TRK-820 in the abdominal constriction test were was antagonized by norBNI, but not by the selective DOR anatagonist naltrindole or a low dose of naloxone that was selective for the MOR, indicating KOR-mediated effect. In addition, in four other antinociceptive assays, 51°C hot plate, thermal tail flick, mechanical tail pressure and tail pinch tests, TRK-820 (s.c.) had A50 values of 129, 62, 9 and 35 μg/kg, respectively, and was 68-fold to 328-fold more potent than U50,488H, and 41-fold to 349-fold more potent than morphine in producing antinociception. However, TRK-820 was inactive in inhibiting the high temperature (55°C) hot plate response. These results are consistent with those of Nagase et al. (1998) in a communication report.
Liu et al. (2019) demonstrated that in CD-1 mice nalfurafine (s.c.) produced dose-dependent antinociception in the late phase of the formalin test with an A50 value of 8.3 μg/kg, which is 70x more potent than U50,488H.
Snyder et al. (2018) demonstrated following an intraplantar (IPL) injection of capsaicin or acetic acid into a hind paw, mice treated with nalfurafine or the peripherally restricted agonists ICI204,448 or FE200665 showed a significant decrease in the time spent licking the injected hind paw, compared to control mice, indicating antinociceptive effects. In mice, none of the KOR agonists had any effects on paw withdrawal threshold to the application of von Frey filaments (mechanical threshold). In contrast, nalfurafine, but not ICI204,448 or FE200665, significantly increased paw withdrawal latency (PWL) as measured by the Hargreaves assay (thermal threshold). Following acute incision of the hind paw, all the three KOR agonists inhibited mechanical hypersensitivity in the Brennan model, however only nalfurafine, but not the peripherally restricted agonists, inhibited thermal hypersensitivity. Thus, nalfurafine acts both centrally and peripherally to produce antinociceptive effects against thermal and mechanical pain via the KOR.
Kaski et al. (2019) observed that in the tail withdrawal assay (a model for spinal nociception) in C57BL/6 mice, nalfurafine induced significant dose-dependent anti-nociception at all doses tested (15, 30 and 60 μg/kg). U50,488 also elicited spinal anti-nociception at 1.25 mg/kg and at 5.00 mg/kg (but not at 2.50 mg/kg). The highest tested dose of nalfurafine (60 μg/kg) was significantly more anti-nociceptive than was the highest dose of U50,488 (5.00 mg/kg).
By single-unit electrophysiological recordings of colonic nociceptive afferents, Snyder et al. (2018) found that inflammatory mediators (histamine, bradykinin, prostaglandin E2, and serotonin) increased the number of action potentials in response to stretch and nalfurafine significantly reduced this sensitization, which was abolished in KOR−/− mice.
Rats
Endoh et al. (2000) reported that TRK-820 produced a potent dose-dependent and KOR-mediated antinociceptive effect in the paw pressure test in male Wistar rats with A50 values of 64 μg/kg (s.c.) and 75 μg/kg (i.m.). Nalfurafine displayed similar potency in normal rats and adjuvant-induced arthritic rats in the paw pressure test. In the second phase of the formalin test, TRK-820 had a potent antinociceptive effect with an A50 value of 9.6 μg/kg (s.c.), similar to mice.
Townsend et al. (2017) reported that nalfurafine (i.v.) produced dose-dependent thermal antinociception using the hot plate test in male Sprague-Dawley rats. More recently, nalfurafine was reported to reverse writhing behavior in rats induced by i.p. administration of lactic acid (Lazenka et al. 2018). However, in this same report, nalfurafine failed to reverse acid-induced suppression of ICSS. Notably, nalfurafine also reduced ICSS in rats in the absence of acid injections, which precludes the ability to make firm conclusions regarding antinociception in this procedure.
Non-human Primates
Endoh et al. (2001) demonstrated that in cynomolgus monkeys, TRK-820 (i.m.) produced a potent antinociceptive effect that was 295-fold and 495-fold more potent than morphine in the 50°C and 55°C hot-water tail-withdrawal tests, respectively, and 40-fold more potent than U50,488H and 1,000-fold more potent than pentazocine in the 50°C hot-water test. The duration of antinociceptive effects of TRK-820 (10 and 30 μg/kg, i.m.) was >6 h, much longer than that of U50,488H. The antinociception induced by a lower dose of TRK-820 (10 μg/kg, i.m.) was inhibited by norBNI (10 mg/kg, s.c.), indicating KOR-mediated actions, whereas that produced by a higher dose of TRK-820 (30 μg/kg, i.m.) was not antagonized by norBNI (3.2 and 10 mg/kg, s.c.) or by naloxone (0.1 mg/kg, s.c.), which effectively inhibited the antinociception induced by U50,488H (1.0 mg/kg, i.m.) and morphine (10 mg/kg, i.m.), respectively, suggesting that there may be other targets at 30 μg/kg of nalfurafine.
3.3 Anti-allodynic and Anti-hyperalgesic Effects
Takasaki et al. (2004) used a mouse model of acute herpetic pain in which percutaneous inoculation with herpes simplex virus type-1 induced tactile allodynia and mechanical hyperalgesia in the hind paw on the inoculated side. In female BALB/c mice, TRK-820 (10–100 μg/kg, p.o.) inhibited the allodynia and hyperalgesia in a dose-dependent manner, similar to morphine (5–20 mg/kg) and the KOR agonist enadoline (1–10 mg/kg). Interestingly, in the effective dose ranges, enadoline greatly reduced spontaneous locomotor activity, but TRK-820 did not. The effects of TRK-820 were blocked by pretreatment with norBNI, but not by a low dose of naltrexone, which antagonized the effects of morphine. Repeated administration of TRK-820 did not attenuate its anti-allodynic and anti-hyperalgesic effects, indicating no tolerance. Intrathecal and intracerebroventricular, but not intraplantar, injections of TRK-820 also inhibited the allodynia and hyperalgesia. Snyder et al. (2018) reported that following surgical incision injury of the hind paw, hypersensitivity to mechanical and thermal stimuli developed. Nalfurafine and peripherally acting KOR agonists ICI204,488 and FE200665 inhibited mechanical hypersensitivity. In contrast, only nalfurafine inhibited thermal hypersensitivity. Thus, the anti-allodynic and anti-hyperalgesic effects of TRK-820 are mediated by the KOR at the spinal and supraspinal levels, an at peripheral sites.
3.4 Inhibition of Neurogenic Inflammation
Snyder et al. (2018) reported that in C57BL/6 mice an IPL injection of capsaicin caused plasma extravasation and an increase in paw temperature, a model of neurogenic inflammation. Nalfurafine or the peripherally restricted KOR agonist ICI204,488 or FE200665 significantly reduced these two reactions. Nalfurafine-induced inhibition of neurogenic inflammation was eliminated by KOR deletion. Similar results were obtained when neurogenic inflammation was induced with a mixture of bradykinin and prostaglandin E2. Thus, activation of KOR in the periphery is sufficient to inhibit neurogenic inflammation.
3.5 Aversive Effects or Lack Thereof
KOR agonists were first shown to produce conditioned place aversion (CPA) in mice and rats in the 1980s (Mucha and Herz 1985; Shippenberg 1986). In both mice and rats, whether nalfurafine causes CPA depends on the dose used. The results in the literature support the notion that at the doses effective in the antinociceptive and anti-scratching tests, nalfurafine did not produce CPA; however, nalfurafine did cause CPA at higher doses. For example, Liu et al. (2019) reported that in CD-1 mice, the A50 values of nalfurafine-induced antinociception in the formalin test and in the anti-scratch test were 5.8 and 8.3 μg/kg, respectively, and that nalfurafine (5–20 μg/kg, s.c.) did not cause CPA or CPP.
Mice
Tsuji et al. (2001) reported that TRK-820 (3–30 μg/kg, s.c.) did not produce a significant CPP or CPA in mice. Liu et al. (2019) demonstrated that in CD-1 mice, nalfurafine (5–20 μg/kg, s.c.) did not cause CPA or CPP. Zhou and Kreek (2019b) also found that nalfurafine at 10 μg/kg (i.p.) did not produce CPA in C57BL/6 J mice. In addition, Liu et al. (2019) found that nalfurafine (10 μg/kg, s.c.) did not induce CPA in MOR−/− mice, ruling out the possibility that nalfurafine did not produce CPA because it acted on both the MOR and KOR. Kaski et al. (2019) showed that in C57BL/6 J mice, nalfurafine at 30 μg/kg produced CPA, but not at 15 or 60 μg/kg, whereas U50,488 was aversive at three doses tested (1.25–5.00 mg/kg). The reason for this inverted U-shaped dose-response relationship is not clear.
Rats
Mori et al. (2002) reported that in male Fischer 344 rats, TRK-820 at 10, 20, 40 μg/kg did not induce CPA or CPP, but at 80 μg/kg it did cause significant CPA. However, a recent study in male Sprague-Dawley rats reported that 0.18 mg/kg nalfurafine (s.c.) did not produce a significant CPA (Zamarripa et al. 2020b).
3.6 Effects on Locomotor Activity and Motor Coordination
The locomotor-suppressing actions of KOR agonists in rodents have long been recognized (Iwamoto 1981; von Voigtlander et al. 1983). Locomotor activity has commonly been assessed with the rotarod test (Iwamoto 1981), measurement with circular actophotometer cages (von Voigtlander et al. 1983) or rectangular locomotor activity chambers (Liu et al. 2019; White et al. 2015) or the wheel running test (Togashi et al. 2002).
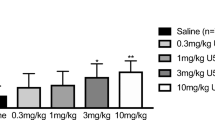
The hypolocomotion induced by KOR agonists generally occurs in the dose ranges that produce antinociceptive and anti-pruritic effects (for example, U50,488H in von Voigtlander et al. 1983; Liu et al. 2019; Kaski et al. 2019, methoxysalvinorin B in Liu et al. 2019). The unique feature of nalfurafine is that in the dose ranges effective for antinociceptive and anti-pruritic effects, nalfurafine either did not cause a hypolocomotor effect (Liu et al. 2019) or produced a small (≤ 25%) but significant effect (Kaski et al. 2019). For example, Liu et al. (2019) reported that the A50 values of nalfurafine-induced antinociception in the formalin test and in the anti-scratch test were 5.8 and 8.3 μg/kg, respectively and at 20 μg/kg, it did not affect total distance traveled and slightly impaired rotarod performance. Zhou and Kreek (2019b) also found that nalfurafine at 10 μg/kg (i.p.) had no effect on spontaneous locomotor activity in C57BL/6 J mice.
In some studies, TRK-820 did produce sedation, but at doses which were higher than those producing antinociception (Endoh et al. 1999; Togashi et al. 2002). Endoh et al. (1999) demonstrated that in male ddY mice TRK-820 (s.c.) produced antinociceptive effect in the acetic acid-induced abdominal constriction assay and impaired rotarod performance with A50 values of 3.3 μg/kg and 27 μg/kg, respectively. Togashi et al. (2002) reported that in male ICR mice TRK-820 (p.o., at −30 min) inhibited histamine-induced scratching and suppressed running activity with A50 values of 7.3 μg/kg and 102.8 μg/kg, respectively. Kaski et al. (2019) found that the inhibitory effect of nalfurafine at 15 and 30 μg/kg on novelty-induced locomotor activity was slight (by ≤25%) but significant, whereas at 60 μg/kg, the drug impaired locomotor activity by >50%.
Mori et al. (2002) reported that in a cocaine drug discrimination test using male Fisher 344 rats, 10–40 μg/kg TRK-820 did not affect the response rates, but at 80 μg/kg significantly decreased the response rates.
3.7 Water Diuretic Effects
Inan et al. (2009b) observed that nalfurafine (5–20 μg/kg, s.c.) caused water diuresis in a dose-dependent manner in male rats, similar to U50,488H and methoxymethyl salvinorin B (MOM-SalB). Nalfurafine, like U50,488H and MOM-SalB, increased urine volume and free water clearance and lowered urine osmolality, without excreting sodium ion. The effect of nalfurafine was blocked by the KOR antagonist 5′-guanidinonaltrindole, indicating KOR-mediated effect. Tolerance did not develop following repeated injection of nalfurafine (once/day, 20 μg/kg, s.c.) for 7 days.
3.8 Nalfurafine and U50,488H Induced Different Phosphoproteomic Changes in Mouse Brains
Liu et al. (2019) observed that in CD-1 mice both U50,488H and nalfurafine produced antinociceptive and anti-scratch effects in a dose-dependent manner. However, in the dose ranges effective in both effects, U50,488H, but not nalfurafine, caused CPA, inhibition of novelty-induced hyperlocomotion and motor incoordination. Phosphoproteomics studies on brains of mice revealed that U50,488H and nalfurafine imparted phosphorylation changes to proteins in different cellular compartments and signaling pathways in different brain regions. Notably, U50,488H, but not nalfurafine, activated the mammalian target of rapamycin (mTOR) pathway in the striatum and cortex. Inhibition of the mTOR pathway by rapamycin abolished U50,488H-induced CPA, without affecting analgesic, anti-scratch, and sedative effects and motor incoordination. The results indicate that the mTOR pathway is involved in KOR agonist-induced aversion. Recently, Zhou et al. (2020) reported that excessive alcohol drinking enhanced gene expression of molecules in the mTORC1 pathway in the mouse ventral striatum, and U50,488H (but not nalfurafine)-induced increases in alcohol intake were attenuated by rapamycin pretreatment, suggesting the mTOR pathway is also involved in KOR activation-induced drug taking behavior. In addition, several pathways were differentially regulated (Liu et al. 2019). For example, U50,488H, but not nalfurafine, activated the Wnt signaling pathway in the cortex and hippocampus, but nalfurafine, not U50,488H, activated pathways involved in tight junction and inositol phosphate metabolism in the cortex. How these pathways are involved in the different pharmacological effects of the two agonists remains to be investigated.
3.9 Effect of Nalfurafine on Pharmacological Actions of Morphine
3.9.1 Effects on Morphine-Induced Itch
Morphine-induced itch is a significant clinical issue. In mice, Umeuchi et al. (2003) showed that intracisternal morphine-induced scratching was significantly and dose-dependently inhibited by TRK-820 (s.c.). Nalfurafine is also active via the intrathecal (i.t.) route. Sakakihara et al. (2016) reported that in mice nalfurafine (i.t.) produced an antipruritic effect against i.t. morphine-induced scratching without affecting sedation scores, and the effect of TRK-820 was blocked by pretreatment with norBNI (i.p.). In rhesus monkeys, TRK-820 (i.v. or p.o.) inhibited systemic skin scratching induced by i.v. or i.t. morphine (Ko and Husbands 2009; Wakasa et al. 2004) without affecting morphine-induced analgesia and respiratory depression.
3.9.2 Effect on Morphine-Induced Antinociception
Endoh et al. (1999) reported that co-administration of TRK-820 (10 or 30 μg/kg, s.c.) with morphine (10 mg/kg) slightly enhanced morphine-induced antinociception in the 55°C hot plate test in male ddY mice. In contrast, pentazocine (3 or 10 mg/kg, s.c.) reduced morphine-induced antinociception.
Co-injection of TRK-820 with morphine (both i.t.) significantly enhanced thermal antinociceptive effects of morphine in mice (Sakakihara et al. 2016).
Kaski et al. (2019) showed that nalfurafine (15 μg/kg, s.c.) co-administered with morphine (5 mg/kg, i.p.) significantly augmented the spinal anti-nociceptive effect of morphine in the 55°C warm water tail withdrawal assay, similar to U50,488H (5 mg/kg, i.p.). In addition, nalfurafine (15, 30, 60 μg/kg, s.c.) significantly increased (~3x) supraspinal anti-nociceptive effect of morphine (5 mg/kg, i.p.) in the 53°C hot plate test (Kaski et al. 2019). A similar augmentation was seen only with 5 mg/kg U50,488H, but not with 1.25 or 2.50 mg/kg.
3.9.3 Effects on Rewarding Properties of Morphine and Oxycodone
Tsuji et al. (2001) found that in ddY mice, an outbred strain, TRK-820 (10 and 30 μg/kg, s.c.) co-injected with morphine (5 mg/kg, s.c.) significantly suppressed morphine-induced CPP, and this effect was antagonized by pretreatment with nor-BNI. TRK-820 alone did not produce CPP or CPA. A similar result was recently reported in male Sprague-Dawley rats with a combination of nalfurafine (0.18 mg/kg; s.c.) and oxycodone (3.2 mg/kg; s.c.) (Zamarripa et al. 2020b). Using C57BL/6 J mice, an inbred strain, Kaski et al. (2019) reported that co-administration of nalfurafine (s.c.) at 15 or 60 μg/kg, but not 30 μg/kg, significantly reduced morphine (5 mg/kg)-elicited CPP. For comparison, U50,488H at 1.25, 2.50 or 5.0 mg/kg inhibited morphine CPP. Nalfurafine alone caused CPA at 30 μg/kg, but not at 15 or 60 μg/kg. The differences between the two studies may be due to genetic backgrounds of the mice. Recently Zhang and Kreek (2020) reported that C57BL/6J mice nalfurafine caused a dose-dependent reduction of oxycodone self-administration and CPP at doses up to 40 μg/kg and the effects of nalfurafine were blocked by pretreatment with nor-BNI.
3.9.4 Effects on Morphine-Induced Tolerance and Dependence
Tsuji et al. (2000b) showed that in mice antinociceptive tolerance to morphine in the 51°C warm plate test following daily injection of morphine (10 mg/kg, s.c.) was suppressed by co-administration of U50,488H (1–10 mg/kg, s.c.) dose-dependently, but not by co-administration of TRK-820 (3–30 μg/kg, s.c.). Tsuji et al. (2000a) also found that co-treatment of mice with nalfurafine (3–30 μg/kg, s.c.) during chronic treatment with escalating doses of morphine blocked naloxone-precipitated withdrawal syndromes, whereas co-treatment with U50,488H (1–10 mg/kg, s.c.) did not. Thus, co-administration of TRK-820 with morphine prevented naloxone-precipitated withdrawal, but had no effect on morphine tolerance.
3.9.5 Effects on Morphine-Induced Hyperlocomotion
Tsuji et al. (2001) reported that in ddY mice, TRK-820 (10, 30 μg/kg, s.c.) suppressed morphine-induced hyperlocomotion, and this suppression was blocked by nor-BNI. Kaski et al. (2019) found that in C57BL/6J mice nalfurafine (15, 30, 60 μg/kg, s.c.) had similar effects in a dose-dependent manner. In comparison, U50,488 significantly reduced morphine-stimulated locomotor activity only at 5 mg/kg (Kaski et al. 2019).
4 Nalfurafine as an Abuse-Deterring Agent for Prescription Opioid Abuse
4.1 Translational Significance of Contingent vs. Non-contingent Administration of KOR Agonists
As indicated above, treating experimental subjects with KOR agonists has been shown to reduce the reinforcing effects of a number of drugs and nondrug reinforcers in the self-administration design (Cosgrove and Carroll 2002; Mello and Negus 1998; Morani et al. 2009; Simonson et al. 2015). From a translational perspective, studies in which the administration of KOR agonists are not contingent on the behavior of the organism are generally focused on the development of treatments for Substance Use Disorder (SUD) because the administration of the test compound is independent of the seeking and taking of the drug reinforcer (as would be the case with a maintenance therapy). An alternative approach is to study the effects of contingently administered KOR agonists on self-administration of a drug of abuse. Under this experimental arrangement, the animal self-administers a drug reinforcer, and each injection of that drug results in the co-administration of a dose of a KOR agonist. Thus, the effects of the KOR agonist are only experienced when the drug reinforcer is self-administered.
The translational focus of this “yoked” arrangement of drug delivery is the development of abuse-deterrent formulations for therapeutics. That is, self-administering the drugs together models the circumstance in which a pill containing a combined formulation of the therapeutic molecule and the KOR agonist would be taken. Contingent administration of prototypical KOR agonists such as salvinorin A and U69,593 has been demonstrated to reduce self-administration of the MOR agonists, fentanyl and remifentanil, and the stimulant, cocaine, in nonhuman primates (Freeman et al. 2014; Negus et al. 2008), providing evidence that MOR/KOR agonist combinations have less abuse potential than MOR agonists alone.
4.2 Behavioral Pharmacology of Nalfurafine/Oxycodone Combinations
4.2.1 Studies in Rats
The “mild” side-effect profile of nalfurafine increases the feasibility of using a KOR agonist as a combination therapy with a MOR agonist because of its potential to reduce the abuse-related effects of MOR agonists without producing KOR-typical side effects. Townsend et al. (2017) reported that nalfurafine (0.32, 1, or 3.2 μg/kg/injection), when co-administered with a highly-reinforcing dose of oxycodone (0.056 mg/kg/injection; i.v.) in male Sprague-Dawley rats, decreased oxycodone self-administration in a dose-dependent manner under a progressive-ratio schedule of reinforcement. The corresponding oxycodone/nalfurafine ratios were 175:1, 56:1, and 18:1, respectively. At 18:1 ratio, oxycodon no longer had reinforcing effect. In addition, combinations of oxycodone and nalfurafine produced additive thermal antinociception, suggesting that nalfurafine may enable a dose-sparing approach for oxycodone as a combined formulation. Moreover, when a dose of oxycodone that produced significant respiratory depression was compared to an equi-analgesic dose combination of oxycodone and nalfurafine, the drug combination produced no significant respiratory depression. Lastly, Zamarripa et al. (2020b) recently reported that a mixture of nalfurafine and oxycodone blocked acquisition of oxycodone self-administration and oxycodone-induced CPP in male Sprague-Dawley rats. Thus, select combinations of oxycodone and nalfurafine have been shown to produce thermal antinociception equivalent to oxycodone alone without producing abuse-related and respiratory-depressant effects.
4.2.2 Studies in Nonhuman Primates
Zamarripa et al. (2020a) reported that nalfurafine, combined with oxycodone, reduced self-administration of oxycodone under a progressive-ratio schedule of reinforcement in male rhesus monkeys. A notable difference between the self-administration results in rats (Townsend et al. 2017) and rhesus monkeys was the dose ratio of nalfurafine:oxycodone required to reduce self-administration to saline levels. The asymptotic dose of oxycodone under a progressive-ratio schedule of reinforcement is approximately 0.05 mg/kg/inj in both species (Townsend et al. 2017; Zamarripa et al. 2020a). However, the dose of nalfurafine required to reduce self-administration of this asymptotic dose of oxycodone to saline levels is ~3 times higher in rats (0.32, 1, or 3.2 μg/kg/injection) than in monkeys (0.1, 0.18 or 0.32 μg/kg/injection), suggesting that species differences affect the potency of nalfurafine to reduce the abuse-related effects of MOR agonists.
A complementary study was recently conducted to investigate the unconditioned behavioral effects of prototypical and atypical KOR agonists, alone and following oxycodone administration, in male rhesus monkeys (Huskinson et al. 2020). The prototypical KOR agonists, salvinorin A and U50,488H, produced KOR-typical effects including disruption of species-typical activity and “lip droop”, a proxy for muscle relaxation. Generally, nalfurafine produced effects that were comparable to the typical KOR agonists at sufficient doses, but the lowest dose required to disrupt species-typical behaviors in rhesus monkeys was three times higher than the dose required to reduce oxycodone self-administration to saline levels in this species (Zamarripa et al. 2020a) and oxycodone-induced scratching (Huskinson et al. 2020), suggesting that the potency of nalfurafine to produce certain therapeutic effects is greater than its potency to produce side effects. Notably, time course assessments revealed a relatively late onset for some of the behavioral effects of nalfurafine when compared to other KOR agonists. As such, it is important when studying nalfurafine to ensure that adequate time is allowed to capture the potential occurrence of late onset of behavioral effects.
5 Effects of Nalfurafine on Alcohol Drinking
5.1 The Dynorphins/KOR System in Alcohol-Related Behaviors
Dynorphins/KOR activation has been found to be associated with the negative reinforcement aspects of alcohol addictions. It has been demonstrated that selective blockade of KOR attenuates excessive drinking and stress- or cue-induced alcohol-seeking in rodents [see (Anderson and Becker 2017) for a recent review]. These findings provide support for a critical role of the dynorphins/KOR system in alcohol addiction, although the literature is not very consistent. Microdialysis studies have revealed that acute alcohol increases extracellular dynorphin A levels in the CeA and NAc (Lam and Gianoulakis 2011). As the CeA is one of critical brain regions involved in depression- and anxiety-like behaviors (Koob 2021), it is a likely site for potential interaction of alcohol with the dynorphins/KOR system. In fact, in Sardinian alcohol-preferring rats, an increase in prodynorphin mRNA levels is found in the CeA after drinking a large amount of alcohol, suggesting that the dynorphins/KOR system in brain regions related to stress responsivity (e.g., CeA) is activated after excessive alcohol consumption (Zhou et al. 2013). It has been confirmed that there are increases in dynorphin peptide levels and KOR signaling in the CeA in alcohol-dependent Wistar rats after chronic intermittent alcohol vapor exposure (D’Addario et al. 2013; Kissler et al. 2014). Together, the enhanced dynorphins/KOR activity in the CeA may present a homeostatic adaptation of the CNS after chronic alcohol exposure or in the negative affective state during alcohol withdrawal (Haun et al. 2020).
5.2 Development of Nalfurafine as a Potential Therapeutic Agent for Alcoholism
Early work revealed that “classic” KOR agonists decreased alcohol drinking and alcohol reward (Lindholm et al. 2001), but they also produced sedation and dysphoria - side effects that limited their potentials for clinical use as discussed in Introduction. Therefore, the development of novel KOR agonists with reduced side effects may produce useful compounds for the treatment of alcoholism.
As mentioned in the Introduction, nalfurafine is the first KOR agonist approved for clinical use as an anti-pruritus medicine in Japan with few side effects (Kozono et al. 2018). Zhou and Kreek (2019b) thus investigated whether nalfurafine alone or in combination with the MOR antagonist naltrexone (having partial KOR agonist activity) would change excessive alcohol drinking in mice. Both male and female C57BL/6J mice subjected to a chronic intermittent-access drinking paradigm (2-bottle choice, 24-h access every other day) for 3 weeks. On the test day, a single administration of nalfurafine (1–10 μg/kg) decreased excessive alcohol intake and alcohol preference in a dose-dependent manner via a KOR-mediated mechanism. In contrast, nalfurafine does not alter sucrose (caloric reinforcer) or saccharin (non-caloric reinforcer) consumption. Of interest and significance, repeated daily nalfurafine administrations for 10 days decreased alcohol consumption without showing any blunted effects, suggesting tolerance did not develop to nalfurafine. Lack of tolerance to nalfurafine effect is similar to the finding that in humans, tolerance to its antipruritic effects is not observed after treatment of patients for 1 year, and also with no evidence of physical or psychological dependency reported (Kozono et al. 2018). Zhou and Kreek (2019b) demonstrated that nalfurafine at 10 μg/kg did not cause any sedation (spontaneous locomotor activity), anhedonia-like (sucrose preference test), anxiety-like (elevated plus maze test), or dysphoria-like (conditioned place aversion test) behaviors, consistent with the rodent literature (e.g. (Liu et al. 2019)), suggesting that nalfurafine had few side effects at clinically relevant doses. In addition, Zhou and Kreek (2019b) found that combinations of nalfurafine and naltrexone, at doses lower than individual effective doses, profoundly decreased excessive alcohol intake in both sexes of mice. Further, acute administration of nalfurafine alone or in combination with low-dose naltrexone significantly reduced relapse-like drinking in an alcohol deprivation effect model (Zhou and Kreek 2019a). Finally, nalmefene (a clinically utilized KOR partial agonist with MOR antagonism) (Bart et al. 2005) combined with nalfurafine also reduced relapse-like drinking in mice of either sex (Zhou and Kreek 2019a). Together, these in vivo results suggest that nalfurafine alone or combined with naloxone or nalmefene may offer a novel approach to treat alcoholism without the dysphoric properties of classic KOR agonists.
Using different chronic alcohol exposure models, several groups have found that classic KOR agonists increase alcohol intake in mice after excessive alcohol drinking (Rose et al. 2016; Zhou et al. 2020), and trigger alcohol-seeking and relapse-like drinking (Anderson and Becker 2017). Therefore, the data of Zhou and Kreek (2019a, b) that the KOR full agonist nalfurafine decreases, rather than increases, excessive or relapse-like drinking may present a different situation. After chronic excessive alcohol consumption, the endogenous dynorphin peptides (G-protein- and β-arrestin-dependent agonists) and KOR system are activated in several brain regions, including the CeA (Bloodgood et al. 2020; D’Addario et al. 2013; Kissler et al. 2014; Zhou et al. 2013). Activation of mTORC1 or β-arrestin/p38 mitogen-activated protein kinase pathway by stress-related dynorphins/KOR stimulation is associated with aversion, dysphoria, anxiety- and depression-like behaviors (Bruchas et al. 2007; Liu et al. 2018, 2019) that can drive excessive and relapse drinking (Koob 2021; Zhou et al. 2020). As discussed above, nalfurafine at doses that produced antinociceptive and anti-pruritic effects, caused fewer side effects (sedation, anhedonia, aversion, dysphoria, anxiety- or depression-like behavior) in mice and monkeys (Huskinson et al. 2020; Liu et al. 2019; Zhou and Kreek 2019b). Thus, nalfurafine, a potent KOR agonist, may compete with the released dynorphins to bind the KOR, thereby reducing undesired effects induced by KOR activation, perhaps by reducing mTORC1 or β-arrestin/p38 mitogen-activated protein kinase signaling pathway (Zhou and Kreek 2019a, b; Zhou et al. 2020). This may be responsible, at least in part, for reducing excessive alcohol intake, as nalfurafine may attenuate the dynorphin-induced dysphoria and anxiety- or depression-like behavior after chronic alcohol exposure or during alcohol withdrawal. Taken together, these studies support the notion that nalfurafine may exhibit different molecular, cellular, and behavioral properties than classic KOR agonists and provide support for development of nalfurafine as an anti-addiction medication.
6 Effect of Nalfurafine on CPP and Discriminative Stimulus Effects of Cocaine
Rats: Pretreatment of male Fischer 344 rats with 20 and 40 μg/kg TRK-820, which itself did not produce CPP or CPA, significantly attenuated cocaine-induced CPP and these effects of TRK-820 were reversed by nor-BNI (Hasebe et al. 2004; Mori et al. 2002).
In drug discrimination test in rats, cocaine produced a dose-related increase in cocaine appropriate responses. Pretreatment with 10 and 20 μg/kg TRK-820 significantly shifted the dose-response curve for cocaine to the right without changing the response rate (Mori et al. 2002).
Mice: Pretreatment of C57BL/6 mice with nalfurafine (3 and 10 μg/kg) and U50,488 (3 mg/kg) for 15 min blocked cocaine CPP, but did not cause sedation in the rotarod assay or aversion in a place-conditioning assay (Dunn et al. 2020).Pretreatment of mice with 10 μg/kg nalfurafine and 3 mg/kg U50,488 immediately before test potentiated cocaine self-administration. Further 10 μg/kg nalfurafine also increased progressive ratio break point, indicating enhanced cocaine-seeking behavior (Dunn et al. 2020).
8 Conclusion
In humans, nalfurafine produces anti-pruritic effects without causing dysphoria or psychotomimesis, side effects associated with typical KOR agonists. Different labs reported different results as to whether in vitro nalfurafine is a biased KOR agonist, depending on the functional end points and assay conditions used. In animal studies, nalfurafine induces anti-scratch effects at doses lower than those producing CPA, hypolocomotion, and motor incoordination, consistent with human data. Thus, it provides a cautionary tale about correlating in vitro biased agonism and in vivo pharmacological effects. Because of its unique side effect profile, nalfurafine may be useful for other clinical uses. Nalfurafine produces antinociceptive effects in experimental animals; however, whether KOR agonists are sufficiently efficacious analgesics in humans is a matter of debate. Nalfurafine may be useful as an adjunct for prescription MOR agonists, which at certain ratios of the two drugs, increases analgesic effects while reducing abuse liability. In addition, nalfurafine may be useful for treatment of opioid and alcohol abuse disorders. Besides being a clinically useful drug, nalfurafine is a very unique experimental tool for elucidating signaling properties underlying KOR-mediated side effects.
References
Akiyama T, Carstens MI, Piecha D, Steppan S, Carstens E (2015) Nalfurafine suppresses pruritogen- and touch-evoked scratching behavior in models of acute and chronic itch in mice. Acta Derm Venereol 95:147–150. https://doi.org/10.2340/00015555-1879
Anderson RI, Becker HC (2017) Role of the dynorphin/kappa opioid receptor system in the motivational effects of ethanol alcohol. Clin Exp Res 41:1402–1418. https://doi.org/10.1111/acer.13406
Bart G, Schluger JH, Borg L, Ho A, Bidlack JM, Kreek MJ (2005) Nalmefene induced elevation in serum prolactin in normal human volunteers: partial kappa opioid agonist activity? Neuropsychopharmacology 30:2254–2262. https://doi.org/10.1038/sj.npp.1300811
Bloodgood DW, Hardaway JA, Stanhope CM, Pati D, Pina MM, Neira S, Desai S, Boyt KM, Palmiter RD, Kash TL (2020) Kappa opioid receptor and dynorphin signaling in the central amygdala regulates alcohol intake. Mol Psychiatry. https://doi.org/10.1038/s41380-020-0690-z
Bruchas MR, Land BB, Aita M, Xu M, Barot SK, Li S, Chavkin C (2007) Stress-induced p38 mitogen-activated protein kinase activation mediates kappa-opioid-dependent dysphoria. J Neurosci 27:11614–11623
Cao D, Huang P, Chiu YT, Chen C, Wang H, Li M, Zheng Y, Ehlert FJ, Zhang Y, Liu-Chen LY (2020) Comparison of pharmacological properties between the kappa opioid receptor agonist nalfurafine and 42B, its 3-dehydroxy analogue: disconnect between in vitro agonist bias and in vivo pharmacological effects. ACS Chem Neurosci 11:3036–3050. https://doi.org/10.1021/acschemneuro.0c00407
Che T, Majumdar S, Zaidi SA, Ondachi P, McCorvy JD, Wang S, Mosier PD, Uprety R, Vardy E, Krumm BE, Han GW, Lee MY, Pardon E, Steyaert J, Huang XP, Strachan RT, Tribo AR, Pasternak GW, Carroll FI, Stevens RC, Cherezov V, Katritch V, Wacker D, Roth BL (2018) Structure of the nanobody-stabilized active state of the kappa opioid receptor cell 172:55–67 e15. https://doi.org/10.1016/j.cell.2017.12.011
Cosgrove KP, Carroll ME (2002) Effects of bremazocine on self-administration of smoked cocaine base and orally delivered ethanol, phencyclidine, saccharin, and food in rhesus monkeys: a behavioral economic analysis. J Pharmacol Exp Ther 301:993–1002. https://doi.org/10.1124/jpet.301.3.993
Cowan A, Gmerek DE (1986) In-vivo studies on kappa opioid receptors. Trends Pharmacol Sci 7:69–72
D’Addario C, Caputi FF, Rimondini R, Gandolfi O, Del Borrello E, Candeletti S, Romualdi P (2013) Different alcohol exposures induce selective alterations on the expression of dynorphin and nociceptin systems related genes in rat brain. Addict Biol 18:425–433. https://doi.org/10.1111/j.1369-1600.2011.00326.x
Dunn AD, Reed B, Erazo J, Ben-Ezra A, Kreek MJ (2019) Signaling properties of structurally diverse kappa opioid receptor ligands: toward in vitro models of in vivo responses. ACS Chem Neurosci 10:3590–3600. https://doi.org/10.1021/acschemneuro.9b00195
Dunn A, Windisch K, Ben-Ezra A, Pikus P, Morochnik M, Erazo J, Reed B, Kreek MJ (2020) Modulation of cocaine-related behaviors by low doses of the potent KOR agonist nalfurafine in male C57BL6 mice. Psychopharmacology 237:2405–2418. https://doi.org/10.1007/s00213-020-05543-7
Elliott G, Vanwersch R, Soeberdt M, Metze D, Lotts T, Stander S, Abels C (2016) Topical nalfurafine exhibits anti-inflammatory and anti-pruritic effects in a murine model of AD. J Dermatol Sci 84:351–354. https://doi.org/10.1016/j.jdermsci.2016.09.008
Endoh T, Matsuura H, Tajima A, Izumimoto N, Tajima C, Suzuki T, Saitoh A, Suzuki T, Narita M, Tseng L, Nagase H (1999) Potent antinociceptive effects of TRK-820, a novel kappa-opioid receptor agonist. Life Sci 65:1685–1694
Endoh T, Tajima A, Suzuki T, Kamei J, Narita M, Tseng L, Nagase H (2000) Characterization of the antinociceptive effects of TRK-820 in the rat. Eur J Pharmacol 387:133–140
Endoh T, Tajima A, Izumimoto N, Suzuki T, Saitoh A, Suzuki T, Narita M, Kamei J, Tseng LF, Mizoguchi H, Nagase H (2001) TRK-820, a selective kappa-opioid agonist, produces potent antinociception in cynomolgus monkeys. Jpn J Pharmacol 85:282–290
Freeman KB, Naylor JE, Prisinzano TE, Woolverton WL (2014) Assessment of the kappa opioid agonist, salvinorin a, as a punisher of drug self-administration in monkeys. Psychopharmacology 231:2751–2758. https://doi.org/10.1007/s00213-014-3436-2
Gillis A, Gondin AB, Kliewer A, Sanchez J, Lim HD, Alamein C, Manandhar P, Santiago M, Fritzwanker S, Schmiedel F, Katte TA, Reekie T, Grimsey NL, Kassiou M, Kellam B, Krasel C, Halls ML, Connor M, Lane JR, Schulz S, Christie MJ, Canals M (2020) Low intrinsic efficacy for G protein activation can explain the improved side effect profiles of new opioid agonists. Sci Signal 13:eaaz3140. https://doi.org/10.1126/scisignal.aaz3140
Hasebe K, Kawai K, Suzuki T, Kawamura K, Tanaka T, Narita M, Nagase H, Suzuki T (2004) Possible pharmacotherapy of the opioid kappa receptor agonist for drug dependence. Ann N Y Acad Sci 1025:404–413. https://doi.org/10.1196/annals.1316.050
Haun HL, Griffin WC, Lopez MF, Becker HC (2020) Kappa opioid receptors in the bed nucleus of the stria terminalis regulate binge-like alcohol consumption in male and female mice. Neuropharmacology 167:107984. https://doi.org/10.1016/j.neuropharm.2020.107984
Huskinson SL, Platt DM, Brasfield M, Follett ME, Prisinzano TE, Blough BE, Freeman KB (2020) Quantification of observable behaviors induced by typical and atypical kappa-opioid receptor agonists in male rhesus monkeys. Psychopharmacology 237:2075–2087. https://doi.org/10.1007/s00213-020-05519-7
Inan S, Cowan A (2004) Kappa opioid agonists suppress chloroquine-induced scratching in mice. Eur J Pharmacol 502:233–237
Inan S, Cowan A (2006) Nalfurafine, a kappa opioid receptor agonist, inhibits scratching behavior secondary to cholestasis induced by chronic ethynylestradiol injections in rats. Pharmacol Biochem Behav 85:39–43
Inan S, Dun NJ, Cowan A (2009a) Nalfurafine prevents 5′-guanidinonaltrindole- and compound 48/80-induced spinal c-fos expression and attenuates 5′-guanidinonaltrindole-elicited scratching behavior in mice. Neuroscience 163:23–33
Inan S, Lee DY, Liu-Chen LY, Cowan A (2009b) Comparison of the diuretic effects of chemically diverse kappa opioid agonists in rats: nalfurafine, U50,488H, and salvinorin a. Naunyn Schmiedeberg's Arch Pharmacol 379:263–270
Iwamoto ET (1981) Locomotor activity and antinociception after putative mu, kappa and sigma opioid receptor agonists in the rat: influence of dopaminergic agonists and antagonists. J Pharmacol Exp Ther 217:451–460
Kardon AP, Polgar E, Hachisuka J, Snyder LM, Cameron D, Savage S, Cai X, Karnup S, Fan CR, Hemenway GM, Bernard CS, Schwartz ES, Nagase H, Schwarzer C, Watanabe M, Furuta T, Kaneko T, Koerber HR, Todd AJ, Ross SE (2014) Dynorphin acts as a neuromodulator to inhibit itch in the dorsal horn of the spinal cord. Neuron 82:573–586. https://doi.org/10.1016/j.neuron.2014.02.046
Kaski SW, White AN, Gross JD, Trexler KR, Wix K, Harland AA, Prisinzano TE, Aube J, Kinsey SG, Kenakin T, Siderovski DP, Setola V (2019) Preclinical testing of nalfurafine as an opioid-sparing adjuvant that potentiates analgesia by the mu opioid receptor-targeting agonist morphine. J Pharmacol Exp Ther 371:487–499. https://doi.org/10.1124/jpet.118.255661
Kissler JL, Sirohi S, Reis DJ, Jansen HT, Quock RM, Smith DG, Walker BM (2014) The one-two punch of alcoholism: role of central amygdala dynorphins/kappa-opioid receptors. Biol Psychiatry 75:774–782. https://doi.org/10.1016/j.biopsych.2013.03.014
Ko MC, Husbands SM (2009) Effects of atypical kappa-opioid receptor agonists on intrathecal morphine-induced itch and analgesia in primates. J Pharmacol Exp Ther 328:193–200. https://doi.org/10.1124/jpet.108.143925
Koob GF (2021) Drug addiction: hyperkatifeia/negative reinforcement as a framework for medications development. Pharmacol Rev 73:163–201. https://doi.org/10.1124/pharmrev.120.000083
Kozono H, Yoshitani H, Nakano R (2018) Post-marketing surveillance study of the safety and efficacy of nalfurafine hydrochloride (Remitch((R)) capsules 2.5 mug) in 3,762 hemodialysis patients with intractable pruritus. Int J Nephrol Renovasc Dis 11:9–24. https://doi.org/10.2147/IJNRD.S145720
Kumagai H, Ebata T, Takamori K, Muramatsu T, Nakamoto H, Suzuki H (2010) Effect of a novel kappa-receptor agonist, nalfurafine hydrochloride, on severe itch in 337 haemodialysis patients: a phase III, randomized, double-blind, placebo-controlled study. Nephrol Dial Transplant 25:1251–1257. https://doi.org/10.1093/ndt/gfp588
Kumagai H, Ebata T, Takamori K, Miyasato K, Muramatsu T, Nakamoto H, Kurihara M, Yanagita T, Suzuki H (2012) Efficacy and safety of a novel k-agonist for managing intractable pruritus in dialysis patients. Am J Nephrol 36:175–183. https://doi.org/10.1159/000341268
Lam MP, Gianoulakis C (2011) Effects of corticotropin-releasing hormone receptor antagonists on the ethanol-induced increase of dynorphin A1-8 release in the rat central amygdala. Alcohol 45:621–630. https://doi.org/10.1016/j.alcohol.2011.05.001
Lazenka ML, Moerke MJ, Townsend EA, Freeman KB, Carroll FI, Negus SS (2018) Dissociable effects of the kappa opioid receptor agonist nalfurafine on pain/itch-stimulated and pain/itch-depressed behaviors in male rats. Psychopharmacology 235:203–213. https://doi.org/10.1007/s00213-017-4758-7
Lindholm S, Werme M, Brené S, Franck J (2001) The selective kappa-opioid receptor agonist U50,488H attenuates voluntary ethanol intake in the rat. Behav Brain Res 120:137–146. https://doi.org/10.1016/s0166-4328(00)00368-5
Liu JJ, Sharma K, Zangrandi L, Chen C, Humphrey SJ, Chiu YT, Spetea M, Liu-Chen LY, Schwarzer C, Mann M (2018) In vivo brain GPCR signaling elucidated by phosphoproteomics. Science 360:eaao4927. https://doi.org/10.1126/science.aao4927
Liu JJ, Chiu YT, DiMattio KM, Chen C, Huang P, Gentile TA, Muschamp JW, Cowan A, Mann M, Liu-Chen LY (2019) Phosphoproteomic approach for agonist-specific signaling in mouse brains: mTOR pathway is involved in kappa opioid aversion. Neuropsychopharmacology 44:939–949. https://doi.org/10.1038/s41386-018-0155-0
Mello NK, Negus SS (1998) Effects of kappa opioid agonists on cocaine- and food-maintained responding by rhesus monkeys. J Pharmacol Exp Ther 286:812–824
Morani AS, Kivell B, Prisinzano TE, Schenk S (2009) Effect of kappa-opioid receptor agonists U69593, U50488H, spiradoline and salvinorin a on cocaine-induced drug-seeking in rats. Pharmacol Biochem Behav 94:244–249. https://doi.org/10.1016/j.pbb.2009.09.002
Mori T, Nomura M, Nagase H, Narita M, Suzuki T (2002) Effects of a newly synthesized kappa-opioid receptor agonist, TRK-820, on the discriminative stimulus and rewarding effects of cocaine in rats. Psychopharmacology (Berlin) 161:17–22
Mucha RF, Herz A (1985) Motivational properties of kappa and mu opioid receptor agonists studied with place and taste preference conditioning. Psychopharmacology 86:274–280
Nagase H, Fujii H (2013) Essential structure of the kappa opioid receptor agonist nalfurafine for binding to the kappa receptor. Curr Pharm Des 19:7400–7414
Nagase H, Hayakawa J, Kawamura K, Kawai K, Takezawa Y, Matsuura H, Tajima C, Endo T (1998) Discovery of a structurally novel opioid kappa-agonist derived from 4,5-epoxymorphinan. Chem Pharm Bull (Tokyo) 46:366–369
Nakao K, Mochizuki H (2009a) Nalfurafine hydrochloride: a new drug for the treatment of uremic pruritus in hemodialysis patients. Drugs Today (Barc) 45:323–329. https://doi.org/10.1358/dot.2009.45.5.1362067
Nakao K, Mochizuki H (2009b) Nalfurafine hydrochloride: a new drug for the treatment of uremic pruritus in hemodialysis patients. Drugs Today (Barc) 45:323–329
Nakao K, Ikeda K, Kurokawa T, Togashi Y, Umeuchi H, Honda T, Okano K, Mochizuki H (2008) Effect of TRK-820, a selective kappa opioid receptor agonist, on scratching behavior in an animal model of atopic dermatitis. Nihon Shinkei Seishin Yakurigaku Zasshi 28:75–83
Negus SS, Schrode K, Stevenson GW (2008) Micro/kappa opioid interactions in rhesus monkeys: implications for analgesia and abuse liability. Exp Clin Psychopharmacol 16:386–399. https://doi.org/10.1037/a0013088
Pande AC, Pyke RE, Greiner M, Wideman GL, Benjamin R, Pierce MW (1996) Analgesic efficacy of enadoline versus placebo or morphine in postsurgical pain. Clin Neuropharmacol 19:451–456
Pfeiffer A, Brantl V, Herz A, Emrich HM (1986) Psychotomimesis mediated by kappa opiate receptors. Science 233:774–776. https://doi.org/10.1126/science.3016896
Rose JH, Karkhanis AN, Chen R, Gioia D, Lopez MF, Becker HC, McCool BA, Jones SR (2016) Supersensitive kappa opioid receptors promotes ethanol withdrawal-related behaviors and reduce dopamine signaling in the nucleus accumbens. Int J Neuropsychopharmacol 19. https://doi.org/10.1093/ijnp/pyv127
Sakakihara M, Imamachi N, Saito Y (2016) Effects of intrathecal kappa-opioid receptor agonist on morphine-induced itch and antinociception in mice. Reg Anesth Pain Med 41:69–74. https://doi.org/10.1097/AAP.0000000000000326
Schattauer SS, Kuhar JR, Song A, Chavkin C (2017) Nalfurafine is a G-protein biased agonist having significantly greater bias at the human than rodent form of the kappa opioid receptor. Cell Signal 32:59–65. https://doi.org/10.1016/j.cellsig.2017.01.016
Seki T, Awamura S, Kimura C, Ide S, Sakano K, Minami M, Nagase H, Satoh M (1999) Pharmacological properties of TRK-820 on cloned mu-, delta- and kappa-opioid receptors and nociceptin receptor. Eur J Pharmacol 376:159–167
Shippenberg TS, Herz A (1986) Differential effects of mu and kappa opioid systems on motivational processes NIDA. Res Monogr 75:563–566
Simonson B, Morani AS, Ewald AW, Walker L, Kumar N, Simpson D, Miller JH, Prisinzano TE, Kivell BM (2015) Pharmacology and anti-addiction effects of the novel κ opioid receptor agonist Mesyl Sal B, a potent and long-acting analogue of salvinorin a. Br J Pharmacol 172:515–531. https://doi.org/10.1111/bph.12692
Snyder LM, Chiang MC, Loeza-Alcocer E, Omori Y, Hachisuka J, Sheahan TD, Gale JR, Adelman PC, Sypek EI, Fulton SA, Friedman RL, Wright MC, Duque MG, Lee YS, Hu Z, Huang H, Cai X, Meerschaert KA, Nagarajan V, Hirai T, Scherrer G, Kaplan DH, Porreca F, Davis BM, Gold MS, Koerber HR, Ross SE (2018) Kappa opioid receptor distribution and function in primary afferents. Neuron 99:1274–1288.e1276. https://doi.org/10.1016/j.neuron.2018.08.044
Takasaki I, Suzuki T, Sasaki A, Nakao K, Hirakata M, Okano K, Tanaka T, Nagase H, Shiraki K, Nojima H, Kuraishi Y (2004) Suppression of acute herpetic pain-related responses by the kappa-opioid receptor agonist (−)-17-cyclopropylmethyl-3,14beta-dihydroxy-4,5alpha-epoxy-beta-[n-methyl-3-trans −3-(3-furyl) acrylamido] morphinan hydrochloride (TRK-820) in mice. J Pharmacol Exp Ther 309:36–41. https://doi.org/10.1124/jpet.103.059816
Togashi Y, Umeuchi H, Okano K, Ando N, Yoshizawa Y, Honda T, Kawamura K, Endoh T, Utsumi J, Kamei J, Tanaka T, Nagase H (2002) Antipruritic activity of the kappa-opioid receptor agonist, TRK-820. Eur J Pharmacol 435:259–264
Townsend EA, Naylor JE, Negus SS, Edwards SR, Qureshi HN, McLendon HW, McCurdy CR, Kapanda CN, do Carmo JM, da Silva FS, Hall JE, Sufka KJ, Freeman KB (2017) Effects of nalfurafine on the reinforcing, thermal antinociceptive, and respiratory-depressant effects of oxycodone: modeling an abuse-deterrent opioid analgesic in rats. Psychopharmacology 234:2597–2605. https://doi.org/10.1007/s00213-017-4652-3
Tsuji M, Takeda H, Matsumiya T, Nagase H, Yamazaki M, Narita M, Suzuki T (2000a) A novel kappa-opioid receptor agonist, TRK-820, blocks the development of physical dependence on morphine in mice. Life Sci 66:L353–L358
Tsuji M, Yamazaki M, Takeda H, Matsumiya T, Nagase H, Tseng LF, Narita M, Suzuki T (2000b) The novel kappa-opioid receptor agonist TRK-820 has no affect on the development of antinociceptive tolerance to morphine in mice. Eur J Pharmacol 394:91–95
Tsuji M, Takeda H, Matsumiya T, Nagase H, Narita M, Suzuki T (2001) The novel kappa-opioid receptor agonist TRK-820 suppresses the rewarding and locomotor-enhancing effects of morphine in mice. Life Sci 68:1717–1725
Umeuchi H, Togashi Y, Honda T, Nakao K, Okano K, Tanaka T, Nagase H (2003) Involvement of central mu-opioid system in the scratching behavior in mice, and the suppression of it by the activation of kappa-opioid system. Eur J Pharmacol 477:29–35
Umeuchi H, Kawashima Y, Aoki CA, Kurokawa T, Nakao K, Itoh M, Kikuchi K, Kato T, Okano K, Gershwin ME, Miyakawa H (2005) Spontaneous scratching behavior in MRL/lpr mice, a possible model for pruritus in autoimmune diseases, and antipruritic activity of a novel kappa-opioid receptor agonist nalfurafine hydrochloride. Eur J Pharmacol 518:133–139. https://doi.org/10.1016/j.ejphar.2005.06.019
von Voigtlander PF, Lahti RA, Ludens JH (1983) U-50,488: a selective and structurally novel non-mu (kappa) opioid agonist. J Pharmacol Exp Ther 224:7–12
Wadenberg ML (2003) A review of the properties of spiradoline: a potent and selective kappa-opioid receptor agonist. CNS Drug Rev 9:187–198
Wakasa Y, Fujiwara A, Umeuchi H, Endoh T, Okano K, Tanaka T, Nagase H (2004) Inhibitory effects of TRK-820 on systemic skin scratching induced by morphine in rhesus monkeys. Life Sci 75:2947–2957. https://doi.org/10.1016/j.lfs.2004.05.033
Walsh SL, Strain EC, Abreu ME, Bigelow GE (2001) Enadoline, a selective kappa opioid agonist: comparison with butorphanol and hydromorphone in humans. Psychopharmacology 157:151–162. https://doi.org/10.1007/s002130100788
Wang Y, Tang K, Inan S, Siebert D, Holzgrabe U, Lee DY, Huang P, Li JG, Cowan A, Liu-Chen L-Y (2005) Comparison of pharmacological activities of three distinct k ligands (Salvinorin a, TRK-820 and 3FLB) on k opioid receptors in vitro and their antipruritic and antinociceptive activities in vivo. J Pharmacol Exp Ther 312:220–230
White KL, Robinson JE, Zhu H, DiBerto JF, Polepally PR, Zjawiony JK, Nichols DE, Malanga CJ, Roth BL (2015) The G protein-biased kappa-opioid receptor agonist RB-64 is analgesic with a unique spectrum of activities in vivo. J Pharmacol Exp Ther 352:98–109. https://doi.org/10.1124/jpet.114.216820
Zamarripa CA, Naylor JE, Huskinson SL, Townsend EA, Prisinzano TE, Freeman KB (2020a) Kappa opioid agonists reduce oxycodone self-administration in male rhesus monkeys. Psychopharmacology 237:1471–1480. https://doi.org/10.1007/s00213-020-05473-4
Zamarripa CA, Patel TR, Williams BC, Pareek T, Schrock HM, Prisinzano TE, Freeman KB (2020b) The kappa-opioid receptor agonist, nalfurafine, blocks acquisition of oxycodone self-administration and oxycodone's conditioned rewarding effects in male rats. Behav Pharmacol. https://doi.org/10.1097/FBP.0000000000000581
Zhang Y, Kreek MJ (2020) Nalfurafine modulates the reinforcing effects of oxycodone in male and female adolescent C57BL/6J mice. Neuropharmacology 176:108244. https://doi.org/10.1016/j.neuropharm.2020.108244
Zhou Y, Kreek MJ (2019a) Clinically utilized kappa-opioid receptor agonist nalfurafine combined with low-dose naltrexone prevents alcohol relapse-like drinking in male and female mice. Brain Res 1724:146410. https://doi.org/10.1016/j.brainres.2019.146410
Zhou Y, Kreek MJ (2019b) Combination of clinically utilized kappa-opioid receptor agonist nalfurafine with low-dose naltrexone reduces excessive alcohol drinking in male and female mice. Alcohol Clin Exp Res 43:1077–1090. https://doi.org/10.1111/acer.14033
Zhou Y, Colombo G, Gessa GL, Kreek MJ (2013) Effects of voluntary alcohol drinking on corticotropin-releasing factor and preprodynorphin mRNA levels in the central amygdala of Sardinian alcohol-preferring rats. Neurosci Lett 554:110–114. https://doi.org/10.1016/j.neulet.2013.08.071
Zhou Y, Liang Y, Kreek MJ (2020) mTORC1 pathway is involved in the kappa opioid receptor activation-induced increase in excessive alcohol drinking in mice. Pharmacol Biochem Behav 195:172954. https://doi.org/10.1016/j.pbb.2020.172954
Acknowledgements
The writing of this manuscript was supported by the NIH grants F30DA044711 (SK), R01DA039167 (KBF), R01DA041359, R21DA045274 and P30DA013429 (LYLC). YZ and MJK thank the support of Dr. Miriam and Sheldon G. Adelson Medical Research Foundation and Robertson Therapeutic Discover Fund at the Rockefeller University. We are grateful to NIDA Division of Drug Supply and Analytical Services for nalfurafine used in the experiments.
Conflict of Interests
The authors declare no conflict of interests.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2021 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Zhou, Y. et al. (2021). Preclinical Studies on Nalfurafine (TRK-820), a Clinically Used KOR Agonist. In: Liu-Chen, LY., Inan, S. (eds) The Kappa Opioid Receptor. Handbook of Experimental Pharmacology, vol 271. Springer, Cham. https://doi.org/10.1007/164_2021_443
Download citation
DOI: https://doi.org/10.1007/164_2021_443
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-89073-5
Online ISBN: 978-3-030-89074-2
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)