
Abstract
Rationale
Agonists of the kappa opioid receptor (KOR) have been shown to block the rewarding effects of drugs of abuse, but with negative side effects. The antipruritic drug nalfurafine, approved in Japan in 2009, is a potent, selective KOR agonist that does not cause significant side effects in humans. Nalfurafine has not been extensively tested for its effect on drug reward and reinforcement in preclinical models.
Objectives
The goal of this study was to compare the effects of nalfurafine and a reference KOR agonist for a variety of KOR-mediated endpoints in male C57BL6 mice. Specifically, we aimed to evaluate the “therapeutic window”—doses of agonists lower than those eliciting negative side effects, while still effective for desired therapeutic effects.
Methods
In this study, several low doses of nalfurafine and U50,488 were tested for serum prolactin release, rotarod-mediated sedation, and place-conditioning in male C57BL6 mice. These agonists were also tested for effects on intravenous cocaine self-administration, both on an FR1 schedule and on a progressive ratio schedule for 0.5 mg/kg/infusion cocaine.
Results
Serum prolactin levels increased following doses of both nalfurafine (3 μg/kg and 10 μg/kg) and U50,488 (3 mg/kg). These doses did not cause sedation in the rotarod assay or aversion in a place-conditioning assay, but blocked conditioned place preference for cocaine. Immediate pretreatment of mice with 10 μg/kg nalfurafine and 3 mg/kg U50,488, however, potentiated cocaine self-administration. Further 10 μg/kg nalfurafine was also observed to potentiate cocaine-seeking behavior as demonstrated by increased progressive ratio break point.
Conclusions
Both nalfurafine and U50,488 showed a separation of negative side effects and the modulation of cocaine reward, suggesting this effect of KOR agonists at low doses may be characteristic of the KOR system in general. At higher doses, nalfurafine had similar effects to traditional KOR agonists like U50,488, indicating that its relative potency, rather than differences in KOR signaling, may be responsible for its unique effects in humans.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The opioid system plays a unique role in mediating mood and reward, as well as pain and other sensations such as pruritus. Activation of the kappa opioid receptor (KOR) in particular negatively regulates striatal dopamine in opposition to the mu opioid receptor (MOR) and also causes analgesia and attenuates itching (Di Chiara and Imperato 1988; Zhang et al. 2004). As such, the KOR is currently under investigation as a potential therapeutic target for a range of diseases, including several kinds of pain, addiction, and depression (Kreek et al. 2002). KOR agonists cause sedation both in preclinical animal studies and in humans, however, and other negative side effects such as anhedonia, aversion, and psychotomimesis have hampered the development of KOR agonists as therapeutics (Pande et al. 1996; Wadenberg 2003; MacLean et al. 2013).
There are currently many ongoing drug discovery efforts aimed at identifying KOR-targeting agonists that maintain their clinically useful properties but do not cause these negative side effects. For example, there are efforts to identify peripherally selective KOR agonists for pain and pruritus that would not cause centrally mediated side effects (Bourgeois et al. 2014; Albert-Vartanian et al. 2016). For CNS use, evidence across several structural types of KOR ligands indicates that β-arrestin-2 signaling downstream of the KOR is significantly correlated with sedation, suggesting that a G-protein biased agonist may help avoid KOR-mediated sedation (reviewed in Mores et al. 2019) (White et al. 2015; Brust et al. 2016; Mores et al. 2019; Dunn et al. 2018; Dunn et al. 2019). However, it is unclear if this signaling pathway is related to other negative side effects. There is recent evidence that the mTOR signaling pathway, for example, is involved in KOR-mediated aversion, indicating that screening KOR compounds for their mTOR activation could provide an additional strategy for decreasing negative side effects (Liu et al. 2019).
The only example of a KOR-selective compound currently in clinical use is nalfurafine. Nalfurafine, which is sold as Remitch®, is approved to treat pruritus, exclusively in Japan. Nalfurafine also binds to the MOR, but with lower affinity than the KOR. Reports of its selectivity vary for different receptor species and assays, from 2.5 times higher affinity (Nagase et al. 2010; Nemoto et al. 2013) for KOR than MOR to 10 times higher (Wang et al. 2005). Its antipruritic effects, however, have been shown to be mediated through the KOR (Togashi et al. 2002). Nalfurafine is also very potent—the recommended dose of Remitch is 2.5 μg once per day for adults (Kozono et al. 2018). The downstream signaling effects of nalfurafine have been studied by several labs, with varying results. Differences in context make it difficult to compare across studies, but nalfurafine has been characterized as a G-protein biased agonist and an unbiased agonist, albeit with varying degrees of bias and with varying endpoints (Schattauer et al. 2017; Liu et al. 2018). In contrast, we recently reported nalfurafine to be β-arrestin-2 biased compared with the KOR reference agonist U69,593 (Dunn et al. 2019). Regardless, all reports of in vitro nalfurafine activity demonstrate that it is a full agonist in all signaling pathways compared with reference agonists such as U50,488 and U69,593—that is, nalfurafine has close to 100% maximal efficacy in both G-protein and β-arrestin-2 signaling pathways.
Since its approval in 2009, post-marketing studies have reported very few negative side effects. The largest study examined over 3500 patients and found very little evidence of typical KOR adverse reactions (e.g., 32 patients reported somnolence, 7 reported hallucinations) (Kozono et al. 2018). Studies in animals are currently exploring nalfurafine in models of other, desirable, KOR-mediated effects such as antinociception and blockade of drug-related behaviors. Nalfurafine has been shown to cause antinociception in several rodent models of pain at doses that did not cause sedative or aversive side effects. In contrast, traditional KOR agonists such as U50,488 and salvinorin A tested in the same paradigms caused both antinociceptive effects and negative side effects at similar doses (Wang et al. 2005; Schattauer et al. 2017; Liu et al. 2019). In these assays, nalfurafine appears to have a better therapeutic window, between desired therapeutic effect and negative side effects, than traditional KOR agonists.
Additionally, nalfurafine has been shown to block effects of alcohol and cocaine, such as reward and locomotor effects, at doses that did not cause sedative or aversive side effects (Mori et al. 2002; Hasebe et al. 2004; Zhou and Kreek 2019). These data, along with some of the in vitro signaling data that has been published on nalfurafine, have led to the hypothesis that nalfurafine is a KOR agonist that could be uniquely useful for the treatment of addictive diseases. There have not been head-to-head comparisons of nalfurafine and traditional KOR agonists in these drug-related behaviors, however. Additionally, there have been limited studies looking at the effect of nalfurafine on drug self-administration.
In this study, we compared nalfurafine and the full, unbiased KOR agonist U50,488. We used prolactin release in C57BL6 mice as a biomarker of KOR activation to find the minimal effective dose of nalfurafine in this system. We then tested low, but KOR-efficacious, doses of nalfurafine and U50,488 in mouse models of KOR-mediated sedation, aversion, and cocaine reward, and reinforcement to compare their therapeutic windows for these drug-related effects. This includes the first study on the effects of nalfurafine pretreatment on cocaine self-administration. The goal of this study was to directly compare nalfurafine and U50,488 to identify if nalfurafine had a unique profile for mediating drug-related behaviors.
Materials and methods
Chemical compounds
U50,488 [trans-(±)-3,4-dichloro-N-methyl-N-[2-(1-pyrrolidinyl)cyclohexyl] benzeneacetamide] was obtained from Sigma (St. Louis, MO, USA). Nalfurafine was obtained from Adooq Bioscience (Irvine, CA, USA). LY2444296 [(S)-3-fluoro-4-(4-((2-(3-fluorophenyl) pyrrolidin-1-yl) methyl)phenoxy)benzamide] was also synthesized by WuXi AppTec (Shanghai, China), with a small portion generously donated by Eli Lilly (Indianapolis, IN), which was used to confirm that the WuXi-synthesized compound was molecularly identical (as determined by reversed-phase high-performance liquid chromatography retention time). Nalfurafine and U50,488 were administered in saline, and LY2444296 was administered in vehicle (10% ethanol, 10% Tween-80, and 80% distilled-deionized water) for in vivo experiments.
Animals
Male C57BL6 mice (8–13 weeks, 20–30 g; Charles River Laboratories, Wilmington, MA, USA) were used in all studies. Mice were housed, four to a cage, in sound attenuated chambers with individual light controls, in stress-minimized rooms, with a reverse 12-h light/dark cycle (lights off 0700, lights on 1900), and food and water provided ad libitum. All animals were habituated to animal facility for at least 1 week, with daily handling, prior to studies. Animals were housed and euthanized in a manner approved by The Rockefeller University Institutional Animal Care and Use Committee.
Prolactin release
Mice were injected intraperitoneally with KOR agonists in saline 30 min prior to sampling. LY2444296 or vehicle (10% ethanol, 10% Tween-80, and 80% distilled-deionized water) pretreatment, if applicable, was given by intraperitoneal injection 60 min prior to sampling. Trunk blood was collected by rapid decapitation, followed within 2 h by preparation of serum. Serum prolactin levels were determined using a commercially available enzyme-linked immunoassay (Abcam, Cambridge, UK) following dilution of serum 5-fold in assay buffer. Because prolactin levels naturally vary depending on environmental stress, including laboratory handling procedures (Balcombe et al. 2004), mice are housed in a stress-minimized facility; however, there can still be significant variability between cohorts. For this reason, each experimental group is compared only with the concurrently run saline or vehicle control group.
Rotarod-mediated sedation
Rotarod experiments were conducted with mice using a dedicated rodent rotarod apparatus, with up to five animals tested concurrently (IITC Life Science, Woodland Hills, CA, USA). Rotarod rotation rate began at 3 rotations per minute and ramped to 30 rotations per minute over the course of 300 s. Upon falling from the rotarod, the times are recorded and animals are returned to their home cage. If animals are still on at the end of the 300-s ramping, the assay was terminated and animals returned to their home cage. Animals were acclimated to the rotarod on at least two occasions prior to the day of the test. On the day of the test, baseline times for each animal to fall off the rotarod were recorded. Mice were then injected intraperitoneally with vehicle or compound, and rotarod measurements conducted, beginning 0–2 min after injection, and then subsequently at select time points thereafter. Animals that failed to remain on the rotarod for at least 150 s during baseline testing were removed from the analysis.
Conditioned place preference (CPP)
Mouse place preference chambers had three distinct compartments, separated by removable doors (ENV-3013; Med Associates, St. Albans, VT). The central chamber had a solid gray floor and gray walls. The larger black and white compartments had stainless-steel rod and mesh floors, respectively. Experiments were performed in dimly lit, sound-attenuated chambers. The studies used a biased, counterbalanced design. During the preconditioning session, each animal was placed in the center compartment with the doors between chambers open. Time spent in each chamber was recorded for 30 min.
In a pretest session, mice had free access to all chambers for 30 min and were assessed for their chamber preference (black or white). Mice were assigned their non-preferred compartment (black or white) as the drug-paired chamber. Mice received two days of conditioning, with two conditioning sessions (treatment-paired and saline-paired) per day. Mice were counterbalanced for AM or PM drug-paired conditioning sessions, and sessions were at least 3 h apart. During each conditioning session, mice were pretreated with an intraperitoneal injection with a KOR agonist (U50,488, nalfurafine, or saline) for 15 min, before saline or cocaine injection. Then, mice were immediately placed in the appropriate chamber (with doors shut) for 30 min. After the two conditioning days, the post-test was performed in which mice were allowed free access to all chambers and time spent in each chamber was recorded for 30 min.
Intravenous self-administration (IVSA)
Adult mice (8–10 weeks) were anesthetized with 5% vaporized isoflurane and maintained on 2–3% isoflurane for the duration of the surgery. A catheter approximately 6 cm in length was inserted into the right jugular vein, up to a silicon ball placed 1.2–1.3 cm from the end of the catheter. Starting 48 h after surgery, catheters were flushed with physiological saline containing heparin and the antibiotic gentamicin every other day to maintain catheter patency. For the intravenous self-administration (IVSA) experiments, mouse self-administration chambers inside sound-attenuating chambers were used (ENV-307W; Med Associates, St. Albans, VT). Each chamber contained a wall with two small holes, one defined as active and one defined as inactive. During infusion, a cue light above the active hole was illuminated and then followed by a 20-s “time-out” period during which no infusions could occur. For the fixed ratio 1 (FR1) experiments (Fig. 8), mice were tested for 2 h daily, with infusions of 0.5 mg/kg/infusion cocaine on an FR1 schedule following each active nose-poke. Pretreatment experiments began when mice reached acquisition criteria, between 6 and 10 days after IVSA sessions began, depending on the animal. Acquisition criteria were > 70% of nose-pokes in the active hole and < 20% variation across two consecutive days. On each day of the pretreatment experiments, mice were given subcutaneous injections of a KOR agonist (U50,488 or nalfurafine) or saline immediately before being placed in self-administration chambers. For the antagonist blockade experiment, mice were pretreated with 3 mg/kg LY2444296 or vehicle by subcutaneous injection and then were given with the KOR agonist or saline injections before being immediately placed in self-administration chambers. For the progressive ratio experiment (Fig. 9), mice from a separate cohort received similar acquisition training (2 h daily IVSA on a FR1 schedule for a 0.5-mg/kg/infusion unit dose of cocaine) for 7 consecutive days followed by three sessions on a partial reinforcement FR3 schedule. The next day (session 11) mice were immediately pretreated with nalfurafine or saline by subcutaneous injection before being placed in the self-administration chambers for the 6-h 0.5-mg/kg/infusion progressive ratio session. An arithmetic PR3 reinforcement schedule was used such that the number of active nose-pokes required for each successive infusion increased by 3 nose-pokes (i.e., 3, 6, 9, 12, …). The break point was defined as the response requirement for the last reinforcer earned after either a 60-min limited hold (i.e., 60-min period without a reinforcer earned) or 6 h (to prevent health hazard). Mice remained in the operant chambers for the full 6 h even if break point was achieved early with nose-poke (active and inactive) response recorded with no programmed consequence (i.e., drug infusion). Catheter patency was checked weekly by infusion of ~ 30 μL ketamine (2 mg/mL), and only data from mice that passed this catheter patency test (defined as loss of muscle tone within a few seconds after ketamine infusion) were included in data analysis.
Statistical analysis
Analysis of variance (ANOVA) tests were used to assess the effects of drug treatments on in vivo endpoints. Post hoc multiple comparison tests were chosen based on relevant comparisons. Dunnett’s post hoc test was used to compare treatment groups to vehicle or saline control groups. The Newman-Keuls post hoc test was used to compare all treatment groups to each other, for antagonist pretreatment experiments for KOR specificity. Finally, Sidak’s multiple comparison post hoc test was used in the case of the intravenous self-administration experiments, in which there were repeated measures on time.
Results
KOR-mediated prolactin release
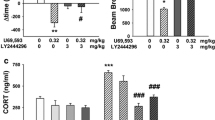
Prolactin release, downstream of the tuberoinfundibular dopaminergic system, is a biomarker of kappa opioid receptor activation in both human and animal models. In male C57BL6 mice, doses of 3 mg/kg and 10 mg/kg U50,488 caused significant prolactin release compared with saline treatment (Fig. 1). Ten micrograms per kilogram of nalfurafine caused a significant increase in serum prolactin levels in trunk blood 30 min after injection, compared with saline (Fig. 2a). This was blocked by a 15-min pretreatment with a KOR-selective dose of the antagonist LY2444296, indicating that this effect was mediated by the KOR (Fig. 2a). Doses of nalfurafine as low as 3 μg/kg caused significant prolactin release (Fig. 2b); however, lower doses of 0.3 μg/kg and 1 μg/kg did not (Fig. 2c). Levels of prolactin remained elevated for at least 60 min after intraperitoneal injection of 10 μg/kg nalfurafine, returning to baseline after 120 min (Fig. 2d).

Prolactin release caused by U50,488. Serum prolactin levels were measured in samples taken 30 min after intraperitoneal injection of varying doses of U50,488. Bars are averages (± SD). One-way ANOVA revealed a significant effect of treatment (F(4,35) = 4.66, p < 0.005). Dunnett’s multiple comparison test revealed significant differences between the saline-treated and both the 3-mg/kg and 10-mg/kg U50,488 groups (*p < 0.05, **p < 0.01)

Prolactin release caused by the morphinan compound nalfurafine. All bar graphs are shown as averages (± SD). a Mice received an injection intraperitoneally of either LY2444296 or vehicle 15 min before an injection of 10 μg/kg nalfurafine. Samples were collected 30 min after the second injection. Two-way ANOVA (LY244296 pretreatment × nalfurafine treatment) revealed a significant effect of both LY2444296 pretreatment (F(1,30) = 86.75, p < 0.0001) and nalfurafine treatment (F(1,30) = 115.3, p < 0.0001) and their interaction (F(1,30) = 96.36, p < 0.0001). The Newman-Keuls multiple comparison test revealed significant differences between the vehicle/nalfurafine group and all other groups (****p < 0.0001). b Serum prolactin levels were measured in samples taken 30 min after intraperitoneal injection of varying doses of nalfurafine. One-way ANOVA revealed a significant effect of treatment (F(4,35) = 9.67, p < 0.0001). Dunnett’s multiple comparison test revealed significant differences between nalfurafine- or U50,488-treated groups and saline-treated control group (**p < 0.005, ****p < 0.0001). c Serum prolactin levels were measured in samples taken 30 min after intraperitoneal injection of varying doses of nalfurafine. One-way ANOVA revealed a significant effect of treatment (F(4,30) = 13.64, p < 0.0001). Dunnett’s multiple comparison test revealed significant differences between nalfurafine- or U50,488 treated groups and saline-treated control group (*p < 0.05, ***p < 0.0005). d Serum prolactin levels were measured in samples taken 15–120 min after intraperitoneal injection of 10 μg/kg nalfurafine or saline. Two-way ANOVA (treatment × time) revealed a significant effect of treatment (F(1,56) = 62.56, p < 0.0001), time (F(3,56) = 7.24, p < 0.0005), and their interaction (F(3,56) = 5.73, p < 0.005). The Newman-Keuls multiple comparison at each time point revealed significant differences between the saline- and nalfurafine-treated groups at early time points (**p < 0.005, ***p < 0.0005, ****p < 0.0001)
KOR-mediated rotarod sedation
The rotarod assay was used to assess the sedative effect of various doses of both U50,488 and nalfurafine in male C57BL6 mice. Rotarod performance was assessed at baseline and then tested again at 0–2 min, 30 min, and 60 min post-injection. Rotarod performance is reported here as a percentage of baseline performance. The lowest dose of U50,488 tested (3 mg/kg) did not cause any impairment in rotarod performance, while doses of 10 mg/kg and 30 mg/kg U50,488 caused significant decreases in rotarod performance at 30 min post-injection (Fig. 3).

Rotarod sedation caused by U50,488. Mice were treated with varying doses of U50,488 or saline, and rotarod performance was measured at selected time points after injection, and compared with baseline. Bars are averages (± SD). Two-way ANOVA (treatment × time) revealed a significant effect of treatment (F(3,24) = 13.77, p < 0.0001), time (F(2,48) = 7.05, p < 0.005), and their interaction (F(6,48) = 3.03, p < 0.05). Dunnett’s multiple comparison test at each time point revealed significant differences between the U50,488- and saline-treated groups (**p < 0.005, ****p < 0.0001)
Nalfurafine treatment at a dose of 100 μg/kg caused significant rotarod sedation at both 30- and 60-min post-injection that was blocked by pretreatment with the KOR antagonist LY2444296 (Fig. 4a). Nalfurafine treatment at higher doses of 300 μg/kg and 3 mg/kg resulted in a significant decrease in rotarod performance, to 13% of baseline, after 30 min (Fig. 4b). After 60 min, the mice that received 300 μg/kg nalfurafine were recovering (rotarod performance of 24% of baseline), while the 3-mg/kg group had decreased performance to 10% of baseline. Despite this significant sedation, after 24 h, mice exhibited normal home-cage behavior with normal mobility (experimenter observation). At lower doses of 3 μg/kg and 10 μg/kg, however, nalfurafine did not cause rotarod sedation compared with baseline (Fig. 4c).

Rotarod sedation caused by nalfurafine. Rotarod performance was measured at select time points after IP injection of nalfurafine, or saline, and compared with baseline rotarod performance in all experiments. Bars are averages (± SD). a Mice were pretreated for 15 min with 3 mg/kg LY2444296 before injection with 100 μg/kg nalfurafine. There was no effect of LY2444296 alone on rotarod sedation (Dunn et al. 2018). Two-way ANOVA (treatment × time) revealed a significant effect of treatment (F(2,21) = 14.38, p < 0.0005), time (F(2,24) = 7.99, p < 0.005), and their interaction (F(4,42) = 2.66, p < 0.05). Dunnett’s multiple comparison test at each time point revealed significant differences between the vehicle/saline control group and vehicle/nalfurafine group only (**p < 0.005, ***p < 0.0005). b Mice were treated with varying concentrations of nalfurafine, followed by assessment of rotarod sedation. Two-way ANOVA (treatment × time) revealed a significant effect of treatment (F(4,31) = 48.72, p < 0.0001), time (F(2,62) = 26.64, p < 0.0001), and their interaction (F(8,62) = 4.0, p < 0.0005). Dunnett’s multiple comparison test at each time point revealed significant differences between nalfurafine treatment groups and saline-treated control group (**p < 0.05, ***p < 0.0005, ****p < 0.0001). c Mice were treated with varying concentrations of nalfurafine, as well as 10 mg/kg U50,488 as a comparison, followed by assessment of rotarod sedation. Two-way ANOVA (treatment × time) revealed a significant effect of treatment (F(4,30) = 3.38), p < 0.05) and time (F(2,60) = 7.12, p < 0.005) but no effect of their interaction (F(8,60) = 1.19). Dunnett’s multiple comparison test at each time point revealed significant differences between the nalfurafine and U50,488 treatment groups and saline-treated control group (**p < 0.05)
Effect of KOR agonists on conditioned place preference and aversion
A place-conditioning paradigm was used to assess both the aversive effects of nalfurafine and U50,488 and their effects on cocaine reward. In a three-chamber conditioned place preference paradigm, 15 mg/kg cocaine caused an increase in the proportion of time spent in the drug-paired chamber indicating cocaine reward as expected. When mice received an injection of a low dose of U50,488 (3 mg/kg) 15 min before the cocaine injection, this preference was attenuated (Fig. 5). This dose of U50,488 had no effect alone compared with saline (Fig. 5). A pretreatment of 10 μg/kg nalfurafine also attenuated cocaine conditioned place preference, and had no effect compared to saline on its own (Fig. 6a). While there was no significant effect of cocaine alone compared with saline in the 3-μg/kg nalfurafine experiment, there was a significant difference between the cocaine group and the 3-μg/kg nalfurafine/cocaine group, indicating this dose of nalfurafine was also able to block cocaine reward without causing aversion on its own (Fig. 6b).

U50,488 blockade of cocaine conditioned place preference. Mice were treated with 3 mg/kg U50,488 or saline, and then 15 min later treated with 15 mg/kg cocaine or saline before immediately being placed in appropriate conditioning chambers for 30 min. Mice received 4 total conditioning sessions, 2 in each box, across 2 days. Bars are averages (± SD). Two-way ANOVA (U50,488 pretreatment × cocaine treatment) revealed a significant effect of cocaine treatment (F(1,36) = 5.11, p < 0.05) but no effect of U50,488 pretreatment (F(1,36) = 1.60) or interaction (F(1,36) = 4.07). The Newman-Keuls multiple comparison test revealed significant differences between the cocaine group and all other groups (*p < 0.05)

Nalfurafine blockade of cocaine conditioned place preference. Bars are averages (± SD). a Mice were treated with 10 μg/kg nalfurafine or saline, and then 15 min later treated with 15 mg/kg cocaine or saline before immediately being placed in appropriate conditioning chambers for 30 min. Mice received 4 total conditioning sessions, 2 in each box, across 2 days. Two-way ANOVA (nalfurafine pretreatment × cocaine treatment) revealed a significant effect of cocaine treatment (F(1,32) = 22.59, p < 0.0001) but not nalfurafine pretreatment (F(1,32) = 1.68), as well as a significant cocaine × nalfurafine interaction (F(1,32) = 9.01, p < 0.01). The post hoc Newman-Keuls analysis revealed significant differences between nalfurafine/cocaine (**p < 0.005), nalfurafine/saline (**p < 0.005), and saline/saline (***p < 0.0005) groups and the saline/cocaine group, and no differences between other groups. b Mice were pretreated with 3 μg/kg nalfurafine or saline, followed by 15 mg/kg cocaine or saline using identical methods. Two-way ANOVA (nalfurafine pretreatment × cocaine treatment) revealed no significant effect of cocaine treatment (F(1,28) = 2.06) or nalfurafine pretreatment alone (F(1,28) = 1.88); however, there was a significant cocaine × nalfurafine interaction (F(1,24) = 5.22, p < 0.05). The post hoc Newman-Keuls analysis revealed a significant difference between saline/cocaine and nalfurafine/cocaine (*p < 0.05)
Effect of KOR agonists on cocaine self-administration
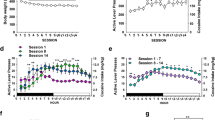
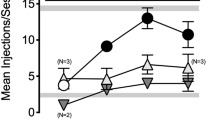
Intravenous self-administration was used to test the effects of these KOR agonists on the reinforcing effects of cocaine, through a traditional operant paradigm. After 7 days of 2-h, FR1 self-administration sessions of 0.5 mg/kg/infusion cocaine, mice were pretreated with an injection of 3 mg/kg U50,488 or saline before sessions on the 8th and 9th days. Pretreatment with U50,488 caused an increase in active nose-pokes compared with saline (Fig. 7). In a similar paradigm, mice that had acquired self-administration of 0.5 mg/kg/infusion cocaine were given daily pretreatments of 10 μg/kg nalfurafine. This also resulted in an increase in active nose-pokes that was sustained over the next 7 days (Fig. 8). This potentiation in responding was blocked by a 15-min pretreatment with a KOR-selective dose of the antagonist LY2444296 (Fig. 8). Pretreatment with 3 μg/kg nalfurafine, however, had no effect on cocaine self-administration (Fig. 8). Similarly, in a progressive ratio paradigm, the 10-μg/kg dose of nalfurafine caused a significant increase in cocaine-seeking behavior as measured by break point (Fig. 9).

Effect of U50,488 on cocaine IVSA. Mice intravenously self-administered 0.5 mg/kg/infusion cocaine under an FR1 schedule during 2-h daily sessions until acquisition was stable (< 20% variation across two consecutive days) and > 70% of total nose-pokes were in the active hole. Mice were injected with 3 mg/kg U50,488 or saline immediately before IVSA sessions on pretreatment days (days 8 and 9). Data points and bars are averages (± SD). a Daily cocaine intake. Two-way ANOVA (treatment × time), with repeated measures on time, revealed a significant effect of treatment (F(1,12) = 5.84, p < 0.05) and time (F(8,96) = 15.78, p < 0.0001) but no interaction (F(8,96) = 1.49). Sidak’s multiple comparison test for each day revealed a significant difference between the saline- and U50,488-treated groups on day 8 (*p < 0.05). b Average cocaine intake during the pretreatment days (days 8 and 9). T test revealed a significant increase in cocaine intake for the U50,488-treated group on days 8 and 9 compared with the saline-treated group (****p < 0.0001)

Effect of nalfurafine on FR1 schedule cocaine IVSA. Mice intravenously self-administered 0.5 mg/kg/infusion cocaine under an FR1 schedule for 2 h daily. Data points and bars are averages (± SD). a Daily cocaine intake. On pretreatment days (days 7–13, shaded), mice were injected with 3 mg/kg LY2444296 or vehicle, and then 15 min later injected with 10 μg/kg nalfurafine or saline immediately before IVSA sessions. Two-way ANOVA (overall treatment condition × time), with repeated measures on time, revealed no overall effect of treatment (F(3,24) = 0.75), but a significant effect of both time (F(12,288) = 7.31, p < 0.0001) and treatment × time interaction (F(36,288) = 1.63, p < 0.05). Sidak’s multiple comparisons test for each day revealed only a significant difference between the vehicle/nalfurafine group and the LY/saline group on day 11 (*p < 0.05). b Average cocaine intake during the pretreatment days (days 7–13). Two-way ANOVA (LY pretreatment × nalfurafine treatment) revealed no significant effect of LY244296 pretreatment alone (F(1,24) = 1.67) or nalfurafine treatment alone (F(1,24) = 2.67); however, there was a significant interaction of LY2444296 × nalfurafine (F(1,24) = 4.71, p < 0.05). The Newman-Keuls post hoc analysis revealed a significant difference between the vehicle/nalfurafine and LY/saline groups (*p < 0.05). c Effect of 3 μg/kg nalfurafine on cocaine self-administration. Mice intravenously self-administered 0.5 mg/kg/infusion cocaine under an FR1 schedule for 2 h daily until responding was stable (< 20% variation across two consecutive days) and > 70% of total nose-pokes were in the active hole. On pretreatment days (days 8–14), mice were injected with 3 μg/kg nalfurafine or saline immediately before IVSA sessions. Two-way ANOVA (treatment × time), with repeated measures on time, showed a significant effect of time (F(13,234) = 9.17), but no effect of treatment (F(1,18) = 0.194) and no interaction

Effect of nalfurafine on progressive ratio schedule of cocaine IVSA. Data points and bars are averages (± SD), with error shown as bars (a) or shaded area (b). a Mice intravenously self-administered 0.5 mg/kg/infusion cocaine under an FR1 schedule during 2-h daily sessions for 7 days. The mice then had 3 sessions on a partial reinforcement schedule (FR3) before the single 6-h progressive ratio (PR3) experiment. a Total cocaine intake and break points during progressive ratio experiment. One-way ANOVA revealed no overall effect of treatment (F(2,26) = 3.07). Dunnett’s multiple comparisons test revealed only a significant difference between the saline group and the 10-μg/kg nalfurafine group (*p < 0.05). b Active nose-pokes during each hour of the progressive ratio experiment. Two-way ANOVA (overall treatment condition × time), with repeated measures on time, revealed an overall effect of treatment (F(2,162) = 6.51, p < 0.005) and time (F(5,162) = 6.08, p < 0.0001) but no effect of their interaction (F(10, 162) = 1.38). Dunnett’s multiple comparisons test for each hour-bin revealed only a significant difference between the saline group and the 10-μg/kg nalfurafine group during the 3rd (*< 0.05) and 4th hours of the session (**p < 0.005)
Discussion
In the search for KOR-targeting therapeutics, nalfurafine can be used as an example of a clinically approved selective KOR agonist that is well-tolerated at low doses. It is unclear, however, which properties of nalfurafine could potentially be emulated in future drug discovery efforts for different KOR-related indications. In this study, we used serum prolactin as a marker of KOR efficacy in vivo to determine doses of nalfurafine and U50,488 that were KOR-efficacious without inducing the common KOR agonist side effects such as aversion and sedation. We then tested these doses of nalfurafine for effects on complementary cocaine-related behaviors and compared them to a low dose of the well-studied, full, unbiased agonist U50,488.
The lowest dose of U50,488 that caused KOR-mediated prolactin release was 3 mg/kg (Fig. 1), while nalfurafine caused prolactin release at doses of 3 μg/kg and higher (Fig. 2b). In the rotarod assay, U50,488 at higher doses caused significant sedation, as expected; however, the dose of 3 mg/kg did not have any apparent rotarod effects (Fig. 3). Nalfurafine caused significant KOR-mediated sedation only at doses of 30 μg/kg and higher (Fig. 4b, c), consistent with previous reports of nalfurafine-induced sedation (Hasebe et al. 2004; Zhou and Kreek 2019). Our lab recently reported that rotarod-mediated sedation in mice significantly correlates with β-arrestin-2 recruitment for KOR agonists (Dunn et al. 2019), and the sedation shown here is consistent with the robust β-arrestin-2 recruitment by nalfurafine previously reported by several groups. Prolactin release, on the other hand, is thought to be mediated by G-protein signaling activity. Nalfurafine caused significant prolactin release too, which is consistent with its well-established robust G-protein activation in vitro. Interestingly, at the extremely low doses used in humans to treat pruritus, increased serum prolactin was observed in a small percentage of patients (Kumagai et al. 2012). Together, these results suggest that nalfurafine and U50,488 activate both the G-protein and β-arrestin-2 signaling pathways downstream of the KOR in mice, but with different dose thresholds. We found both agonists to be less potent for sedative effects than other KOR-mediated effects, in this case prolactin release.
At both 3 μg/kg and 10 μg/kg, nalfurafine blocked cocaine conditioned place preference without causing aversion (Fig. 6). Three milligrams per kilogram of U50,488 had a similar effect blocking conditioned place preference (Fig. 5). Here, nalfurafine is three orders of magnitude more potent than U50,488 in vivo. While reports vary in the literature, nalfurafine’s increased potency is not as dramatic in in vitro studies ([35S]GTPγS EC50 of 1.3 nM (± 0.7) for nalfurafine compared with 0.6 nM (± 0.2) for U50,488 (Dunn et al. 2019); [35S]GTPγS EC50 of 0.025 nM (± 0.003) for nalfurafine compared with 2.2 nM (± 0.3) for U50,488 (Wang et al. 2005)).
As in previous studies, nalfurafine was more potent for the therapeutically advantageous effect than the sedative and aversive side effects; however in this case, this property was not unique to nalfurafine. In fact, the dose of U50,488 that blocked cocaine conditioned place preference (3 mg/kg) did not cause sedation or aversion, demonstrating that the reward-blockade effects of KOR agonists might be especially sensitive compared with these other effects. This suggests that this therapeutic window, between modulation of cocaine reward and sedative/aversive side effects, could be characteristic of KOR agonists in general. The mechanism of this uncoupling could be related to different signaling mechanisms, as has been suggested for the uncoupling of therapeutic effects and sedation; however, it is also possible that both antireward and aversion are mediated by dopamine inhibition downstream of the KOR and their uncoupling is a result of different thresholds.
In the IVSA experiments using 0.5 mg/kg/infusion cocaine, an immediate pretreatment of 3 mg/kg U50,488 or 10 μg/kg nalfurafine caused a significant increase in drug intake (Figs. 7 and 8). Prolactin studies indicated that the effects of nalfurafine last for the majority of the 120-min IVSA sessions (Fig. 2d). In fact, mice that were treated with 10 μg/kg nalfurafine before the progressive ratio experiment increased their active responses during the first several hours of the PR session, even as the requirements for earning infusions increased (Fig. 9b). Additionally, while there was no effect of 3 μg/kg in the FR1 schedule, there was a trend towards an increase in both break point and total active nose-pokes with the PR3 schedule (Fig. 9). This data is consistent with some previous studies of KOR agonists like U50,488 that have also shown a potentiation of cocaine reward, using both conditioned place preference (CPP) (McLaughlin et al. 2006) and IVSA, at certain doses of cocaine (Kuzmin et al. 1997). Additionally, in a food vs cocaine-choice paradigm in rhesus monkeys, chronic U50,488 treatment increased the response for cocaine over food, even as it decreased response rates overall (Mello and Negus 1998; Stevens Negus 2004). Future studies investigating the effects of U50,488 and nalfurafine on the self-administration of varying doses of cocaine, as well as dopamine release in the brain throughout these experiments, would help to clarify these mechanisms.
The divergent effects of 3 mg/kg U50,488 and 10 μg/kg nalfurafine in cocaine CPP and cocaine IVSA also highlight the differences between these assays. While both assays are commonly used to measure drug reward/reinforcement, studies have suggested that they do not measure the same reward processes in the brain (reviewed in Bardo and Bevins 2000). Certain classes of drugs, such as the hallucinogen lysergic acid diethylamide (LSD), cause CPP in rats but are not self-administered (Meehan and Schechter 1998), and others, such as pentobarbital, are self-administered but do not cause CPP (Collins et al. 1983; Lew and Parker 1998). Additionally, pretreatment of animals with D2-receptor antagonists blocks the self-administration of cocaine, but not cocaine CPP, suggesting that these behaviors involve different mechanisms (Caine and Koob 1994; Cervo and Samanin 1995).
A lower dose of 3 μg/kg nalfurafine, however, blocked cocaine conditioned place preference (Fig. 6b) and did not cause any measurable change in cocaine self-administration (Fig. 8), suggesting that these behaviors could have different thresholds of sensitivity to KOR agonist effects. Low-dose nalfurafine (as low as 0.3 μg/kg) has also been effective in blocking alcohol intake in animal models, in combination with naltrexone, a MOR antagonist and KOR G-protein biased partial agonist (Zhou and Kreek 2019). It is also possible that other pharmacological strategies, such as KOR partial agonists first suggested as a treatment for cocaine addiction in the late 1990s, could have similar effects (Kreek 1997). Overall, these data suggest that low doses of potent KOR agonists may be able to successfully modulate drug-related behaviors at doses that do not cause the side effects that have hampered the development of KOR agonists previously, and that this therapeutic window is not unique to nalfurafine. KOR compounds with different downstream signaling preferences may show different therapeutic windows, and this possibility awaits further investigation. It appears that cocaine reward-blockade effects, even at very low doses, could be characteristic of KOR activation, and future studies could be aimed at exploiting this property for therapeutic use.
References
Albert-Vartanian A, Boyd MR, Hall AL et al (2016) Will peripherally restricted kappa-opioid receptor agonists (pKORAs) relieve pain with less opioid adverse effects and abuse potential? J Clin Pharm Ther 41:371–382. https://doi.org/10.1111/jcpt.12404
Balcombe JP, Barnard ND, Sandusky C (2004) Laboratory routines cause animal stress. Contemp Top Lab Anim Sci 43(6):42–51
Bardo MT, Bevins RA (2000) Conditioned place preference: what does it add to our preclinical understanding of drug reward? Psychopharmacology 153:31–43
Bourgeois C, Werfel E, Galla F et al (2014) Synthesis and pharmacological evaluation of 5-pyrrolidinylquinoxalines as a novel class of peripherally restricted κ-opioid receptor agonists. J Med Chem 57:6845–6860. https://doi.org/10.1021/jm500940q
Brust TF, Morgenweck J, Kim SA et al (2016) Biased agonists of the kappa opioid receptor suppress pain and itch without causing sedation or dysphoria. Sci Signal 9:ra117–ra117. https://doi.org/10.1126/scisignal.aai8441
Caine SB, Koob GF (1994) Effects of dopamine D-1 and D-2 antagonists on cocaine self-administration under different schedules of reinforcement in the rat. J Pharmacol Exp Ther 270
Cervo L, Samanin R (1995) Effects of dopaminergic and glutamatergic receptor antagonists on the acquisition and expression of cocaine conditioning place preference. Brain Res 673:242–250. https://doi.org/10.1016/0006-8993(94)01420-M
Collins RJ, Weeks JR, Cooper MM et al (1983) Prediction of abuse liability of drugs using IV self-administration by rats. Psychopharmacology 82:6–13. https://doi.org/10.1007/BF00426372
Di Chiara G, Imperato A (1988) Opposite effects of mu and kappa opiate agonists on dopamine release in the nucleus accumbens and in the dorsal caudate of freely moving rats. J Pharmacol Exp Ther 244
Dunn AD, Reed B, Guariglia C et al (2018) Structurally related kappa opioid receptor agonists with substantial differential signaling bias: neuroendocrine and behavioral effects in C57BL6 Mice. Int J Neuropsychopharmacol 21:847–857. https://doi.org/10.1093/ijnp/pyy034
Dunn AD, Reed B, Erazo J et al (2019) Signaling properties of structurally diverse kappa opioid receptor ligands: toward in vitro models of in vivo responses. ACS Chem Neurosci 10:3590–3600. https://doi.org/10.1021/acschemneuro.9b00195
Hasebe K, Kawai K, Suzuki T et al (2004) Possible pharmacotherapy of the opioid κ receptor agonist for drug dependence. Ann N Y Acad Sci 1025:404–413. https://doi.org/10.1196/annals.1316.050
Kozono H, Yoshitani H, Nakano R (2018) Post-marketing surveillance study of the safety and efficacy of nalfurafine hydrochloride (Remitch® capsules 2.5 μg) in 3,762 hemodialysis patients with intractable pruritus. Int J Nephrol Renovasc Dis Volume 11:9–24. https://doi.org/10.2147/IJNRD.S145720
Kreek MJ (1997) Opiate and cocaine addictions: challenge for pharmacotherapies. Pharmacol Biochem Behav 57:551–569. https://doi.org/10.1016/S0091-3057(96)00440-6
Kreek MJ, LaForge KS, Butelman E (2002) Pharmacotherapy of addictions. Nat Rev Drug Discov 1:710–726. https://doi.org/10.1038/nrd897
Kumagai H, Ebata T, Takamori K et al (2012) Efficacy and safety of a novel ĸ-agonist for managing intractable pruritus in dialysis patients. Am J Nephrol 36:175–183. https://doi.org/10.1159/000341268
Kuzmin AV, Semenova S, Gerrits MAF et al (1997) κ-Opioid receptor agonist U50,488H modulates cocaine and morphine self-administration in drug-naive rats and mice. Eur J Pharmacol 321:265–271. https://doi.org/10.1016/S0014-2999(96)00961-2
Lew G, Parker LA (1998) Pentobarbital-induced place aversion learning. Anim Learn Behav 26:219–224. https://doi.org/10.3758/BF03199214
Liu JJ, Sharma K, Zangrandi L et al (2018) In vivo brain GPCR signaling elucidated by phosphoproteomics. Science (80- ) 360:eaao4927. https://doi.org/10.1126/science.aao4927
Liu JJ, Chiu Y-T, DiMattio KM et al (2019) Phosphoproteomic approach for agonist-specific signaling in mouse brains: mTOR pathway is involved in κ opioid aversion. Neuropsychopharmacology 44:939–949. https://doi.org/10.1038/s41386-018-0155-0
MacLean KA, Johnson MW, Reissig CJ et al (2013) Dose-related effects of salvinorin A in humans: dissociative, hallucinogenic, and memory effects. Psychopharmacology 226:381–392. https://doi.org/10.1007/s00213-012-2912-9
McLaughlin JP, Land BB, Li S et al (2006) Prior activation of kappa opioid receptors by U50,488 mimics repeated forced swim stress to potentiate cocaine place preference conditioning. Neuropsychopharmacology 31:787–794. https://doi.org/10.1038/sj.npp.1300860
Meehan SM, Schechter MD (1998) LSD produces conditioned place preference in male but not female fawn hooded rats. Pharmacol Biochem Behav 59:105–108. https://doi.org/10.1016/S0091-3057(97)00391-2
Mello NK, Negus SS (1998) Effects of kappa opioid agonists on cocaine- and food-maintained responding by rhesus monkeys. J Pharmacol Exp Ther 286:812–824
Mores KL, Cummins BR, Cassell RJ, van Rijn RM (2019) A review of the therapeutic potential of recently developed G protein-biased kappa agonists. Front Pharmacol 10:407. https://doi.org/10.3389/fphar.2019.00407
Mori T, Nomura M, Nagase H et al (2002) Effects of a newly synthesized κ-opioid receptor agonist, TRK-820, on the discriminative stimulus and rewarding effects of cocaine in rats. Psychopharmacology 161:17–22. https://doi.org/10.1007/s00213-002-1028-z
Nagase H, Watanabe A, Nemoto T et al (2010) Drug design and synthesis of a novel kappa opioid receptor agonist with an oxabicyclo[2.2.2]octane skeleton and its pharmacology. Bioorg Med Chem Lett 20:121–124. https://doi.org/10.1016/j.bmcl.2009.11.027
Nemoto T, Yamamoto N, Wada N et al (2013) The effect of 17-N substituents on the activity of the opioid κ receptor in nalfurafine derivatives. Bioorg Med Chem Lett 23:268–272. https://doi.org/10.1016/J.BMCL.2012.10.100
Pande AC, Pyke RE, Greiner M et al (1996) Analgesic efficacy of enadoline versus placebo or morphine in postsurgical pain. Clin Neuropharmacol 19:451–456. https://doi.org/10.1097/00002826-199619050-00009
Schattauer SS, Kuhar JR, Song A, Chavkin C (2017) Nalfurafine is a G-protein biased agonist having significantly greater bias at the human than rodent form of the kappa opioid receptor. Cell Signal 32:59–65. https://doi.org/10.1016/j.cellsig.2017.01.016
Stevens Negus S (2004) Effects of the kappa opioid agonist U50,488 and the kappa opioid antagonist nor-binaltorphimine on choice between cocaine and food in rhesus monkeys. Psychopharmacology 176:204–213. https://doi.org/10.1007/s00213-004-1878-7
Togashi Y, Umeuchi H, Okano K et al (2002) Antipruritic activity of the kappa-opioid receptor agonist, TRK-820. Eur J Pharmacol 435:259–264. https://doi.org/10.1016/s0014-2999(01)01588-6
Wadenberg M-LG (2003) A review of the properties of spiradoline: a potent and selective kappa-opioid receptor agonist. CNS Drug Rev 9:187–198
Wang Y, Tang K, Inan S et al (2005) Comparison of pharmacological activities of three distinct kappa ligands (Salvinorin A, TRK-820 and 3FLB) on kappa opioid receptors in vitro and their antipruritic and antinociceptive activities in vivo. J Pharmacol Exp Ther 312:220–230. https://doi.org/10.1124/jpet.104.073668
White KL, Robinson JE, Zhu H et al (2015) The G protein-biased κ-opioid receptor agonist RB-64 is analgesic with a unique spectrum of activities in vivo. J Pharmacol Exp Ther 352:98–109
Zhang Y, Butelman ER, Schlussman SD et al (2004) Effect of the endogenous κ opioid agonist dynorphin A(1-17) on cocaine-evoked increases in striatal dopamine levels and cocaine-induced place preference in C57BL/6J mice. Psychopharmacology 172:422–429. https://doi.org/10.1007/s00213-003-1688-3
Zhou Y, Kreek MJ (2019) Combination of clinically utilized kappa-opioid receptor agonist nalfurafine with low-dose naltrexone reduces excessive alcohol drinking in male and female mice. Alcohol Clin Exp Res 43:1077–1090. https://doi.org/10.1111/acer.14033
Acknowledgments
We gratefully acknowledge Dr. Eduardo Butelman for intellectual contributions and Dr. Yong Zhang for valuable guidance on mouse self-administration experiments. We thank Dr. Linda Rorick-Kehn for facilitating the generous provision by Eli Lilly and Co. of a small portion of LY2444296. We thank the Rockefeller University Comparative Biosciences Center and Jonathon Negron for animal husbandry.
Conflict of interest
The authors declare that they have no conflicts of interest.
Funding
This work was supported by the Dr. Miriam and Sheldon Adelson Medical Research Foundation.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Animals were housed and euthanized in a manner approved by The Rockefeller University Institutional Animal Care and Use Committee.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Dunn, A., Windisch, K., Ben-Ezra, A. et al. Modulation of cocaine-related behaviors by low doses of the potent KOR agonist nalfurafine in male C57BL6 mice. Psychopharmacology 237, 2405–2418 (2020). https://doi.org/10.1007/s00213-020-05543-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00213-020-05543-7