
Abstract
New experimental breast cancer therapies directed against novel targets are currently in clinical These experimental agents are likely to be effective for a niche of breast cancers with specific “driver mutations”. The ability to perform comprehensive molecular profiling of individual tumors has rapidly expanded over the last few years, and new DNA sequencing technologies require relatively limited quantities of fresh or archived paraffin-embedded or snap-frozen tumor tissue and provide rapid turnaround of sequencing results within a few weeks or less. All these technologies provide an unprecedented opportunity to identify patients with rare “driver” molecular alternations that are candidates for proof-of-concept clinical trials with matched targeted therapy, in the context of basket trials. The aim of this chapter on molecular profiling is to summarize the known recurrent molecular alterations in breast cancer that are potentially amenable to investigational targeted therapy, to provide an overview of the existing technological platforms for molecular profiling and ongoing or planned institutional/national screening initiatives and to outline a vision for molecular screening that may be integrated into the future activities of breast cancer research.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

Keywords
1 Introduction
There is an exciting array of experimental breast cancer therapies directed against novel targets that are currently in clinical development. These investigational agents are likely to be effective for small subsets of breast cancers with specific “driver mutations”. The ability to perform comprehensive molecular profiling of individual tumours has rapidly expanded over the last few years, as the cost of DNA sequencing technologies that allow for targeted multiplex “hotspot” mutation testing or deeper targeted exome and whole genome DNA sequencing has become cheaper than traditional Sanger-based DNA sequencing methods. New DNA sequencing technologies require relatively limited quantities of fresh or archived paraffin-embedded or snap-frozen tumour tissue and provide rapid turnaround of sequencing results within a few weeks or less. These technological advances allow for the prospect of point-of-care molecular profiling that can be used to guide the development of personalized breast cancer medicine therapy. For an international collective of academic breast cancer researchers, this provides an unprecedented opportunity to identify patients with rare “driver” molecular alternations that are candidates for proof-of-concept clinical trials with matched targeted therapy. The aim of this report on molecular profiling is to review the known recurrent molecular alterations in breast cancer that are potentially amenable to investigational targeted therapy, to provide an overview of the existing technological platforms for molecular profiling and ongoing or planned institutional/national screening initiatives and to outline a vision for molecular screening that may be integrated into the future activities of breast cancer research.
2 Background and Rationale
Personalized Medicine and New Drug Development
The “oncogene revolution” has led to an explosion of molecularly targeted therapeutics in preclinical and clinical development over the last decade (Hanahan and Weinberg 2000). It is estimated that there are more than 800 targeted anticancer therapies currently in various stages of clinical development. Disappointingly, historical data indicate that only 5% of these investigational therapies will ultimately progress to registration for widespread use. These high attrition rates have multiple causes, including lack of efficacy and excessive toxicity (Kola and Landis 2004). In particular, when patients are selected for phase III trials based on histopathology alone, a targeted drug with a 5–10% single-agent response rate runs a high risk of failure (Stewart and Kurzrock 2009). Recent efforts to systematically sequence cancer genomes have revealed that individual tumours frequently harbour multiple “driver” somatic mutations that confer growth advantage and positive selection (Stratton et al. 2009).
The increasing identification of specific somatic mutations and other genetic aberrations that drive cancers leaves us on the threshold of a new era of “personalized cancer medicine”, in which specific biomarkers will be used to direct targeted agents only to those patients deemed most likely to respond. The potential medical, scientific and economic benefits of such a personalized approach to cancer therapy are immense and self-evident. Yet despite some important advances, only a limited number of approved targeted agents have had their approvals predicated on specific biomarkers of sensitivity or resistance. The premises behind personalized cancer medicine include (1) genetic aberrations exist in human malignancies; (2) a subset of these aberrations, often present across multiple cancer types, have functional relevance as “drivers” for oncogenesis and tumour progression; (3) such genetic aberrations are potentially “druggable” targets; and (4) there are tolerable medicinal compounds that can effectively modulate such targets (Greenman et al. 2007). A key requirement of this new, personalized approach to anticancer therapy is that specific patients must be matched to a particular drug or combination of drugs. Molecular profiling of tumours to identify somatic mutations and/or other genetic aberrations are examples of enrichment strategies to assist in matching patients to drugs or treatments that have gained increasing interest in the oncology community (Callaway 2010). The true merits of such personalized medicine strategies remain to be established. However, proof-of-concept clinical trials that establish the value of matching targeted treatments to rare molecular alterations in breast cancer and other malignancies are beyond the scope of any single pharmaceutical sponsor, cancer treatment facility or national cancer agency and will ultimately require international collaboration. Recent examples demonstrate that sequential testing of infrequent genomic alterations to identify candidates for clinical trials with matched targeted is inefficient, expensive and wasteful of scarce archived tumour tissue resources. Comprehensive molecular screening programs, which provide simultaneous testing of multiple biomarkers early in the course of a patient’s natural history of disease, are most likely to advance personalized cancer medicine.
Genomic Alterations in Breast Cancer
Somatic mutations are responsible for approximately 90% of breast cancers. Although data from comprehensive, large-scale breast cancer DNA sequencing projects are still awaited (Ellis et al. 2007), key features of the genomic breast cancer landscape have begun to emerge. First, although multiple regions of copy gain are observed, none occurs as frequently as 17q12 which harbours ERBB2/HER2; second, there are high-frequency somatic point mutations in three “gene mountains” (Greenman et al. 2007) – TP53 (44%), PIK3CA (26%) and CDH1 (19%) – but low-frequency recurrent point mutations (<5%) are also seen in genes that are validated drug targets in other types of cancer (i.e. KRAS, BRAF and EGFR); third, genes with somatic point mutations are also frequently regions of copy number gain in independent tumour samples (i.e. PIK3CA, ERBB2), highlighting their importance as oncogenes; and fourth, point mutations are observed in multiple components of a signalling pathway at a higher rate than expected by chance alone (i.e. PIK3CA, PTEN, AKT1) indicating the relevance of the signalling pathway as a therapeutic target in mutated tumours. Additional data from large-scale sequencing projects, such as the Cancer Genome Atlas (TGCA) and the International Cancer Genome Consortium (ICGC), should provide additional insight with regard to the characteristic genome alterations that define the intrinsic molecular subtypes of breast cancer.
3 Molecular Screening Programs
Clinical Application of Targeted Genomic Sequencing
Recent advances in DNA sequencing technology allow for rapid testing of multiple hotspot mutations using limited quantities of tumour DNA isolated from archival paraffin-embedded tumour material at an affordable cost (MacConaill et al. 2009; Dias-Santagata et al. 2010; Thomas et al. 2007). Studies by Thomas et al., MacConaill et al. and Dias-Santagata et al. examined between 250 and 1,000 individual tumour specimens for 120–400 mutations in 13–33 known oncogenes and tumour suppressor genes. These studies found at least one mutation in 30–37% of tumour samples (Thomas et al. 2007; Dias-Santagata et al. 2010; MacConaill 2013). Recently, Sequist et al. published their experience at Massachusetts General Hospital (MGH) with molecular screening of 552 non-small cell lung cancer patients using the multiplex PCR-based SNaPshot assays, which detects ~50 mutations and 14 genes, and FISH for ALK translocations (Sequist et al. 2011). They identified ≥1 mutation in 51% of patients who underwent successful profiling and directed 70 (22%) of 353 patients with advanced disease to a genotype-directed therapy. There are two reported studies that have investigated if therapy matched to molecular profile (MP) improves outcome. Von Hoff et al. conducted a study of matching treatments to MP in 86 patients across 9 different centres in the United States (Von Hoff et al. 2010). Only 66 patients proceeded to MP, wherein 64 targets were examined using a combination of immunohistochemistry (IHC), FISH and gene expression microarrays. Each aberration was matched to a predefined treatment. In 18 of 66 patients, they demonstrated progression free survival (PFS) for matched treatment to be 1.3 times greater than PFS for the treatment patients received immediately prior. Tsimberidou et al. performed molecular analysis on 1,283 patients, with success in 1,144 (89%) (Tsimberidou et al. 2011). They used polymerase chain reaction (PCR), fluorescence in situ hybridization (FISH) and immunohistochemistry (IHC) in examining for 11 separate molecular aberrations. In their cohort, 40% of patients had at least one aberration. They matched each aberration to a targeted treatment when available and demonstrated that patients who received matched targeted therapy had better response rates and improved time to treatment failure.
Molecular Screening Platforms
The advantage of multiplex PCR-based platforms such as Sequenom’s Oncocarta or OncoMap and Applied Biosystem’s SNaPshot assay is that they provide excellent coverage of frequently mutated “druggable” oncogenes when mutations cluster in a limited number of DNA sequence regions, such as KRAS (9 bases account for >99% of all mutations), BRAF (15–18 bases account for >90% of all mutations) and PIK3CA (12–15 bases account for >80% of all mutations). However, for clinically relevant tumour suppressor genes, such as TP53, PTEN, BRCA1 or BRCA2, where mutations are more widely distributed across a much larger DNA coding region, the ability to detect mutations is limited to a few selected hotspots. In addition, the published molecular screening panels using these platforms are only able to detect known base-pair substitutions and limited deletions or insertions (indels) and gene amplification. They do not include translocations, larger indels or novel base-pair substitutions. The Sequenom MassARRAY Analyzer has developed methods to evaluate copy number variation (CNV); however, this has not been validated for point-of-care molecular profiling using human tumour samples.
Next-Generation Sequencing
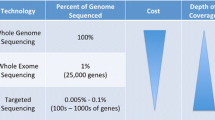
Sequenom, SNaPshot and other PCR-based multiplex assays are constrained by bandwidth and throughput. Next-generation sequencing (NGS) refers to technological platforms that allow for massive parallel sequencing of millions of DNA templates. “Second”-generation deep sequencing refers to clonal amplification of DNA templates on a solid support matrix followed by cyclical sequencing with short reads. These instruments are currently used to sequence entire genomes, exomes, transcriptomes and methylomes that often require weeks for sample template preparation, sequence generation and data analyses. As a result, their use is largely confined to large genome centres. Since “second-generation” DNA sequencing instruments are not employed in diagnostic settings, additional validation of potential candidate mutations is required using clinical-grade sequencing assays in certified diagnostic laboratories. The advent of “third”-generation sequencers such as Pacific Biosciences PacBio RS and Life Technologies’ Ion Torrent Personal Genome Machine (PGM) provides increased speed of sequencing due to their use of sensors that detect nucleotides as they are added to DNA molecules in synthesis, although parallelization and machine throughput currently is much lower than with second-generation technologies. In addition to the Ion Torrent PGM, other so-called “bench sequencing” machines have recently been released by Illumina (MiSeq) and Roche/454 (GS Junior), which are moderate-throughput platforms with fast run times, long DNA reads and automated library preparation that are well-suited to clinical applications. The appeal of these low-cost (≤125,000€ per instrument) “bench sequencing” platforms is that they offer the opportunity to comprehensively test a large targeted panel of relevant cancer genes (1,000 or more) with 30–50× or greater coverage to identify rare (<5% prevalence) mutations and copy number alterations that are potentially relevant to clinical care with a rapid turnaround time to results of 1 week or less. One of the major obstacles to NGS for cancer diagnostics is the ability to assess DNA extracted from limited formalin-fixed paraffin-embedded (FFPE) material, such as archival tumour blocks or small core tumour biopsies. Preliminary experience suggests that NGS is feasible from FFPE core tumour biopsies, although the quality DNA isolated from archival tumour material that is routinely stored for >5 years and the robustness of methods of sequence enrichments remain questionable.
Ongoing Molecular Screening Programs
Recognizing that cancer genome sequencing is likely to be integrated in routine clinical decision-making in the near future, many leading cancer research institutions and national cancer agencies have recently launched or are soon to launch broadscale molecular screening programs for solid tumours, including breast cancer (Tuma 2011). Massachusetts General Hospital (MGH) has implemented a phased roll out of the SNaPshot testing (which now includes ~120 mutations in 16 oncogenes) using archival tumour tissue in four tumour types: lung, colon, breast and glioblastoma multiforme (GBM). The Vanderbilt-Ingram Cancer Center (VICC) also initiated a similar program of SNaPshot screening of archival tumour tissue in non-small cell lung cancer and melanoma in 2010 including ~40 mutations in 6–8 genes. They integrated the molecular screening results into the patient’s electronic medical record. Their “My Cancer Genome” (www.mycancergenome.org) website includes information about common activating mutations in “druggable” oncogenes and includes links to clinical trials with molecular selection based upon molecular profiling. In July 2011, they expanded their program to include PI3-kinase pathway-specific mutation panel for breast cancer. The Dana-Farber Cancer Institute (DFCI) in partnership with the Brigham and Women’s Hospital has recently announced an ambitious US$43-million program (PROFILE) to perform mutation profiling using OncoMap (which includes ~470 mutations in 41 genes) in selected tumours, including colon, lung, breast and some sarcomas and leukaemias. Their project will include patients with early stage and advanced disease, linking genomic information with clinical outcomes and response to matched targeted therapies. It has been estimated that the program will include up to 10,000 patients annually (Tuma 2011). In Canada, the Ontario Institute for Cancer Research (OICR) and Princess Margaret Hospital (PMH) opened a pilot feasibility with biopsy of metastatic lesions involving patients with advanced solid tumours for profiling using the Sequenom Oncocarta (v1.0) and the third-generation NGS platform PacBio RS analyser for the same 19 genes as are included on the Oncocarta v1.0 panel. The initial results for the first 30 patients accrued were presented at the 2011 AACR-NCI-EORTC Molecular Target and Cancer Therapeutics Meetings (Tran et al. 2011). PMH will soon launch its own internal program entitled the Integrated Molecular Profiling in Advanced Cancers Trial (IMPACT) to perform mutation profiling using a customized Sequenom panel that includes ~277 mutations in 25 genes for patients with advanced non-small cell lung cancer, colorectal cancer, ovarian cancer, breast cancer and patients considered for phase I clinical trials. The IMPACT study will initially include 500 patients annually and will be expanded to include additional disease sites and NGS technology.
Investigators at the University of Michigan also recently published their pilot experience with real-time high-throughput whole exome sequencing for two patients enrolled in the MI-ONCOSEQ protocol (Roychowdhury et al. 2011). They successfully performed whole exome sequencing of fresh tumour biopsies from two patients – with colorectal cancer and melanoma – on the Illumina HiSeq platform and reviewed the results at a Sequencing Tumour Board within 4 weeks from the time of tumour biopsy. There are plan to perform deep whole exome sequencing of approximately 100 patients with advanced solid tumours per year, with the aim of matching patients to investigational clinical trials with targeted therapies. In Europe, there are also molecular screening programs that are underway. At the Institut Gustavy Roussy (IGR) in Paris, the ongoing MOSCATO (Molecular Screening for Cancer Clinical Trial Optimization) clinical trial protocol will perform molecular profiling using array comparative genome hybridization (aCGH) and Sanger sequencing for selected mutation hotspots in 600 patients over 3 years who are candidates for phase I clinical trials. Similarly, the ZAFIR01 clinical trial protocol at IGR will perform aCGH and targeted Sanger sequencing (PIK3CA and AKT1) in 400 patients with advanced breast cancer who undergo tumour biopsies for molecular screening. Cancer Research UK has recently launched the “Stratified Medicine Program” across seven cancer research hospitals in the United Kingdom which will perform molecular profiling for ~20 alterations in 8 genes using archival tumour material from 9,000 patients with advanced melanoma, breast, prostate, ovarian, colorectal and non-small cell lung cancer over 2 years. The details of the platform that will be used for molecular profiling have not been publicly disclosed. In the Netherlands, hospitals from Amsterdam, Rotterdam and Utrecht have launched a molecular screening to perform next-generation sequencing of fresh tumour biopsies from patients who are candidates for phase 1 clinical trials. Approximately 1,200 patients will be enrolled over the next 3 years, with plans to profile approximately 2,000 genes per patient using targeted sequencing on the Illumina HiSeq platform. The Breast International Group is also running a molecular screening program in metastatic breast cancer named AURORA project. AURORA has two broad purposes:
-
1.
To analyse breast cancer samples using techniques including but not limited to targeted DNA sequencing and RNA sequencing, in order to better understand the genetic aberrations related to breast cancer. This part of AURORA could help us understand breast cancer disease evolution (this will be done in all patients) and determine why some patients respond well to a certain treatment while others don’t (this will only be done in a minority of patients). This may not provide you with any benefit directly, but your participation is likely to help us find answers to questions which could help to improve the treatment and/or quality of life of future breast cancer patients.
-
2.
To identify patients potentially eligible to participate in approved studies testing new therapeutic strategies based on known breast cancer-related molecular aberrations found in the breast cancer samples. Such identification is done when the aberrations of your primary and/or metastatic tumour DNA found by targeted sequencing match an ongoing therapeutic clinical trial testing a drug against the aberration. These trials might not be available at the time being, but your treating physician shall inform you in case they become available. If you are found to be eligible for an ongoing trial, your treating physician will give you more information and an additional informed consent form to sign, specific to that particular trial. Note that enrolment in one of these candidate trials is completely up to you. Please note that aberrations for which therapeutic clinical trials are available may be found only in a minority of patients. To start with, this research project will involve 1,300 patients from hospitals mainly located in Europe.
4 Future Perspectives
It is likely that future clinical trials in breast cancer with targeted therapies will be conducted in molecularly defined subpopulations of disease. Advances in high-throughput DNA sequencing technology allow for screening a large number of genes simultaneously at a relatively low cost to molecularly characterize individual tumours for triage of clinical trials with targeted therapies. These molecular screening programs are rapidly being developed by large cancer research hospitals and national cancer societies in North America and Europe. It is very unlikely that a single pharmaceutical sponsor will be able to support the large-scale molecular screening programs to identify relatively rare subpopulations (≤5%) of breast cancer that are amenable to clinical trials with matched targeted therapies. The existing model of sequential prescreening for individual clinical trials – with separate informed consent forms, processes of tumour material retrieval and shipping and methods of laboratory testing and reporting – is expensive, inefficient and not well-suited to the current era of molecularly targeted drug development. We need to find new paths to access innovations to clinical research and daily practice. To ensure that continued innovation meets the needs of patients, the therapeutic alliance between patients and academic-led research should to be extended to include relevant pharmaceutical companies and drug regulators with a unique effort to bring innovation into clinical practice. We need to bring together major players from the world of breast cancer research to map out a coordinated strategy on an international scale, to address the disease fragmentation, to share financial resources and to integrate scientific data. The final goal will be to improve access to an affordable, best standard of care for all patients in each country.
References
Callaway E (2010) Cancer-gene testing ramps up. Nature 467(7317):766–767
Dias-Santagata D et al (2010) Rapid targeted mutational analysis of human tumours: a clinical platform to guide personalized cancer medicine. EMBO Mol Med 2(5):146–158
Ellis MJ et al (2007) A luminal breast cancer genome atlas: progress and barriers. J Steroid Biochem Mol Biol 106(1–5):125–129
Greenman C et al (2007) Patterns of somatic mutation in human cancer genomes. Nature 446(7132):153–158
Hanahan D, Weinberg RA (2000) The hallmarks of cancer. Cell 100(1):57–70
Kola I, Landis J (2004) Can the pharmaceutical industry reduce attrition rates? Nat Rev Drug Discov 3(8):711–715
MacConaill LE (2013) Existing and emerging technologies for tumor genomic profiling. J Clin Oncol 31(15):1815–1824
MacConaill LE et al (2009) Profiling critical cancer gene mutations in clinical tumor samples. PLoS One 4(11):e7887
Roychowdhury S et al (2011) Personalized oncology through integrative high-throughput sequencing: a pilot study. Sci Transl Med 3(111):111ra121
Sequist LV et al (2011) Implementing multiplexed genotyping of non-small-cell lung cancers into routine clinical practice. Ann Oncol 22:2616–2624
Stewart DJ, Kurzrock R (2009) Cancer: the road to Amiens. J Clin Oncol 27(3):328–333
Stratton MR, Campbell PJ, Futreal PA (2009) The cancer genome. Nature 458(7239):719–724
Thomas RK et al (2007) High-throughput oncogene mutation profiling in human cancer. Nat Genet 39(3):347–351
Tran B et al (2011) Feasibility study of molecular profiling (MP) in patients (Pts) with advanced solid cancers using targeted mutation analysis and targeted exome sequencing. Mol Cancer Ther 10(Suppl 11):B48
Tsimberidou AM et al (2011) Personalized medicine in a phase I clinical trials program: the M. D. Anderson Cancer Center Initiative. J Clin Oncol 29(Suppl):Abstract CRA2500
Tuma RS (2011) Large-scale genome projects enter the clinic on both sides of the Atlantic. J Natl Cancer Inst 103(23):1730–1731
Von Hoff DD et al (2010) Pilot study using molecular profiling of patients’ tumors to find potential targets and select treatments for their refractory cancers. J Clin Oncol Off J Am Soc Clin Oncol 28(33):4877–4883
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer International Publishing AG, part of Springer Nature
About this chapter

Cite this chapter
Curigliano, G. (2018). Cancer Evolution as the New Frontier of Precision Medicine. In: Mandalà, M., Romano, E. (eds) Mechanisms of Drug Resistance in Cancer Therapy. Handbook of Experimental Pharmacology, vol 249. Springer, Cham. https://doi.org/10.1007/164_2018_147
Download citation
DOI: https://doi.org/10.1007/164_2018_147
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-10506-8
Online ISBN: 978-3-030-10507-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)