
Abstract
The application of population genomics to the understanding of speciation has led to the emerging field of speciation genomics. This has brought new insight into how divergence builds up within the genome during speciation and is also revealing the extent to which species can continue to exchange genetic material despite reproductive barriers. It is also providing powerful new approaches for linking genotype to phenotype in admixed populations. In this chapter, we give an overview of some of the methods that have been used and some of the novel insights gained. We also outline some of the pitfalls of the most commonly used methods and possible problems with interpretation of the results.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
Speciation is a fundamental process in evolution, giving rise to biological diversity (Box 1). It involves the divergence of populations, with the establishment of reproductive isolation (RI) being an essential feature for maintaining distinctive characteristics of the incipient species (Coyne and Orr 2004). The emerging field of speciation genomics makes use of dense genome-wide markers to understand how genetic differences build up within the genome and to identify genetic loci that contribute to speciation (Butlin 2008; Nosil and Feder 2012; Seehausen et al. 2014). Gene flow between diverging populations slows down genome divergence by homogenising genetic variation. Establishment of barrier loci involved in RI is also difficult in the face of gene flow because such loci may be quickly eliminated by selection, and therefore, key questions in speciation research are how differences accumulate and how RI mechanisms are established (Coyne and Orr 2004). Speciation genomics studies have shown that divergence can persist in the face of gene flow, with sharing of alleles being detected to a greater or lesser extent between a wide range of taxa, including those that are considered good species (Payseur and Rieseberg 2016). By studying species at different points along the ‘speciation’ or ‘divergence continuum’, from those that have diverged little to species that may not even be sister to one another, we are starting to understand how genetic differentiation has accumulated within the genome (Feulner et al. 2015; Nadeau et al. 2013; Riesch et al. 2017; Seehausen et al. 2014). However, species may currently maintain distinctive features despite some ongoing gene flow, but this does not mean that these differences were accumulated initially in the face of gene flow (i.e. in sympatry, Box 1). Hybridisation can happen in different spatial contexts, from narrow hybrid zones where parapatric populations meet, to complete sympatry (Abbott et al. 2013). Similarly, there can be a diversity of different temporal contexts, ranging from brief periods of secondary contact to continuous contact with divergent selection. Distinguishing these different scenarios from genomic data is not straightforward (Payseur and Rieseberg 2016). Therefore, empirical studies need to be interpreted in the light of a sound theoretical understanding of how differences accumulate in the genome under different scenarios (Nosil and Feder 2012; Payseur and Rieseberg 2016).
Box 1 Definitions and Concepts
-
Speciation – the separation of populations, originally able to interbreed, into distinct species, no longer able to interbreed freely. This definition depends on the species definition being used. The strict biological species concept demands complete reproductive isolation between species, while other definitions may relax this (Coyne and Orr 2004). Speciation genomics studies often consider taxa below the species level, with the idea being that these may be in the early stages of speciation (Seehausen et al. 2014) (see ‘The speciation continuum’).
-
Sympatry – occurring together or with overlapping geographic areas. Sympatric speciation occurs without any physical barriers to gene flow.
-
Allopatry – occurring in separate, nonoverlapping geographic areas. Allopatric speciation occurs when populations are physically isolated and so unable to exchange genetic material.
-
Parapatry – occurring in partially overlapping geographic areas or areas with a partial barrier between them.
-
Reproductive isolation (RI) – a reduction or absence of gene flow between populations beyond that caused by geographic barriers, usually due to incompatibilities in the reproductive systems of the organisms, either before fertilisation (prezygotic, e.g. timing of reproduction, courtship, mate choice or physical incompatibilities) or after fertilisation (post-zygotic, e.g. inviable or infertile offspring, offspring with reduced fitness).
-
The speciation continuum – the idea that speciation proceeds gradually and so it should be possible to observe populations with different levels of divergence that are at different points long the continuum. By studying these populations, we can understand how speciation proceeds. One possible problem with this paradigm is that some of the populations with low levels of divergence may be at a stable point and not in fact proceeding towards becoming full species.
-
Genome scan – an analysis of genome-wide genetic markers to detect loci with elevated genetic differentiation. In this chapter we are mostly referring to scans of FST between two populations in order to detect loci that are under divergent selection or exhibit reduced gene flow between populations.
-
Speciation/differentiation islands – regions of the genome showing increased levels of differentiation between two populations. These are usually inferred to contain genetic loci responsible for maintaining differences between the populations.
-
Admixture – mixing of genetically distinct populations through interbreeding.
-
Hybridisation – mating between individuals of different species or distinct populations.
-
Introgression – the transfer of genetic loci from one species to another following hybridisation and repeated backcrossing.
-
Gene flow – the movement of genetic material between populations, usually by migration and interbreeding.
-
Hybrid zone – a restricted geographic region where phenotypically or genetically distinct populations or species meet and interbreed, forming hybrids.
-
Cline – a spatial transition from one genotypic or phenotypic form to another, or a change in allele frequency across a geographical region.
-
F ST (also known as Wright’s fixation index) – a measure of genetic differentiation between populations varying between zero (no difference) and one (a fixed genetic difference). It involves comparing how similar two individuals from the same subpopulation are as compared to the total population, so giving a measure of the amount of genetic variance that can be explained by population structure. The formula normally used for DNA sequence data is: \( {\mathrm{F}}_{\mathrm{ST}}=\frac{\uppi_{\mathrm{Between}}-{\uppi}_{\mathrm{Within}}}{\uppi_{\mathrm{Between}}} \) where πBetween and πWithin are the pairwise genetic differences between individuals sampled from within a (sub)population (πWithin) or from different populations (πBetween).
-
Hard sweep – a selective sweep by positive selection acting on a new mutation. This results in advantageous variants reaching fixation in a population. Genetic variation at sites that are tightly linked to the selected sites is eliminated by genetic hitchhiking.
-
Soft sweep – selection acting on variants that segregate in a population as standing genetic variation. These variants may not confer a selective advantage in one population or under one set of conditions but do so in another population under different conditions. Because the selected variants are present in a variety of different genetic backgrounds, variation at linked sites is not reduced to the same extent as in a hard sweep.
-
Linkage disequilibrium (LD) – the non-random association of alleles at different loci within a population. This is most often due to physical linkage between loci but can also be found between unlinked loci. For example, unlinked loci under divergent selection between two populations will tend to be in LD. LD is also elevated in admixed populations because of associations between loci coming from the same parental population.
-
Barrier loci – positions in the genome that contribute to restriction of gene flow between diverging populations. These loci may be involved in various types of reproductive isolation, including divergent ecological selection (extrinsic reproductive isolation), mate choice (pre-mating reproductive isolation), egg-sperm incompatibility (post-mating-prezygotic reproductive isolation) and hybrid sterility/inviability (post-zygotic reproductive isolation).
Studies of speciation have long made use of hybrid zones (Box 1), where distinct populations or species come into contact and interbreed (Kawakami and Butlin 2012). When high-resolution genomic tools were not available, studying phenotypic variation and few loci within and across hybrid zones provided useful insight into the nature of barriers to gene exchange and the selective forces at play in keeping distinct populations from fully mixing (Barton and Hewitt 1985). Building on this solid foundation, population genomic analyses of hybrid zones can bring new insights at a much finer scale, for example, determining the extent and nature of barriers to gene flow by characterising how much of the genome is being exchanged (Gompert et al. 2017; Harrison and Larson 2016). In this chapter, we explore the new insights that population genomics approaches are bringing to the field of speciation research, as well as how population genomics of admixed populations and hybrid zones can help to identify the genetic basis of phenotypic differences more broadly. Key systems in the speciation genomics literature are summarised in Table 1.
2 Genomic Signatures of Speciation and Reproductive Isolation
High-throughput sequencing technologies allow biologists to investigate genome-wide patterns of genetic differentiation between diverging populations (Seehausen et al. 2014; Wolf and Ellegren 2016). Speciation can be driven by extrinsic (environmental) factors where divergent selection reduces gene flow between populations or by intrinsic factors where genes incompatible in a foreign genomic background result in reduced fitness in hybrids (Coyne and Orr 2004). The former is known as ecological speciation, where locally adapted populations are exposed to divergent ecological selection in different environments, leading to the establishment of barriers to gene flow (Nosil 2012). The latter cases are formulated by theoretical models where epistatic interactions of incompatible alleles at multiple loci have evolved in diverging populations, resulting in a reduction in hybrid fitness (Dobzhansky-Muller incompatibility) (Dobzhansky 1936; Muller 1940; Orr 1995). However, this binary classification does not fit all situations; for example, local adaptation may be mediated by epistatically interacting alleles that could also give rise to reduced hybrid fitness. Regardless of the types of reproductive barriers, the genic model of speciation predicts that genetic differentiation is initially accumulated at a small number of genomic regions that are under selection associated with RI. These barrier loci are resistant to gene flow, either by ecological divergent selection or intrinsic incompatibility, while the rest of the genome is homogenised by gene flow (Wu 2001).
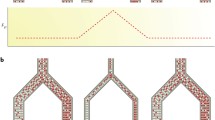
Barrier loci could be established in the face of gene flow or in geographically isolated populations. In the case of gene flow, the level of genetic differentiation is kept low in regions unlinked to the barrier loci, whereas under geographic isolation, genetic differentiation accumulated during the allopatric period may be eroded by gene flow following secondary contact occurring across the genome except in regions containing barrier loci. In both cases, at the very onset of speciation, the genomes of two diverging populations may be characterised as a small number of regions with elevated differentiation surrounded by regions of low differentiation (hence often referred to as ‘differentiation islands’ or ‘genomic islands of divergence’ as an oceanic island metaphor) (Nosil and Feder 2012; Turner et al. 2005). In addition, it has been proposed that restricted gene flow near differentiation islands can allow for the sequential accumulation of additional barrier loci at neighbouring sites, and as a result, these differentiation islands can increase in height and width as speciation proceeds (Via 2012). As additional barrier loci accumulate in a genome, either at proximal or distal regions of existing differentiation islands, the strength of RI increases and genetic differentiation would increase across the whole genome (Fig. 1). There is a suggestion from both theory (Feder et al. 2012; Flaxman et al. 2014) and empirical evidence (Riesch et al. 2017) that this increase does not occur linearly and that there may be a ‘tipping point’ in either the strength of RI or the number of differentiated regions, at which point populations transition from having a small number of differentiation islands to effectively genome-wide differentiation (Nosil et al. 2017). Nevertheless, the idea of differentiation islands has motivated a number of researchers to characterise genome-wide patterns of genetic differentiation between closely related species and between diverging lineages, aiming to characterise underlying genetic mechanisms of RI.

Three models for the evolution of differentiation islands. (a) Speciation island model without geographic isolation. Gene flow maintains low genetic differentiation throughout the speciation continuum by homogenising genetic materials elsewhere in genomes of diverging populations except loci involved in divergent selection and reproductive isolation (red triangles). As speciation progresses (from the top to the bottom panels), new reproductive isolation loci are accumulated either at proximal region to the existing differentiation islands or at novel regions. This makes the differentiation islands higher and wider. (b) Speciation island model with geographic isolation. Genetic differentiation is initially accumulated in geographically isolated populations (top panel). Reproductive isolation loci are also accumulated during this period. Upon secondary contact (middle panel), gene flow erodes genetic differentiation elsewhere in a genome except reproductive isolation loci. Additional reproductive isolation loci may be accumulated, further strengthening the barrier to gene flow. (c) Incidental island model. Because of the heterogeneity in recombination rate and gene density (purple line and green rectangles, respectively, in the top panel), shared ancestral polymorphisms between diverging populations are removed more extensively at regions of low recombination rate and high gene density. This results in non-uniform reduction of nucleotide diversity (π) in each population (middle panel) and heterogeneous differentiation landscape (bottom panel)
2.1 Genome Divergence Scans to Identify Barrier Loci
There are an increasing number of studies reporting heterogeneous patterns of genomic differentiation (Ellegren et al. 2012; Nadeau et al. 2012, 2014; Renaut et al. 2013; Turner et al. 2005; Via et al. 2012), but interpretation of these differentiation islands is not as straightforward as one might think based on the genic model of speciation. Specifically, it remains challenging to determine whether the differentiation islands evolved as a result of speciation (i.e. ‘speciation islands’) or by other processes independent of the evolution of RI mechanisms (i.e. ‘incidental islands’) (Cruickshank and Hahn 2014). Under the genic model of speciation, gene flow plays a critical role in the formation of differentiation islands by homogenising genetic diversity between species at the vast majority of genomic regions that do not harbour loci involved in RI. However, there are several studies reporting similar patterns of heterogeneous differentiation between geographically isolated populations, which have no apparent contemporary gene flow between them (Martin et al. 2013; Renaut et al. 2013; Vijay et al. 2016). Incomplete lineage sorting of ancestral polymorphisms and stochasticity in allele frequency changes can result in heterogeneity in genetic variation between closely related species even in the absence of current gene flow, especially when selection acts on linked neighbouring sites (Cruickshank and Hahn 2014; Nachman and Payseur 2012; Noor and Bennett 2009). Selection in this case can be either positive or negative (background/purifying selection) and does not have to be directly associated with RI mechanisms. The rationale is that recurrent positive and negative selection removes polymorphisms not only at target sites of selection but also at neighbouring sites in linkage disequilibrium (LD). This process, collectively referred to as ‘linked selection’ (Cutter and Payseur 2013), can create regions with locally reduced effective population size (N e), which in turn accelerates lineage sorting, decreases genetic diversity (π), and increases differentiation (FST). Because the effect of linked selection is a function of the density of target sites for selection (e.g. gene density) and local recombination rate, the magnitude of lineage sorting and, hence, genetic differentiation is inherently heterogeneous across a genome. Therefore, it is necessary to understand the underlying genetic mechanisms and evolutionary processes in the formation of differentiation islands.
One way of distinguishing these two scenarios is to compare patterns of genetic differentiation along a genome between multiple pairs of closely related species with different divergence times. An important assumption is that conserved recombination rate and genome structure among closely related species, such as gene density and distribution, result in shared patterns of genetic differentiation by linked selection. There are several studies showing that differentiation islands observed both in very early stages of speciation (i.e. less divergent pairs of species) and more advanced stages of speciation (i.e. more divergent pairs of species) likely represent ‘incidental islands’, while population-specific differentiation islands represent candidate ‘speciation islands’ (Andrew and Rieseberg 2013; Burri et al. 2015; Poelstra et al. 2014; Renaut et al. 2013; Vijay et al. 2016). In addition, at the early stage of speciation, ‘speciation islands’ can be distinguished from heterogeneous genomic differentiation shaped by linked selection unrelated to RI, because strong divergent selection can create a small number of extremely differentiated regions with long haplotype blocks (Andrew and Rieseberg 2013; Poelstra et al. 2014).
Due to the increasing accessibility of genome-wide polymorphism data in various organisms, the genome scan approach is a tractable first step towards the understanding of the genetic basis of reproductive isolation by characterising genetic differentiation along a genome. One advantage of this approach is that phenotypic differences do not need to have been previously characterised, meaning that it has the potential to identify loci underlying novel divergently selected traits. In addition, it can be a powerful tool for detecting divergently selected regions between readily interbreeding taxa, because it makes use of the genomic signatures left by both selection and gene flow. However, deciphering underlying mechanisms for the formation of differentiation islands (i.e. divergent selection related to reproductive isolation vs. linked selection) remains a challenge, not only because these two processes can take place simultaneously but also because these two processes would leave very similar signals (Yeaman et al. 2016). One way forward is to combine trait information with genome scan analysis, by which one can further narrow down the candidate genomic regions from numerous differentiation islands identified by the genome scan. In practice this has rarely been done for traits that were not already well characterised or genetically mapped. A study by Toews et al. (2016) on warblers is one of the few examples to use an outlier approach to identify anonymous outlier loci and to then link these to phenotypic differences between populations (Fig. 2), although, even in this case, the phenotypes were well-characterised differences in colouration. The process of linking anonymous loci to phenotypes necessarily starts with an informed guess, which makes it difficult for the genome scan approach to identify really novel or unexpected divergently selected traits (but see Bosse et al. 2017). Although undoubtedly useful for characterising the patterns of divergence across the genome, genome scan analysis alone may have a limited power to identify causal genes for reproductive isolation. In section 3, we describe approaches that gain additional power from the information present in admixed populations to identify barrier loci and those loci underlying divergent traits more broadly.

FST outlier scan between golden-winged and blue-winged warblers (Vermivora chrysoptera and V. cyanoptera) identified six divergent regions between species, four of which contained candidate plumage colour genes. Associations between these loci and particular plumage colour elements were then confirmed by characterising particular SNPs in a larger number of individuals, including hybrids. Reprinted from Toews et al. (2016), with permission from Elsevier
2.2 Key Examples of Applications of Genome Divergence Scans in Speciation Population Genomics
Here we outline ‘genome scans’ performed on the three-spine stickleback (Gasterosteus aculeatus) to show how genetic differentiation accumulates along a genome at different stages of the ‘speciation continuum’. Two avian examples, highlighting some of the issues with interpretation of divergence scans, are also presented. Additional examples are summarised in Table 1 and have been reviewed elsewhere (Haasl and Payseur 2016; Ravinet et al. 2017; Wolf and Ellegren 2016).
2.2.1 Three-Spine Stickleback
The three-spine stickleback provides a powerful model system for studying the genetic basis of adaptation and ecological speciation. This small fish is widely distributed in the Northern hemisphere and shows a remarkable history of independent colonisation from the marine environment to freshwater ecosystems after the glacial retreat (ca. 12,000 years ago) (Bell and Foster 1994). Freshwater and marine ecotypes show marked differences in body size and shape, colouration, courtship behaviour, trophic specialisation, the number of skeletal armour plates, and spine length (Fig. 1a) (McKinnon and Rundle 2002). The repeated observation of these morphological and behavioural shifts at multiple locations in North America and Europe suggests that the selection pressures associated with the colonisation of freshwaters have been instrumental in driving recurrent/parallel evolution. In fact, parallel evolution of freshwater-adapted phenotypes has likely been facilitated through repeated selection of rare genetic variants segregating in the marine ancestor (Colosimo et al. 2005; Jones et al. 2012b; Roesti et al. 2015). After the colonisation of freshwaters, populations have further diversified into several distinctive ecotypes. For example, populations in open water lake habitat show ecologically distinctive life history traits by having pelagic lifestyle feeding on zooplankton (‘lake ecotypes’ or ‘limnetic ecotypes’), whereas populations in rivers and small stream habitat show a benthic lifestyle by feeding on macroinvertebrates (‘stream ecotypes’ or ‘benthic ecotypes’) (Berner et al. 2010; Moser et al. 2015). In both cases, increases in allele frequency of adaptive variants in newly colonised habitat may leave a specific signature in their genomes, and genome scan analysis, in theory, can detect such a signature as an elevated differentiation relative to the surrounding genomic regions. Moreover, repeated occurrence of differentiation islands at the same genomic location between multiple, independent pairs of ecotypes is commonly taken as evidence of parallel evolution at the molecular level (Hohenlohe et al. 2010). However, linked selection unrelated to adaptive divergence could also contribute to the parallel evolution of differentiation islands because these ecotypes likely share common genomic features important to the magnitude of linked selection across a genome, such as variation in gene density and recombination rate, which would then result in positive correlation in the magnitude of differentiation between ecotype comparisons.
Several studies have identified key genes associated with phenotypic traits that confer adaptation to the newly colonised habitat in sticklebacks (Chan et al. 2010; Colosimo et al. 2005). For example, higher predation pressure in open-water habitat (either in marine populations or lake populations) than small stream populations, has resulted in more complete lateral armour plates (Bell and Foster 1994; Berner et al. 2010; Roesti et al. 2015). Allelic variation at the Ectodysplasin (Eda) gene on chromosome 4 is strongly associated with phenotypic variation in this trait (Berner et al. 2014; Colosimo et al. 2005), representing a prime candidate for selection. Another well-studied candidate gene for adaptive evolution is Pituitary homeobox transcription factor 1 (Pitx1) gene, whose regulatory mutations resulted in partial or complete loss of pelvic spines in freshwater ecotypes (Chan et al. 2010). While EDA represents a classic case for adaptation from standing genetic variation, the evolution of Pitx1 has involved repeated de novo mutations in multiple populations. Therefore, these loci offer an opportunity to test a predicted genomic pattern, in which divergent selective sweeps increase genetic differentiation at these loci, while ongoing gene flow maintains low differentiation at the genomic background.
To identify putative genomic regions under divergent selection, several studies took a population genomics approach to characterise genome-wide patterns of genetic differentiation between marine and freshwater ecotypes and between lake and stream ecotypes by using RAD-sequencing approaches and whole-genome re-sequencing approaches (Deagle et al. 2011; Feulner et al. 2015; Hohenlohe et al. 2010, 2012; Jones et al. 2012a, b; Roesti et al. 2012, 2014, 2015). The first genome-wide survey of genetic differentiation identified nine differentiation islands in three comparisons of ancestral oceanic populations versus derived freshwater populations in Alaska by using over 45,000 RAD sequencing markers (Hohenlohe et al. 2010). Jones et al. (2012b) further expanded sampling populations (21 locations across Northern hemisphere) as well as the number of SNPs covering the entire assembled genome and identified 174 regions of elevated differentiation between marine and freshwater ecotypes with median size of 3 kb with 5% false discovery rate (FDR). Consistent with the a priori expectation, the EDA locus showed elevated differentiation in both studies, confirming that genome scan analysis can recover signatures of divergent selection; however, Ptx1 was not located at differentiation islands despite the apparent phenotypic differences between the ecotypes (Hohenlohe et al. 2012; Jones et al. 2012b). The insignificant genetic difference at Ptx1 could be interpreted as (1) weak or no selection on Ptx1 or (2) difficulties in detecting a selective signature by this approach if adaptive causal variants are found in multiple haplotype backgrounds (i.e. ‘soft sweeps’, see Box 1) (Hohenlohe et al. 2010). In addition to these a priori candidate genes, other differentiation islands contained a number of genes with functions related to skeletal traits, response to osmotic stress, signal response, behavioural interaction between organisms, amine and fatty acid metabolism, cell–cell junctions, WNT developmental signalling, epithelial barrier, and immune functions (Jones et al. 2012a, b), which represent candidate genes for functional analysis. Importantly, there are many other differentiation islands distributed in intergenic regions, implying that adaptive divergence can involve changes in both protein coding genes and non-coding regulatory regions (Jones et al. 2012b).
Lake and stream population pairs also provide a useful system for parallel evolution of differentiation. For instance, after colonisation to Lake Constance in Central Europe, small creeks and streams connected to the lake were subsequently colonised by stickleback populations (Roesti et al. 2015), thus possibly representing much more recent divergence than the marine-freshwater comparison. Despite the short evolutionary time window, Marques et al. (2016) identified 37 differentiation islands that consisted of 1–26 SNPs. Importantly, 19 out of these 37 differentiation islands were consistently identified in two pairs of stream and lake ecotypes, indicating potential parallel change in allele frequency driven by ecological adaptation. Three other tributaries of Lake Constance also showed heterogeneous genetic differentiation with 2–25 highly differentiated SNPs scattered across the genome in at least one of the three comparisons of lake versus stream populations (Roesti et al. 2015). All three comparisons showed a similar shift in allele frequency at these loci, supporting the parallel action of similar ecological pressure at the genomic level.
An important difference from the marine-freshwater comparison is that signature of selection at the Eda locus is much weaker in the lake-stream comparison as represented by the inconsistent elevation of genetic differentiation at this locus among population pairs (Roesti et al. 2015). This may possibly be because of recent recolonisation history in the lake-stream system where the selective sweep is likely incomplete. An additional complication with regard to the genetic differentiation at the Eda locus is that adaptive alleles can be unconditionally favoured in both stream and lake populations on Vancouver Island in Canada, which generates a peculiar pattern where genetic differentiation is reduced at Eda locus due to the fixation of shared ancestral haplotypes, while the surrounding neutral regions of the Eda locus are characterised as elevated differentiation (Roesti et al. 2012, 2014). Unconditional selection on the adaptive alleles at the Eda locus, if any, could be due to similar ecological selective pressure in lakes and streams on Vancouver Island, whereas selective pressure may be more contrasting in the Lake Constance system in Central Europe because of its larger size. Altogether, this highlights difficulties and challenges in using genome scans to detect signatures of selective sweeps, even at genomic regions with strong candidate genes under ecological selection.
Since stickleback recolonisation has likely taken place independently at different times, multiple pairs of lake-stream populations can also provide an opportunity to test how differentiation islands emerge and increase in number and size along the speciation continuum (Fig. 1). If an increase in differentiation in the background genomic region is accompanied by increase in the number and size of differentiation islands as predicted by the genic model of speciation, then population pairs with higher genome-wide differentiation should have more and wider differentiation islands than population pairs with lower genome-wide differentiation. Feulner et al. (2015) compared genetic differentiation (measured as FST) among five pairs of lake-stream populations in the Northern Hemisphere, with varying degrees of genome-wide FST, ranging from 0.10 to 0.28 (Fig. 3b). They found no apparent growth of differentiation islands despite the significant difference in the background FST, which may partly be due to population-specific selection for each locality and/or differences in the extent of divergent selection. Similar patterns were also found in Timema stick insects (Riesch et al. 2017), although evidence for the growth of differentiation islands has been reported in Heliconius butterflies (Nadeau et al. 2013). Theoretical studies have suggested that the differentiation islands could grow in size by accumulating additional RI loci in the presence of gene flow, but their growth may require specific conditions composed of rather narrow parameter space, such as low migration, strong selection, low level of differentiation in background regions, and locally reduced recombination rate (Feder and Nosil 2010; Yeaman et al. 2016). In addition, since a transition from an early stage of speciation with detectable differentiation islands to an advanced stage with genome-wide differentiation (Fig. 1) may happen rapidly, detecting signals for the growth of differentiation islands may be challenging (Feder and Nosil 2010). Additional empirical studies may refine these theoretical models to predict necessary conditions for broadening the regions of differentiation under various demographic scenarios.

Divergent phenotypes of three-spine stickleback (G. aculeatus) and genome-wide patterns of genetic differentiation between ecotypes. (a) Freshwater (top) and marine (bottom) ecotypes. Reprinted by permission from Macmillan Publishers Ltd: Nature (Jones et al. 2012b), copyright 2012 (b) Distribution of FST along a genome in five pairs of stream and lake ecotypes with different levels of genome average FST (smallest at the top and biggest at the bottom panels). Note that location of loci that are exceptionally different (i.e. elevated FST, coloured dots) is not always conserved between population pairs, and the number and intensity of these high differentiation regions are not correlated with background level of FST. Reprinted from Feulner et al. (2015) under the Creative Commons Attribution License
Linked selection also plays a critical role in the formation of heterogeneous differentiation landscape along the stickleback genome by removing genetic variation, particularly at low recombination regions. Like many other species (Auton et al. 2012; Kawakami et al. 2014), recombination rate is highly variable along the stickleback genome with the rate generally increasing towards the ends of chromosomes (Roesti et al. 2013). This ‘U-shape’ distribution of recombination events along a chromosome is inversely correlated with genetic differentiation at a global genomic scale (Roesti et al. 2012, 2013), supporting an action of linked selection where lineage sorting takes place much more extensively at low recombination regions by the removal of shared ancestral genetic variation by positive selection or negative (background) selection (Fig. 1). The strong influence of linked selection at low recombination regions is consistent with the pattern reported in a wide variety of species (Burri et al. 2015; Martin et al. 2016; Vijay et al. 2016; Wang et al. 2016). These studies also show that the effect of linked selection is stronger at gene dense regions because the extent of the removal of genetic variation at physically linked sites is proportional to gene density. Given the significant correlation between genetic diversity and recombination rate, it is important to take into account the variation in recombination rate between diverging populations, which can potentially create population-specific patterns of diversity landscape along a genome (Kawakami et al. 2017; Smukowski and Noor 2011).
2.2.2 Flycatchers
Collared flycatcher (Ficedula albicollis) and pied flycatcher (F. hypoleuca) have been intensively studied to identify divergence in life history traits, traits under sexual selection (e.g. plumage colour and song), and hybrid fitness reduction (Qvarnström et al. 2010). Both species are small migratory passerine birds that overwinter in sub-Saharan Africa but return to their breeding ranges in summer in Europe. Occasional hybridisation has been reported at regions where two species overlap in central Europe (Svedin et al. 2008), but reproductive isolation is near complete despite their relatively recent divergence (<1 million years) (Nadachowska-Brzyska et al. 2013). By sequencing whole-genomes of 20 individuals (10 individuals/species), Ellegren et al. (2012) discovered that the pattern of genetic differentiation was highly heterogeneous along the genome with about 50 regions with elevated differentiation between species (measured by FST). To further investigate underlying mechanisms for the formation of these ‘differentiation islands’, Burri et al. (2015) expanded the samples to other flycatcher species. These multi-population and multi-species comparisons revealed similar patterns of genetic differentiation both within species and between species, regardless of their divergence time (Fig. 4), indicating that shared genomic features among these Ficedula species are likely responsible for the emergence of differentiation islands. In fact, recombination rate estimated based on the linkage map in collared flycatcher (Kawakami et al. 2014) and the density of coding sequence were significantly correlated with genetic diversity (π) and genetic differentiation (FST and dXY), suggesting that ‘linked selection’ plays a more predominant role than gene flow in the formation of differentiation islands in flycatcher.

(a) About 20 genomes per population were sequenced (collared flycatcher [F. albicollis], pied flycatcher [F. hypoleuca], atlas flycatcher [F. speculigera], and semicollared flycatcher [F. semitorquata]). Outgroup species were red-breasted flycatcher (F. parva) and snowy-browed flycatcher (F. hyperythra) (not shown). Four populations each of collared flycatcher and pied flycatcher were sampled across Europe (E Spain, CZ Czech Republic, B Baltic, S Sweden, H Hungary, I Italy), which allowed within-species comparisons. (b) Genetic difference (FST) along an example chromosome (chromosome 11). Differentiation islands observed in collared-pied comparison (green) were also observed in collared-atlas comparison (orange), collared-semicollared comparison (red), collared-red-breasted comparison (dark red), and collared-snowy-browed comparison (black). Importantly, the differentiation island starts emerging within species comparisons (I-H collared flycatcher populations [dark blue] and I-B collared flycatcher populations [light blue]). Modified from Burri et al. (2015) with permission
2.2.3 Crows
The Corvus crow species complex in Eurasia (Corvus [corone] corone, C. [c]. cornix, C. [c]. orientalis and C. [c]. pectoralis) represents another classic example of speciation in birds (Mayr 1942). This species complex has been extensively studied to understand genetic mechanisms of the traits under divergent selection, which are the key in the maintenance of stable hybrid zones (Fig. 5) (Randler 2007). Because RI between carrion crow (C. [c]. corone) and hooded crow (C. [c]. cornix) is incomplete with frequent backcrossing of hybrids, this pair of taxa may be at an earlier stage of the speciation continuum than the flycatcher species pair. By using the whole genome sequencing approach, Poelstra et al. (2014) identified five ‘differentiation islands’ based on FST outlier analysis. The largest differentiation island, identified on chromosome 18, harboured genes associated with colour pigmentation and visual perception, which are likely responsible for differences in plumage colour and assortative mating. In addition, long-range sequencing analysis using PacBio and Nanopore optical mapping revealed that this region coincided with putative centromeric region, suggesting that the combined effect of low recombination and positive selection resulted in the elevated genetic differentiation (Weissensteiner et al. 2017). In addition, Vijay et al. (2016) identified several differentiation islands in the other species pairs (Siberian hybrid zone between C. [c] cornix and C. [c] orientaris and Asian hybrid zone between C. [c] orientaris and C. [c] pectolaris) (Fig. 5). Importantly, the locations of these islands were mostly different from the ones identified in the corone-cornix hybrid zone, and consequently, genes identified on the differentiation islands hardly overlapped between three species pairs. Nevertheless, these differentiation islands also contained genes involved in pigmentation and melanogenesis, suggesting that parallel divergent selection acts on plumage colour at multiple independent hybrid zones but on different genes in the same melanogenesis pathways. The pattern found in the crow species complex is quite contrasting to that found in Heliconius butterflies, in which the parallel patterns of phenotypic divergence are largely based on selection acting on the same genomic regions (Nadeau et al. 2014).

(a) Distribution of Corvus crow species complex. (b) The corone-cornix hybrid zone in central Europe was used in Poelstra et al. (2014), revealing a strong genetic difference on chromosome 18 (top panel). The cornix-orientalis comparison (middle panel) and the orientalis-pectoralis comparison (bottom panel) showed differentiation islands that are at different genomic regions. Standardised genetic differentiation FST′ (black, positive axis) and net genetic differentiation ΔFST′ (blue, mirrored to the negative axis) in 50 kb windows across the genome. Genomic regions of extreme differentiation (499th percentile) are shown in red for both FST′ and ΔFST′. Modified from Vijay et al. (2016) with permission
3 Using Admixture to Identify Genes Underlying Divergent Traits
Hybrid zones or other situations in which phenotypically distinct populations meet and interbreed provide a valuable opportunity to study the genetic basis of divergent traits. It has long been recognised that hybrid zones can act as natural laboratories in which many generations of crossing generate novel genetic combinations and the potential to identify loci contributing to adaptive phenotypic differences (Barton and Hewitt 1985). However, it is only relatively recently, with the advent of population genomics approaches, that this potential has begun to be realised.
3.1 Clines
The rate of change in allele frequency across a cline can be used to infer the strength of divergent selection acting on that locus if the average dispersal distance for the organism is also known (Barton and Hewitt 1985). This approach has been used extensively for single loci or phenotypes but has rarely been used with population genomic data. Nevertheless, it does have the potential to narrow down lists of candidates identified through outlier scan approaches by identifying the loci with the steepest allele frequency changes and with cline centres corresponding to the centre of the hybrid zone or phenotypic transition. Stankowski et al. (2017) applied this approach to a hybrid zone between monkey-flowers (Mimulus aurantiacus) with different floral traits and found that just 130 out of the 426 most differentiated loci had clines similar to that of the phenotypic trait (Fig. 6).

Geographic clines across a hybrid zone between yellow and red forms of monkey-flowers (Mimulus aurantiacus) for 426 highly differentiated loci (top 1% of the FCT distribution). The red line shows the cline at the colour controlling locus, MaMyb2. The dashed line is the average cline across all 426 loci. Most markers have cline slopes shallower than those seen at the known divergently selected locus, suggesting that only a subset are under divergent selection, despite all showing high differentiation. Reproduced from Stankowski et al. (2017), with permission
It is also possible to use sets of hybrid individuals to infer ‘genomic clines’ that can be independent of geographical clines. The method, developed by Gompert and Buerkle (2009) and Gompert and Alex Buerkle (2010), uses multiple loci to estimate a genomic background level of admixture for each individual and then detects loci that deviate significantly from this neutral background rate across the population. These loci can either show increased rates of introgression, indicating that they are under positive selection and sweeping through both populations (or spreading from one to the other), or reduced introgression, indicating that they are under divergent selection and not spreading between the populations (Fig. 7). The admixture proportions generated by this method can also be useful for inferring the age of the hybrid zone and the strength of barriers to gene flow, by establishing the proportions of early versus late generation hybrids that are present (Gompert et al. 2014; Nadeau 2014). However, unlike the geographic cline approach, where populations may differ at only a small number of loci, the genomic cline approach requires the parental populations to have marked allele frequency differences in order to reconstruct a background genomic cline.

Genomic clines from a simulated data set. (a) Across all loci and individuals. Loci are ordered based on map locations, and individuals are ordered based on their hybrid index (fraction of alleles coming from population 1). Each block in the plot denotes an individual’s genotype at that locus (dark green, homozygous population 1; green, heterozygous population 1/population 2; light green, homozygous population 2). (b) Hybrid index of each individual. (c) Clines at three individual loci (black lines, proportion homozygous population 1; dashed lines, proportion heterozygous) compared to the 95% confidence intervals for the genomic background (dark green and light green). The left plot shows a locus under selection, the middle plot is a locus linked to this, and the right plot is a locus not under selection. The circles show the raw genotype data for each individual. Reproduced from Gompert and Alex Buerkle (2010), with permission
3.2 Admixture Mapping
The most widely used approach for identifying genetic loci underlying a particular trait is to perform controlled laboratory crosses. Offspring from F2 or backcross generations can be genotyped with a relatively small number of parentally informative markers to identify the inheritance of large chromosomal blocks and to characterise where recombination breaks have occurred. This is then used to generate a dense linkage map and identify the genomic location of either Mendelian loci or quantitative trait loci (QTL) (many descriptions of these methods have been published previously, e.g. Liu 1997). This approach has been extensively and successfully used but is limited to taxa that can be reared in captivity and can usually only identify fairly large genomic intervals because of the limited number of recombination events that occur within a limited number of offspring and a few generations.
Admixture mapping has the same underlying rational as linkage mapping using crosses, but instead uses naturally admixed populations (Winkler et al. 2010). This also relies on the mixing populations being sufficiently genetically distinct that they will consistently differ at many positions across the genome, allowing blocks of the genome in admixed individuals to be assigned to one or other of the parental populations (Fig. 8). It then looks for statistical associations between inheriting a particular chromosomal block from one parental population and a trait found in that population. The main applications of admixture mapping have been to map phenotypic and disease traits in admixed human populations, for example, African Americans who can trace their ancestry to both African and European populations (Shriver et al. 2003). However, it can also be applied to other species, particularly where genetically distinct populations meet and mix in hybrid zones. For example, QTL for leaf morphological traits have been mapped in naturally occurring hybrids of white poplar (Populus alba) and European aspen (Populus tremula) tree species (Lindtke et al. 2013). The Populus system is ideally suited to admixture mapping because the parental populations (species in this case) show marked allele frequency differences and natural hybrid zones occur at the boundaries of the preferred habitat (flood plain vs. upland) of each species.

Schematic representation of admixture between two starting (parental) populations, on one pair of chromosomes, over a limited number of generations (F1–F6). After many generations (Fx), the genotypes of the two populations have become homogenised, except for regions tightly linked to those under divergent selection, which resembles the situation of divergence in sympatry. Populations in which distinct genomic blocks can be assigned to one or other parental population are suitable for admixture mapping, while those that are more genetically homogenous are more suitable for genome-wide association mapping
A major advantage of admixture mapping over traditional QTL mapping using crosses is that there are likely to have been many generations of hybridisation and recombination, leading to small ancestry blocks, giving the potential to map loci to narrow genomic intervals. However, in reality the power of admixture mapping to identify QTL decreases with the number of generations of admixture (Lindtke et al. 2013), because the genomic blocks inherited from each parental population become too small to be identified. Ultimately this comes down to the same issue as low genetic differentiation between the parental populations; many generations of hybridisation will erode the genetic differentiation between the parental populations, leading to an inability to assign genetic markers to a population of origin (Fig. 8).
3.3 Genome-Wide Association Mapping
In situations in which genetic differentiation between parental populations is too low to allow admixture mapping, a suitable alternative approach can be to use genome-wide association (GWA) mapping. This method is dealt with in detail in another chapter, but it is worth highlighting some of the considerations when applying this technique to hybrid zone populations. Like admixture mapping, GWA mapping was first developed for human populations, with the idea of being able to map loci linked to disease susceptibility. Although this approach has been reasonably successful, a major limitation has been that the traits being mapped are usually due to rare alleles, after all, alleles causing disease will tend to be removed by purifying selection. This is compounded in cases of complex phenotypes, where individual loci often have small effect sizes (Fig. 9). Together, these factors mean that extremely large sample sizes are needed in order to have the power to detect loci (Bush and Moore 2012; Kardos et al. 2016).

The power of genome-wide association studies increases with both the minor allele frequency and the effect size of the underlying loci. Traits that segregate across hybrid zones will tend to have a balanced allele frequency and in many cases are also controlled by large-effect loci
In contrast, loci controlling traits that differ across hybrid zones will usually have alleles at high frequency on either side of the hybrid zone. Therefore, sampling evenly from across the hybrid zone will tend to sample each allele at around 50%, making these potentially extremely powerful situations in which to use GWA mapping (Fig. 9). In addition, many traits that differ across hybrid zones have been found to be controlled by major-effect loci (Nadeau et al. 2014; Scordato and Safran 2016). In these situations, relatively small numbers of individuals (less than 100) can be sufficient to identify loci underlying phenotypic differences using a GWA framework. For example, just 30 individuals sampled from across a natural hybrid zone were successfully used to map major effect loci controlling colour pattern differences in the butterfly Heliconius melpomene (Nadeau et al. 2014) (Fig. 10). Hybridising populations will also tend to have relatively high linkage disequilibrium (LD) between loci within the genome (Box 1), with the extent depending on the level of genetic differentiation between the parental populations (as well as general factors such as population size and recombination rate). High LD will also tend to increase the power of GWA studies, particularly when reduced representation sequencing is used, due to larger numbers of loci being in LD with the causative site (Kardos et al. 2016). However, high LD between loci, especially the long-range LD that can occur in contact zones, will also increase the false positive rate and make fine mapping of functional variants more difficult.

Identifying loci underlying divergently selected traits in the butterfly Heliconius melpomene: a comparison of genomic differentiation and phenotypic association methods. (a) Butterflies from high elevation (left) and low elevation (right) near Tarapoto in Peru have very different wing colour patterns (photographs courtesy of Mathieu Joron). A narrow hybrid zone exists between these populations, which are maintained by strong positive frequency dependent selection, due to predator recognition of particular warning colour patterns (Mallet and Barton 1989). (b) Genome-wide differentiation (FST) between high and low elevation populations shows little background differentiation and few regions of high differentiation predicted to be under divergent selection (red points). Each point represents one SNP. (c) Genome-wide association mapping of red colour pattern elements (red points) and yellow colour pattern elements (yellow points) from 30 individuals, including 10 with hybrid phenotypes, from across the hybrid zone. This clearly identifies distinct loci for each trait, which correspond to the two most prominent divergently selected loci. The patterns of phenotypic association are less noisy than the patterns of genomic differentiation. Produced with data from Nadeau et al. (2014)
Population structure, causing genome-wide LD between unlinked loci, is a consideration for all GWA studies (Segura et al. 2012). However, it can be particularly problematic for hybrid zones. Even if there is little genetic differentiation between parental populations, a trait that changes along a linear transect will tend to be correlated with genome-wide genetic differences due to isolation by distance. For traits controlled by large-effect loci that change rapidly over short geographical distances, such as wing pattern in H. melpomene (Fig. 10), the problem is reduced because loci tightly linked to those controlling the traits will tend to show much stronger associations with phenotype than other loci in the genome. Similarly, a GWA study identified a major-effect locus controlling colour pattern in the stick insect Timema cristinae (Comeault et al. 2015). In this case the colour pattern morphs are cryptic on different host plants, which occur in mosaic patches within the landscape. In mosaic hybrid zones with high gene flow, such as this, background genetic structure is more likely to be decoupled from divergently selected loci (Nosil et al. 2002), making GWA mapping a potentially powerful tool. However, for traits that change gradually and linearly with distance and have a polygenic architecture, disentangling real versus correlated genetic associations is likely to be difficult. Although many ecologically relevant traits are likely to follow this pattern, there have been few attempts so far to apply GWA mapping to polygenic traits with broad geographic clines, perhaps because of the inherent challenge this poses. However, efficient mixed model approaches have been successfully used to control for complex population structure in GWA studies of humans and plants (Berg and Coop 2014; Segura et al. 2012; Zhou and Stephens 2012), demonstrating the potential of GWA mapping to identify the genetic basis of complex traits that show clinal variation.
In summary, the presence of admixture between populations or species provides a valuable opportunity to identify loci that are divergently selected or control particular phenotypes. There are several methods for detecting these loci, and their applicability depends partly on the extent of gene flow between species. Methods that test for associations between genotype and phenotype are the most powerful (Crawford and Nielsen 2013) and arguably also the most informative in terms of understanding the underlying selective pressures.
4 Detecting Hybridisation and Gene Flow Between Species
A major insight from population genomics studies has been the extent and prevalence of gene flow between species at multiple levels of divergence. Genome-wide markers allow introgressed variation to be identified, quantified and the history of hybridisation modelled through time, as never before. A large number of methods have emerged for identifying, quantifying and/or characterising gene flow between species, which are summarised in Table 2 taken from a thorough review of the topic by Payseur and Rieseberg (2016). Some of these methods overlap with those described in the previous sections for characterising divergently selected loci. Detecting gene flow is in some regards the inverse of this, and for populations where gene flow is high, these methods can be appropriate. However, for situations in which gene flow is rare or more ancient, more sensitive methods are needed.
Studies of humans have again largely paved the way in these approaches, motivated by the question of whether modern humans hybridised with Neanderthals during their colonisation of Europe. Sequencing of Neanderthal mitochondrial DNA did not reveal any evidence for hybridisation, with all Neanderthal sequences forming a cluster distinct from that of modern humans (Caramelli et al. 2006). However, genome-wide sequencing revealed an excess of genetic variants shared between Neanderthals and present-day Eurasian populations as compared to present-day African populations, suggesting gene flow may have occurred between Neanderthals and Eurasian modern humans (Green et al. 2010). This analysis was formalised as the D-statistic (or ABBA-BABA test), which uses an outgroup to test for an excess of shared derived SNPs between two putatively hybridising taxa (Fig. 8). Unfortunately this analysis has some problems, the most significant being that similar patterns of shared derived SNPs can be found if spatial population structure is present in the ancestral populations that both species diverged from, which is likely to have been the case in these archaic hominins (Durand et al. 2011; Eriksson and Manica 2012).
Nevertheless, subsequent studies using other approaches have also found evidence for gene flow between Neanderthals and modern humans. Sankararaman et al. (2012) used the extent of LD within the genomes of present-day Europeans to confirm and date the periods of gene flow with Neanderthals. LD is expected to break down with time, so if shared genetic variants were due to ancient population structure then blocks of LD would be shorter than if these were due to introgression events. Based on the size of the LD blocks containing variants shared between Europeans and Neanderthals, they concluded that introgression occurred between 37,000 and 86,000 years ago, long after the split between modern humans and Neanderthals. Subsequently, Lohse and Frantz (2014), estimated the maximum likelihood fit to models of admixture or ancestral population structure, using small non-recombining blocks of the genomes of two modern humans and a Neanderthal. They found strong support for Neanderthal admixture and obtained higher estimates of the rate of admixture (3.4–7.3%) than previous methods.
The Heliconius butterflies are another system in which population genomics has been used to characterise the extent and timing of gene flow between species. It had long been suspected that species within this genus did hybridise occasionally in the wild, with hybrids even found between fairly distantly related species (Dasmahapatra et al. 2007). The first population genomic evidence for gene flow between species again used ABBA-BABA D-statistics to show an excess of shared derived variants between sympatric sister species as opposed to allopatric populations of these species (Heliconius Genome Consortium 2012) (Fig. 11). In this case it was hard to envisage a scenario under which ancestral population structure could have given rise to these shared variants, because increased levels of shared variants were found in multiple sympatric population pairs in different geographic locations (Martin et al. 2013; Nadeau et al. 2013). However, other problems with the D-statistic were revealed. In particular, D-statistics do not reliably give the location of introgressed variants in the genome because genome-wide patterns are strongly correlated with nucleotide diversity, and simulations revealed that they could not reliably be used to compare the extent of gene flow between loci (Martin et al. 2014). Instead a different statistic, f, has been proposed, which also makes use of ABBA-BABA patterns but was found to be more robust to variation in nucleotide diversity and a better estimator of localised gene flow within the genome (Fig. 11).

ABBA-BABA methods for detecting gene flow between species. Initially formulated to test for introgression from Neanderthals (P3) into modern European humans (P2), by comparison to an outgroup (O) and an ingroup that would not have experienced gene flow (P1, Africans in this case) (Green et al. 2010). The coloured lines show the situation of incomplete lineage sorting, where ABBA and BABA patterns can arise due to polymorphism in the ancestor of P1, P2 and P3, which is sorted between the species. Without gene flow, an equal number of ABBA and BABA sites should be present, while gene flow will increase the number of ABBA sites. The D-statistic measures the relative proportion of ABBA to BABA sites, with CABBA and CBABA being counts of the number of sites showing ABBA and BABA patterns, respectively. The f statistic was initially proposed to quantify the fraction of the genome shared through introgression, by comparing the difference between CABBA and CBABA to the maximum difference possible by substituting P2 for P3 (Green et al. 2010). This statistic and variations thereof were also proposed to be more suitable for identifying introgressed regions of the genome, for example, to test if colour pattern controlling loci had introgressed between sympatric populations of the butterflies Helcionius melpomene and H. timareta (Martin et al. 2014). Butterfly photographs courtesy of Mathieu Joron
A wide range of population genomic methods for inferring gene flow between species now exist. While some of these, such as ABBA-BABA and FST, are attractive because of their intuitive simplicity, they can be influenced by factors other than migration and do not provide estimates of the rate or timing of gene flow. Undoubtedly better are methods that test the fit of population genomic models, which can include varying amounts and timings of gene flow and can also incorporate factors such as population structure and varying population size to either patterns of nucleotide variation (Lohse and Frantz 2014) or the frequency spectrum of genetic variants (Gutenkunst et al. 2009). Roux et al. (2016) used an approximate Bayesian computation (ABC) framework to assess the extent of gene flow between 61 pairs of diverse animal species/populations from across the divergence continuum. They found a strong relationship between a simple divergence metric, Da (relative divergence, corrected for within-species diversity, which is strongly correlated with FST) and the extent of gene flow. However, both distinct species with virtually no gene flow and populations with high gene flow were present within a ‘grey zone’ between 0.5 and 2% net synonymous divergence, demonstrating the increased power of model-based approaches to detect and quantify gene flow.
5 Future Perspectives
As with all areas of population genomics, the field is moving quickly, with new methods and approaches continually being developed. The field of speciation genomics essentially started with genome scans for divergence or differentiation outliers, but the challenges in this approach are now widely appreciated (Ravinet et al. 2017). Comparative genome scan approaches using multiple pairs of species can provide a powerful framework to distinguish differentiation islands containing barrier loci from high divergence regions not directly associated with barriers to gene flow (incidental islands) (Burri 2017). Nevertheless, we also need an explicit null model, to understand how baseline genetic diversity varies under background selection at linked sites (Comeron 2017; Ravinet et al. 2017). Current differentiation outlier analyses implicitly assume uniform Ne along a genome and stable Ne over evolutionary time (i.e. uniform and stable recombination rate and gene density), but we are increasingly aware of the heterogeneity of these parameters associated with variation in the effect of background selection. Signatures of selection at barrier loci can be detected by comparing the observed patterns of genetic diversity with those expected under a null model with background selection. Ideally, detailed recombination maps for the organism in question would be used to simulate baseline genetic diversity, but such maps are rarely available. However, a broad ‘U-shape’ recombination landscape (i.e. higher recombination rate at the both ends of chromosomes) appears to be a general pattern in various species (Berner and Roesti 2017) and can be used as a proxy for species without detailed recombination maps.
The problems raised by variable Ne across the genome are not unique to divergence measures and will also influence other metrics such as cline shape and ABBA-BABA D-statistics (Gompert et al. 2017; Martin et al. 2014). Neutral processes (drift) and background selection combined with variation in recombination and mutation rates across the genome will produce variation in cline shapes. Therefore, to reliably detect either barrier loci or adaptively introgressed loci between species, null distributions for genomic clines and admixture proportions are needed. These should again ideally take into account recombination rate variation across the genome (Payseur and Rieseberg 2016). In addition, if the ultimate goal is to understand the role of natural selection in speciation, outlier loci detected by any method need to be linked to the phenotypes they control. Therefore, studies of genomes alone can only take us so far and need to be partnered with a detailed understanding of the phenotypes and ecology of the organisms in question.
The recent advances in long-read sequencing (e.g. PacBio and Oxford Nanopore), linked read sequencing (e.g. 10× Genomics) and long-range scaffolding technologies (e.g. optical mapping and Hi-C chromosome conformation capture) are beginning to substantially improve the contiguity of reference genomes. For instance, recently published reference genomes of mosquito (Aedes aegypti), grey mouse lemur (Microcebus murinus) and hooded crow (Corvus [corone] cornix) cover almost entire chromosomes, including highly repetitive regions, such as centromeres and pericentromeric regions (Dudchenko et al. 2017; Larsen et al. 2017; Weissensteiner et al. 2017). These regions tend to have low recombination rate due to their heterochromatic nature and likely coincide with elevated differentiation (Ellegren et al. 2012), possibly due to the effect of selection at linked sites. Importantly, one of these low recombination regions in crows contained several genes associated with plumage colour difference, which are likely to be under divergent selection (Fig. 5) (Poelstra et al. 2014).
Long-read and long-range sequencing technologies are also key tools for the identification of large structural variants, such as inversions and translocations (Peichel et al. 2017). Chromosomal rearrangements have been suggested to play a key role in speciation by suppressing recombination and extending the effects of linked barrier loci (Kirkpatrick and Barton 2006; Navarro and Barton 2003; Noor et al. 2001; Rieseberg 2001). These models suggest that selection can facilitate the establishment and spread of new chromosomal mutations that harbour combinations of alleles contributing to local adaptation, or that rearrangements protect combinations of alleles that contribute to reproductive isolation from being disrupted by recombination. There are a growing number of examples showing an association between inversions and segregating phenotypes under divergent selection (Feder et al. 2003; Lowry and Willis 2010; McGaugh and Noor 2012; Turner et al. 2005). However, in other systems, such as Heliconius, divergence at many loci can be maintained in the absence of major structural variants or suppression of recombination (Davey et al. 2017). It is therefore not clear whether recombination modifiers, such as inversions, or more generally regions of low recombination (as found in crows), are necessary for the process of divergence with gene flow. New sequencing technologies will provide new insights into the frequencies of structural polymorphisms and their potential roles in speciation.
6 Conclusions
As high-throughput sequencing technologies have become accessible to many evolutionary biologists, there are a number of empirical studies published every year, describing genetic differences between genomes of diverging species and quantifying the level of gene flow between hybridising taxa. Nevertheless, despite the prediction based on the genic model of speciation (Wu 2001), genomic regions of elevated differentiation do not always harbour genes involved in RI or divergent selection. This does not necessarily mean that the model or analytical approaches are incorrect, but we need to develop an analytically tractable null model to predict genome-wide pattern of genetic diversity. Hybrid zones and admixed populations have been known as powerful model systems in speciation research for decades, but the advent of big population genomic data allows to fully exploit the power of these research systems by applying both traditional cline analysis and GWA. A combination of emerging new sequencing technologies and the development of analytical models will further provide a clearer picture of species divergence in the face of gene flow and identify barrier loci and their relative roles in the process of speciation.
References
Abbott R, Albach D, Ansell S, Arntzen JW, Baird SJE, Bierne N, et al. Hybridization and speciation. J Evol Biol. 2013;26(2):229–46.
Alexander DH, Novembre J, Lange K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009;19(9):1655–64.
Andrew RL, Rieseberg LH. Divergence is focused on few genomic regions early in speciation: incipient speciation of sunflower ecotypes. Evolution. 2013;67:2468–82.
Ané C, Larget B, Baum DA, Smith SD, Rokas A. Bayesian estimation of concordance among gene trees. Mol Biol Evol. 2007;24(2):412–26.
Auton A, Fledel-Alon A, Pfeifer S, Venn O, Segurel L, Street T, et al. A fine-scale chimpanzee genetic map from population sequencing. Science. 2012;336:193–8.
Barton NH, Hewitt GM. Analysis of hybrid zones. Annu Rev Ecol Syst. 1985;16(1):113–48.
Beaumont MA, Balding DJ. Identifying adaptive genetic divergence among populations from genome scans. Mol Ecol. 2004;13(4):969–80.
Beaumont MA, Zhang W, Balding DJ. Approximate Bayesian computation in population genetics. Genetics. 2002;162(4):2025–35.
Becquet C, Przeworski M. A new approach to estimate parameters of speciation models with application to apes. Genome Res. 2007;17(10):1505–19.
Begun DJ, Holloway AK, Stevens K, Hillier LW, Poh YP, Hahn MW, et al. Population genomics: whole-genome analysis of polymorphism and divergence in Drosophila simulans. PLoS Biol. 2007;5(11):2534–59.
Bell MA, Foster SA. The evolutionary biology of the threespine stickleback. Oxford: Oxford University Press; 1994.
Berg JJ, Coop G. A population genetic signal of polygenic adaptation. PLoS Genet. 2014;10(8):e1004412.
Berner D, Roesti M. Genomics of adaptive divergence with chromosome-scale heterogeneity in crossover rate. Mol Ecol. 2017;26(22):6351–69.
Berner D, Roesti M, Hendry AP, Salzburger W. Constraints on speciation suggested by comparing lake-stream stickleback divergence across two continents. Mol Ecol. 2010;19:4963–78.
Berner D, Moser D, Roesti M, Buescher H, Salzburger W. Genetic architecture of skeletal evolution in European lake and stream stickleback. Evolution. 2014;68:1792–805.
Bosse M, Spurgin LG, Laine VN, Cole EF, Firth JA, Gienapp P, et al. Recent natural selection causes adaptive evolution of an avian polygenic trait. Science. 2017;358(6361):365–8.
Brandvain Y, Kenney AM, Flagel L, Coop G, Sweigart AL. Speciation and introgression between Mimulus nasutus and Mimulus guttatus. PLoS Genet. 2014;10(6):e1004410.
Burri R. Interpreting differentiation landscapes in the light of long-term linked selection. Evol Lett. 2017;1(3):118–31.
Burri R, Nater A, Kawakami T, Mugal CF, Olason PI, Smeds L, et al. Linked selection and recombination rate variation drive the evolution of the genomic landscape of differentiation across the speciation continuum of Ficedula flycatchers. Genome Res. 2015;25:1656–65.
Bush WS, Moore JH. Genome-wide association studies. PLoS Comput Biol. 2012;8(12):e1002822.
Butlin RK. Population genomics and speciation. Genetica. 2008;138(4):409–18.
Caputo B, Pichler V, Mancini E, Pombi M, Vicente JL, Dinis J, et al. The last bastion? X chromosome genotyping of Anopheles gambiae species pair males from a hybrid zone reveals complex recombination within the major candidate ‘genomic island of speciation’. Mol Ecol. 2016;25(22):5719–31.
Caramelli D, Lalueza-Fox C, Condemi S, Longo L, Milani L, Manfredini A, et al. A highly divergent mtDNA sequence in a Neandertal individual from Italy. Curr Biol. 2006;16(16):R630–2.
Carneiro M, Albert FW, Afonso S, Pereira RJ, Burbano H, Campos R, et al. The genomic architecture of population divergence between subspecies of the European rabbit. PLoS Genet. 2014;10(8):e1003519.
Chan YF, Marks ME, Jones FC, Villarreal G, Shapiro MD, Brady SD, et al. Adaptive evolution of pelvic reduction in sticklebacks by recurrent deletion of a Pitx1 enhancer. Science. 2010;327(5963):302–5.
Colosimo PF, Hosemann KE, Balabhadra S, Villarreal G, Dickson M, Grimwood J, et al. Widespread parallel evolution in sticklebacks by repeated fixation of Ectodysplasin alleles. Science. 2005;307(5717):1928–33.
Comeault AA, Flaxman SM, Riesch R, Curran E, Soria-Carrasco V, Gompert Z, et al. Selection on a genetic polymorphism counteracts ecological speciation in a stick insect. Curr Biol. 2015;25(15):1975–81.
Comeron JM. Background selection as null hypothesis in population genomics: insights and challenges from Drosophila studies. Philos Trans R Soc Lond B Biol Sci. 2017;372(1736).
Coyne JA, Orr HA. Speciation. New York: W. H. Freeman; 2004.
Crawford JE, Nielsen R. Detecting adaptive trait loci in nonmodel systems: divergence or admixture mapping? Mol Ecol. 2013;22(24):6131–48.
Crawford JE, Riehle MM, Guelbeogo WM, Gneme A, Sagnon N, Vernick KD, et al. Reticulate speciation and barriers to introgression in the Anopheles gambiae species complex. Genome Biol Evol. 2015;7(11):3116–31.
Cruickshank TE, Hahn MW. Reanalysis suggests that genomic islands of speciation are due to reduced diversity, not reduced gene flow. Mol Ecol. 2014;23(13):3133–57.
Cutter AD, Payseur BA. Genomic signatures of selection at linked sites: unifying the disparity among species. Nat Rev Genet. 2013;14(4):262–74.
Dasmahapatra KK, Silva-Vásquez A, Chung J-W, Mallet J. Genetic analysis of a wild-caught hybrid between non-sister Heliconius butterfly species. Biol Lett. 2007;3(6):660–3.
Davey JW, Barker SL, Rastas PM, Pinharanda A, Martin SH, Durbin R, et al. No evidence for maintenance of a sympatric Heliconius species barrier by chromosomal inversions. Evol Lett. 2017;1(3):138–54.
Deagle BE, Jones FC, Chan YF, Absher DM, Kingsley DM, Reimchen TE. Population genomics of parallel phenotypic evolution in stickleback across stream–lake ecological transitions. Proc R Soc B Biol Sci. 2011. http://rspb.royalsocietypublishing.org/content/early/2011/10/04/rspb.2011.1552.abstract. Cited 13 Dec 2011.
Dobzhansky T. Studies on hybrid sterility. II. Localization of sterility factors in Drosophila pseudoobscura hybrids. Genetics. 1936;21:113–35.
Dudchenko O, Batra SS, Omer AD, Nyquist SK, Hoeger M, Durand NC, et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science. 2017;356(6333):92–5.
Durand EY, Patterson N, Reich D, Slatkin M. Testing for ancient admixture between closely related populations. Mol Biol Evol. 2011;28(8):2239–52.
Duvaux L, Belkhir K, Boulesteix M, Boursot P. Isolation and gene flow: inferring the speciation history of European house mice. Mol Ecol. 2011;20(24):5248–64.
Egan SP, Ragland GJ, Assour L, Powell THQ, Hood GR, Emrich S, et al. Experimental evidence of genome-wide impact of ecological selection during early stages of speciation-with-gene-flow. Ecol Lett. 2015;18(8):817–25.
Elgvin TO, Trier CN, Tørresen OK, Hagen IJ, Lien S, Nederbragt AJ, et al. The genomic mosaicism of hybrid speciation. Sci Adv. 2017;3(6):e1602996.
Ellegren H, Smeds L, Burri R, Olason PI, Backström N, Kawakami T, et al. The genomic landscape of species divergence in Ficedula flycatchers. Nature. 2012;491(7426):756–60.
Eriksson A, Manica A. Effect of ancient population structure on the degree of polymorphism shared between modern human populations and ancient hominins. Proc Natl Acad Sci. 2012;109(35):13956–60.
Falush D, Stephens M, Pritchard JK. Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics. 2003;164(4):1567–87.
Feder JL, Nosil P. The efficacy of divergence hitchhiking in generating genomic islands during ecological speciation. Evolution. 2010;64(6):1729–47.
Feder JL, Roethele JB, Filchak K, Niedbalski J, Romero-Severson J. Evidence for inversion polymorphism related to sympatric host race formation in the apple maggot fly, Rhagoletis pomonella. Genetics. 2003;163(3):939–53.
Feder JL, Gejji R, Yeaman S, Nosil P. Establishment of new mutations under divergence and genome hitchhiking. Philos Trans R Soc Lond B Biol Sci. 2012;367(1587):461–74.
Feulner PGD, Chain FJJ, Panchal M, Huang Y, Eizaguirre C, Kalbe M, et al. Genomics of divergence along a continuum of parapatric population differentiation. PLoS Genet. 2015;11:e1004966.
Fitzpatrick BM. Alternative forms for genomic clines. Ecol Evol. 2013;3(7):1951–66.
Flaxman SM, Wacholder AC, Feder JL, Nosil P. Theoretical models of the influence of genomic architecture on the dynamics of speciation. Mol Ecol. 2014;23(16):4074–88.
Gagnaire P-A, Pavey SA, Normandeau E, Bernatchez L. The genetic architecture of reproductive isolation during speciation-with-gene-flow in lake whitefish species pairs assessed by rad sequencing. Evolution. 2013;67(9):2483–97.
Garrigan D, Kingan SB, Geneva AJ, Andolfatto P, Clark AG, Thornton KR, et al. Genome sequencing reveals complex speciation in the Drosophila simulans clade. Genome Res. 2012;22(8):1499–511.
Gompert Z, Alex Buerkle C. Introgress: a software package for mapping components of isolation in hybrids. Mol Ecol Resour. 2010;10(2):378–84.
Gompert Z, Buerkle CA. A powerful regression-based method for admixture mapping of isolation across the genome of hybrids. Mol Ecol. 2009;18(6):1207–24.
Gompert Z, Buerkle CA. Bayesian estimation of genomic clines. Mol Ecol. 2011;20(10):2111–27.
Gompert Z, Lucas LK, Buerkle CA, Forister ML, Fordyce JA, Nice CC. Admixture and the organization of genetic diversity in a butterfly species complex revealed through common and rare genetic variants. Mol Ecol. 2014;23(18):4555–73.
Gompert Z, Mandeville EG, Buerkle CA. Analysis of population genomic data from hybrid zones. Annu Rev Ecol Evol Syst. 2017;48(1):207–29.
Gravel S. Population genetics models of local ancestry. Genetics. 2012;191(2):607–19.
Green RE, Krause J, Briggs AW, Maricic T, Stenzel U, Kircher M, et al. A draft sequence of the neandertal genome. Science. 2010;328(5979):710–22.
Gutenkunst RN, Hernandez RD, Williamson SH, Bustamante CD. Inferring the joint demographic history of multiple populations from multidimensional SNP frequency data. PLoS Genet. 2009;5(10):e1000695.
Haasl RJ, Payseur BA. Fifteen years of genomewide scans for selection: trends, lessons and unaddressed genetic sources of complication. Mol Ecol. 2016;25(1):5–23.
Han F, Lamichhaney S, Grant BR, Grant PR, Andersson L, Webster MT. Gene flow, ancient polymorphism, and ecological adaptation shape the genomic landscape of divergence among Darwin’s finches. Genome Res. 2017. http://genome.cshlp.org/content/early/2017/04/25/gr.212522.116. Cited 10 May 2017.
Harris K, Nielsen R. Inferring demographic history from a spectrum of shared haplotype lengths. PLoS Genet. 2013;9(6):e1003521.
Harrison RG, Larson EL. Heterogeneous genome divergence, differential introgression, and the origin and structure of hybrid zones. Mol Ecol. 2016;25(11):2454–66.
Heliconius Genome Consortium. Butterfly genome reveals promiscuous exchange of mimicry adaptations among species. Nature. 2012;487(7405):94–8.
Hey J, Nielsen R. Multilocus methods for estimating population sizes, migration rates and divergence time, with applications to the divergence of Drosophila pseudoobscura and D. persimilis. Genetics. 2004;167(2):747–60.
Hey J, Nielsen R. Integration within the Felsenstein equation for improved Markov chain Monte Carlo methods in population genetics. Proc Natl Acad Sci. 2007;104(8):2785–90.
Hohenlohe PA, Bassham S, Etter PD, Stiffler N, Johnson EA, Cresko WA. Population genomics of parallel adaptation in threespine stickleback using sequenced RAD tags. PLoS Genet. 2010;6(2):e1000862.
Hohenlohe PA, Bassham S, Currey M, Cresko WA. Extensive linkage disequilibrium and parallel adaptive divergence across threespine stickleback genomes. Philos Trans R Soc Lond B Biol Sci. 2012;367:395–408.
Hubisz MJ, Falush D, Stephens M, Pritchard JK. Inferring weak population structure with the assistance of sample group information. Mol Ecol Resour. 2009;9(5):1322–32.
Huelsenbeck JP, Andolfatto P. Inference of population structure under a Dirichlet process model. Genetics. 2007;175(4):1787–802.
Janoušek V, Wang L, Luzynski K, Dufková P, Vyskočilová MM, Nachman MW, et al. Genome-wide architecture of reproductive isolation in a naturally occurring hybrid zone between Mus musculus musculus and M. m. domesticus. Mol Ecol. 2012;21(12):3032–47.
Jones FC, Chan YF, Schmutz J, Grimwood J, Brady SD, Southwick AM, et al. A genome-wide SNP genotyping array reveals patterns of global and repeated species-pair divergence in sticklebacks. Curr Biol. 2012a;22:83–90.
Jones FC, Grabherr MG, Chan YF, Russell P, Mauceli E, Johnson J, et al. The genomic basis of adaptive evolution in threespine sticklebacks. Nature. 2012b;484(7392):55–61.
Kardos M, Husby A, McFarlane SE, Qvarnström A, Ellegren H. Whole-genome resequencing of extreme phenotypes in collared flycatchers highlights the difficulty of detecting quantitative trait loci in natural populations. Mol Ecol Resour. 2016;16(3):727–41.
Kawakami T, Butlin RK. Hybrid zones. eLS. 2012. http://onlinelibrary.wiley.com/doi/10.1002/9780470015902.a0001752.pub2/abstract. Cited 20 Jul 2012.
Kawakami T, Smeds L, Backstrom N, Husby A, Qvarnstrom A, Mugal CF, et al. A high-density linkage map enables a second-generation collared flycatcher genome assembly and reveals the patterns of avian recombination rate variation and chromosomal evolution. Mol Ecol. 2014;23:4035–58.
Kawakami T, Mugal CF, Suh A, Nater A, Burri R, Smeds L, et al. Whole-genome patterns of linkage disequilibrium across flycatcher populations clarify the causes and consequences of fine-scale recombination rate variation in birds. Mol Ecol. 2017;26:4158–72.
Kirkpatrick M, Barton N. Chromosome inversions, local adaptation and speciation. Genetics. 2006;173(1):419–34.
Lamichhaney S, Berglund J, Almén MS, Maqbool K, Grabherr M, Martinez-Barrio A, et al. Evolution of Darwin’s finches and their beaks revealed by genome sequencing. Nature. 2015;518(7539):371.
Lamichhaney S, Han F, Webster MT, Andersson L, Grant BR, Grant PR. Rapid hybrid speciation in Darwin’s finches. Science. 2017:eaao4593. https://doi.org/10.1126/science.aao4593.
Larsen PA, Harris RA, Liu Y, Murali SC, Campbell CR, Brown AD, et al. Hybrid de novo genome assembly and centromere characterization of the gray mouse lemur (Microcebus murinus). BMC Biol. 2017;15:110.
Lawson DJ, Hellenthal G, Myers S, Falush D. Inference of population structure using dense haplotype data. PLoS Genet. 2012;8(1):e1002453.
Lindtke D, Buerkle CA, Barbará T, Heinze B, Castiglione S, Bartha D, et al. Recombinant hybrids retain heterozygosity at many loci: new insights into the genomics of reproductive isolation in Populus. Mol Ecol. 2012;21(20):5042–58.
Lindtke D, González-Martínez SC, Macaya-Sanz D, Lexer C. Admixture mapping of quantitative traits in Populus hybrid zones: power and limitations. Heredity. 2013;111(6):474–85.
Liu BH. Statistical genomics: linkage, mapping, and QTL analysis. Boca Raton: CRC Press; 1997.
Liu KJ, Dai J, Truong K, Song Y, Kohn MH, Nakhleh L. An HMM-based comparative genomic framework for detecting introgression in eukaryotes. PLoS Comput Biol. 2014a;10(6):e1003649.
Liu S, Lorenzen ED, Fumagalli M, Li B, Harris K, Xiong Z, et al. Population genomics reveal recent speciation and rapid evolutionary adaptation in polar bears. Cell. 2014b;157(4):785–94.
Lohse K, Frantz LAF. Neandertal admixture in Eurasia confirmed by maximum-likelihood analysis of three genomes. Genetics. 2014;196(4):1241–51.
Lohse K, Harrison RJ, Barton NH. A general method for calculating likelihoods under the coalescent process. Genetics. 2011;189(3):977–87.
Lohse K, Chmelik M, Martin SH, Barton NH. Efficient strategies for calculating blockwise likelihoods under the coalescent. Genetics. 2016;202(2):775–86.
Lowry DB, Willis JH. A widespread chromosomal inversion polymorphism contributes to a major life-history transition, local adaptation, and reproductive isolation. PLoS Biol. 2010;8(9):e1000500.
Mailund T, Halager AE, Westergaard M, Dutheil JY, Munch K, Andersen LN, et al. A new isolation with migration model along complete genomes infers very different divergence processes among closely related great ape species. PLoS Genet. 2012;8(12):e1003125.
Mallet J, Barton NH. Strong natural selection in a warning-color hybrid zone. Evolution. 1989;43(2):421–31.
Marques DA, Lucek K, Meier JI, Mwaiko S, Wagner CE, Excoffier L, et al. Genomics of rapid incipient speciation in sympatric threespine stickleback. PLoS Genet. 2016;12:e1005887.
Martin SH, Dasmahapatra KK, Nadeau NJ, Salazar C, Walters JR, Simpson F, et al. Genome-wide evidence for speciation with gene flow in Heliconius butterflies. Genome Res. 2013;23(11):1817–28.
Martin SH, Davey JW, Jiggins CD. Evaluating the use of ABBA–BABA statistics to locate introgressed loci. Mol Biol Evol. 2014;32(1):244–57.
Martin SH, Möst M, Palmer WJ, Salazar C, McMillan WO, Jiggins FM, et al. Natural selection and genetic diversity in the butterfly Heliconius melpomene. Genetics. 2016;203:525–41.
Mayr E. Systematics and the origin of species, from the viewpoint of a zoologist. London: Columbia University Press; 1942.
McGaugh SE, Noor MAF. Genomic impacts of chromosomal inversions in parapatric Drosophila species. Philos Trans R Soc Lond B Biol Sci. 2012;367(1587):422–9.
McKechnie SW, Blacket MJ, Song SV, Rako L, Carroll X, Johnson TK, et al. A clinally varying promoter polymorphism associated with adaptive variation in wing size in Drosophila. Mol Ecol. 2010;19(4):775–84.
McKinnon JS, Rundle HD. Speciation in nature: the threespine stickleback model systems. Trends Ecol Evol. 2002;17:480–8.
Meng C, Kubatko LS. Detecting hybrid speciation in the presence of incomplete lineage sorting using gene tree incongruence: a model. Theor Popul Biol. 2009;75(1):35–45.
Michel AP, Sim S, Powell THQ, Taylor MS, Nosil P, Feder JL. Widespread genomic divergence during sympatric speciation. Proc Natl Acad Sci. 2010;107(21):9724–9.
Moser D, Kueng B, Berner D. Lake-stream divergence in stickleback life history: a plastic response to trophic niche differentiation? Evol Biol. 2015;42:328–38.
Muller HJ. Bearing of the Drosophila work on systematics. In: Huxley J, editor. New systematics. Oxford: Oxford University Press; 1940. p. 185–268.
Nachman MW, Payseur BA. Recombination rate variation and speciation: theoretical predictions and empirical results from rabbits and mice. Philos Trans R Soc Lond B Biol Sci. 2012;367:409–21.
Nadachowska-Brzyska K, Burri R, Olason PI, Kawakami T, Smeds L, Ellegren H. Demographic divergence history of pied flycatcher and collared flycatcher inferred from whole-genome re-sequencing data. PLoS Genet. 2013;9(11):e1003942.
Nadeau N. Butterfly genomics sheds light on the process of hybrid speciation. Mol Ecol. 2014;23(18):4441–3.
Nadeau NJ, Whibley A, Jones RT, Davey JW, Dasmahapatra KK, Baxter SW, et al. Genomic islands of divergence in hybridizing Heliconius butterflies identified by large-scale targeted sequencing. Philos Trans R Soc Lond B Biol Sci. 2012;367(1587):343–53.
Nadeau NJ, Martin SH, Kozak KM, Salazar C, Dasmahapatra KK, Davey JW, et al. Genome-wide patterns of divergence and gene flow across a butterfly radiation. Mol Ecol. 2013;22(3):814–26.
Nadeau NJ, Ruiz M, Salazar P, Counterman B, Medina JA, Ortiz-Zuazaga H, et al. Population genomics of parallel hybrid zones in the mimetic butterflies, H. melpomene and H. erato. Genome Res. 2014;24(8):1316–33.
Navarro A, Barton NH. Accumulating postzygotic isolation genes in parapatry: a new twist on chromosomal speciation. Evol Int J Org Evol. 2003;57(3):447–59.
Noor MAF, Bennett SM. Islands of speciation or mirages in the desert? Examining the role of restricted recombination in maintaining species. Heredity. 2009;103:439–44.
Noor MAF, Grams KL, Bertucci LA, Reiland J. Chromosomal inversions and the reproductive isolation of species. Proc Natl Acad Sci. 2001;98(21):12084–8.
Nosil P. Ecological speciation. Oxford: Oxford University Press; 2012.
Nosil P, Feder JL. Genomic divergence during speciation: causes and consequences. Philos Trans R Soc Lond B Biol Sci. 2012;367:332–42.
Nosil P, Crespi BJ, Sandoval CP. Host-plant adaptation drives the parallel evolution of reproductive isolation. Nature. 2002;417(6887):440–3.
Nosil P, Feder JL, Flaxman SM, Gompert Z. Tipping points in the dynamics of speciation. Nat Ecol Evol. 2017;1(2):0001.
Orr HA. The population-genetics of speciation – the evolution of hybrid incompatibilities. Genetics. 1995;139:1805–13.
Patterson N, Price AL, Reich D. Population structure and eigenanalysis. PLoS Genet. 2006;2(12):e190.
Patterson N, Moorjani P, Luo Y, Mallick S, Rohland N, Zhan Y, et al. Ancient admixture in human history. Genetics. 2012;192(3):1065–93.
Payseur BA, Rieseberg LH. A genomic perspective on hybridization and speciation. Mol Ecol. 2016;25(11):2337–60.
Pease JB, Hahn MW. Detection and polarization of introgression in a five-taxon phylogeny. Syst Biol. 2015;64(4):651–62.
Peichel CL, Sullivan ST, Liachko I, White MA. Improvement of the threespine stickleback genome using a Hi-C-based proximity-guided assembly. J Hered. 2017;108(6):693–700.
Pickrell JK, Pritchard JK. Inference of population splits and mixtures from genome-wide allele frequency data. PLoS Genet. 2012;8(11):e1002967.
Picq S, McMillan WO, Puebla O. Population genomics of local adaptation versus speciation in coral reef fishes (Hypoplectrus spp, Serranidae). Ecol Evol. 2016;6(7):2109–24.
Poelstra JW, Vijay N, Bossu CM, Lantz H, Ryll B, Müller I, et al. The genomic landscape underlying phenotypic integrity in the face of gene flow in crows. Science. 2014;344:1410–4.
Pool JE, Nielsen R. Inference of historical changes in migration rate from the lengths of migrant tracts. Genetics. 2009;181(2):711–9.
Pool JE, Corbett-Detig RB, Sugino RP, Stevens KA, Cardeno CM, Crepeau MW, et al. Population genomics of sub-Saharan Drosophila melanogaster: African diversity and non-African admixture. PLoS Genet. 2012;8(12):e1003080.
Porter AH, Wenger R, Geiger H, Scholl A, Shapiro AM. The pontia daplidice-edusa hybrid zone in northwestern Italy. Evolution. 1997;51(5):1561–73.
Powell THQ, Hood GR, Murphy MO, Heilveil JS, Berlocher SH, Nosil P, et al. Genetic divergence along the speciation continuum: the transition from host race to species in Rhagoletis (Diptera: Tephritidae). Evolution. 2013;67(9):2561–76.
Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006;38(8):904–9.
Price AL, Tandon A, Patterson N, Barnes KC, Rafaels N, Ruczinski I, et al. Sensitive detection of chromosomal segments of distinct ancestry in admixed populations. PLoS Genet. 2009;5(6):e1000519.
Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155(2):945–59.
Puebla O, Bermingham E, McMillan WO. Genomic atolls of differentiation in coral reef fishes (Hypoplectrus spp., Serranidae). Mol Ecol. 2014;23(21):5291–303.
Qvarnström A, Rice AM, Ellegren H. Speciation in Ficedula flycatchers. Philos Trans R Soc Lond B Biol Sci. 2010;365(1547):1841–52.
Raj A, Stephens M, Pritchard JK. fastSTRUCTURE: variational inference of population structure in large SNP data sets. Genetics. 2014;197(2):573–89.
Randler C. Assortative mating of Carrion Corvus corone and Hooded Crows C. cornix in the hybrid zone in eastern Germany. Ardea. 2007;95(1):143–9.
Ravinet M, Westram A, Johannesson K, Butlin R, André C, Panova M. Shared and nonshared genomic divergence in parallel ecotypes of Littorina saxatilis at a local scale. Mol Ecol. 2016;25(1):287–305.
Ravinet M, Faria R, Butlin RK, Galindo J, Bierne N, Rafajlović M, et al. Interpreting the genomic landscape of speciation: a road map for finding barriers to gene flow. J Evol Biol. 2017;30(8):1450–77.
Renaut S, Maillet N, Normandeau E, Sauvage C, Derome N, Rogers SM, et al. Genome-wide patterns of divergence during speciation: the lake whitefish case study. Philos Trans R Soc Lond B Biol Sci. 2012;367(1587):354–63.
Renaut S, Grassa CJ, Yeaman S, Moyers BT, Lai Z, Kane NC, et al. Genomic islands of divergence are not affected by geography of speciation in sunflowers. Nat Commun. 2013;4:1827.
Riesch R, Muschick M, Lindtke D, Villoutreix R, Comeault AA, Farkas TE, et al. Transitions between phases of genomic differentiation during stick-insect speciation. Nat Ecol Evol. 2017;1:0082.
Rieseberg LH. Chromosomal rearrangements and speciation. Trends Ecol Evol. 2001;16(7):351–8.
Rieseberg LH, Whitton J, Gardner K. Hybrid zones and the genetic architecture of a barrier to gene flow between two sunflower species. Genetics. 1999;152(2):713–27.
Robinson JD, Bunnefeld L, Hearn J, Stone GN, Hickerson MJ. ABC inference of multi-population divergence with admixture from unphased population genomic data. Mol Ecol. 2014;23(18):4458–71.
Roesti M, Hendry AP, Salzburger W, Berner D. Genome divergence during evolutionary diversification as revealed in replicate lake–stream stickleback population pairs. Mol Ecol. 2012;21(12):2852–62.
Roesti M, Moser D, Berner D. Recombination in the threespine stickleback genome – patterns and consequences. Mol Ecol. 2013;22:3014–27.
Roesti M, Gavrilets S, Hendry AP, Salzburger W, Berner D. The genomic signature of parallel adaptation from shared genetic variation. Mol Ecol. 2014;23:3944–56.
Roesti M, Kueng B, Moser D, Berner D. The genomics of ecological vicariance in threespine stickleback fish. Nat Commun. 2015;6:8767.
Rogers SM, Campbell D, Baird SJE, Danzmann RG, Bernatchez L. Combining the analyses of introgressive hybridisation and linkage mapping to investigate the genetic architecture of population divergence in the lake whitefish (Coregonus clupeaformis, Mitchill). Genetica. 2001;111(1–3):25–41.
Roux C, Fraïsse C, Romiguier J, Anciaux Y, Galtier N, Bierne N. Shedding light on the grey zone of speciation along a continuum of genomic divergence. PLoS Biol. 2016;14(12):e2000234.
Sankararaman S, Patterson N, Li H, Pääbo S, Reich D. The date of interbreeding between Neandertals and modern humans. PLoS Genet. 2012;8(10):e1002947.
Scordato ESC, Safran RJ. Evolutionary genetics: small genomic regions make a big impact. Curr Biol. 2016;26(21):R1155–7.
Seehausen O, Butlin RK, Keller I, Wagner CE, Boughman JW, Hohenlohe PA, et al. Genomics and the origin of species. Nat Rev Genet. 2014;15:176–92.
Segura V, Vilhjálmsson BJ, Platt A, Korte A, Seren Ü, Long Q, et al. An efficient multi-locus mixed-model approach for genome-wide association studies in structured populations. Nat Genet. 2012;44(7):825–30.
Sethuraman A, Hey J. IMa2p – parallel MCMC and inference of ancient demography under the isolation with migration (IM) model. Mol Ecol Resour. 2016;16(1):206–15.
Shriver MD, Parra EJ, Dios S, Bonilla C, Norton H, Jovel C, et al. Skin pigmentation, biogeographical ancestry and admixture mapping. Hum Genet. 2003;112(4):387–99.
Smukowski CS, Noor MAF. Recombination rate variation in closely related species. Heredity. 2011;107(6):496–508.
Soria-Carrasco V, Gompert Z, Comeault AA, Farkas TE, Parchman TL, Johnston JS, et al. Stick insect genomes reveal natural selection’s role in parallel speciation. Science. 2014;344(6185):738–42.
Sousa VC, Carneiro M, Ferrand N, Hey J. Identifying loci under selection against gene flow in isolation-with-migration models. Genetics. 2013;194(1):211–33.
Stankowski S, Sobel JM, Streisfeld MA. Geographic cline analysis as a tool for studying genome-wide variation: a case study of pollinator-mediated divergence in a monkeyflower. Mol Ecol. 2017;26(1):107–22.
Steinrücken M, Kamm JA, Song YS. Inference of complex population histories using whole-genome sequences from multiple populations. bioRxiv. 2015:026591. https://doi.org/10.1101/026591.
Svedin N, Wiley C, Veen T, Gustafsson L, Qvarnström A. Natural and sexual selection against hybrid flycatchers. Proc R Soc Lond B Biol Sci. 2008;275(1635):735–44.
Szymura JM, Barton NH. Genetic analysis of a hybrid zone between the fire-bellied toads, Bombina bombina and B. variegata, near Cracow in southern Poland. Evolution. 1986;40(6):1141–59.
Tang H, Peng J, Wang P, Risch NJ. Estimation of individual admixture: analytical and study design considerations. Genet Epidemiol. 2005;28(4):289–301.
Teeter KC, Payseur BA, Harris LW, Bakewell MA, Thibodeau LM, O’Brien JE, et al. Genome-wide patterns of gene flow across a house mouse hybrid zone. Genome Res. 2008;18(1):67–76.
Toews DPL, Taylor SA, Vallender R, Brelsford A, Butcher BG, Messer PW, et al. Plumage genes and little else distinguish the genomes of hybridizing warblers. Curr Biol. 2016;26(17):2313–8.
Turner TL, Hahn MW, Nuzhdin SV. Genomic islands of speciation in Anopheles gambiae. PLoS Biol. 2005;3:e285.
Turner TL, Levine MT, Eckert ML, Begun DJ. Genomic analysis of adaptive differentiation in Drosophila melanogaster. Genetics. 2008;179(1):455–73.
Vallejo-Marín M, Buggs RJA, Cooley AM, Puzey JR. Speciation by genome duplication: repeated origins and genomic composition of the recently formed allopolyploid species Mimulus peregrinus. Evolution. 2015;69(6):1487–500.
Via S. Divergence hitchhiking and the spread of genomic isolation during ecological speciation-with-gene-flow. Philos Trans R Soc Lond B Biol Sci. 2012;367(1587):451–60.
Via S, Conte G, Mason-Foley C, Mills K. Localizing F(ST) outliers on a QTL map reveals evidence for large genomic regions of reduced gene exchange during speciation-with-gene-flow. Mol Ecol. 2012;21:5546–60.
Vijay N, Bossu CM, Poelstra JW, Weissensteiner MH, Suh A, Kryukov AP, et al. Evolution of heterogeneous genome differentiation across multiple contact zones in a crow species complex. Nat Commun. 2016;7:13195.
Wang Y, Hey J. Estimating divergence parameters with small samples from a large number of loci. Genetics. 2010;184(2):363–79.
Wang J, Street NR, Scofield DG, Ingvarsson PK. Variation in linked selection and recombination drive genomic divergence during allopatric speciation of European and American Aspens. Mol Biol Evol. 2016;33:1754–67.
Wegmann D, Kessner DE, Veeramah KR, Mathias RA, Nicolae DL, Yanek LR, et al. Recombination rates in admixed individuals identified by ancestry-based inference. Nat Genet. 2011;43(9):847–53.
Weissensteiner MH, Pang AWC, Bunikis I, Höijer I, Vinnere-Pettersson O, Suh A, et al. Combination of short-read, long-read and optical mapping assemblies reveals large-scale tandem repeat arrays with population genetic implications. Genome Res. 2017;27(5):697–708.
Winkler CA, Nelson GW, Smith MW. Admixture mapping comes of age. Annu Rev Genomics Hum Genet. 2010;11(1):65–89.
Wolf JBW, Ellegren H. Making sense of genomic islands of differentiation in light of speciation. Nat Rev Genet. 2016;18:87–100.
Wu CI. The genic view of the process of speciation. J Evol Biol. 2001;14:851–65.
Yang Z. A likelihood ratio test of speciation with gene flow using genomic sequence data. Genome Biol Evol. 2010;2:200–11.
Yeaman S, Aeschbacher S, Bürger R. The evolution of genomic islands by increased establishment probability of linked alleles. Mol Ecol. 2016;25(11):2542–58.
Yu Y, Degnan JH, Nakhleh L. The probability of a gene tree topology within a phylogenetic network with applications to hybridization detection. PLoS Genet. 2012;8(4):e1002660.
Yu Y, Barnett RM, Nakhleh L. Parsimonious inference of hybridization in the presence of incomplete lineage sorting. Syst Biol. 2013;62(5):738–51.
Yu Y, Dong J, Liu KJ, Nakhleh L. Maximum likelihood inference of reticulate evolutionary histories. Proc Natl Acad Sci. 2014;111(46):16448–53.
Zhou X, Stephens M. Genome-wide efficient mixed-model analysis for association studies. Nat Genet. 2012;44(7):821–4.
Acknowledgements
We would like to thank Marius Roesti and Roger Butlin for their helpful comments on this chapter.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer International Publishing AG, part of Springer Nature
About this chapter

Cite this chapter
Nadeau, N.J., Kawakami, T. (2018). Population Genomics of Speciation and Admixture. In: Rajora, O. (eds) Population Genomics. Population Genomics. Springer, Cham. https://doi.org/10.1007/13836_2018_24
Download citation
DOI: https://doi.org/10.1007/13836_2018_24
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-04587-6
Online ISBN: 978-3-030-04589-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)