
Abstract
The utility of native chemical ligation (NCL) in the solution or solid phase synthesis of peptides, cyclic peptides, glycopeptides, and neoglycoconjugates is reviewed. In addition, the mechanistic details of inter- or intra-molecular NCLs are discussed from experimental and computational points of view.
✠Katritzky was deceased at the time of publication.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Acyl migration
- Acyl transfer
- Chemical ligation
- Computational studies
- Cyclic peptides
- Glycopeptides
- Neoglycoconjugates
- SPPS
1 Introduction to Native Chemical Ligation
Native chemical ligation (NCL) has shown great utility and is a proven method for the preparation of peptides, cyclic peptides, proteins, and glycoproteins via synthetic or semi-synthetic pathways [1–5]. The concept of NCL dates back to the pioneering work of Wieland et al. [6], but it was not until 1994 that this method gained widespread attention when Kent et al. reported its application in the synthesis of interleukin-8, a cytokine responsible for the proliferation of B cells during immune response [7].
Approaches to ligation of two peptide segments include prior thiol capture [8], NCL [1], conformationally assisted ligation [9], and Staudinger ligation [10], of which NCL is the most widely used method. NCL involves the chemoselective coupling of two protein or peptide fragments, one containing a C-terminal thioester and the other, typically, an N-terminal Cys residue (Fig. 1). The two components combine via an intramolecular S- to N-acyl shift to produce, irreversibly, the ligated amide bond (a native peptide bond) at the point of ligation. The fact that this reaction occurs in aqueous solution and in the absence of protecting groups has placed this powerful technology at the forefront of protein synthesis. The driving force for NCL is the formation of the thermodynamically stable amide link, at around neutral pH [3].

Intermolecular chemical ligation (NCL)
The rate of the ligation reaction depends on the C-terminal amino acid. Dawson et al. reported that all 20 amino acids could be used, but amino acids such as Pro, Val, and Ile gave reduced reaction rates (Fig. 2) [10]. Additives such as urea and guanidinium chloride can be added to the buffer solution to prevent the aggregation of amino acids or peptides in the solution phase synthesis [11].

NCL with all amino acids
The requirement of a Cys residue (Xaa-Cys, where Xaa=any amino acid) at the ligation site can be problematic because not all proteins contain a Cys residue and the Cys residues may be present at locations in the protein which are not appropriate for NCL [12]. This has resulted in efforts to carry out ligations at Xaa-Xaa sites in which a thiol group is attached to a side chain of the N-terminal amino acid. Subsequent desulfurization then affords the desired peptide (Fig. 3) [13]. Desulfurization of thiol-modified amino acids can be achieved using Raney nickel or Pd/Al2O3, or under free-radical-based conditions [12, 14]. To date, the ligation-desulfurization technique has been achieved at Ala [12], Phe [15], Val [16], Lys [17, 18], Thr [19], Leu [20], Pro [21], Gln [22], and, most recently, Arg residues [23]. The design of thiol-containing amino acids was recently reviewed by He et al. [24]. However, where cysteine residues are present in the peptide sequence at positions other than the ligation junction, protection of the side chain is required to prevent unselective desulfurization. In this instance the acetamidomethyl group is typically used [25], and Pentelute and Kent were the first to show its application in NCL [26].

NCL and subsequent desulfurization
Dawson et al. demonstrated that the addition of thiophenol or benzyl mercaptan can increase the rate of ligation [27] and Johnson and Kent studied the effect that various thiols may have on NCL [28]. It was found that aryl thiols exchange with peptide alkyl thioesters to form peptide aryl thioesters, which then act as efficient leaving groups, thus facilitating the ligation. Mercaptophenylacetic acid (MPPA) was found to be a more effective thiol additive than those previously used.
2 Solid Phase vs Solution Phase Chemical Ligation
The most successful method of fragment condensation for the synthesis of polypeptides and proteins in solution phase is NCL, reported by Dawson for the first time in 1994 [7]. This was a significant contribution because NCL overcomes one of the main limitations of solid phase peptide synthesis (SPPS), namely the production of long peptide sequences (>50 amino acid residues) [1, 3, 29]. NCL may be used in both solution and solid phases; solution phase NCL has been used for the synthesis of small peptides and cyclic peptides [30–32] whereas SPPS is more widely applied in polypeptide and protein synthesis.
SPPS has proved more useful than solution phase techniques in terms of ligation reactions for long peptides or polypeptides [33], because both N-terminal Cys-containing peptides and C-terminal thioester-containing peptides can be efficiently prepared on solid supports. Zhang et al. used 3-mercaptopropionyl MBHA resin for the preparation of thioesters using Boc chemistry [34]. However, harsh acidic conditions (HF/anisole, 9:1) were required to cleave the peptide thioester from the solid support. Clippingdale et al. reported the synthesis of peptide thioesters via Fmoc SPPS in order to avoid the use of HF [35]. However, piperidine, widely used for Fmoc-deprotection, may cause hydrolysis of peptide-α-thioesters, and thus methods to overcome this difficulty have been the subject of a number of studies [36].
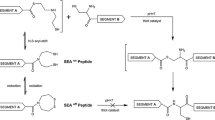
Raibaut et al. demonstrated the advantages of SPPS for preparing peptide sequences for use in NCL. An efficient solid-phase synthesis of large polypeptides was achieved by iterative ligations of bis(2-sulfanylethyl)amido (SEA) peptide segments [37]. Sequential NCL by N- to C-elongation cycles between the supported peptide thioester (blocked with SEA) and a free C-terminal thiol group-SEA activated N-terminus peptide (Fig. 4) allowed the synthesis of peptide thioesters containing 60 amino acids and the assembly of five peptide segments to give a 15-kDa polypeptide [38].

N- to C-assembly of peptides
3 Intramolecular Chemical Ligation (Acyl Migration)
In general, NCL means intermolecular ligation but the phrase has also been applied to intramolecular ligation, often called acyl migration, which occurs when an acyl group migrates from X→N (X=S, O, N) within an isopeptide (Fig. 5). Recently, Panda et al. investigated chemical ligation from isopeptides in the solution phase via different cyclic transition states [39].

Intramolecular chemical ligation
3.1 S- to N-Acyl Migration
Isopeptide ligation is an alternative method for the synthesis of cysteine peptides via an intramolecular chemical ligation by an entropically favored mechanism. S- to N-Acyl migration via various cyclic transition states was investigated by carrying out the ligation with mono-isopeptides under microwave irradiation (50°C, 50 W, 1–3 h) using 1 M NaH2PO4/Na2HPO4 phosphate buffer to maintain pH 7.3 (Scheme 1). The feasibility of intramolecular acyl migrations via 5- to 19-membered cyclic transition states was demonstrated and the yields of long-range S- to N-acyl transfers were found to depend on the size of the macrocyclic transition state (TS). Thus the relative rates, based on yields, depended on the ring size of the TS in the order, 5 >10 >11 >14, 16, 17 >12 >13, 15, 19 >18 >>>9 >8 [40–44].

Intramolecular ligation studies via S- to N-acyl migration
3.2 O- to N-Acyl Migrations
Chemical ligation of serine isopeptides via O- to N-acyl transfer with 8- and 11-membered TSs occurs without the use of an auxiliary group (Scheme 2) [45]. This is in contrast to cysteine isopeptides, in which the 8-membered TS was disfavored even under basic conditions. Intramolecular acyl transfer of Thr isopeptide through 5- and 9-membered TSs was favored over 8- and 11-membered TSs (Scheme 2) [46].

Ligation studies via O- to N-acyl migration
Chemical ligation studies of Tyr isopeptides under microwave irradiation (50°C, 50 W, 3 h) using 1 M phosphate buffer and a DMF-piperidine medium showed that intramolecular O- to N-acyl transfer occurs via 11- to 13-membered TSs under basic conditions and with 14- to 18-membered TSs in aqueous media (Scheme 3) [47].

Ligation studies via O- to N-acyl migration
3.3 N- to N-Acyl Migrations
The intramolecular chemical ligation of tryptophan isopeptides via N- to N-acyl migration occurs through 7- to 18-membered cyclic TSs forming the native peptides in basic, non-aqueous media rather than aqueous buffered conditions (Scheme 4) [48].

Ligation studies via N- to N-acyl migration
4 Applications of Native Chemical Ligation
4.1 Synthesis of Cyclic Peptides
In 1944 Gause and Brazhnikova discovered Gramicidin S, a cyclic peptide [49], and used it in the treatment of septic gunshot wounds during the Second World War. Since then, both natural and unnatural cyclic peptides have become important synthetic targets because of their potential applications as antibiotics and other therapeutic agents [50–54]. Selected examples include anticancer agents (ADH-1), antibiotics (colistin), growth hormone inhibitors (octreotide), and immunosuppressant agents (cyclosporine A) (Fig. 6) [55].

Selected cyclic peptides
The constrained conformation of cyclic peptides often results in increased exo- and endopeptidase resistance, enhanced binding affinity, and in certain cases, increased cell penetration compared to their linear counterparts. Numerous strategies, both in solution and solid-phase, have been reported for the synthesis of cyclic peptides [56–58]; NCL, the reaction of a C-terminal peptide thioester with an N-terminal cysteine peptide, is now an established method for production of peptides with a cyclized backbone [59–61].
Dawson demonstrated ligation strategies on 1,2-aminothiols which depend on a capture/rearrangement mechanism to link two peptide fragments under mild conditions [7]. The cyclization process involves a reaction between a weakly activated C-terminal thioester and an unprotected N-terminal cysteine residue. The thermodynamic stability of an amide bond over a thioester is again the driving force behind this reaction, made possible through a proximity-driven S- to N-acyl migration. Zhang and Tam used the above methodology to synthesize cyclic peptides in a head-to-tail fashion (Fig. 7) [62].

Native chemical ligation applied to the head-to-tail cyclization of peptides
Hackenberger and Kleineweischede reported a traceless Staudinger ligation for the head-to-tail macrocyclization of peptides without a deprotection step (Fig. 8) [63]. In this strategy, a phosphine tethered to a thioester at the C-terminus of a peptide reacts intramolecularly with an azide at the N-terminus to form the cyclic peptide.

The head-to-tail macrocyclization of peptides through a traceless Staudinger ligation strategy
Recently, cLac (cyclic peptide-mimicking lactadherin) was synthesized using NCL and studied for phosphatidylserine (PS) recognition [64, 65]. The linear precursors for the synthesis of cLac derivatives were prepared following standard protocols for automated Fmoc-peptide synthesis and the cyclic peptides (cLac variants) were obtained in the presence of 4-mercaptophenylacetic acid and isolated via HPLC in yields of 60–70% (Fig. 9). All the synthesized cLac variants were labeled with the thiol-reactive fluorescein-5-maleimide in DMF containing 2% N-methylmorpholine (NMM) by taking advantage of the free cysteine side chain [65].

cLac and a cLac variant
The phosphatidylserine (PS) recognition study suggests that the cLac peptide effectively mimics the PS binding mechanism of lactadherin, in which multiple polar residues are conformationally preorganized by the protein or the cyclic peptide to balance various noncovalent forces (desolvation, hydrogen bonding, and salt bridges) for specific PS recognition [65].
Liu et al. reported a modified version of NCL to synthesize the native peptide from a C-terminal peptide hydrazide and an N-terminal Cys under NaNO2-mediated activation [66, 67]. This method (hydrazide ligation) was also applied in the synthesis of cyclic peptides. One of its important advantages is that peptide hydrazides can be easily prepared through routine Fmoc SPPS. The linear hydrazide peptides were synthesized from hydrazine-Trt(2-Cl) by following standard Fmoc SPPS. The peptide hydrazides cyclize in two steps, in a one-pot fashion, in the presence of NaNO2 and the thio-additive MPAA. Aqueous phosphate buffer containing 6.0 M guanidinium chloride was used as the solvent system. A mixture of an organic solvent with aqueous phosphate buffer also works as medium for this transformation (Scheme 5). A number of cyclic peptides were prepared by following this methodology in 18–65% yields (Table 1) [68].

Synthesis of cyclic peptides on a solid support
SPPS is often used for the preparation of linear precursors required for NCL. Barany and Tulla-Puche reported NCL on resin to avoid tedious purification steps, usually necessary after each step. The linear precursor was prepared using 1-hydroxy-7-aza-benzotriazole (HOAt) or 1-hydroxybenzotriazole (HOBt) and N,N′-diisopropylcarbodiimide (DIPCDI) as coupling agents. Key aspects of on-resin NCL include Fmoc/tBu chemistry, side-chain anchoring, allyl protection of the penultimate residue to allow introduction of the C-terminal thioester later in the synthetic sequence, a new derivative, Trt-Cys(Xan)-OH, which facilitates selective and mild removal of both protecting groups. The synthesis of cyclo(Cys-Thr-Abu-Gly-Gly-Ala-Arg-Pro-Asp-Phe) using on-resin NCL is illustrated in Scheme 6 [69].

Synthesis of cyclic peptides via on-resin NCL
Fukuzumi et al. introduced a strategy in which linear peptides bearing side chains with unprotected functional groups could be cyclized under high dilution conditions [70]. This strategy, known as α-ketoacid-hydroxylamine amide-ligation, is achieved by the Fmoc-based SPPS of linear peptides bearing a C-terminal sulfur ylide linker, which acts as a ‘masked’ α-ketoacid (Fig. 10). The introduction of the N-hydroxylamine can also be carried out on a solid support. Subsequent global deprotection and cleavage from the solid support affords the linear peptide which, following sulfur ylide oxidation, undergoes the desired cyclization. The general applicability of this strategy was demonstrated by the preparation of five natural product cyclic peptides (Table 2).

General strategy for the preparation of unprotected linear peptides with N-terminal hydroxylamine and C-terminal α-ketoacids for direct, reagent-less cyclizations
van de Langemheen et al. synthesized cyclic peptides containing a thioester handle using a ‘sulfo-click’ linker. In this approach the ‘sulfo-click’ linker used for the synthesis of the linear precursor was prepared by Fmoc SPPS (Scheme 7) [71]. Three different cyclic peptide sequences were synthesized, corresponding to the loops present in HIV protein gp120 interacting with CD4 as found in the X-ray structure of the gp120-CD4 complex. On the basis of this structure, the 365SGGDPEIVT373, 424INMWQEVGKA433, and 454LTRDGGN460 peptide sequences were selected for the preparation of cyclic peptide thioesters (Fig. 11) [72]. HIV-gp120 plays a crucial role in the first steps of HIV-infection through its attachment to the CD4 receptor [73]. Preventing attachment of gp120 to cells and/or using gp120 as a starting point to develop a vaccine may offer alternative approaches to avoid the further spread of HIV [74, 75].

Synthesis of linear precursors for NCL by Fmoc SPPS

365SGGDPEIVT373, 424INMWQEVGKA433, and 454LTRDGGN460 loops
Chen et al. developed a strategy for preparing cyclic peptides via in situ generation of a thioester resulting from disulfide reduction; subsequent NCL results in the desired peptide (Fig. 12) [76]. This strategy was used to synthesize linear glycopeptides, which, after thioester formation, resulted in cyclization to form a model glycopeptide in 73% yield (Scheme 8).

Proposed mechanism for the in situ generation of thioesters and subsequent ligation

Synthesis of cyclic glycopeptides following the in situ generation of thioesters
The synthesis of branched peptides using masked side-chain thioester derivatives of Asp and Glu which are compatible with Fmoc-SPPS is an important goal. Boll et al. synthesized cyclic and branched chain peptides using bis(2-sulfanylethyl)amido (SEA) side-chain derivatives of Asp and Glu via Fmoc SPPS [77]. The tail-to-side-chain cyclization via an in situ reduction of both acyclic and cyclic disulfides with tris(2-carboxyethyl)phosphine (TCEP) triggered the SEA intramolecular ligation. Glu derivatives cyclized more readily than the Asp analogues and without formation of side products (Scheme 9).

Tail to side-chain cyclization using bis(2-sulfanylethyl)amido (SEA) ligation
There are several drawbacks associated with the use of thioester surrogates such as SEA, in SPPS, especially when the thioester surrogate is attached to the resin via an MeCys linker [78]. These limitations include the need for multiple coupling reactions to attach the C-terminal amino acid to the resin and the possibility that the protected MeCys linker can, during peptide elongation, undergo β-elimination and then piperidine conjugate addition to form MeAla(Pip) as a side product [79]. In order to overcome these limitations, Taichi et al. developed the thioethylbutylamido (TEBA) group as an alternative to SEA-derived thioester surrogates (Fig. 13). The utility of the TEBA-thioester surrogate was demonstrated by the synthesis of cysteine-rich cyclic peptides [78, 80].

Thioester surrogates
Li et al. developed an efficient method for the synthesis of peptides and proteins. In this method, an O-salicylaldehyde ester at the C-terminus reacts with N-terminal serine or threonine to realize peptide ligations via an O- to N-acyl transfer (Scheme 10) [81].

NCL via an O- to N-acyl transfer
The utility of this ligation approach has been demonstrated through the convergent syntheses of therapeutic peptides (ovine-corticoliberin and Forteo) and the human erythrocyte acylphosphatase protein (∼11 kDa) [82]. The requisite peptide salicylaldehyde ester precursor is prepared in an epimerization-free manner via Fmoc–solid-phase peptide synthesis. This approach was also used for the synthesis of Daptomycin (Fig. 14), a lipodepsipeptide isolated from Streptomyces roseoporus which was obtained from a soil sample from Mount Ararat (Turkey) [83].

Daptomycin
4.2 Synthesis of Glycopeptides
Post-translational modifications of proteins and peptides can have profound effects on the overall biological and chemical properties of these biopolymers. In particular, glycosylation of proteins and peptides results in highly complex structures known as glycoproteins [84, 85]. The presence of glycans can ensure the stability and activity of glycoproteins, with biological functions such as cell adhesion, differentiation, and growth [86–88]. Carbohydrates are linked to proteins via either a N-glycosidic bond at an asparagine residue or an O-glycosidic bond at a serine or threonine residue [89]. N-Linked glycoproteins are biosynthesized in a process commencing in the endoplasmic reticulum (ER) in which a 14-mer oligosaccharide is transferred to the amide nitrogen of an asparagine residue, in an Asn-Xxx-Ser/Thr sequon, where Xxx is any amino acid apart from proline, from a dolichol phosphate. Glycosidases then truncate the 14-mer to a pentasaccharide core fragment which is further modified to afford the final N-glycoprotein, which is in turn either transported to the cell surface or secreted (Fig. 15) [90].

Pentasaccharide core fragment present on N-linked glycopeptides where R=Asp
The most prevalent class of O-linked glycoproteins are the mucins, in which a galactosamine (GalNAc) monosaccharide is linked to the peptide backbone via an α-glycosidic bond from GalNAc to a Ser or Thr residue [91]. The GalNAc monosaccharide, often known as the TN antigen (Fig. 14), is then modified at the C-3 or C-6 positions with GalNAc, galactose, or glucosamine via α- or β-glycosidic bonds, resulting in a series of di- and trisaccharides which are known as the core structures (Fig. 16).

Core structures of mucin O-linked glycans where R is a Ser or Thr residue [91]
O-Linked glycoproteins are biosynthesized in a continuous process that occurs in the ER and the Golgi apparatus. This is not a template driven process, but rather is subject to numerous sequential and competitive enzymatic pathways, and the O-glycans vary according to the cell lineage, tissue location, and developmental stage of the cell [91, 92]. Furthermore, as the pattern of O-glycosylation alters in response to mucosal infection and inflammation, O-linked glycoproteins have important implications in a wide range of diseases, including cystic fibrosis, Crohn’s disease, and cancers [93–99]. Aberrant glycosylation can result in truncated core structures, lacking backbone motifs, which leave the TN and T antigens or their sialylated versions exposed, providing disease markers which might have important implications in the design and synthesis of anti-cancer vaccines (Fig. 17) [100, 101].

Common tumor-associated carbohydrate antigens (TACAs) where R=H (Ser) or CH3 (Thr)
To determine the exact role of glycoproteins and glycopeptides in biological systems, it is necessary to access pure samples of these compounds. Furthermore, glycoproteins and glycopeptides are important compounds in drug discovery because of their potential therapeutic benefits [102, 103]. However, because of the dynamic and heterogeneous nature of biological systems, progress in glycobiology has been hindered, and it is not currently possible to isolate single glycoforms from natural sources [104]. Thus chemical synthesis of glycoproteins and glycopeptides is an important area of research [105, 106].
Chemical ligation is a very attractive method for the synthesis of glycoproteins [107], and NCL, traceless Staudinger ligation [108], and sugar-assisted ligation (SAL) (Fig. 18) [109–111] have all been used for this purpose. SAL has been used for the synthesis of a large number of glycopeptides [112–114], but difficulties were encountered in the presence of larger glycans [115].

Sugar-assisted ligation (SAL)
The synthesis of glycoproteins has been reviewed recently [84, 85, 106, 116, 117], and hence the focus here is on the use of NCL for the synthesis of glycopeptides and glycopeptide mimetics, the so-called ‘neoglycopeptides’ [118, 119]. Although NCL has been applied to the synthesis of glycoproteins and glycopeptides [120], the synthesis of glycopeptides is inherently challenging [121].
Boc-SPPS, traditionally used to prepare peptides required for NCL, is not used for the synthesis of glycopeptides because it is incompatible with acid labile glycosidic linkages such as sialyl- or fucosyl-glycosidic bonds. However, Murakami et al. were able to prepare a sialylglycopeptide by substituting the harsh acidic conditions (HF) typically used to remove Boc protecting groups for an acidic deprotection cocktail of TFA/TfOH/DMS/m-cresol (5:1:3:1) [122]. Alternatively, Fmoc-SPPS, used in glycopeptide synthesis, is not compatible with the synthesis of peptide-α-thioesters, as the thioester may be cleaved by the piperidine used to remove Fmoc protecting groups. Because the Fmoc strategy is more widely used than Boc strategies, especially in automated SPPS, methods to overcome difficulties associated with Fmoc-protected peptide-α-thioesters have received a great deal of attention [36].
The requirement of a Cys residue at the ligation junction for successful NCL can be problematic when attempting to synthesize glycopeptides because of the low natural abundance of Cys residues in nature (only 1.7%). This results in a lack of Cys residues in the target glycoprotein or the need to synthesize glycopeptide sequences of 30–50 amino acid residues. It is worth mentioning that the synthesis of N-linked glycopeptide chains is more challenging than the corresponding amino acid sequence without an attached glycan, which can lead to difficulties in glycopeptide synthesis [106, 123, 124]. Consequently, a large number of techniques have been developed, including Ser or Thr ligation strategies [125] and the addition of thiol groups to amino acids [24] which, subject to successful desulfurization, might afford the target glycopeptide. Desulfurization of peptides was originally achieved with either Raney nickel or Pd/Al2O3 [12], but these conditions are not always compatible with desulfurization of glycopeptides [16, 126].
The in situ generation of thioesters has been applied to the synthesis of cyclic glycopeptides bearing a single carbohydrate moiety (Fig. 12) [76]. Wan and Danishefsky applied this strategy to the synthesis of glycopeptides containing strategically placed Cys residues to facilitate NCL. Following NCL, a radical-based, metal-free desulfurization process using TCEP and tBuSH enabled the Cys residue to be converted into the desired Ala (Scheme 11) [14]. The NCL free-radical-based desulfurization was subsequently reported to be applicable to thiol-modified Val residues [127] and thiol-protected Thr residues [19].

Free-radical desulfurization of a glycopeptide
Efforts have been made to improve free-radical desulfurization strategies and in this respect one-pot strategies are particularly attractive because they avoid time-consuming purification after each reaction. Moyal et al. [128] recently reported a one-pot ligation-desulfurization protocol and subsequently Thompson et al. developed a one-pot ligation-desulfurization protocol, using a novel thiol additive, 2,2,2-trifluoroethanethiol, for the synthesis of short peptide fragments [129]. Neither of these strategies has yet been applied to glycopeptide synthesis.
To achieve a more widely applicable method for glycopeptide synthesis, Okamoto and Kajihara developed a strategy for the conversion of Cys to a Ser residue after NCL. Thus, S-methylation of Cys, followed by an intramolecular rearrangement activated by CNBr, results in an O- to N-acyl shift, which affords an O-ester peptide intermediate. A second O- to N-acyl shift generates the desired glycopeptide (Fig. 19) [130]. This strategy was used to access a fragment (residues 79–98) of EPO, N-linked glycopeptide and a repeat sequence of MUC-1, an O-linked glycoprotein. Furthermore, this procedure can be carried out in the presence of methionine residues, when protected as the sulfoxide. Subsequently, Okamoto et al. reported that the substitution of Cys to Ser could be accomplished in the presence of acid-labile sialyl-glycosidic linkages such as those found on sialyl-TN antigens [131].

Conversion of Cys to Ser following NCL
NCL has been achieved at unprotected Ser and Thr residues via an O- to N-acyl transfer [81]. Hojo et al. employed a slightly different strategy in which a mercaptomethyl group was used to protect the Ser and Thr residues. In this instance the ligation step occurs through an S- to N-acyl transfer via a seven-membered ring and this approach was used to access O-linked glycoprotein contulakin-G (Fig. 20) [132].

Synthesis of contulakin-G via an S- to N-acyl transfer
N- to S-acyl shifts have been used to access peptide thioesters (Fig. 21) [133, 134]. Macmillan et al. used this strategy to prepared thioesters, which were subsequently used in NCL to access model glycopeptides based on the erythropoietin (EPO) amino acid sequence [135, 136].

Synthesis of peptide thioesters via an N- to S-acyl shift
Hsieh et al. prepared S-glycosylated peptides using NCL (Scheme 12) [137]. In this instance, the glycopeptides fragments were prepared using Fmoc SPPS and the subsequent NCL proceeded in yields of over 80% for the three examples. Disulfide-bridge formation at key cysteine residues achieved the natural product, bacteriocin glycopeptide Sublancin 168, and two derivatives bearing alternative sugars.

Synthesis of S-linked glycopeptides via NCL
Glycoconjugate mimetics, known as neoglycoconjugates, which bear an ‘unnatural’ linkage between the carbohydrate and aglycon moieties, have been widely explored, as this allows access to novel glycopeptides which might have enhanced activity relative to the naturally occurring glycopeptide [119]. The triazole unit is one of the most widely studied ‘unnatural’ glycosidic linkages and, in this context, Macmillan and Blanc synthesized a neoglycopeptide, using Boc-SPPS, in which the peptide sequence was based upon human erythropoietin. Cleavage of the neoglycopeptide from the solid support afforded an 11-mer peptide sequence which was able to undergo NCL with a suitable thioester to afford the target neoglycoconjugate (Scheme 13), neatly demonstrating that NCL is compatible with triazole moieties [138].

NCL of neoglycopeptides containing a triazole motif
Lee et al. subsequently synthesized similar neoglycopeptides using a one-pot strategy in which two propargyl peptides were coupled via NCL; subsequent 1,3-dipolar cycloaddition afforded the glycosylated peptide (Fig. 22) [139].

One-pot NCL followed by 1,3-dipolar cycloaddition
Finally, it has been shown that NCL has applications in the context of synthesizing RNA mimics [140], and selective desulfurization again plays an important role in producing target molecules [141].
5 Computational Rationalization of Chemical Ligation
Chemical ligation has been widely reported and discussed, and numerous attempts have been made to increase reactivity and improve yields to make the peptide ligation method more effective. In particular, mechanistic studies of the reaction have played an important role in rationalizing the rate of ligation.
Wang et al. investigated the mechanism of NCL and calculated energy barriers for various C-terminal amino acids in order to compare their reactivity [142]. In the reaction between a peptide thioalkyl ester and a Cys-peptide in the presence of an aryl thiol catalyst, it is alleged that both the thiol–thioester exchange step and the trans-thioesterification step proceed by a concerted SN2 displacement, whereas the intramolecular rearrangement occurs by an addition-elimination mechanism (Scheme 14).

Hypothetical routes for thioesterification
The energy barrier for the thiol–thioester exchange step depends on steric hindrance associated with the side-chain of C-terminal amino acids, whereas that of the acyl-transfer step depends on steric hindrance caused by the side-chain of the N-terminal amino acid. In auxiliary-mediated peptide ligation, between a peptide thiophenyl ester and an N-2-mercaptobenzyl peptide, the thiol–thioester exchange step and intramolecular acyl-transfer step proceed by a concerted SN2-type displacement mechanism (Scheme 15). For N-terminal Gly, the thioester exchange is rate limiting, whereas the acyl transfer is the rate-limiting step for N-terminal non-Gly amino acids (Table 3) [142]. When the difference in ΔG ‡ between the two transition states is, for example, 3 kcal mol−1, then the reaction with the lower activation energy is 150 times faster (Table 3).

Mechanism of auxiliary-mediated peptide ligation
When compared to intermolecular NCL, there have been far more computational studies into the intramolecular chemical ligation of isopeptides. Monbaliu et al. discussed the computational approach and developed the first systematic theoretical background for an n-exo-trig intramolecular S-to N-acyl transfer [143]. Cyclic TSs in the ring size range n = 5–10 were controlled by enthalpic factors and a classical range of ΔG ‡ values were found. The calculations also emphasized that the substituents at R1 and R2 (Fig. 23b) had little impact on the nature of the TS. The preorganization of the structures and, in particular, the emergence of stabilizing hydrogen bonds in intramolecular TSs appeared as major factors governing the variations of ΔG ‡ with ring size. In isomeric structures, the presence of an internal non-natural amino acid (β-Ala or GABA) directly after the Cys residue favored hydrogen bonds (5.7–8.4 kcal mol−1), which stabilize the TS. The competition between the intra- and intermolecular acyl transfers is driven by parameters which govern the approach of the reactive termini, for example, the ring strain (Fig. 23) [143].

(a) 5-Exo-trig S- to N-acyl transfer as commonly encountered in classical NCL and (S)-isopeptide rearrangements (left) and n-exo-trig S- to N-acyl transfer in internal Cys NCL and extended isopeptides rearrangements (right). (b) Isomerization of (S)-acyl isopeptides to native peptide analogues if m = 0, n = 5
The feasibility and rate of intramolecular ligation depends on various factors such as preorganization energy, hydrogen bonding, and the distance between the reaction sites. Oliferenko and Katritzky rationalized the curious behavior of the intramolecular ligation of Cys-containing isopeptides by computational chemistry. Preorganization is an important factor in chemical ligation because it occurs through a cyclic TS [144]. The more easily the starting material achieves an appropriate cyclic conformation, the higher the probability of intramolecular reaction. Hydrogen bonding and NH–π interactions play a major role in the stabilization of a preorganized conformer [44]. Intramolecular ligation via eight-membered cyclic TSs of Cys-containing isopeptides was disfavored, whereas, surprisingly, Ser-containing isopeptides showed an efficient O- to N-acyl migration via eight-membered cyclic TSs. The O- to N-acyl migration of Tyr-containing isopeptides showed intramolecular chemical ligation in the presence of a base with 12- to 14-membered cyclic TSs and in a buffered medium with 15- to 19-membered cyclic TSs. The reactivity of Tyr isopeptides is described in terms of preorganization energy, hydrogen bonding and bond distance (Table 4) [145].
Biswas et al. reported, the design of a predictive model using statistical techniques to correlate the relative abundance (ligated product percentage) with quantitative structural activity/property relationship (QSAR/QSPR) [146]. The genetic algorithm linear regression method was performed using QSARINS software [147], which establishes a correlation between the dependent variable (property/response relative to the abundance of ligated peptide) and independent variables (molecular descriptors or factors) (Table 5). It was found that the percentage of ligated peptides increases with both a shorter spatial b(N-C) distance and a higher Balaban index (Fig. 24).

(a) Correlation plot for relative abundance model of ligated product. (b) Correlation between relative abundance and distance b(N-C). (c) Correlation between relative abundance and Balaban index
6 Conclusions
The increasing demand for peptides and chemically modified peptides has produced a significant expansion in alternative synthetic routes for peptide and protein synthesis. Native chemical ligation has proved to be a very useful tool for the synthesis of proteins, peptides, cyclic-peptides, glycopeptides, and neoglycoconjugates. In spite of ligation-desulfurization techniques, the general requirement of an N-terminal cysteine and a C-terminal thioester remains a limitation. However, considerable efforts have been made to generalize NCL. Both solution and solid phase peptide synthesis have been used for NCL, and NCL is the more acceptable method for preparing peptides because of the high-yielding isolation of long-chain peptides and cyclic peptides with a lower likelihood of thioester racemization. Because thioesterification can occur at mildly acidic or neutral pH, NCL promises to have a major impact on the synthesis of peptides and peptide mimetics and is particularly suitable for the synthesis of natural products, cyclic peptides, and glycopeptides. The rate of acyl transfer in chemical ligation reactions depends on various factors and these have been analyzed by computational studies. This theoretical rationalization should assist in the preparation of new cyclic peptides and glycopeptides via chemical ligation.
References
Dawson PE, Kent SBH (2000) Synthesis of native proteins by chemical ligation. Annu Rev Biochem 69:923–960
Hackenberger CPR, Schwarzer D (2008) Chemoselective ligation and modification strategies for peptides and proteins. Angew Chem Int Ed 47:10030–10074
Kent SBH (2009) Total chemical synthesis of proteins. Chem Soc Rev 38:338–351
Boltje TJ, Buskas T, Boons GJ (2009) Opportunities and challenges in synthetic oligosaccharide and glycoconjugate research. Nat Chem 1:611–622
Wang P, Danishefsky SJ (2010) Promising general solution to the problem of ligating peptides and glycopeptides. J Am Chem Soc 132:17045–17051
Wieland T, Bokelmann E, Bauer L, Lang HU, Lau H (1953) Über Peptidsynthesen. 8. Bildung von S-haltigen Peptiden durch intramolekulare Wanderung von Aminoacylresten. Liebigs Ann Chem 583:129–149
Dawson PE, Muir TW, Clark-Lewis I, Kent SBH (1994) Synthesis of proteins by native chemical ligation. Science 266:776–779
Kemp DS, Buckler DR (1991) Highly enantioselective amide ligation by prior thiol capture. Tetrahedron Lett 32:3013–3016
Beligere GS, Dawson PE (1999) Conformationally assisted protein ligation using C-terminal thioester peptides. J Am Chem Soc 121:6332–6333
Kohn M, Breinbauer R (2004) The Staudinger ligation – a gift to chemical biology. Angew Chem Int Ed 43:3106–3116
Millevoi S, Chiaraluce R, Consalvi V, Giangiacomo L, Pasquo A, Linda Britton K, Stillman TJ, Rice DW, Engel PC (1998) A monomeric mutant of Clostridium symbiosum glutamate dehydrogenase: comparison with a structured monomeric intermediate obtained during refolding. Protein Sci 7:966–974
Yan LZ, Dawson PE (2001) Synthesis of peptides and proteins without cysteine residues by native chemical ligation combined with desulfurization. J Am Chem Soc 123:526–533
Dawson PE (2011) Native chemical ligation combined with desulfurization and deselenization: a general strategy for chemical protein synthesis. Isr J Chem 51:862–867
Wan Q, Danishefsky SJ (2007) Free-radical-based, specific desulfurization of cysteine: a powerful advance in the synthesis of polypeptides and glycopolypeptides. Angew Chem Int Ed 46:9248–9252
Crich D, Banerjee A (2007) Native chemical ligation at phenylalanine. J Am Chem Soc 129:10064–10065
Haase C, Rohde H, Seitz O (2008) Native chemical ligation at valine. Angew Chem Int Ed 47:6807–6810
Kumar KSA, Haj-Yahya M, Olschewski D, Lashuel HA, Brik A (2009) Highly efficient and chemoselective peptide ubiquitylation. Angew Chem Int Ed 48:8090–8094
Yang R, Pasunooti KK, Li F, Liu X-W, Liu C-F (2009) Dual native chemical ligation at lysine. J Am Chem Soc 131:13592–13593
Chen J, Wang P, Zhu JL, Wan Q, Danishefsky SJ (2010) A program for ligation at threonine sites: application to the controlled total synthesis of glycopeptides. Tetrahedron 66:2277–2283
Harpaz Z, Siman P, Kumar KSA, Brik A (2010) Protein synthesis assisted by native chemical ligation at leucine. ChemBioChem 11:1232–1235
Shang SY, Tan ZP, Dong SW, Danishefsky SJ (2011) An advance in proline ligation. J Am Chem Soc 133:10784–10786
Siman P, Karthikeyan SV, Brik A (2012) Native chemical ligation at glutamine. Org Lett 14:1520–1523
Malins LR, Cergol KM, Payne RJ (2013) Peptide ligation-desulfurization chemistry at arginine. ChemBioChem 14:559–563
He Q-Q, Fang G-M, Liu L (2013) Design of thiol-containing amino acids for native chemical ligation at non-Cys sites. Chin Chem Lett 24:265–269
Veber D, Milkowski J, Varga S, Denkewalter R, Hirschmann R (1972) Acetamidomethyl. A novel thiol protecting group for cysteine. J Am Chem Soc 94:5456–5461
Pentelute BL, Kent SBH (2007) Selective desulfurization of cysteine in the presence of Cys(Acm) in polypeptides obtained by native chemical ligation. Org Lett 9:687–690
Dawson PE, Churchill MJ, Ghadiri MR, Kent SBH (1997) Modulation of reactivity in native chemical ligation through the use of thiol additives. J Am Chem Soc 119:4325–4329
Johnson ECB, Kent SBH (2006) Insights into the mechanism and catalysis of the native chemical ligation reaction. J Am Chem Soc 128:6640–6646
Kent SBH (1988) Chemical synthesis of peptides and proteins. Annu Rev Biochem 57:957–989
Offer J (2010) Native chemical ligation with N α acyl transfer auxiliaries. Biopolymers (Pept Sci) 94:530–541
Payne RJ, Wong C-H (2010) Advances in chemical ligation strategies for the synthesis of glycopeptides and glycoproteins. Chem Commun 46:21–43
Macmillan D, De Cecco M, Reynolds NL, Santos LFA, Barran PE, Dorin JR (2011) Synthesis of cyclic peptides through an intramolecular amide bond rearrangement. ChemBioChem 12:2133–2136
Palomo JM (2014) Solid-phase peptide synthesis: an overview focused on the preparation of biologically relevant peptides. RSC Adv 4:32658–32672
Zhang LS, Tam JP (1999) Lactone and lactam library synthesis by silver ion-assisted orthogonal cyclization of unprotected peptides. J Am Chem Soc 121:3311–3320
Clippingdale AB, Barrow CJ, Wade JD (2000) Peptide thioester preparation by Fmoc solid phase peptide synthesis for use in native chemical ligation. J Pept Sci 6:225–234
Mende F, Seitz O (2011) 9-Fluorenylmethoxycarbonyl-based solid-phase synthesis of peptide α-thioesters. Angew Chem Int Ed 50:1232–1240
Raibaut L, Adihou H, Desmet R, Delmas AF, Aucagne V, Melnyk O (2013) Highly efficient solid phase synthesis of large polypeptides by iterative ligations of bis(2-sulfanylethyl)amido (SEA) peptide segments. Chem Sci 4:4061–4066
Boll E, Drobecq H, Ollivier N, Raibaut L, Desmet R, Vicogne J, Melnyk O (2014) A novel PEG-based solid support enables the synthesis of >50 amino-acid peptide thioesters and the total synthesis of a functional SUMO-1 peptide conjugate. Chem Sci 5:2017–2022
Panda SS, Hall CD, Oliferenko AA, Katritzky AR (2014) Traceless chemical ligation from S-, O-, and N-acyl isopeptides. Acc Chem Res 47:1076–1087
Katritzky AR, Abo-Dya NE, Tala SR, Abdel-Samii ZK (2010) The chemical ligation of selectively S-acylated cysteine peptides to form native peptides via 5-, 11- and 14-membered cyclic transition states. Org Biomol Chem 8:2316–2319
Katritzky AR, Tala SR, Abo-Dya NE, Ibrahim TS, El-Feky SA, Gyanda K, Pandya KM (2011) Chemical ligation of S-scylated cysteine peptides to form native peptides via 5-, 11-, and 14-membered cyclic transition states. J Org Chem 76:85–96
Panda SS, El-Nachef C, Bajaj K, Al-Youbi AO, Oliferenko A, Katritzky AR (2012) Study of chemical ligation via 17-, 18- and 19-membered cyclic transition states. Chem Biol Drug Des 80:821–827
Ha K, Chahar M, Monbaliu JCM, Todadze E, Hansen FK, Oliferenko AA, Ocampo CE, Leino D, Lillicotch A, Stevens CV, Katritzky AR (2012) Long-range intramolecular S→N acyl migration: a study of the formation of native peptide analogues via 13-, 15-, and 16-membered cyclic transition states. J Org Chem 77:2637–2648
Bol’shakov O, Kovacs J, Chahar M, Ha K, Khelashvili L, Katritzky AR (2012) S- to N-acyl transfer in S-acylcysteine isopeptides via 9-, 10-, 12-, and 13-membered cyclic transition states. J Pept Sci 18:704–709
El Khatib M, Elagawany M, Jabeen F, Todadze E, Bol’shakov O, Oliferenko A, Khelashvili L, El-Feky SA, Asiri A, Katritzky AR (2012) Traceless chemical ligations from O-acyl serine sites. Org Biomol Chem 10:4836–4838
Panda SS, Elagawany M, Marwani HM, Caliskan E, El Khatib M, Oliferenko A, Almary KA, Katritzky AR (2014) Chemical ligation from O-acyl isopeptides via 8- and 11-membered cyclic transition states. Arkivoc iv:91–106
Popov V, Panda SS, Katritzky AR (2013) Ligations from tyrosine isopeptides via 12-to 19-membered cyclic transition states. J Org Chem 78:7455–7461
Popov V, Panda SS, Katritzky AR (2013) Ligations of N-acyl tryptophan units to give native peptides via 7-, 10-, 11- and 12-membered cyclic transition states. Org Biomol Chem 11:1594–1597
Gause GF, Brazhnikova MG (1944) Gramicidin S and its use in the treatment of infected wounds. Nature 154:703–703
White CJ, Yudin AK (2011) Contemporary strategies for peptide macrocyclization. Nat Chem 3:509–524
Tyndall JDA, Nall T, Fairlie DP (2005) Proteases universally recognize beta strands in their active sites. Chem Rev 105:973–999
Craik DJ (2006) Seamless proteins tie up their loose ends. Science 311:1563–1564
Hamada Y, Shioiri T (2005) Recent progress of the synthetic studies of biologically active marine cyclic peptides and depsipeptides. Chem Rev 105:4441–4482
Daly NL, Craik DJ (2011) Bioactive cystine knot proteins. Curr Opin Chem Biol 15:362–368
Sewald N, Jakube H-D (2009) Peptides: chemistry and biology, 2nd edn. Wiley, Weinheim
Kates SA, Solé NA, Albericio F, Barany G (1994) In: Basava C, Anantharamaiah GM (eds) Peptides: design, synthesis and biological activity. Birkhäuser, Boston, pp 39–58
Davies JS (2003) The cyclization of peptides and depsipeptides. J Pept Sci 9:471–501
Gilon C, Mang C, Lohof E, Friedler A, Kessler H (2002) In: Goodman M, Felix AM, Moroder L, Toniolo C (eds) Synthesis of peptides and peptidomimetics (Houben-Weyl E22b: Methods of organic chemistry). Georg Thieme, Stuttgart/New York
Camarero JA, Muir TW (1997) Chemoselective backbone cyclization of unprotected peptides. Chem Commun 1369–1370
Shao Y, Lu WY, Kent SBH (1998) A novel method to synthesize cyclic peptides. Tetrahedron Lett 39:3911–3914
Clark RJ, Craik DJ (2012) In: Wittrup KD, Gregory LV (eds) Methods enzymol. Academic Press, Maryland Heights, pp 57–74
Zhang LS, Tam JP (1997) Synthesis and application of unprotected cyclic peptides as building blocks for peptide dendrimers. J Am Chem Soc 119:2363–2370
Kleineweischede R, Hackenberger CPR (2008) Chemoselective peptide cyclization by traceless Staudinger ligation. Angew Chem Int Ed 47:5984–5988
Zheng H, Wang F, Wang Q, Gao J (2011) Cofactor-free detection of phosphatidylserine with cyclic peptides mimicking lactadherin. J Am Chem Soc 133:15280–15283
Hosseini AS, Zheng H, Gao J (2014) Understanding lipid recognition by protein-mimicking cyclic peptides. Tetrahedron 70:7632–7638
Fang G-M, Li Y-M, Shen F, Huang Y-C, Li J-B, Lin Y, Cui H-K, Liu L (2011) Protein chemical synthesis by ligation of peptide hydrazides. Angew Chem Int Ed 50:7645–7649
Fang G-M, Wang J-X, Liu L (2012) Convergent chemical synthesis of proteins by ligation of peptide hydrazides. Angew Chem Int Ed 51:10347–10350
Zheng J-S, Tang S, Guo Y, Chang H-N, Liu L (2012) Synthesis of cyclic peptides and cyclic proteins via ligation of peptide hydrazides. ChemBioChem 13:542–546
Tulla-Puche J, Barany G (2004) On-resin native chemical ligation for cyclic peptide synthesis. J Org Chem 69:4101–4107
Fukuzumi T, Ju L, Bode JW (2012) Chemoselective cyclization of unprotected linear peptides by α-ketoacid-hydroxylamine amide-ligation. Org Biomol Chem 10:5837–5844
van de Langemheen H, Brouwer AJ, Kemmink J, Kruijtzer JAW, Liskamp RMJ (2012) Synthesis of cyclic peptides containing a thioester handle for native chemical ligation. J Org Chem 77:10058–10064
van de Langemheen H, van Hoeke M, Quarles van Ufford HC, Kruijtzer JAW, Liskamp RMJ (2014) Scaffolded multiple cyclic peptide libraries for protein mimics by native chemical ligation. Org Biomol Chem 12:4471–4478
Kwong PD, Wyatt R, Robinson J, Sweet RW, Sodroski J, Hendrickson WA (1998) Structure of an HIV gp120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody. Nature 393:648–659
Franke R, Hirsch T, Overwin H, Eichler J (2007) Synthetic mimetics of the CD4 binding site of HIV-1 gp120 for the design of immunogens. Angew Chem Int Ed 46:1253–1255
Chamorro C, Kruijtzer JAW, Farsaraki M, Balzarini J, Liskamp RMJ (2009) A general approach for the non-stop solid phase synthesis of TAC-scaffolded loops towards protein mimics containing discontinuous epitopes. Chem Commun 821–823
Chen JH, Warren JD, Wu B, Chen G, Wan Q, Danishefsky SJ (2006) A route to cyclic peptides and glycopeptides by native chemical ligation using in situ derived thioesters. Tetrahedron Lett 47:1969–1972
Boll E, Dheur J, Drobecq H, Melnyk O (2012) Access to cyclic or branched peptides using bis(2-sulfanylethyl)amido side-chain derivatives of Asp and Glu. Org Lett 14:2222–2225
Taichi M, Hemu X, Qiu YB, Tam JP (2013) A thioethylalkylamido (TEA) thioester surrogate in the synthesis of a cyclic peptide via a tandem acyl shift. Org Lett 15:2620–2623
Lukszo J, Patterson D, Albericio F, Kates SA (1996) 3-(1-Piperidinyl)alanine formation during the preparation of C-terminal cysteine peptides with the Fmoc/t-Bu strategy. Lett Pept Sci 3:157–166
Hemu X, Taichi M, Qiu YB, Liu DX, Tam JP (2013) Biomimetic synthesis of cyclic peptides using novel thioester surrogates. Biopolymers 100:492–501
Li X, Lam HY, Zhang Y, Chan CK (2010) Salicylaldehyde ester-induced chemoselective peptide ligations: enabling generation of natural peptidic linkages at the serine/threonine sites. Org Lett 12:1724–1727
Zhang YF, Xu C, Lam HY, Lee CL, Li XC (2013) Protein chemical synthesis by serine and threonine ligation. Proc Natl Acad Sci U S A 110:6657–6662
Lam HY, Zhang YF, Liu H, Xu JC, Wong CTT, Xu C, Li XC (2013) Total synthesis of daptomycin by cyclization via a chemoselective serine ligation. J Am Chem Soc 135:6272–6279
Davis BG (2002) Synthesis of glycoproteins. Chem Rev 102:579–602
Gamblin DP, Scanlan EM, Davis BG (2009) Glycoprotein synthesis: an update. Chem Rev 109:131–163
Dwek RA (1996) Glycobiology: toward understanding the function of sugars. Chem Rev 96:683–720
Varki A (1993) Biological roles of oligosaccharides: all of the theories are correct. Glycobiology 3:97–130
Dube DH, Bertozzi CR (2005) Glycans in cancer and inflammation. Potential for therapeutics and diagnostics. Nat Rev Drug Discov 4:477–488
Haase C, Seitz O (2007) In: Wittmann V (ed) Glycopeptides and glycoproteins. Springer, Berlin/Heidelberg, pp 1–36
Unverzagt C, Kajihara Y (2013) Chemical assembly of N-glycoproteins: a refined toolbox to address a ubiquitous posttranslational modification. Chem Soc Rev 42:4408–4420
Marcaurelle LA, Bertozzi CR (2002) Recent advances in the chemical synthesis of mucin-like glycoproteins. Glycobiology 12:69r–77r
Varki A (2006) Nothing in glycobiology makes sense, except in the light of evolution. Cell 126:841–845
Therkildsen MH, Mandel U, Christensen M, Dabelsteen E (1993) Simple mucin-type Tn and sialosyl-Tn carbohydrate antigens in salivary gland carcinomas. Cancer 72:1147–1154
Brockhausen I, Yang J-M, Burchell J, Whitehouse C, Taylor-Papadimitriou J (1995) Mechanisms underlying aberrant glycosylation of MUC1 mucin in breast cancer cells. Eur J Biochem 233:607–617
Hanisch FA (2001) O-Glycosylation of the mucin type. Biol Chem 382:143–149
Burchell J, Mungul A, Taylor-Papadimitriou J (2001) O-Linked glycosylation in the mammary gland: changes that occur during malignancy. J Mammary Gland Biol Neoplasia 6:355–364
Larsson JMH, Karlsson H, Crespo JG, Johansson MEV, Eklund L, Sjövall H, Hansson GC (2011) Altered O-glycosylation profile of MUC2 mucin occurs in active ulcerative colitis and is associated with increased inflammation. Inflamm Bowel Dis 17:2299–2307
Stuchlová Horynová M, Raška M, Clausen H, Novak J (2013) Aberrant O-glycosylation and anti-glycan antibodies in an autoimmune disease IgA nephropathy and breast adenocarcinoma. Cell Mol Life Sci 70:829–839
Remmers N, Anderson JM, Linde EM, DiMaio DJ, Lazenby AJ, Wandall HH, Mandel U, Clausen H, Yu F, Hollingsworth MA (2013) Aberrant expression of mucin core proteins and O-linked glycans associated with progression of pancreatic cancer. Clin Cancer Res 19:1981–1993
Anish C, Schumann B, Pereira Claney L, Seeberger Peter H (2014) Chemical biology approaches to designing defined carbohydrate vaccines. Chem Biol 21:38–50
Gaidzik N, Westerlind U, Kunz H (2013) The development of synthetic antitumour vaccines from mucin glycopeptide antigens. Chem Soc Rev 42:4421–4442
Kajihara Y, Izumi M, Hirano K, Murase T, Macmillan D, Okamoto R (2011) Elucidating the function of complex-type oligosaccharides by use of chemical synthesis of homogeneous glycoproteins. Isr J Chem 51:917–929
Galan MC, Benito-Alifonso D, Watt GM (2011) Carbohydrate chemistry in drug discovery. Org Biomol Chem 9:3598–3610
Ficht S, Payne RJ, Guy RT, Wong C-H (2008) Solid-phase synthesis of peptide and glycopeptide thioesters through side-chain-anchoring strategies. Chem Eur J 14:3620–3629
Kiessling LL, Splain RA (2010) Chemical approaches to glycobiology. Annu Rev Biochem 79:619–653
Kajihara Y, Yamamoto N, Okamoto R, Hirano K, Murase T (2010) Chemical synthesis of homogeneous glycopeptides and glycoproteins. Chem Rec 10:80–100
Hojo H, Katayama H, Nakahara Y (2010) Progress in the ligation chemistry for glycoprotein synthesis. Trends Glycosci Glycotechnol 22:269–279
Liu L, Hong Z-Y, Wong C-H (2006) Convergent glycopeptide synthesis by traceless Staudinger ligation and enzymatic coupling. ChemBioChem 7:429–432
Brik A, Yang Y-Y, Ficht S, Wong C-H (2006) Sugar-assisted glycopeptide ligation. J Am Chem Soc 128:5626–5627
Brik A, Ficht S, Yang Y-Y, Wong C-H (2006) Sugar-assisted ligation of N-linked glycopeptides with broad sequence tolerance at the ligation junction. J Am Chem Soc 128:15026–15033
Brik A, Wong C-H (2007) Sugar-assisted ligation for the synthesis of glycopeptides. Chem Eur J 13:5670–5675
Ficht S, Payne RJ, Brik A, Wong C-H (2007) Second-generation sugar-assisted ligation: a method for the synthesis of cysteine-containing glycopeptides. Angew Chem Int Ed 46:5975–5979
Yang Y-Y, Ficht S, Brik A, Wong C-H (2007) Sugar-assisted ligation in glycoprotein synthesis. J Am Chem Soc 129:7690–7701
Payne RJ, Ficht S, Tang S, Brik A, Yang Y-Y, Case DA, Wong C-H (2007) Extended sugar-assisted glycopeptide ligations: development, scope, and applications. J Am Chem Soc 129:13527–13536
Bennett CS, Dean SM, Payne RJ, Ficht S, Brik A, Wong C-H (2008) Sugar-assisted glycopeptide ligation with complex oligosaccharides: scope and limitations. J Am Chem Soc 130:11945–11952
Wang L-X, Amin MN (2014) Chemical and chemoenzymatic synthesis of glycoproteins for deciphering functions. Chem Biol 21:51–66
Westerlind U (2012) Synthetic glycopeptides and glycoproteins with applications in biological research. Beilstein J Org Chem 8:804–818
Peri F, Nicotra F (2004) Chemoselective ligation in glycochemistry. Chem Commun 623–627
Specker D, Wittmann V (2007) In: Wittmann V (ed) Glycopeptides and glycoproteins. Springer, Berlin/Heidelberg, pp 65–107
Shin Y, Winans KA, Backes BJ, Kent SBH, Ellman JA, Bertozzi CR (1999) Fmoc-based synthesis of peptide-(alpha)thioesters: application to the total chemical synthesis of a glycoprotein by native chemical ligation. J Am Chem Soc 121:11684–11689
Buskas T, Ingale S, Boons G-J (2006) Glycopeptides as versatile tools for glycobiology. Glycobiology 16:113R–136R
Murakami M, Okamoto R, Izumi M, Kajihara Y (2012) Chemical synthesis of an erythropoietin glycoform containing a complex-type disialyloligosaccharide. Angew Chem Int Ed 51:3567–3572
Liu L, Bennett CS, Wong CH (2006) Advances in glycoprotein synthesis. Chem Commun 21–33
Hojo H, Nakahara Y (2007) Recent progress in the field of glycopeptide synthesis. Biopolymers 88:308–324
Monbaliu JCM, Katritzky AR (2012) Recent trends in Cys- and Ser/Thr-based synthetic strategies for the elaboration of peptide constructs. Chem Commun 48:11601–11622
Garner J, Jolliffe KA, Harding MM, Payne RJ (2009) Synthesis of homogeneous antifreeze glycopeptides via a ligation-desulfurisation strategy. Chem Commun 6925–6927
Chen J, Wan Q, Yuan Y, Zhu JL, Danishefsky SJ (2008) Native chemical ligation at valine: a contribution to peptide and glycopeptide synthesis. Angew Chem Int Ed 47:8521–8524
Moyal T, Hemantha HP, Siman P, Refua M, Brik A (2013) Highly efficient one-pot ligation and desulfurization. Chem Sci 4:2496–2501
Thompson RE, Liu XY, Alonso-Garcia N, Pereira PJB, Jolliffe KA, Payne RJ (2014) Trifluoroethanethiol: an additive for efficient one-pot peptide ligation-desulfurization chemistry. J Am Chem Soc 136:8161–8164
Okamoto R, Kajihara Y (2008) Uncovering a latent ligation site for glycopeptide synthesis. Angew Chem Int Ed 47:5402–5406
Okamoto R, Souma S, Kajihara Y (2009) Efficient substitution reaction from cysteine to the serine residue of glycosylated polypeptide: repetitive peptide segment ligation strategy and the synthesis of glycosylated tetracontapeptide having acid labile sialyl-T-N antigens. J Org Chem 74:2494–2501
Hojo H, Ozawa C, Katayama H, Ueki A, Nakahara Y, Nakahara Y (2010) The mercaptomethyl group facilitates an efficient one-pot ligation at Xaa-Ser/Thr for (glyco)peptide synthesis. Angew Chem Int Ed 49:5318–5321
Kang J, Macmillan D (2010) Peptide and protein thioester synthesis via N→S acyl transfer. Org Biomol Chem 8:1993–2002
Macmillan D, Adams A, Premdjee B (2011) Shifting native chemical ligation into reverse through N→S acyl transfer. Isr J Chem 51:885–899
Ackrill T, Anderson DW, Macmillan D (2010) Towards biomolecular assembly employing extended native chemical ligation in combination with thioester synthesis using an N→S acyl shift. Biopolymers 94:495–503
Premdjee B, Adams AL, Macmillan D (2011) Native N-glycopeptide thioester synthesis through N→S acyl transfer. Bioorg Med Chem Lett 21:4973–4975
Hsieh YSY, Wilkinson BL, O’Connell MR, Mackay JP, Matthews JM, Payne RJ (2012) Synthesis of the bacteriocin glycopeptide sublancin 168 and S-glycosylated variants. Org Lett 14:1910–1913
Macmillan D, Blanc J (2006) A novel neoglycopeptide linkage compatible with native chemical ligation. Org Biomol Chem 4:2847–2850
Lee DJ, Mandal K, Harris PWR, Brimble MA, Kent SBH (2009) A one-pot approach to neoglycopeptides using orthogonal native chemical ligation and click chemistry. Org Lett 11:5270–5273
Geiermann AS, Polacek N, Micura R (2011) Native chemical ligation of hydrolysis-resistant 3′-peptidyl-tRNA mimics. J Am Chem Soc 133:19068–19071
Geiermann AS, Micura R (2012) Selective desulfurization significantly expands sequence variety of 3′-peptidyl-tRNA mimics obtained by native chemical ligation. ChemBioChem 13:1742–1745
Wang C, Guo Q-X, Fu Y (2011) Theoretical analysis of the detailed mechanism of native chemical ligation reactions. Chem Asian J 6:1241–1251
Monbaliu JCM, Dive G, Stevens CV, Katritzky AR (2013) Governing parameters of long-range intramolecular S-to-N acyl transfers within (S)-acyl isopeptides. J Chem Theory Comput 9:927–934
Oliferenko AA, Katritzky AR (2011) Alternating chemical ligation reactivity of S-acyl peptides explained with theory and computations. Org Biomol Chem 9:4756–4759
Panda SS, Oliferenko AA, Marwani HM, Katritzky AR (2014) Effects of preorganization and hydrogen bonding on intramolecular chemical ligation of (N)- and (O)-acyl isopeptides. Mendeleev Commun 24:75–77
Biswas S, Kayaleh R, Pillai GG, Seon C, Roberts I, Popov V, Alamry KA, Katritzky AR (2014) Long-range chemical ligation from N→N acyl migrations in tryptophan peptides via cyclic transition states of 10-to 18-members. Chem Eur J 20:8189–8198
Gramatica P, Chirico N, Papa E, Cassani S, Kovarich S (2013) QSARINS: a new software for the development, analysis, and validation of QSAR MLR models. J Comput Chem 34:2121–2132
Acknowledgements
We thank the University of Florida and The Kenan Foundation for financial support.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Panda, S.S., Jones, R.A., Dennis Hall, C., Katritzky, A.R. (2014). Applications of Chemical Ligation in Peptide Synthesis via Acyl Transfer. In: Liu, L. (eds) Protein Ligation and Total Synthesis I. Topics in Current Chemistry, vol 362. Springer, Cham. https://doi.org/10.1007/128_2014_608
Download citation
DOI: https://doi.org/10.1007/128_2014_608
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-19185-0
Online ISBN: 978-3-319-19186-7
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)