
Abstract
Living with limits. Getting more from less. Producing commodities and high-value products from renewable resources including waste. What is the driving force and quintessence of bioeconomy outlines the lifestyle and product portfolio of Aspergillus, a saprophytic genus, to which some of the top-performing microbial cell factories belong: Aspergillus niger, Aspergillus oryzae and Aspergillus terreus. What makes them so interesting for exploitation in biotechnology and how can they help us to address key challenges of the twenty-first century? How can these strains become trimmed for better growth on second-generation feedstocks and how can we enlarge their product portfolio by genetic and metabolic engineering to get more from less? On the other hand, what makes it so challenging to deduce biological meaning from the wealth of Aspergillus -omics data? And which hurdles hinder us to model and engineer industrial strains for higher productivity and better rheological performance under industrial cultivation conditions? In this review, we will address these issues by highlighting most recent findings from the Aspergillus research with a focus on fungal growth, physiology, morphology and product formation. Indeed, the last years brought us many surprising insights into model and industrial strains. They clearly told us that similar is not the same: there are different ways to make a hypha, there are more protein secretion routes than anticipated and there are different molecular and physical mechanisms which control polar growth and the development of hyphal networks. We will discuss new conceptual frameworks derived from these insights and the future scientific advances necessary to create value from Aspergillus Big Data.
Graphical Abstract

Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Aspergillus
- Protein secretion
- Morphology
- Modelling
- Physiology
- -omics
- Database
- Genome-scale model
- Metabolism
- Secondary metabolite
1 Introduction
Because Aspergilli, like many other filamentous fungi, first digest and then ingest their food, they have astounding capabilities in secreting enzymes hydrolysing biopolymers such as starch, (ligno-)cellulose, pectin, xylan, proteins and lipids. This feature is exploited by many biotech companies to manufacture enzymes for use in different industries such as paper and pulp, food and feed, laundry and textile, and biofuel. Moreover, Aspergilli produce and secrete a variety of bioactive compounds including organic acids and secondary metabolites which outcompete cohabitant bacteria and fungi and which are beneficial for human health and consumption. Many recent reviews have comprehensively discussed the wealth and variety of marketed Aspergillus products and listed the respective manufacturers [1–4].
What more is to come? New products! Commodities which are competitive with current petrochemical-derived products, smarter enzyme mixtures which faster and fully degrade lignocellulosic biomass and pharmaceuticals which have not been produced with Aspergilli so far. Better strains! Which produce more from cheaper substrates and which are genetically streamlined for better rheological performance under industrial fermentation conditions? These are very challenging goals, which need to be accomplished to help to reduce the heavy dependence of the modern world on fossil resources and to move it towards a post-petroleum future. In the following chapters, we will discuss current concepts, tools and approaches to reach these ambitious goals. To come to the point, some recent publications refuted long-lasting dogmas: (i) ‘the key to the fungal hypha lies in the apex’, i.e. protein secretion occurs exclusively at the hyphal apex [5, 6], (ii) industrial fungi such as Penicillium chrysogenum and Trichoderma reesei do not have sex [7, 8]. Others paved the way for new product classes which can be manufactured with A. niger: (i) nonribosomal peptides for medical applications [9], (ii) itaconic acid for use in bioplastics and biofuel industry [10, 11].
Although the post-genomic era has offered us holistic insights into the genomic and physiological landscape of Aspergillus species, it left us also with a flood of data. Decisions that were previously intuitive or based on data from laborious trial-and-error experimental approaches can now be made based on bioinformatics data itself. However, the ‘Big Data tsunami’ [12] poses major challenges. Managing and unbiased mining of the sheer size of -omics data is one. Not leaving them under-analysed in databases is the other. But deducing meaningful knowledge and new hypothesis is the most difficult one. What are the success stories in the field of systems biology of industrial filamentous fungi? Which hurdles hinder the move from intuitive to knowledge-based strain optimisation strategies? We will highlight in this review some examples to illustrate the current state of the art of Aspergillus systems biology with a focus on fungal growth, morphology, physiology and product formation. For further information, the reader is directed to recent reviews [13, 14]. Last but not least, we will summarise the state of the art of micro- or macro-morphological models for filamentous growing fungi such as Aspergillus to describe growth and geometrical appearance of pellets and mycelia. Recent approaches and mathematical concepts will be highlighted and the challenges discussed to develop biologically better founded models.
2 More from Less: Enlarging the Product Portfolio of Aspergillus
The pioneering role of Aspergillus species for microbial biotechnology is undisputed. Since almost 100 years, citric acid is produced on an industrial scale with A. niger and used as preservative and flavour enhancer in food and beverages. Due to its long history of safe use and its capability to withstand harsh industrial fermentation conditions, it later became leading for fermentation-derived enzyme production [4]. A. oryzae is the exclusive producer of another acid, kojic acid, which is applied as bleaching agent in cosmetics and as building block for biodegradable plastics. More influential, A. oryzae has traditionally been used to ferment starch-rich grains, like rice and soybeans, for the production of Asian food and beverages. The importance of A. terreus is attributed to its product lovastatin, a cholesterol-lowering agent which became a blockbuster after Merck released it for marketing in 1987 (trade name Mevacor, [15]). There are legitimate reasons to believe that industrial Aspergillus strains could keep and extend their pioneering role in microbial biotechnology. The following examples substantiate this prognosis.
2.1 Aspergillus Trimmed for Second-Generation Feedstocks
To achieve economic and ecological sustainable microbial production processes, it is crucial that feedstocks do not compete with the food supply chain but rely on non-edible crop residues such as lignocellulose. Current commercial cellulase cocktails for conversion of lignocellulosic biomass to bioethanol are derived from filamentous fungi (e.g. Cellic CTec2 from Novozymes and Accellerase from DuPont) and are a blend of three classes of enzymes: cellulases, hemicellulases and β-glucosidases. Whereas the first two are produced by T. reesei fermentation, the latter is a product of A. niger. Although T. reesei is the ‘undisputable champion’ in cellulose hydrolysis (T. reesei secretes cellulases up to 100 g/l), its cellulose preparations have to be supplemented with A. niger β-glucosidases to achieve efficient cellulose saccharification ([16] and references therein). Recently, a two-stage biorefining strategy was reported which utilised wheat straw as lignocellulosic raw material and only A. niger enzymes for saccharification [17]. Note that wheat straw is the second most abundant lignocellulosic crop residue in the world [18]. In the first stage, a solid-state fermentation of A. niger was performed on wheat straw for the production of (hemi)cellulases and β-glucosidases. These enzymes were extracted and used in the second stage to hydrolyse the fermented wheat straw. Remarkably, the freshly extracted enzymes of A. niger performed the same or even better than the commercial CTec2 solution [17]. Another report demonstrated that enzyme cocktails derived from submerged co-cultivation of T. reesei with Aspergillus species can compete with blended extracts from corresponding single cultures. This is an interesting alternative as it lowers production cost due to the integration of two fermentations into a single bioreactor and due to the elimination of an enzyme blending step [19]. A. oryzae is a poor secretor of cellulases. However, Yamada and co-workers demonstrated that overexpression of the kojic acid gene cluster specific transcription factor kojT in combination with heterologous expression of three fungal cellulases in A. oryzae led the fungus to produce kojic acid directly from cellulose. Although the yield was very low, the proof of concept was given that A. oryzae can be trimmed to produce kojic acid when cultivated on second-generation feedstocks [20].
2.2 A. niger Engineered for New Products
Itaconic acid stems from the citric acid cycle and is one of the most promising and flexible new platform chemicals derived from cellulosic biomass (for a review see [21]). Although it is currently produced by A. terreus with yields up to 76 % of the theoretical maximum, there are efforts to establish A. niger as itaconate producer. Several considerations make A. niger interesting as an alternative production host: first, its enormous citric acid production capacity could potentially be redirected to itaconate synthesis. Second, the fermentation can be performed at low pH which allows autosterile production and simplifies downstream processing. However, it is currently impossible to predict whether A. terreus can indeed be replaced by A. niger. Although the transfer of itaconate synthesis genes and reduction of by-product formation has been achieved, limited intracellular transport has been identified as severe bottleneck [10, 21–23]. Further studies are necessary to fully lift the potential of A. niger as heterologous itaconate producer.
Most recently, it was demonstrated for the first time that A. niger is a potent expression host for heterologous nonribosomal peptides [9]. For the proof of concept, the nonribosomal peptide synthetase gene esyn1 from Fusarium oxysporum was expressed under control of the tunable bacterial–fungal hybrid promoter Tet-On [24] in A. niger. In F. oxysporum, ESYN catalyses the synthesis of the cyclic depsipeptide enniatin, an antibiotic used to treat bacterial throat infections. The strong inducibility of the Tet-On system combined with controlled bioreactor cultivation allowed impressively high enniatin yields in A. niger (about 5 g/l), not reported so far for any heterologous enniatin production host. As also an autonomous A. niger expression host was engineered, being independent from feeding with the enniatin precursor d-2-hydroxyvaleric acid, the production cost was lowered considerably [9].
2.3 Aspergillus is Becoming Transparent
As of 2010, ten genomes of different Aspergillus species were publically accessible [25]. As of 2014, the number increased to 21 fully sequenced Aspergillus species, which are accessible through MycoCosm, a fungal genomics portal (http://jgi.doe.gov/fungi) developed by the US Department of Energy Joint Genome Institute [26]. In addition, dozens of Aspergillus strains are becoming currently resequenced including A. niger strains which have been transformed with bacterial cellulases, which are enzyme hyperproducers or spontaneous mutants (http://jgi.doe.gov/fungi). Subsequent comparative genome analyses will likely make Aspergillus one of the best studied and understood fungal genera. A tremendous opportunity, which will help to distinguish between common and strain-specific genomic features as exemplarily shown for two A. niger strains, the citric acid-producing strain ATCC 1015 and the enzyme-producing strain CBS 513.88 [27] (see Chap. 5).
3 The Life Cycle of Aspergillus and Its Link to Product Formation
To fully lift the potential of Aspergillus as a multi-purpose cell factory, a key question needs to be addressed: which metabolites, proteins and biopolymer-degrading enzymes are predominantly produced during which phase of its life cycle? As nature has evolutionary optimised Aspergillus already to produce certain compounds under specific habitat and nutrient conditions, one ‘simply’ needs to understand how the native metabolic properties of Aspergillus are linked to its lifestyle and life cycle in order to further optimise Aspergillus by rational means. Clearly, certain product classes are associated with different stages of its life cycle, such as secondary metabolite synthesis in A. niger with sporulation [28]. Likewise, its saprophytic lifestyle is the basis for the tight regulation of polysaccharide hydrolase expression and secretion [29].
3.1 Morphogenesis and Product Formation
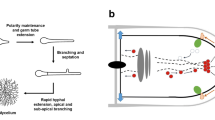
The life cycle of Aspergillus and filamentous fungi starts with breaking dormancy of (a)sexual spores (Fig. 1). In A. niger, the resulting onset of spore germination, which is characterised by isotropic spore swelling, becomes only initiated when a carbon source, a nitrogen source and a germination trigger are present in the environment [30]. d-glucose, 2-deoxy-d-glucose, d-mannose, d-xylose and l-amino acids such as l-asparagine, l-glutamine and l-tryptophan were identified as germination triggers. A full list of triggers and non-triggers can be found in [30, 31]. Interestingly, dormant spores (named conidia) of A. niger contain a high level of transcripts from about 40 % of A. niger genes. These transcripts include genes involved in protein synthesis and respiration and probably prime the conidia for the onset of germination [32, 33]. Note that the A. niger genome has been predicted to contain about 14,000 genes [34]. Interestingly, also a considerable amount of antisense transcripts (10 %) were detected, which are assumed to prevent expression of proteins that are not required during spore swelling [32].

The life cycle of Aspergillus niger. The different stages of spore swelling, germ tube outgrowth, germ tube elongation and branch formation are schematically shown. These processes lead to the formation of a true circular colony, which appears brownish black due to brownish-black-coloured conidia. The conidia become constricted from a conidiophore (schematically shown)
The first stage of spore swelling is followed by the outgrowth of a germ tube. This break in cell symmetry results from the establishment of a polarity axis. If this polarity axis becomes maintained, sustained germ tube elongation and the formation of a hypha can be observed. In higher fungi, such as Ascomycetes and Basidiomycetes, this hypha becomes compartmentalised by septa, which contain a central pore that allows inter-compartmental streaming [35]. Lateral and/or apical branch formation in this hypha eventually leads to the formation of an interconnected hyphal network, the mycelium (Fig. 1, [36]). In consequence, the age of the mycelium increases from the periphery towards the central zone, whereas substrate concentrations decrease. This spatial heterogeneity results in profound differences on the level of the transcriptome [37] and secretome [38] in different zones of A. niger colonies and supports the idea that medium substrates become sequentially degraded. Notably, a large fraction of the secreted proteins remains trapped in the fungal cell wall from which they become only slowly released. In turn, a remarkable fraction of proteins secreted within a more central zone of a mycelium was synthesised earlier during growth, i.e. when the respective protein-producing hyphae were at the periphery of the colony. As an example, partial degradation of fungal cell walls in the central zones allowed the detection of ~120 secreted proteins, but of only ~60 in the untreated control colony [38]. Such retention phenomena bring additional complexity to the heterogeneous spatio-temporal substrate degradation pattern observed in A. niger colonies and most likely also occur in submerged pellet-forming mycelia of A. niger during industrial fermentation processes.
Beside this interzonal heterogeneity, intrazonal hyphal heterogeneity is also a characteristic attribute of A. niger. Although the periphery of A. niger colonies or pellets is the active growth zone and the primary zone of protein secretion [39], neighbouring hyphae differ with respect to RNA and protein levels [40–42]. Whilst it may well be assumed that such heterogeneities could be beneficial for A. niger under natural habitat conditions, an increase of the actively secreting hyphal population would be a constructive approach to improve the productivity of submerged industrial processes [43]. However, heterogeneity of neighbouring hyphae is less pronounced in submerged pellets compared to surface cultures, which is likely linked to a more homogeneous substrate distribution in liquid compared to solid media [42]. Last but not least, carbon starvation-induced heterogeneity can be observed in dispersed mycelia of A. niger when cultivated under submerged conditions. Two morphologically different hyphal populations appear when the carbon source is depleted (Fig. 2), on the one hand thick but non-growing hyphae with many empty compartments and on the other hand thin but growing hyphae. Growth of the latter is likely fuelled by carbon recycling via autophagy of the older hyphae [44, 45]. This is in fact an elegant strategy of filamentous fungi, to make use of internal resources, i.e. degradation products, before a compartment collapses for the survival of a neighbouring compartment [46].

Two hyphal populations can be observed during carbon starvation of A. niger when cultivated under submerged conditions. Hyphal diameters can be used to distinguish both populations: old hyphae (formed during exponential growth phase) and young hyphae (formed during starvation). An image analysis algorithm developed by [44] analyses microscopic pictures from samples of various cultivation time points (a, b) and plots probability density curves for the distributions of hyphal diameters. Diameters from exponentially growing hyphae (a) display a normal distribution with a mean of approximately 3 μm. The second population of thinner hyphae with a mean diameter of approximately 1 μm start to emerge during starvation (b). During prolonged starvation, the ratio of thin/thick hyphae gradually increases [44]. Bar, 10 μm
3.2 Asexual Development and Product Formation
Surface cultures of Aspergillus start to form aerial hyphae and conidiophores after achieving developmental competence (Fig. 1), a process which can be triggered by the liquid–air interface, chemical signals and light. At the end of this developmental process, conidia are formed via mitotic events. For details on key morphogenetic genes and regulatory networks involved, the reader is directed to [47, 48]. Notably, conidiophore structures are morphologically informative enough to distinguish between the genera Aspergillus and Penicillium [49]. Although the process of conidiation is repressed under submerged conditions, it is possible to induce sporulation by severe carbon limitation in retentostats, where growth rates approaching zero can be adjusted [50].
Several studies analysed the transcriptomes of conidiating A. niger and T. reesei cultures during liquid [44, 50] and solid-state cultivations [51, 52] and demonstrated that nutrient starvation and conidiation are tightly linked. The fraction of differentially expressed genes ranged between 10 and 50 %, depending on the platform and statistics applied, and clearly highlighted that the transition from vegetative hyphae to developmental structures is accompanied by complex physiological adaptations. A major starvation-induced response linked with asexual development is activation of secondary metabolite genes and gene clusters, which are under control of different signalling pathways including epigenetic control mechanisms [53–55]. These processes can easily be tracked and studied in retentostat cultivations [44, 50].
Another major response during carbon starvation is induction of carbohydrate-active enzymes (CAZymes), including enzyme classes degrading the fungal cell wall and plant-derived polysaccharides. Both are of major interest for industrial applications. Whereas the first class is unwanted as it supposedly reduces the fraction of actively growing hyphae, the second class contains (un)known candidate enzymes for new biorefining strategies (see Chap. 1 and [17]). Delmas et al. [29] and Van Munster et al. [56] uncovered that carbon starvation induces de-repression of genes encoding sugar transporters and a comprehensive set of CAZymes, which become secreted as ‘scouting enzymes’. Depending on the actual polysaccharide(s) present in the surroundings, they catalyse the liberation of specific sugar monomers, which in turn trigger expression and secretion of the specific enzyme set necessary to fully degrade the polysaccharide(s) present in the environment. The requirement of complex enzyme mixtures for efficient saccharification of lignocellulosic biomass underlines the importance of genome-wide and systematic approaches to comprehensively decipher the polysaccharide degradation potential of Aspergillus. Recently, structural data from 16 major plant-derived polysaccharides have been combined with a list of 188 predicted CAZymes from A. niger [57]. The resulting analysis framework can now be queried systematically with data from genome-wide expression profiling analyses such as obtained from A. niger cultivated on sugarcane bagasse [58] or wheat straw [29].
4 Polarised Growth of Aspergillus and Its Link to Protein Production
4.1 There Are More Ways to Make a Hypha
Although sustained polarised growth is the defining attribute of filamentous fungi, hyphal filaments exhibit a striking diversity in organisation. In some fungi, the nuclei are distributed dot-like along the hypha (e.g. Aspergillus nidulans); in others, they appear in bulk (e.g. A. niger). Some fungi form filaments, which are dikaryotic and largely composed of vacuoles (e.g. Ustilago maydis, [59]), some form hyphal networks which are remarkably heterogeneous with respect to gene expression and physiology as described in Chap. 2 (e.g. A. niger and Neurospora crassa, [60, 61]). Recently, Woronin bodies, originally thought to be emergency patches which seal septal pores of filamentous ascomycetes upon mechanical injury, were shown to maintain hyphal heterogeneity in intact hyphae by impeding cytoplasmic streaming. Cytoplasmic GFP expression was heterogeneous in neighbouring hyphae of A. oryzae which formed Woronin bodies but was homogeneous in a strain that could not form these organelles. Closure of septa is a dynamic and reversible process, resulting on average in 40 % closed apical septa of exploring A. oryzae hyphae [62].
4.2 Microscale and Macroscale Polarity
Why does a hypha grow in a polarised manner? Because of pressure! On the one hand, cytoskeletal structures (microtubuli, actin) form the polarised architecture of growing hyphae. Along these, secretory vesicles, containing lipids, cell wall proteins, building blocks and proteins destined for secretion, are actively transported by motor proteins towards the hyphal tip (Fig. 3, [63]). On the other hand, internal hydrostatic pressure (turgor) is created. Water flows into the growing hyphal tip due to the uptake of solutes and the biosynthesis of osmotically active solutes within the cell. According to the model after Lee [64], water inflow is greatest at the tip and declines exponentially behind the tip, thus causing the cell wall to expand. Stretching of the cell membrane and cell wall provokes a tip-localised calcium gradient that eventually mediates vesicle fusion and thus cell expansion. Next to this microscale polarity, macroscale polarity exists as well. Mycelia can be viewed as ‘hydraulic networks’ because of a laminar mass flow of cytoplasm towards the colony edge. The driving force for this cytoplasmic movement is thought to result from maximum uptake of nutrients behind the foraging hyphae, leading to increased water inflow and increased pressure [64]. It has been proposed that this intrahyphal transport is regulated by the closure of septa, which become plugged by Woronin bodies [65]. It is very likely that this pressure-driven long-distance transport complements the short-distance transport mediated by cytoskeletal motor proteins [66].

Subcellular structures define and maintain the architecture of a growing hypha. Cytoskeletal-based transport of vesicles and endosomes is realised by different motor proteins (kinesins, dyneins and myosins) which move along microtubules and actin cables in different directions. For the sake of brevity, the endocytotic actin ring is not shown. For details, see text
4.3 Microscale Polarity and the Secretory Route of Aspergillus
Our current knowledge on the implementation and regulation of vesicle flux towards growing hyphal tips of filamentous fungi in general and Aspergillus in particular has recently been reviewed [4, 67, 68] and will be summarised here in a very condensed and simplified manner: Proteins and enzymes destined for a function at the cell surface or for secretion into the environment become translocated into the lumen of the endoplasmic reticulum (ER) (Fig. 3). Here, several chaperons and foldases including the binding protein BipA, protein disulphide isomerase PdiA and calnexin ClxA assist secretory proteins in their folding. Many secretory proteins also become glycosylated, after which they are packed into vesicles and transported to and through the Golgi complex via COPII-vesicles and are then delivered to the Spitzenkörper via a microtubule-mediated transport. The Spitzenkörper is a multi-component, vesicle-rich, pleomorphic structure at the hyphal tip and viewed as the main choreographer of hyphal tip growth (for a detailed review see [69]). It translocates secretory vesicles to actin cables, which transport them to the plasma membrane. Vesicles fuse with the plasma membrane, thus releasing their cargo. Whilst secretory proteins become released into the environment, GPI-anchored proteins and cell wall-synthesising proteins (e.g. chitin and glucan synthases) become retained at the membrane to catalyse cell wall polymers such as chitin, glucans and galactomannans, which eventually expand the cell wall (for see review [70]). The vectorial flux of vesicles from the Spitzenkörper to the plasma membrane is orchestrated by the polarisome, which mediates the nucleation of actin cables and together with cell end markers directs the vesicle flux towards the apex (Fig. 3, [63, 68, 71]).
It is currently a matter of debate, whether exocytosis and endocytosis are spatially coupled. Retrieval of excess membrane material resulting from vesicle fusion with the membrane and removal of cell wall synthetising enzymes from subapical membrane regions by endocytosis are thought to be essential to maintain the fungal shape and its polarity [63, 72–74]. In A. nidulans, empty vesicles are proposed to become recycled by endocytosis at a subapical actin collar, transported back to the Golgi, where they become reloaded with new protein cargo destined for secretion [63]. Hence, protein secretion should be viewed as a concerted and balanced activity of exo- and endocytotic events. Furthermore, restriction of cell wall enzymes to the very tip in C. albicans has been shown to ensure polarised cell wall expansion as deduced from experimental and 3D modelling data [74]. However, a mathematical model proposed for the vesicle flux to and from the Spitzenkörper in Rhizoctonia solani predicts that removal of membrane material is not significant to compensate a potential excess of membrane added by exocytosis [75]. Is this further evidence that there are more ways to make a hypha? So far, experimental data from industrial relevant Aspergilli are not available which do illuminate any potential link between their exocytotic and endocytotic machineries and which could refute or validate either model. Demonstrated so far is that tubulin and actin cytoskeletal elements are key for the transport of secretory vesicles to the hyphal tips of A. niger and A. oryzae, whereby secretory vesicles were also occasionally observed at septa, suggesting that secretion can also occur at sites of septa [6, 76].
In addition to protein and membrane turnover via endocytosis, pioneering work in the model systems A. nidulans and U. maydis has shown that bi-directional movement of early endosomes along actin cables and microtubuli actively disperses mRNA and ribosomes, i.e. the protein translation machinery along hyphal compartments, thus also contributing to hyphal growth (Fig. 3, for details see reviews [77, 78]). Hence, a complex interplay of cytoskeletal structures, polarity factors, cell end markers, and exocytotic and endocytotic events assure the secretion of proteins and polarised hyphal growth [68, 79].
4.4 A Holistic View on the Secretory Route of Aspergillus
Filamentous fungi are extraordinary in their capacities to secrete endogenous proteins, whereby A. niger (up to 20 g/l) and T. reesei (up to 100 g/l) are outstanding [80, 81]. We and others have most recently reviewed the current strategies to exploit these capacities for the expression of heterologous proteins in Aspergillus and the systems biology approaches that have been undertaken to study the physiological and regulatory aspects related to homologous and heterologous protein production. In order to not exceed the framework of this review, the reader is directed to these reviews [4, 14, 25, 82]. What have we learnt so far? First, A. niger and A. oryzae flexibly adapt the protein flux through the secretory pathway. If genetically forced protein overexpression causes an overload of the secretory route, both ER-associated degradation and the unfolded proteins response UPR aim to restore cellular homeostasis. In addition, many genes become transcriptionally down-regulated, which are obviously less important for growth and survival under the specific growth condition. This phenomenon is called repression under secretion stress (RESS) and was first observed in T. reesei [83]. Second, under identical specific growth rates realised in chemostat cultivations, a glucoamylase-overproducing strain of A. niger has to modify the expression of about 1,500 genes compared to the wild-type strain in order to secrete about seven times more glucoamylase. The up-regulated gene set contained statistically enriched genes functioning in ER translocation, protein glycosylation, vesicle transport and ion homeostasis [84]. A comparative transcriptomics analysis of six independent overexpression and UPR stress studies in A. niger uncovered that a key set of about 40 genes is crucial for A. niger to overexpress a protein and cope with this cellular burden. The complete gene list can be found in [84] and includes genes encoding ER-resident chaperones and foldases (e.g. clxA, pdiA, bipA), as well as genes important for ER translocation, protein glycosylation and COPII-based vesicle trafficking to name but a few. Third, the amino acid sequence of secretory proteins somehow affects the level of secretion. By comparing the extracellular protein yield of about 600 homologous and 2,000 heterologous proteins in A. niger, van den Berg and colleagues identified a positive effect for tyrosine and asparagine, whereas methionine and lysine hampered high protein yields [85]. Fourth, we are far away from rationally and predictively designing A. niger or A. oryzae as cell factories for protein production. Despite more than 30 years of research on these strains, the quantitative biology behind protein secretion is completely unknown. For example, how many vesicles can a hyphal tip of A. niger accommodate per unit of time? How many vesicles are necessary to ensure maximum transport of the protein of interest whilst not interfering with the transport of other vesicles carrying proteins and building blocks for normal growth? How long does the protein of interest need to become properly folded? What does this mean with respect to how many proteins can be channelled through the secretory pathway in order to provide each protein molecule sufficient time to become correctly folded? What would then be the optimum transcription rate for the respective encoding gene and the half-life of its mRNA? And finally, how is the capacity for protein secretion related to primary metabolism which provides the secretory route with ATP, redox equivalents and amino acids? Although Aspergillus already entered the post-genomic era, we are far away from answering these questions and still work on the inventory of its secretory machinery. We have no idea yet, who guides the secretory route, who keeps all players in homeostatic balance and what might be the mechanistic explanation(s) for the phenomenon that A. niger is such an outstanding protein secretor.
4.5 There Are More Ways to Secrete a Protein
There is accumulating evidence that filamentous fungi secrete proteins not only at hyphal tips via the secretory pathway described above. These unconventional secretion pathways (or ‘unconventional membrane trafficking pathways’) have also been postulated for plant and mammalian systems (for reviews see [86–90]). The first report for filamentous fungi goes back to 2011, where it was demonstrated that exocytosis constitutively occurs in A. oryzae at hyphal septa [6]. Originally, it was thought that cytoskeleton-mediated transport of enzymes and materials for cell wall synthesis towards septa is required only during their formation. However, it was shown that exocytosis continues to occur after septation. Although the representative secretory enzyme of A. oryzae α-amylase was mainly transported to the hyphal apex, plasma membrane transporters involved in purine and amino acid uptake were mainly transported to septa [6]. Similarly, a plasma membrane proton pump of N. crassa is incorporated in distal hyphal regions independently of the Spitzenkörper at the hyphal tip [91]. Although these observations were unexpected and refute the dogma that ‘the key to the fungal hypha lies in the apex’, i.e. protein secretion occurs exclusively at the hyphal apex [5], they basically link microscale polarity with macroscale polarity. If nutrients are preferentially taken up in regions more central in the colony, they will generate the small pressure gradient necessary to establish the mass flow of cytoplasmic material. We are coming closer to unveil the secret of fungal micro-fluidics!
Other examples for unconventional secretion mechanisms in filamentous fungi have been reviewed most recently in [86] and are schematically summarised in Fig. 4. First, a brefeldin A-insensitive protein secretion pathway is likely to exist. Brefeldin A is a compound which inhibits vesicle trafficking between the ER and the Golgi. However, addition of brefeldin A does not inhibit chitin synthase secretion in N. crassa [92], suggesting that secretory proteins can leave the ER and reach the plasma membrane independently of the Golgi apparatus [86]. Another example is secretion of proteins which do not contain a signal peptide and thus do not enter the ER. This has for example been shown for an endochitinase from U. maydis [93]. A third example for unconventional secretion is based on the multi-vesicular bodies, which are late endosomal compartments which take up Golgi-derived vesicles. Upon fusion with the plasma membrane, they release these vesicles into the extracellular space [86]. Hence, secretion in filamentous fungi can be realised by more than a single pathway, it would not be surprising if additional routes will become uncovered in the future.

Different protein secretion routes executed in eukaryotic cell systems including fungi (F), plants (P) and mammalian cells (M). For details, see text
5 The Genetics and Physiology Underlying Aspergillus Morphology and Productivity
5.1 From Genomes to Gene Function
The Aspergillus Genome Database (AspGD; http://www.aspgd.org/) is a freely accessible Web resource providing an outstanding comparative genomics toolbox for the fungal community. AspGD houses sequence data from currently 20 Aspergillus genomes and improves gene annotation using publically available transcriptomics data [94, 95]. As of September 2014, 14,056 genes have been predicted for A. niger strain CBS 513.88 (232 of which have a verified function), 11,902 for A. oryzae (199 of which have a verified function) and 10,678 for the model species A. nidulans (1,149 of which have a verified function). Hence, the industrial species have 98 % uncharacterised genes. However, comparative genomics in tandem with the reconstruction of genome-scale metabolic networks can successfully be used for an initial assignment of gene functions as shown for A. oryzae and A. niger. This resulted in the tentative assignment of gene functions to 1,314 and 871 genes, respectively [96–98]. Comparative genomics also revealed that Aspergilli have evolutionary well adapted for naturally performing both lignocellulose degradation and utilisation of the pentoses xylose and arabinose [99].
Genome-scale metabolic models are very useful for simulating maximum growth rate with different carbon sources or for the analysis of -omics data [13]. Most recently, the functional protein secretory component list of A. oryzae was compiled using the secretory model for conventional secretion in the yeast Saccharomyces cerevisiae as scaffold to which known A. nidulans and A. niger secretory genes were added [100]. Comparative analysis of transcriptomics data from A. oryzae strains expressing different levels of amylase deciphered similar key processes important for protein overexpression as reported earlier for A. niger ([84], see Chap. 4). In addition, the secretome of A. oryzae was defined by using the Fungal Secretome Database (FSD; [101]). More than 2,200 putative genes of A. niger are predicted to become potentially secreted in this species [100].
Paradoxically, the majority of characterised genes in A. niger and A. oryzae (50 and 80 %, respectively) are not in primary metabolism [98], although primary metabolism defines the material and energetic foundation for growth, development and reproduction. Instead, the majority of characterised genes in A. niger and A. oryzae are related to products interesting for commercial exploitation: secondary metabolites [54, 102] and extracellular enzymes (see above).
5.2 Citric Acid or Protein Production—What Makes the Difference?
The genome sequence of the A. niger strain CBS 513.88 used for industrial glucoamylase production was published in 2007 [34]. Four years later, the genome of the acidogenic wild-type strain ATCC 1015 was made publically available and compared to strain CBS 513.88 [27]. Comparative genomics and transcriptomics uncovered a remarkable diversity between both strains. First, about 400/500 genes are unique in CBS 513.88/ATCC 1015, the most notable difference is two alpha-amylases which are only present in strain CBS 513.88. Both genes originate from A. oryzae and were most likely acquired through horizontal gene transfer. Second, a high number of polymorphisms were found (8 ± 16 SNPs/kb) between both strains, many of which were clustered in hypervariable regions. Third, transcription of about 4,800 genes differed in both strains (with an almost equal number up-regulated in each strain) when cultivated under the same growth condition in glucose-based batch cultures. Increased transcription of genes in strain CBS 513.88 was related to glycolysis, amino acid synthesis (including the entire biosynthetic pathways of amino acids which are over-represented in the glucoamylase, i.e. threonine, serine and tryptophan) and tRNA-aminoacyl synthase formation. In contrast, the citric acid-producing strain ATCC 1015 displayed higher expression of genes with a function in the transport of electrons, carbohydrates and organic acids. Interestingly, nitrogen source utilisation is impaired in strain CBS 513.88, an observation which could also be relevant to protein production. This study by Andersen and colleagues thus elegantly showed the power of comparative -omics analysis for the identification of factors contributing to the different product portfolio of A. niger strains.
5.3 Moulding the Mould—The Genetic Basis of Aspergillus Morphology
The number of genes functionally implicated in hyphal morphogenesis and development of the model fungus A. nidulans has been estimated to be about 2,000 [36], a number which most likely can be assumed also for other filamentous fungi including the industrial Aspergillus strains. However, the function of only about 5 % of the assumed morphogenetic genes has been understood so far, mainly from studies in the model organisms A. nidulans, N. crassa or U. maydis. It is beyond the scope of this review to provide a comprehensive overview on the function and regulation of the morphogenetic genes and modifiers known so far. For the interested reader, we recommended the following most recent reviews for further reading: polarity factors and cell end markers [67, 69], membrane trafficking [78, 103], septum function [104], calcium regulation [104, 105], cytoskeletal structures [68], cell wall formation [70, 106], asexual development [48, 49] and fungal microfluidics [64]. Instead, we will discuss in the following some recent findings which could help to illuminate the connection between hyphal morphology, vesicle trafficking and protein secretion in the industrial strains A. niger and A. oryzae.
When cultivated under submerged conditions, filamentous fungi adopt two macroscopic morphologies—a pelleted morphology or a dispersed morphology. As both interfere with the productivity, rheology and downstream processing of industrial fermentation processes (see Chap. 7), a mechanistic framework is sought since a long time in order to describe and control the optimum macroscopic morphology under industrial conditions. A so far unsolved problem as too many process variables including type and concentration of carbon and nitrogen source, pH, temperature, fermenter geometry, agitation systems, culture mode [107] as well as biological parameters including conidial adhesion capacity [108] and branching frequency [109] affect the macroscopic morphology of filamentous fungi. With respect to the genetic basis of filamentous fungal morphology and its link to protein secretion, two hypotheses are currently under discussion: (i) the higher the branching frequency, the higher the probability that macroscopic pellets and not dispersed mycelia will be formed. Often, the tendency to form pellets under submerged conditions can be assessed during solid-state cultivations on agar plates—the colonies formed are more compact, have a reduced colony diameter and are sometimes delayed in sporulation (Fig. 5), (ii) the higher the number of hyphal tips, the more the proteins become secreted. The latter hypothesis has been addressed in several studies in A. niger and A. oryzae; however, only contradictory results have been reported. Some studies proposed a positive correlation between branching frequency and protein secretion yield; others did not [110–114]. It is therefore still a matter of debate which is the best macroscopic morphology of Aspergillus with respect to productivity and rheology during protein production.

Branching frequency determines macroscopic morphology of A. niger. Shown are young hyphae from a wild-type strain (wt) and from the conditional hyperbranching mutant ramosa-1 of the same age. In this mutant, a point mutation within the TORC2 component RmsA provokes excessive apical branching and increased septation under restrictive conditions, leading to drastically reduced colony growth rate on agar plates and the formation of a very compact colony [113]. Note that ramosa-1 is also impaired in sporulation—a defect, which is not a general feature of hyperbranching mutants
Hyphal branching is the basis for mycelial development. Whereas branches usually arise from basal regions (lateral branching), branch formation can also occur via tip splitting (apical branching). Tip splitting is thought to result from the accumulation of secretory vesicles at the tip because the capacity for vesicle fusion with the membrane has been exceeded. To accommodate the abnormal accumulation of the vesicles, the tip divides into two new branches [115]. What, then, are the underlying cellular events and signalling networks of (sub)apical branch formation? Several transcriptomics and functional genomics approaches undertaken in A. niger have shed some light on these processes. Central to these studies was controlled manipulation of the actin cytoskeleton, which resulted in defined mutants with a hyperbranching or apolar growth phenotype [109, 114, 116, 117]. The transcriptomic signatures of these actin (hyper- or de-) polarisation mutants uncovered that several regulatory and metabolic pathways likely participate in morphogenetic events of A. niger: phospholipid signalling, sphingolipid signalling, TORC2 signalling, calcium signalling and cell wall integrity signalling. These pathways supposedly induce different physiological changes during branch formation including changes in sterol metabolism, amino acid metabolism, ion transport (Na+, K+, Ca2+, Fe2+, Zn2+, Co2+) and protein trafficking [109, 114]. A key conclusion of these comparative transcriptomics analyses is the prediction that lipid molecules such as phosphatidic acid, diacylglycerol, phosphatidylinositol-4,5-bisphosphate, inositolphosphates and sphingosin-1-phosphate occupy a central position in the polar growth control of A. niger. Their function as secondary messengers has been proven for many model eukaryotes; however, almost nothing is known about their cellular roles in Aspergilli. Thus, only an integrated systems biology approach which combines transcriptomics data with lipidomics, proteomics and metabolomics data is inevitable to comprehensively inventory the morphogenetic network of A. niger.
These recent studies in A. niger also carved out further very interesting findings: First, the function of some molecular switches controlling actin polarisation (e.g. Rho GTPases) differs in A. niger and A. nidulans [117, 118], giving further evidence that members of the same genus employ alternative strategies to make a hypha and that caution is thus required when extrapolating findings of one (model) organism to another [119]. Second, simply increasing the number of hyphal tips does not per se result in increased protein secretion. If the amount of secretory vesicles remains unchanged, the vesicles become merely distributed to more tips, and the amount of vesicles per individual hyphal tip lowers accordingly [114]. Third, it is possible to generate hyperbranching mutants (e.g. by deleting the Rho GTPase gene racA) which form a more compact macroscopic morphology but pellets and display the same growth rate as the wild-type strain—thus representing ideal systems for future studies to investigate the relationship between intracellular protein secretion capacities and rheological properties under controlled conditions [114].
What more is to come? Although comparative genomics and transcriptomics are so far the dominant approaches studying the genetic basis of Aspergillus morphology, we can soon expect more insights into the translatome and phosphoproteome of Aspergilli during polarised growth. First reports have already been published which pave the way to a more holistic understanding of translational control mechanisms during protein secretion stress [120] and which will help to decode phosphorylation-mediated regulatory networks controlling fungal morphogenesis and secretion [121].
5.4 Moulding the Mould—Linking Aspergillus Morphology with Primary Metabolism
Although we are far away from comprehensively deciphering transcriptomic signatures of A. niger morphological mutants, it is apparent that they suggest a connection between polar growth control and primary metabolism. For example, the A. niger TORC2 complex is not only central for the polarisation of actin at the hyphal tip but also of vital importance for energy metabolism, viability and salt balance of A. niger [109, 116], the function of the Rho GTPase RacA lies not only in the control of actin polarisation but is also somehow linked to carbon and nitrogen source utilisation [114], the transcriptional repressor TupA links morphogenesis, development and nitrogen metabolism [122]. What might be the connection between these different processes?
Of course, cells do only grow if sufficient amount of nutrients are present in the medium; hence, the most obvious interdependence lies between the nutritional/energetic status of a cell which is the prerequisite for any kind of growth. Our current understanding of the main fungal signalling pathways balancing growth and nutritional status is based upon the paradigms established in the models S. cerevisiae and A. nidulans. From these systems, we know that the cAMP-protein kinase A and Snf1 signalling pathways are key for carbon sensing [123], whereas TORC1 signalling is important for nitrogen sensing [124]. However, a very recent publication on the regulation of dimorphism (i.e. the switch between a yeast-like apolar growth mode to a filamentous polar growth mode) in U. maydis has provided for the first time the evidence of a physical interaction of a high-affinity ammonium transporter (Ump2) with a Rho GTPase (Rho1) at the plasma membrane of a filamentous fungus. This interaction occurs under low-ammonium conditions and inhibits Rho1 from negatively regulating Rac1 (the homologue of the A. niger Rho GTPase RacA), thereby inducing the switch to filamentous growth [125]. This is a very compelling study which without any doubt clearly demonstrates that the nutritional status can also directly control polarised growth.
6 Integration and Analysis of Aspergillus Genomic Data
The fungal scientific community witnessed a revolution during the last two decades. Since the publication of the first fungal genome (S. cerevisiae) in 1996 [126], the number of publically available fungal genome sequences increased exponentially reaching more than 250 by 2014 [26, 127]. For the genus Aspergillus, the first sequencing projects started in the late 1990s leading to the publication of the first Aspergillus genome, namely A. nidulans in 2005 [128] followed by other milestone publications for A. oryzae [129], Aspergillus fumigatus [130] and A. niger [34]. As of 2014, the number of fully sequenced Aspergillus species reached 21, all being publically accessible via the database MycoCosm (http://jgi.doe.gov/fungi; [26]). Thanks to increased throughput, reduced cost and improved accuracy of next-generation sequencing technologies, there is no end of this trend within sight. A project with the ambitious title ‘1,000 Fungal Genomes’ is currently running at the US Department of Energy Joint Genome Institute to sequence all major branches of the entire fungal tree of life [26], an initiative which can only be successful if the fungal scientific community continues to jointly develop powerful Web-based and wet-lab tools and pipelines for gene annotation and gene function analyses [131].
6.1 Opportunities and Challenges of Computational Genomics for Aspergillus
The availability of different genome sequences constitutes a rich resource for comparative genomics that generates new knowledge linking genotypes with phenotypic traits. Such comparative genomic studies have provided insights into the genetic basis for citric acid or protein production of A. niger strains as discussed in Chap. 5 [27]. Other comparative genomic studies disclosed CAZymes relevant for lignocellulose degradation in A. nidulans, A. niger and A. oryzae [132] and the closely related white-rot and brown-rot basidiomycetes Ceriporiopsis subvermispora and Phanerochaete chrysosporium [133] or predicted secondary metabolite gene clusters in Aspergilli and plant pathogenic filamentous fungi [134, 135].
The wealth of sequence data will without doubt further significantly enrich the portfolio of proteins, enzymes and metabolites to become produced by industrial Aspergilli or other industrial filamentous fungal production hosts. However, the challenge for the next decade is definitely no longer in sequence generation but data interpretation, data integration and methods for the high-throughput functional characterisation of predicted genes [136]. In the near future, next-generation sequencing approaches will be important for the correction of structural gene annotations ‘just’ by sequencing fungal transcriptomes [95], in addition, microarray-assisted transcriptomics will continue to be a powerful technology for the study or prediction of gene function as recently shown for the accurate assignment of secondary metabolite gene clusters in A. nidulans [137].
A list of currently available databases dedicated to the analysis of fungal genomes with a focus on Aspergillus projects is summarised in Table 1. These resources structure sequence information, provide annotations at different levels and connect gene and protein predictions with functional genomics data. What is important to make these databases sustainable? An important issue here is data conformity, in particular for functional annotations, which should have been generated with the same annotation pipeline in order to provide meaningful results in comparative genomics studies [26]. User-friendly intuitive interfaces are required to extend the acceptance and frequent use of already existing resources. Clearly, conversion of data types, identifiers and formats is a formidable task for scientists with a limited computational background, which will of course limit the use of existing powerful resources. Besides, a major risk for genomic resources can be seen in funding reductions. Expiration of funded projects periods or the lack of long-term community support will result in diversified and redundant resources, which in turn makes data conformity difficult.
In order to move towards conceptual models describing and predicting the link between Aspergillus growth, morphology and product formation, the integration of multiple -omics data types is key. Examples of association studies linking transcript profiles with proteomics, metabolomics data and genome-scale models have been given in the chapters above, some of which also applied guilt-by-association and enrichment/overrepresentation analyses to identify co-expression and gene regulatory networks in A. niger and A. oryzae. Further examples can be found in [14, 109, 138–141] to name but a few.
Computational methods guiding protein redesign have also been successfully used to improve protein production levels in A. niger. The proof-of-concept study was recently provided for two enzymes by van den Berg and colleagues [142, 143], who applied a machine-learning approach to identify DNA and protein features which discriminate between low- and high-level secretion rates based on the data for over 2,600 homologous or heterologous proteins expressed in A. niger. Using this strategy, both proteins were redesigned according to the calculated features of high-level secreted proteins. Up to 45 mutations were introduced which eventually led to a 10-fold increase in extracellular enzyme concentrations. A very impressive work, which can certainly become transferred to other proteins or features as the underlying methodology is generic [143].
6.2 Opportunities and Challenges of Functional Genomics for Aspergillus
98 % of the A. niger and A. oryzae genes are uncharacterised [98], urgently calling for high-throughput experimental techniques to elucidate their function. The systematic inactivation of predicted open reading frames by gene deletion or gene disruption and/or their controlled overexpression are two complementing approaches to study the function of genes. A respective genome-wide deletion project has been initiated for the model fungus N. crassa [144] and knockout mutants have been developed for more than two-third of the 10,000 N. crassa genes [145]. The basis for this achievement was high-throughput production of gene deletion cassettes, high transformation rates via electroporation of conidia and levels of homologous recombination equal or higher than 90 % [145]. In 2009, an effort from within the Aspergillus community was initiated to generate deletion constructs for all A. nidulans genes as well. Very recently, gene knockout constructs for about 93 % of the 10,560 predicted A. nidulans genes were provided to the community [146]. To prove the utility if these constructs, gene deletion strains were established for 128 predicted protein kinases which uncovered a function for previously uncharacterised kinases in vesicle trafficking, polar growth, septation and secondary metabolism [146].
Similar community-wide efforts for genome-deletion projects for other Aspergilli will hopefully become initiated soon as they are one of the most essential and invaluable resources for comprehensively understanding the genetic basis of their lifestyle. In fact, we have all necessary tools in hand for the industrial Aspergillus strains: (i) fast molecular cloning methods including fusion PCR, bipartite, in vivo yeast recombination, Golden Gate and Gibson assembly (for review see [147]), (ii) a broad collection of suitable promoters, selection systems and transformation methods including protoplast-mediated transformation, electroporation and shock wave assisted transformation (for details see [1, 4, 148–153]), and (iii) recipient strains with most efficient homologous recombination frequencies [154–158], (iv) a promoter system with which the function of essential genes can be studied [24]. The availability of these tools has made it now possible to lift functional genomics approaches to the higher, i.e. high-throughput level.
7 Mathematical Modelling of Aspergillus Cultivations
7.1 What Good Are Models?
As in many other disciplines as well, mathematical modelling of Aspergillus cultivations offers a substantial benefit in various aspects. In the most ambitious approach, the huge amount of physiological data obtained could be potentially lumped together to draw a comprehensive picture of morphology elucidating microscopic morphology, colony/pellet growth and interaction with the environment. This would finally allow for a directed control of morphology. Today, this goal seems to be too ambitious not only for Aspergillus, but for all fungi. However, less challenging objectives can be and have to be tackled with mathematical models.
By the very nature of living cells, experimental data are obtained from a highly dynamical system where the behaviour of a cell does not only depend on the actual stimuli but what had happened to the cell in the past. On a rational basis, such data can only be interpreted and condensed in the context of a dynamical model. For fungal organisms, interpretation of physiological data is even more challenging. Besides the compartmentalisation of biological functions in distinct organelles, space- and time-dependent distributions of organelles, other compounds of the cytoplasm, and of stimuli in and around a mycelium occur (see Chap. 4). Models may help here in analysing biochemical data which depends on spatial distribution as well. For technical applications, morphology influences culture broth viscosity, productivity and downstream processing. With image analysis, data quantifying morphology from A. niger can be linked to biomass-independent rheological data see [159, 160]. In solid-state cultivation, aerial hyphae can give rise to a non-uniform oxygen supply or an increased pressure drop in packed-bed cultivations of A. oryzae or Rhizopus oligosporus [161]. In this area, mathematical models are needed to predict rheological and other process data from morphology. If this is possible, such rheological data could be estimated at a very early stage of a process development before performing image analysis. Liu et al. [162] propose an interesting approach for R. oryzae where they predict the probability of pellet formation with a multiple logistic regression model. This method could be transferred to Aspergillus. Details and recent approaches to control morphology and productivity in liquid cultures by the addition of inorganic microparticles, changes of the conidia adhesion capacity, spore concentration, osmolality, pH, mechanical stress, etc., with A. niger, A. terreus and other strains can be found in [107, 108, 163–165].
In addition to the interpretation and compression of biological data, models can be finally exploited in powerful and versatile methods for process design, optimisation, closed-loop control and supervision. In this respect, cultivation processes have not yet reached the same maturity as traditional chemical processes [166]. This huge area of model-based applications, although very important, will not be considered in this short paragraph as it would deserve a separate review (see [167, 168] for an introduction). Here, we rather focus on the challenges coming from an interpretation of physiological data in light of a complex morphology. To this end, some more general comments concerning modelling of mycelial organisms seem appropriate before we direct our attention to some recent developments and necessary future investigations. The references given in what follows mainly cover publications of the last couple of years. Morphological modelling of fungi, however, goes back, at least, to the year 1967 [169]. Excellent reviews of the period in between can be found, e.g. in [107, 170, 171] or in [172] for filamentous bacteria such as Streptomyces species. The cited newer articles, moreover, give credit to other important results from literature which cannot be repeated here for sake of brevity.
7.2 What Can be Modelled?
Mathematical modelling of the morphology of mycelial organisms can focus on scales ranging from nanometers to meters or more [173, 174]. A variety of mechanisms and effects can be elucidated by means of mathematical models. Examples, ordered by a growing length scale, could be the study of exo- and endocytosis for polar growth, of tropism, the onset of new branches, anastomosis, organelle distribution in hyphae, or growth of hyphae over long inert habitats by intrahyphal translocation of nutrients, to name just a few potential applications. Typically, processes involved on a biological level will depend on time- and space-dependent environmental factors as well. A mycelium growing on a solid substrate will deplete it after a while, leading, e.g. to autolysis. A submerged, pellet-forming organism such as A. niger will see in its pellet centres a reduced substrate concentration, especially with respect to oxygen, when the size reaches a certain level [159, 172, 175–177]. This may lead to autolysis, enhanced susceptibility to mechanical stress, or improved productivity due to favourable nutrient concentrations inside the pellet. Here, mathematical models have to describe the particle, i.e. pellet size distribution [175] to predict its effect on productivity [178], and, for example, on viscosity, and, hence, on important parameters such as heat and mass transfer coefficients (see, e.g. [163] and references therein).
All morphological details are the result of highly regulated biochemical reactions on a metabolic level. Due to polarised, i.e. apical growth, and apical or subapical branching, metabolic fluxes will be space-dependent. Additionally, in fungi, a coordinated interplay between various organelles inside a hypha has to be considered to determine, e.g. the timing and location of branch initiation. This is far beyond what has been tackled so far with mathematical models for Aspergillus or other fungi. Even a comprehensive mathematical model of a subset of this multi-scale problem is still out of reach although more and more biological details on a molecular level are discovered.
7.3 Which Model When?
Before continuing this discussion, a classification of models seems necessary. In contrast to unstructured models, structured approaches do not describe the biotic phase by just one variable but by a number of selected compounds, real or hypothetical compartments, morphological compartments, or by a detailed description, e.g. of metabolic fluxes in the realm of systems biology [13]. For fungal cultivations, as for actinomycetes, growth and production described by unstructured or structured models can be further refined accounting for the space-dependent nutrient situation inside large mycelia and pellets [172, 179], and, equally important, in non-ideally mixed large-scale fermenters [166, 180]. For fungi, rather very few attempts with respect to morphologically structured approaches have been done so far. An old example goes back to Megee et al. [181] who have built up a model for hyphae of A. awamori including active, inactive hyphae, tips and substrates. Based on it, refinements were formulated later from different researchers, for example for P. chrysogenum, Geotrichum candidum and the actinomycete S. hygroscopicus. Examples and more references are given in [182–185].
Micro- or macro-morphological models to describe growth, branching and geometrical appearance of small and large mycelia and of pellets, respectively, can be formulated in a continuous or discrete manner or a combination of both (see [174, 183] for an extended discussion). Continuous models go back to the work of Edelstein and co-workers [184] who combined discrete elements into a continuous description of the evolution of the biomass density by the introduction of a tip density and apical growth. This approach, leading to a set of partial differential equations, is especially suited for studying larger mycelia or pellets in homogenous environments. It was subsequently refined by different researchers [173, 174, 185, 186]. However, the inclusion of -omics data in this kind of continuous approach is hardly possible.
Discrete models, on the other hand, often start from predefined lattices along which growth can occur (lattice-based models) [161, 185], from vector-based approaches [187], or from a geometrically unconstrained description [175]. Discrete models can be readily extended to hybrid ones to include continuous descriptions of nutrient transport outside the mycelium. If stochastic elements are included, appealing images are obtained from simulations that are almost indistinguishable from real microscopic pictures. A first model including deterministic and stochastic parts was already formulated in the year 1992 by Yang et al. [188]. It showed a striking similarity to microscopic images after they had been processed by image analysis algorithms. The model was calibrated with data from S. tendae and G. candidum. Later, Lejeune and Baron used it as a basis for T. reesei [177]. More recent examples describing the geometric form from different fungi or streptomycetes can be found in [179, 183, 189], to name just a few. Typically, the rules and kinetics used in these models do not have to rely on detailed biological knowledge to still produce ’realistic’ images, i.e. they do not start from biological data from, e.g. A. niger. To give an example, the simulations shown in Fig. 6 just use the information that the apical growth rate in three dimensions is constant and that septa and branches are formed when a critical length of a hyphal compartment is obtained. Including a random growth direction in the simulation produces the results shown. Such basic model structures can now be used to include biological information to get more from the model than just a nice visual impression. Moreover, such models are needed to predict the macroscopic implications with respect to nutrient transport, rheology, etc. when, e.g. the branching rate is changed by a genetic manipulation [179].

2D projections of four simulated 3D mycelia using a very simple model. All mycelia start as a spore, grow with the same apical growth rate and only differ in age
A last group of models concentrates on individual hyphae to more accurately describe its growth rate or geometric form. Data from the -omics initiatives are missing here as well. A well-known example goes back to Bartnicki-Garcia et al. [190]. They describe the geometrical form of an apex of various fungi with a simple model. It is based on a set of hypotheses how vesicles are transported from the Spitzenkörper to the wall. The model has been refined later to better account for the three-dimensional shape of a tip and the way vesicles are transported to the wall (see, e.g. [72, 191–193]). Recently, a quantitative model to describe the growth of an apex was introduced by Caballero-Lima et al. [74]. They assume that enzyme carrying vesicles fuse with the plasma membrane at the apex proportional to the local exocyst concentration. Realistic shapes of a hypha, however, are only obtained when these enzymes, e.g. (1,3)-ß-glucan synthase, are removed from the plasma membrane by endocytosis at some distance from the apex. This is one of the very few models which are built upon well-established cell biological processes and are supported by quantitative measurements. For the explanation of another micro-morphological appearance, Sugden et al. [194] argue that the long-range transport of material in hyphae is important for length growth. They explain it by a particle transport along a single, hypothetical microtubule extending over the whole length of a hypha. Monte Carlo simulation reveals an interesting result showing a raising concentration of the particles towards the tip as it is observed by laser scanning microscopy from other groups for organelles in fungi. In contrast, [195] explains length growth of Phanerochaete velutina mathematically by a turgor driven intrahyphal flow towards the tip. The model accounts for increasing radii of hyphae when the flow increases which can be shown experimentally.
7.4 What More Is to Come?
As in these last examples, morphological models are mostly based on a very simplified description of the underlying biochemical processes. It is our believe that such unstructured or phenomenologically structured models of small size describing individual hyphae, mycelia or pellets have already reached a high level of maturity. One of the main challenges for future work, in contrast, lies in the exploitation and explanation of Big Data information in view of space-dependent processes in hyphae leading to specific morphologies. A gap exists today between what is known and what can be used in mathematical models. When we proposed the first ‘mechanistic’ model more than 20 years ago [188], we had to assume hypothetical compounds in a hypha to describe an initial exponential and then constant growth rate of an individual branch as biological details for this process were unknown. Today, for fungi and for actinomycetes, a whelm of data is available. Interpreting this molecular and cell biological data on a mechanistic level and combining it with a morphological description seems to be a promising, though intricate route to follow. It is understood, however, that such a detailed description, for example on an organelle-basis, cannot be used to describe growth of larger mycelia and address process control issues. Therefore, as a next step, detailed models have to be reduced again. This reduction, however, will be based on more detailed, biologically better founded models. In the long run, such an approach might help to replace heuristic rules exploited today to model morphology by more physics- or bio-based expressions and close the circle towards a rational control of morphology.
8 Conclusions
The Aspergillus Big Data era creates entirely new opportunities and challenges. In order to make industrial Aspergillus strains completely transparent and productive for us, we need to address the challenges on different levels: (i) the Big Data analysis pipeline—data acquisition, extraction, integration, modelling and interpretation [12]—has to be approached entirely not only with a focus on data acquisition. We should not leave -omics data under-analysed but should constantly re-analyse and re-interpret them in view of ‘old’ and newly incoming -omics data from database repositories. Most importantly, data noisiness and heterogeneity of Aspergillus populations have to be taken into account when analysing these data. Likewise, co-regulation analyses are key to identify sub networks, to map transcriptional networks or to identify connections between signalling pathways. (ii) -Omics data have to be combined with in vivo life imaging studies to link time with space. In addition, functional genomics studies and ‘good old’ enzyme assays are key to ascribe functions to predicted proteins and enzymes. Furthermore, we should not lose the use of electromobility shift assays out of sight, to study and prove binding of predicted transcription factors to predicted promoter binding sites. (iii) And finally, there is a clear bottleneck in the number of fungal scientists working on Aspergillus and empowered to ask questions and finding the right experiments to answer them. We thus need to get many more students—biologist, bioinformaticians and engineers—interested and educated in fungal research. What we can offer? Exciting times for gold diggers!
References
Meyer V (2008) Genetic engineering of filamentous fungi–progress, obstacles and future trends. Biotechnol Adv 26:177–185
Lubertozzi D, Keasling JD (2009) Developing Aspergillus as a host for heterologous expression. Biotechnol Adv 27:53–75
Ward OP (2011) Production of recombinant proteins by filamentous fungi. Biotechnol Adv 30:1119–1139
Fiedler MRM, Nitsche BM, Franziska W, Meyer V (2013) Aspergillus: a cell factory with unlimited prospects. In: Gupta VK, Schmoll M, Maki M (eds) Appl Microb Eng. CRC Press, Taylor & Francis Group, Boca Raton, London, pp 1–51
Read ND (2011) Exocytosis and growth do not occur only at hyphal tips. Mol Microbiol 81:4–7
Hayakawa Y, Ishikawa E, Shoji J-Y, Nakano H, Kitamoto K (2011) Septum-directed secretion in the filamentous fungus Aspergillus oryzae. Mol Microbiol 81:40–55
Seidl V, Seibel C, Kubicek CP, Schmoll M (2009) Sexual development in the industrial workhorse Trichoderma reesei. Proc Natl Acad Sci USA 106:13909–13914
Böhm J, Hoff B, O’Gorman CM, Wolfers S, Klix V, Binger D, Zadra I, Kürnsteiner H, Pöggeler S, Dyer PS, Kück U (2013) Sexual reproduction and mating-type-mediated strain development in the penicillin-producing fungus Penicillium chrysogenum. Proc Natl Acad Sci USA 110:1476–1481
Richter L, Wanka F, Boecker S, Storm D, Kurt T, Vural Ö, Süßmuth R, Meyer V (2014) Engineering of Aspergillus niger for the production of secondary metabolites. Fungal Biol Biotechnol 1:4
Li A, van Luijk N, ter Beek M, Caspers M, Punt P, van der Werf M (2011) A clone-based transcriptomics approach for the identification of genes relevant for itaconic acid production in Aspergillus. Fungal Genet Biol 48:602–611
Van der Straat L, Vernooij M, Lammers M, van den Berg W, Schonewille T, Cordewener J, van der Meer I, Koops A, de Graaff LH (2014) Expression of the Aspergillus terreus itaconic acid biosynthesis cluster in Aspergillus niger. Microb Cell Fact 13:11
Challenges and opportunities with big data challenges and opportunities with big data. http://www.cra.org/ccc/files/docs/init/bigdatawhitepaper.pdf
Caspeta L, Nielsen J (2013) Toward systems metabolic engineering of Aspergillus and Pichia species for the production of chemicals and biofuels. Biotechnol J 8:534–544
Nitsche BM, Meyer V (2014) Transcriptomics of industrial filamentous fungi: a new view on regulation, physiology, and application. In: Nowrousian M (ed) Fungal genomics, vol 13. Berlin, Springer, pp 209–232
Statins: a success story involving FDA, academia and industry. http://www.fda.gov/AboutFDA/WhatWeDo/History/ProductRegulation/SelectionsFromFDLIUpdateSeriesonFDAHistory/ucm082054.htm
Gusakov AV (2011) Alternatives to Trichoderma reesei in biofuel production. Trends Biotechnol 29:419–425
Pensupa N, Jin M, Kokolski M, Archer DB, Du C (2013) A solid state fungal fermentation-based strategy for the hydrolysis of wheat straw. Bioresour Technol 149:261–267
Kim S, Dale BE (2004) Global potential bioethanol production from wasted crops and crop residues. Biomass Bioenerg 26:361–375
Kolasa M, Ahring BK, Lübeck PS, Lübeck M (2014) Co-cultivation of Trichoderma reesei RutC30 with three black Aspergillus strains facilitates efficient hydrolysis of pretreated wheat straw and shows promises for on-site enzyme production. Bioresour Technol 169:143–148
Yamada R, Yoshie T, Wakai S, Asai-Nakashima N, Okazaki F, Ogino C, Hisada H, Tsutsumi H, Hata Y, Kondo A (2014) Aspergillus oryzae-based cell factory for direct kojic acid production from cellulose. Microb Cell Fact 13:71
Klement T, Büchs J (2013) Itaconic acid–a biotechnological process in change. Bioresour Technol 135:422–431
Li A, Caspers M, Punt P (2013) A systems biology approach for the identification of target genes for the improvement of itaconic acid production in Aspergillus species. BMC Res Notes 6:505
Van der Straat L, de Graaff LH (2014) Pathway transfer in fungi: transporters are the key to success. Bioengineered 5
Meyer V, Wanka F, van Gent J, Arentshorst M, van den Hondel CAMJJ, Ram AFJ (2011) Fungal gene expression on demand: an inducible, tunable, and metabolism-independent expression system for Aspergillus niger. Appl Environ Microbiol 77:2975–2983
Meyer V, Wu B, Ram AFJ (2011) Aspergillus as a multi-purpose cell factory: current status and perspectives. Biotechnol Lett 33:469–476
Grigoriev I V, Nikitin R, Haridas S, Kuo A, Ohm R, Otillar R, Riley R, Salamov A, Zhao X, Korzeniewski F, Smirnova T, Nordberg H, Dubchak I, Shabalov I (2014) MycoCosm portal: gearing up for 1000 fungal genomes. Nucleic Acids Res 42(Database issue):D699–D704
Andersen MR, Salazar MP, Schaap PJ, van de Vondervoort PJI, Culley D, Thykaer J, Frisvad JC, Nielsen KF, Albang R, Albermann K, Berka RM, Braus GH, Braus-Stromeyer SA, Corrochano LM, Dai Z, van Dijck PWM, Hofmann G, Lasure LL, Magnuson JK, Menke H, Meijer M, Meijer SL, Nielsen JB, Nielsen ML, van Ooyen AJJ, Pel HJ, Poulsen L, Samson RA, Stam H, Tsang A et al (2011) Comparative genomics of citric-acid-producing Aspergillus niger ATCC 1015 versus enzyme-producing CBS 513.88. Genome Res 21:885–897
Jørgensen TR, Nielsen KF, Arentshorst M, Park J, van den Hondel C a, Frisvad JC, Ram AF (2011) Submerged conidiation and product formation by Aspergillus niger at low specific growth rates are affected in aerial developmental mutants. Appl Environ Microbiol 77:5270–5277
Delmas S, Pullan ST, Gaddipati S, Kokolski M, Malla S, Blythe MJ, Ibbett R, Campbell M, Liddell S, Aboobaker A, Tucker GA, Archer DB (2012) Uncovering the genome-wide transcriptional responses of the filamentous fungus Aspergillus niger to lignocellulose using RNA sequencing PLoS Genet 8:e1002875
Hayer K, Stratford M, Archer DB (2014) Germination of Aspergillus niger conidia is triggered by nitrogen compounds related to L-amino acids. Appl Environ Microbiol 80:6046–6053
Hayer K, Stratford M, Archer DB (2013) Structural features of sugars that trigger or support conidial germination in the filamentous fungus Aspergillus niger. Appl Environ Microbiol 79:6924–6931
Novodvorska M, Hayer K, Pullan ST, Wilson R, Blythe MJ, Stam H, Stratford M, Archer DB (2013) Trancriptional landscape of Aspergillus niger at breaking of conidial dormancy revealed by RNA-sequencing. BMC Genomics 14:246
Van Leeuwen MR, Krijgsheld P, Bleichrodt R, Menke H, Stam H, Stark J, Wösten HAB, Dijksterhuis J (2013) Germination of conidia of Aspergillus niger is accompanied by major changes in RNA profiles. Stud Mycol 74:59–70
Pel HJ, de Winde JH, Archer DB, Dyer PS, Hofmann G, Schaap PJ, Turner G, de Vries RP, Albang R, Albermann K, Andersen MR, Bendtsen JD, Benen JAE, van den Berg M, Breestraat S, Caddick MX, Contreras R, Cornell M, Coutinho PM, Danchin EGJ, Debets AJM, Dekker P, van Dijck PWM, van Dijk A, Dijkhuizen L, Driessen AJM, D’Enfert C, Geysens S, Goosen C, Groot GSP et al (2007) Genome sequencing and analysis of the versatile cell factory Aspergillus niger CBS 513.88. Nat Biotechnol 25:221–231
Jedd G, Pieuchot L (2012) Multiple modes for gatekeeping at fungal cell-to-cell channels. Mol Microbiol 86:1291–1294
Harris SD, Turner G, Meyer V, Espeso EA, Specht T, Takeshita N, Helmstedt K (2009) Morphology and development in Aspergillus nidulans: a complex puzzle. Fungal Genet Biol 46(Suppl 1):S82–S92
Levin AM, de Vries RP, Conesa A, de Bekker C, Talon M, Menke HH, van Peij NNME, Wösten HAB (2007) Spatial differentiation in the vegetative mycelium of Aspergillus niger. Eukaryot Cell 6:2311–2322
Krijgsheld P, Altelaar AFM, Post H, Ringrose JH, Müller WH, Heck AJR, Wösten HAB (2012) Spatially resolving the secretome within the mycelium of the cell factory Aspergillus niger. J Proteome Res 11:2807–2818
Gordon CL, Khalaj V, Ram a F, Archer DB, Brookman JL, Trinci a P, Jeenes DJ, Doonan JH, Wells B, Punt PJ, van den Hondel CA, Robson GD (2000) Glucoamylase::green fluorescent protein fusions to monitor protein secretion in Aspergillus niger. Microbiology 146(2):415–426
Vinck A, de Bekker C, Ossin A, Ohm RA, de Vries RP, Wösten HAB (2011) Heterogenic expression of genes encoding secreted proteins at the periphery of Aspergillus niger colonies. Environ Microbiol 13:216–225
De Bekker C, van Veluw GJ, Vinck A, Wiebenga LA, Wösten HAB (2011) Heterogeneity of Aspergillus niger microcolonies in liquid shaken cultures. Appl Environ Microbiol 77:1263–1267
Van Veluw GJ, Teertstra WR, de Bekker C, Vinck A, van Beek N, Muller WH, Arentshorst M, van der Mei HC, Ram AFJ, Dijksterhuis J, Wösten HAB (2013) Heterogeneity in liquid shaken cultures of Aspergillus niger inoculated with melanised conidia or conidia of pigmentation mutants. Stud Mycol 74:47–57
Wösten HAB, van Veluw GJ, de Bekker C, Krijgsheld P (2013) Heterogeneity in the mycelium: implications for the use of fungi as cell factories. Biotechnol Lett 35:1155–1164
Nitsche BM, Jørgensen TR, Akeroyd M, Meyer V, Ram AFJ (2012) The carbon starvation response of Aspergillus niger during submerged cultivation: insights from the transcriptome and secretome. BMC Genomics 13:380
Nitsche BM, Burggraaf-van Welzen AM, Lamers G, Meyer V, Ram AFJ (2013) Autophagy promotes survival in aging submerged cultures of the filamentous fungus Aspergillus niger. Appl Microbiol Biotechnol 97:8205–8218
Shoji J, Craven KD (2011) Autophagy in basal hyphal compartments: A green strategy of great recyclers. Fungal Biol Rev 25:79–83
Adams TH, Wieser JK, Yu JH (1998) Asexual sporulation in Aspergillus nidulans. Microbiol Mol Biol Rev 62:35–54
Krijgsheld P, Bleichrodt R, van Veluw GJ, Wang F, Müller WH, Dijksterhuis J, Wösten HAB (2013) Development in Aspergillus. Stud Mycol 74:1–29
Harris SD (2012) Evolution of modular conidiophore development in the aspergilli. Ann N Y Acad Sci 1273:1–6
Jørgensen TR, Nitsche BM, Lamers GE, Arentshorst M, van den Hondel CA, Ram AF (2010) Transcriptomic insights into the physiology of Aspergillus niger approaching a specific growth rate of zero. Appl Environ Microbiol 76:5344–5355
Metz B, Seidl-Seiboth V, Haarmann T, Kopchinskiy A, Lorenz P, Seiboth B, Kubicek CP (2011) Expression of biomass-degrading enzymes is a major event during conidium development in Trichoderma reesei. Eukaryot Cell 10:1527–1535
Van Munster JM, Nitsche BM, Krijgsheld P, van Wijk A, Dijkhuizen L, Wösten HA, Ram AF, van der Maarel MJEC (2013) Chitinases CtcB and CfcI modify the cell wall in sporulating aerial mycelium of Aspergillus niger. Microbiology 159(9):1853–1867
Brakhage AA, Schroeckh V (2011) Fungal secondary metabolites—strategies to activate silent gene clusters. Fungal Genet Biol 48:15–22
Brakhage AA (2013) Regulation of fungal secondary metabolism. Nat Rev Microbiol 11:21–32
Wiemann P, Keller NP (2014) Strategies for mining fungal natural products. J Ind Microbiol Biotechnol 41:301–313
Van Munster JM, Daly P, Delmas S, Pullan ST, Blythe MJ, Malla S, Kokolski M, Noltorp ECM, Wennberg K, Fetherston R, Beniston R, Yu X, Dupree P, Archer DB (2014) The role of carbon starvation in the induction of enzymes that degrade plant-derived carbohydrates in Aspergillus niger. Fungal Genet Biol 72:34–47
Andersen MR, Giese M, de Vries RP, Nielsen J (2012) Mapping the polysaccaride degradation potential of Aspergillus niger. BMC Genomics 13:313
De Souza WR, de Gouvea PF, Savoldi M, Malavazi I, de Souza Bernardes LA, Goldman MHS, de Vries RP, de Castro Oliveira J V, Goldman GH (2011) Transcriptome analysis of Aspergillus niger grown on sugarcane bagasse. Biotechnol Biofuels 4:40
Banuett F, Herskowitz I (1989) Different a alleles of Ustilago maydis are necessary for maintenance of filamentous growth but not for meiosis. Proc Natl Acad Sci USA 86:5878–5882
Tey WK, North AJ, Reyes JL, Lu YF, Jedd G (2005) Polarized gene expression determines woronin body formation at the leading edge of the fungal colony. Mol Biol Cell 16:2651–2659
Levin AM, de Vries RP, Conesa A, de Bekker C, Talon M, Menke HH, van Peij NNME, Wösten HAB (2007) Spatial differentiation in the vegetative mycelium of Aspergillus niger. Eukaryot Cell 6:2311–2322
Bleichrodt R-J, van Veluw GJ, Recter B, Maruyama J-I, Kitamoto K, Wösten HAB (2012) Hyphal heterogeneity in Aspergillus oryzae is the result of dynamic closure of septa by Woronin bodies. Mol Microbiol 86:1334–1344
Taheri-Talesh N, Horio T, Araujo-baza L, Dou X, Espeso EA, Pen MA, Osmani SA, Oakley BR (2008) The tip growth apparatus of Aspergillus nidulans. Mol Biol Cell 19:1439–1449
Lew RR (2011) How does a hypha grow? The biophysics of pressurized growth in fungi. Nat Rev Microbiol 9:509–518
Plamann M (2009) Cytoplasmic streaming in Neurospora: disperse the plug to increase the flow? PLoS Genet 5:e1000526
Abadeh A, Lew RR (2013) Mass flow and velocity profiles in Neurospora hyphae: partial plug flow dominates intra-hyphal transport. Microbiology 159(11):2386–2394
Riquelme M (2013) Tip growth in filamentous fungi: a road trip to the apex. Annu Rev Microbiol 67:587–609
Takeshita N, Manck R, Grün N, de Vega SH, Fischer R (2014) Interdependence of the actin and the microtubule cytoskeleton during fungal growth. Curr Opin Microbiol 20C:34–41
Riquelme M, Sánchez-León E (2014) The Spitzenkörper: a choreographer of fungal growth and morphogenesis. Curr Opin Microbiol 20C:27–33
Free SJ (2013) Fungal cell wall organization and biosynthesis. Adv Genet 81:33–82
Hettema EH, Erdmann R, van der Klei I, Veenhuis M (2014) Evolving models for peroxisome biogenesis. Curr Opin Cell Biol 29C:25–30
Gierz G, Bartnicki-Garcia S (2001) A three-dimensional model of fungal morphogenesis based on the vesicle supply center concept. J Theor Biol 208:151–164
Bartnicki-Garcia S, Bracker CE, Gierz G, López-Franco R, Lu H (2000) Mapping the growth of fungal hyphae: orthogonal cell wall expansion during tip growth and the role of turgor. Biophys J 79:2382–2390
Caballero-Lima D, Kaneva IN, Watton SP, Sudbery PE, Craven CJ (2013) The spatial distribution of the exocyst and actin cortical patches is sufficient to organize hyphal tip growth. Eukaryot Cell 12:998–1008
Dijksterhuis J, Molenaar D (2013) Vesicle trafficking via the Spitzenkörper during hyphal tip growth in Rhizoctonia solani. Antonie Van Leeuwenhoek 103:921–931
Kwon MJ, Arentshorst M, Fiedler M, de Groen FLM, Punt PJ, Meyer V, Ram AFJ (2014) Molecular genetic analysis of vesicular transport in Aspergillus niger reveals partial conservation of the molecular mechanism of exocytosis in fungi. Microbiology 160(2):316–329
Higuchi Y, Ashwin P, Roger Y, Steinberg G (2014) Early endosome motility spatially organizes polysome distribution. J Cell Biol 204:343–357
Steinberg G (2014) Endocytosis and early endosome motility in filamentous fungi. Curr Opin Microbiol 20C:10–18
Sudbery P (2011) Fluorescent proteins illuminate the structure and function of the hyphal tip apparatus. Fungal Genet Biol 48:849–857
Durand H, Clanet M, Tiraby G (1988) Genetic improvement of Trichoderma reesei for large scale cellulase production. Enzyme Microb Technol 10:341–346
Finkelstein DB (1987) Improvement of enzyme production in Aspergillus. Antonie Van Leeuwenhoek 53:349–352
Su X, Schmitz G, Zhang M, Mackie RI, Cann IKO (2012) Heterologous gene expression in filamentous fungi. Adv Appl Microbiol 81:1–61
Pakula TM, Laxell M, Huuskonen A, Uusitalo J, Saloheimo M, Penttilä M (2003) The effects of drugs inhibiting protein secretion in the filamentous fungus Trichoderma reesei. Evidence for down-regulation of genes that encode secreted proteins in the stressed cells. J Biol Chem 278:45011–45020
Kwon MJ, Jørgensen TR, Nitsche BM, Arentshorst M, Park J, Ram AFJ, Meyer V (2012) The transcriptomic fingerprint of glucoamylase over-expression in Aspergillus niger. BMC Genomics 13:701
Van den Berg BA, Reinders MJT, Hulsman M, Wu L, Pel HJ, Roubos JA, de Ridder D (2012) Exploring sequence characteristics related to high-level production of secreted proteins in Aspergillus niger. PLoS One 7:e45869
Shoji J-Y, Kikuma T, Kitamoto K (2014) Vesicle trafficking, organelle functions, and unconventional secretion in fungal physiology and pathogenicity. Curr Opin Microbiol 20C:1–9
Le Bihan M-C, Bigot A, Jensen SS, Dennis JL, Rogowska-Wrzesinska A, Lainé J, Gache V, Furling D, Jensen ON, Voit T, Mouly V, Coulton GR, Butler-Browne G (2012) In-depth analysis of the secretome identifies three major independent secretory pathways in differentiating human myoblasts. J Proteomics 77:344–356
Steringer JP, Müller H-M, Nickel W (2014) Unconventional secretion of fibroblast growth factor 2-a novel type of protein translocation across membranes? J Mol Biol. doi: 10.1016/j.jmb.2014.07.012
Ding Y, Robinson DG, Jiang L (2014) Unconventional protein secretion (UPS) pathways in plants. Curr Opin Cell Biol 29:107–115
De Marchis F, Bellucci M, Pompa A (2013) Unconventional pathways of secretory plant proteins from the endoplasmic reticulum to the vacuole bypassing the Golgi complex. Plant Signal Behav 8:piie25129
Fajardo-Somera RA, Bowman B, Riquelme M (2013) The plasma membrane proton pump PMA-1 is incorporated into distal parts of the hyphae independently of the Spitzenkörper in Neurospora crassa. Eukaryot Cell 12:1097–1105
Riquelme M, Bartnicki-García S, González-Prieto JM, Sánchez-León E, Verdín-Ramos JA, Beltrán-Aguilar A, Freitag M (2007) Spitzenkorper localization and intracellular traffic of green fluorescent protein-labeled CHS-3 and CHS-6 chitin synthases in living hyphae of Neurospora crassa. Eukaryot Cell 6:1853–1864
Stock J, Sarkari P, Kreibich S, Brefort T, Feldbrügge M, Schipper K (2012) Applying unconventional secretion of the endochitinase Cts1 to export heterologous proteins in Ustilago maydis. J Biotechnol 161:80–91
Arnaud MB, Cerqueira GC, Inglis DO, Skrzypek MS, Binkley J, Chibucos MC, Crabtree J, Howarth C, Orvis J, Shah P, Wymore F, Binkley G, Miyasato SR, Simison M, Sherlock G, Wortman JR (2012) The Aspergillus Genome Database (AspGD): recent developments in comprehensive multispecies curation, comparative genomics and community resources. Nucleic Acids Res 40(Database issue):D653–9
Cerqueira GC, Arnaud MB, Inglis DO, Skrzypek MS, Binkley G, Simison M, Miyasato SR, Binkley J, Orvis J, Shah P, Wymore F, Sherlock G, Wortman JR (2014) The Aspergillus Genome database: multispecies curation and incorporation of RNA-Seq data to improve structural gene annotations. Nucleic Acids Res 42(Database issue):D705–10
Andersen MR, Nielsen ML, Nielsen J (2008) Metabolic model integration of the bibliome, genome, metabolome and reactome of Aspergillus niger. Mol Syst Biol 4:178
Vongsangnak W, Olsen P, Hansen K, Krogsgaard S, Nielsen J (2008) Improved annotation through genome-scale metabolic modeling of Aspergillus oryzae. BMC Genomics 9:245
Andersen MR (2014) Elucidation of primary metabolic pathways in Aspergillus species: Orphaned research in characterizing orphan genes. Brief Funct Genomics 13:451–455
Battaglia E, Visser L, Nijssen A, van Veluw GJ, Wösten HAB, de Vries RP (2011) Analysis of regulation of pentose utilisation in Aspergillus niger reveals evolutionary adaptations in Eurotiales. Stud Mycol 69:31–38
Liu L, Feizi A, Österlund T, Hjort C, Nielsen J (2014) Genome-scale analysis of the high-efficient protein secretion system of Aspergillus oryzae. BMC Syst Biol 8:73
Choi J, Park J, Kim D, Jung K, Kang S, Lee Y-H (2010) Fungal secretome database: integrated platform for annotation of fungal secretomes. BMC Genomics 11:105
Sanchez JF, Somoza AD, Keller NP, Wang CCC (2012) Advances in Aspergillus secondary metabolite research in the post-genomic era. Nat Prod Rep 29:351–371
Peñalva MA, Galindo A, Abenza JF, Pinar M, Calcagno-Pizarelli AM, Arst HN, Pantazopoulou A (2012) Searching for gold beyond mitosis: Mining intracellular membrane traffic in Aspergillus nidulans. Cell Logist 2:2–14
Juvvadi PR, Lamoth F, Steinbach WJ (2014) Calcineurin-mediated regulation of hyphal growth, septation, and virulence in Aspergillus fumigatus. Mycopathologia 178:341–348
Martín JF (2014) Calcium-containing phosphopeptides pave the secretory pathway for efficient protein traffic and secretion in fungi. Microb Cell Fact 13:117
Malavazi I, Goldman GH, Brown NA (2014) The importance of connections between the cell wall integrity pathway and the unfolded protein response in filamentous fungi. Brief Funct Genomics 13:456–470
Papagianni M (2004) Fungal morphology and metabolite production in submerged mycelial processes. Biotechnol Adv 22:189–259
Colin VL, Baigorí MD, Pera LM (2013) Tailoring fungal morphology of Aspergillus niger MYA 135 by altering the hyphal morphology and the conidia adhesion capacity: biotechnological applications. AMB Express 3:27
Meyer V, Arentshorst M, Flitter SJ, Nitsche BM, Kwon MJ, Reynaga-Peña CG, Bartnicki-Garcia S, van den Hondel C a MJJ, Ram AFJ (2009) Reconstruction of signaling networks regulating fungal morphogenesis by transcriptomics. Eukaryot Cell 8:1677–1691
McIntyre M, Müller C, Dynesen J, Nielsen J (2001) Metabolic engineering of the morphology of Aspergillus. Adv Biochem Eng Biotechnol 73:103–128
Muller C, McIntyre M, Hansen K, Nielsen J (2002) Metabolic Engineering of the Morphology of Aspergillus oryzae by altering chitin synthesis. Appl Environ Microbiol 68:1827–1836
Te Biesebeke R, Record E, van Biezen N, Heerikhuisen M, Franken A, Punt PJ, van den Hondel CAMJJ (2005) Branching mutants of Aspergillus oryzae with improved amylase and protease production on solid substrates. Appl Microbiol Biotechnol 69:44–50
Wongwicharn A, McNeil B, Harvey LM (1999) Effect of oxygen enrichment on morphology, growth, and heterologous protein production in chemostat cultures of Aspergillus niger B1-D. Biotechnol Bioeng 65:416–424
Kwon MJ, Nitsche BM, Arentshorst M, Jørgensen TR, Ram AFJ, Meyer V (2013) The transcriptomic signature of RacA activation and inactivation provides new insights into the morphogenetic network of Aspergillus niger. PLoS One 8:e68946
Harris SD (2008) Branching of fungal hyphae: regulation, mechanisms and comparison with other branching systems. Mycologia 100:823–832
Meyer V, Minkwitz S, Schütze T, Hondel CAMJJ Van Den, Ram AFJ (2010) The Aspergillus niger RmsA protein. A node in a genetic network? Commun Integr Biol 3:195–197
Kwon MJ, Arentshorst M, Roos ED, van den Hondel C a MJJ, Meyer V, Ram AFJ (2011) Functional characterization of Rho GTPases in Aspergillus niger uncovers conserved and diverged roles of Rho proteins within filamentous fungi. Mol Microbiol 79:1151–1167
Harris SD (2011) Cdc42/Rho GTPases in fungi: variations on a common theme. Mol Microbiol 79:1123–1127
Scazzocchio C (2014) Fungal biology in the post-genomic era. Fungal Biol Biotechnol 2014:1
Krishnan K, Ren Z, Losada L, Nierman WC, Lu LJ, Askew DS (2014) Polysome profiling reveals broad translatome remodeling during endoplasmic reticulum (ER) stress in the pathogenic fungus Aspergillus fumigatus. BMC Genomics 15:159
Ramsubramaniam N, Harris SD, Marten MR (2014) The phosphoproteome of Aspergillus nidulans reveals functional association with cellular processes involved in morphology and secretion. Proteomics 14:2454–2459
Schachtschabel D, Arentshorst M, Nitsche BM, Morris S, Nielsen KF, van den Hondel CAMJJ, Klis FM, Ram AFJ (2013) The transcriptional repressor TupA in Aspergillus niger is involved in controlling gene expression related to cell wall biosynthesis, development, and nitrogen source availability. PLoS One 8:e78102
Brown NA, Ries LNA, Goldman GH (2014) How nutritional status signalling coordinates metabolism and lignocellulolytic enzyme secretion. Fungal Genet Biol 72:48–63
Conrad M, Schothorst J, Kankipati HN, Van Zeebroeck G, Rubio-Texeira M, Thevelein JM (2014) Nutrient sensing and signaling in the yeast Saccharomyces cerevisiae. FEMS Microbiol Rev 38:254–299
Paul JA, Barati MT, Cooper M, Perlin MH (2014) Physical and genetic interaction between ammonium transporters and the signaling protein, Rho1, in the plant pathogen Ustilago maydis. Eukaryot Cell
Goffeau A, Barrell BG, Bussey H, Davis RW, Dujon B, Feldmann H, Galibert F, Hoheisel JD, Jacq C, Johnston M, Louis EJ, Mewes HW, Murakami Y, Philippsen P, Tettelin H, Oliver SG (1996) Life with 6000 genes. Science 274(546):563–567
Aguilar-Pontes MV, de Vries RP, Zhou M (2014) (Post-)Genomics approaches in fungal research. Brief Funct Genomics 72:73–81
Galagan JE, Calvo SE, Cuomo C, Ma L-J, Wortman JR, Batzoglou S, Lee S-I, Baştürkmen M, Spevak CC, Clutterbuck J, Kapitonov V, Jurka J, Scazzocchio C, Farman M, Butler J, Purcell S, Harris S, Braus GH, Draht O, Busch S, D’Enfert C, Bouchier C, Goldman GH, Bell-Pedersen D, Griffiths-Jones S, Doonan JH, Yu J, Vienken K, Pain A, Freitag M et al (2005) Sequencing of Aspergillus nidulans and comparative analysis with A. fumigatus and A. oryzae. Nature 438:1105–1115
Machida M, Asai K, Sano M, Tanaka T, Kumagai T, Terai G, Kusumoto K-I, Arima T, Akita O, Kashiwagi Y, Abe K, Gomi K, Horiuchi H, Kitamoto K, Kobayashi T, Takeuchi M, Denning DW, Galagan JE, Nierman WC, Yu J, Archer DB, Bennett JW, Bhatnagar D, Cleveland TE, Fedorova ND, Gotoh O, Horikawa H, Hosoyama A, Ichinomiya M, Igarashi R et al (2005) Genome sequencing and analysis of Aspergillus oryzae. Nature 438:1157–1161
Nierman WC, Pain A, Anderson MJ, Wortman JR, Kim HS, Arroyo J, Berriman M, Abe K, Archer DB, Bermejo C, Bennett J, Bowyer P, Chen D, Collins M, Coulsen R, Davies R, Dyer PS, Farman M, Fedorova N, Fedorova N, Feldblyum TV, Fischer R, Fosker N, Fraser A, García JL, García MJ, Goble A, Goldman GH, Gomi K, Griffith-Jones S et al (2005) Genomic sequence of the pathogenic and allergenic filamentous fungus Aspergillus fumigatus. Nature 438:1151–1156
Hibbett DS, Stajich JE, Spatafora JW (2013) Toward genome-enabled mycology. Mycologia 105:1339–1349
Coutinho PM, Andersen MR, Kolenova K, vanKuyk PA, Benoit I, Gruben BS, Trejo-Aguilar B, Visser H, van Solingen P, Pakula T, Seiboth B, Battaglia E, Aguilar-Osorio G, de Jong JF, Ohm RA, Aguilar M, Henrissat B, Nielsen J, Stålbrand H, de Vries RP (2009) Post-genomic insights into the plant polysaccharide degradation potential of Aspergillus nidulans and comparison to Aspergillus niger and Aspergillus oryzae. Fungal Genet Biol 46(1):S161–S169
Fernandez-Fueyo E, Ruiz-Dueñas FJ, Ferreira P, Floudas D, Hibbett DS, Canessa P, Larrondo LF, James TY, Seelenfreund D, Lobos S, Polanco R, Tello M, Honda Y, Watanabe T, Watanabe T, Ryu JS, San RJ, Kubicek CP, Schmoll M, Gaskell J, Hammel KE, St John FJ, Vanden Wymelenberg A, Sabat G, Splinter BonDurant S, Syed K, Yadav JS, Doddapaneni H, Subramanian V, Lavín JL, et al (2012) Comparative genomics of Ceriporiopsis subvermispora and Phanerochaete chrysosporium provide insight into selective ligninolysis. Proc Natl Acad Sci USA 109:5458–5463
Wiemann P, Guo C-J, Palmer JM, Sekonyela R, Wang CCC, Keller NP (2013) Prototype of an intertwined secondary-metabolite supercluster. Proc Natl Acad Sci USA 110:17065–17070
Takeda I, Umemura M, Koike H, Asai K, Machida M (2014) Motif-independent prediction of a secondary metabolism gene cluster using comparative genomics: application to sequenced genomes of Aspergillus and ten other filamentous fungal species. DNA Res 21:447–457
Ohm RA, Riley R, Salamov A, Min B, Choi I-G, Grigoriev IV (2014) Genomics of wood-degrading fungi. Fungal Genet Biol 72:82–90
Andersen MR, Nielsen JB, Klitgaard A, Petersen LM, Zachariasen M, Hansen TJ, Blicher LH, Gotfredsen CH, Larsen TO, Nielsen KF, Mortensen UH (2013) Accurate prediction of secondary metabolite gene clusters in filamentous fungi. Proc Natl Acad Sci USA 110:E99–107
Andersen MR, Lehmann L, Nielsen J (2009) Systemic analysis of the response of Aspergillus niger to ambient pH. Genome Biol 10:R47
Nitsche BM, Crabtree J, Cerqueira GC, Meyer V, Ram AFJ, Wortman JR, Fj A (2011) New resources for functional analysis of omics data for the genus Aspergillus. BMC Genomics 12:486
Van den Berg RA, Braaksma M, van der Veen D, van der Werf MJ, Punt PJ, van der Oost J, de Graaff LH (2010) Identification of modules in Aspergillus niger by gene co-expression network analysis. Fungal Genet Biol 47:539–550
Vongsangnak W, Hansen K, Nielsen J (2011) Integrated analysis of the global transcriptional response to α-amylase over-production by Aspergillus oryzae. Biotechnol Bioeng 108:1130–1139
Van den Berg BA, Reinders MJT, Hulsman M, Wu L, Pel HJ, Roubos JA, de Ridder D (2012) Exploring sequence characteristics related to high-level production of secreted proteins in Aspergillus niger. PLoS One 7:e45869
Van den Berg BA, Reinders MJT, van der Laan J-M, Roubos JA, de Ridder D (2014) Protein redesign by learning from data. Protein Eng Des Sel 27:281–288
Dunlap JC, Borkovich KA, Henn MR, Turner GE, Sachs MS, Glass NL, McCluskey K, Plamann M, Galagan JE, Birren BW, Weiss RL, Townsend JP, Loros JJ, Nelson MA, Lambreghts R, Colot HV, Park G, Collopy P, Ringelberg C, Crew C, Litvinkova L, DeCaprio D, Hood HM, Curilla S, Shi M, Crawford M, Koerhsen M, Montgomery P, Larson L, Pearson M et al (2007) Enabling a community to dissect an organism: overview of the Neurospora functional genomics project. Adv Genet 57:49–96
Park G, Colot H V, Collopy PD, Krystofova S, Crew C, Ringelberg C, Litvinkova L, Altamirano L, Li L, Curilla S, Wang W, Gorrochotegui-Escalante N, Dunlap JC, Borkovich KA (2011) High-throughput production of gene replacement mutants in Neurospora crassa. Methods Mol Biol 722:179–189
De Souza CP, Hashmi SB, Osmani AH, Andrews P, Ringelberg CS, Dunlap JC, Osmani SA (2013) Functional analysis of the Aspergillus nidulans kinome. PLoS One 8:e58008
Jiang D, Zhu W, Wang Y, Sun C, Zhang K-Q, Yang J (2013) Molecular tools for functional genomics in filamentous fungi: recent advances and new strategies. Biotechnol Adv 31:1562–1574
Meyer V, Ram AFJ, Punt PJ (2010) Genetics, genetic manipulation, and approaches to strain improvement of filamentous fungi. Man Ind Microbiol Biotechnol 1:318–329
Schuster A, Bruno KS, Collett JR, Baker SE, Seiboth B, Kubicek CP, Schmoll M (2012) A versatile toolkit for high throughput functional genomics with Trichoderma reesei. Biotechnol Biofuels 5:1
Delmas S, Llanos A, Parrou J-L, Kokolski M, Pullan ST, Shunburne L, Archer DB (2014) Development of an unmarked gene deletion system for the filamentous fungi Aspergillus niger and Talaromyces versatilis. Appl Environ Microbiol 80:3484–3487
Magaña-Ortíz D, Coconi-Linares N, Ortiz-Vazquez E, Fernández F, Loske AM, Gómez-Lim MA (2013) A novel and highly efficient method for genetic transformation of fungi employing shock waves. Fungal Genet Biol 56:9–16
Loske AM, Fernández F, Magaña-Ortíz D, Coconi-Linares N, Ortíz-Vázquez E, Gómez-Lim MA (2014) Tandem shock waves to enhance genetic transformation of Aspergillus niger. Ultrasonics 54:1656–1662
Blumhoff M, Steiger MG, Marx H, Mattanovich D, Sauer M (2012) Six novel constitutive promoters for metabolic engineering of Aspergillus niger. Appl Microbiol Biotechnol 97:253–267
Ninomiya Y, Suzuki K, Ishii C, Inoue H (2004) Highly efficient gene replacements in Neurospora strains deficient for nonhomologous end-joining. Proc Natl Acad Sci USA 101:12248–12253
Krappmann S, Sasse C, Braus GH (2006) Gene targeting in Aspergillus fumigatus by homologous recombination is facilitated in a nonhomologous end joining-deficient genetic background. Eukaryot Cell 5:212–215
Nayak T, Szewczyk E, Oakley CE, Osmani A, Ukil L, Murray SL, Hynes MJ, Osmani SA, Oakley BR (2006) A versatile and efficient gene-targeting system for Aspergillus nidulans. Genetics 172:1557–1566
Meyer V, Arentshorst M, El-Ghezal A, Drews A-C, Kooistra R, van den Hondel CAMJJ, Ram AFJ (2007) Highly efficient gene targeting in the Aspergillus niger kusA mutant. J Biotechnol 128:770–775
Carvalho NDSP, Arentshorst M, Kwon MJ, Meyer V, Ram AFJ (2010) Expanding the ku70 toolbox for filamentous fungi: establishment of complementation vectors and recipient strains for advanced gene analyses. Appl Microbiol Biotechnol 87:1463–1473
Krull R, Cordes C, Horn H, Kampen I, Kwade A, Neu TR, Nörtemann B (2010) Morphology of filamentous fungi: linking cellular biology to process engineering using Aspergillus niger. Adv Biochem Eng Biotechnol 121:1–21
Wucherpfennig T, Lakowitz A, Krull R (2013) Comprehension of viscous morphology—evaluation of fractal and conventional parameters for rheological characterization of Aspergillus niger culture broth. J Biotechnol 163:124–132
Coradin JH, Braun A, Viccini G, Fernando de Lima Luz L, Krieger N, Mitchell DA (2011) A three-dimensional discrete lattice-based system for modeling the growth of aerial hyphae of filamentous fungi on solid surfaces: A tool for investigating micro-scale phenomena in solid-state fermentation. Biochem Eng J 54:164–171
Liu Y, Liao W, Chen S (2008) Study of pellet formation of filamentous fungi Rhizopus oryzae using a multiple logistic regression model. Biotechnol Bioeng 99:117–128
Wucherpfennig T, Kiep KA, Driouch H, Wittmann C, Krull R (2010) Morphology and rheology in filamentous cultivations. Adv Appl Microbiol 72:89–136
Krull R, Wucherpfennig T, Esfandabadi ME, Walisko R, Melzer G, Hempel DC, Kampen I, Kwade A, Wittmann C (2013) Characterization and control of fungal morphology for improved production performance in biotechnology. J Biotechnol 163:112–123
Gao Q, Liu J, Liu L (2014) Relationship between morphology and itaconic acid production by Aspergillus terreus. J Microbiol Biotechnol 24:168–176
Formenti LR, Nørregaard A, Bolic A, Hernandez DQ, Hagemann T, Heins A-L, Larsson H, Mears L, Mauricio-Iglesias M, Krühne U, Gernaey KV (2014) Challenges in industrial fermentation technology research. Biotechnol J 9:727–738
Dochain D (2010) Automatic Control of Bioprocesses. vol 28. Wiley, New York
Mandenius C-F, Titchener-Hooker NJ (2013) Measurement and control of bioprocesses. Adv Biochem Eng Biotechnol 132:5–8
Cohen D (1967) Computer simulation of biological pattern generation processes. Nature 216:246–248
Prosser JI (1995) Mathematical modelling of fungal growth. In: Gow NAR, Gadd GM (eds) Grow fungus. Springer, Dordrecht, pp 319–335
Davidson FA (2007) Mathematical modelling of mycelia: a question of scale. Fungal Biol Rev 21:30–41
Celler K, Picioreanu C, van Loosdrecht MCM, van Wezel GP (2012) Structured morphological modeling as a framework for rational strain design of Streptomyces species. Antonie Van Leeuwenhoek 102:409–423
Davidson FA, Boswell GP, Fischer MWF, Heaton L, Hofstadler D, Roper M (2011) Mathematical modelling of fungal growth and function. IMA Fungus 2:33–37
Hopkins SM (2012) A hybrid mathematical model of fungal mycelia : tropisms, polarised growth and application to colony competition. PhD thesis, University of Glamorgan
King R (1998) Mathematical modelling of the morphology of streptomyces species. In: Schügerl K (ed) Advances in biochemical engineering, vol 60. Springer, Berlin, pp 95–119
Wittler R, Baumgartl H, Lübbers DW, Schügerl K (1986) Investigations of oxygen transfer into Penicillium chrysogenum pellets by microprobe measurements. Biotechnol Bioeng 28:1024–1036
Lejeune R, Baron GV (1997) Simulation of growth of a filamentous fungus in 3 dimensions. Biotechnol Bioeng 53:139–150
Nieminen L, Webb S, Smith MCM, Hoskisson PA (2013) A flexible mathematical model platform for studying branching networks: experimentally validated using the model actinomycete Streptomyces coelicolor. PLoS One 8:e54316
Rinas U, El-Enshasy H, Emmler M, Hille A, Hempel DC, Horn H (2005) Model-based prediction of substrate conversion and protein synthesis and excretion in recombinant Aspergillus niger biopellets. Chem Eng Sci 60:2729–2739
Werner S, Kaiser SC, Kraume M, Eibl D (2014) Computational fluid dynamics as a modern tool for engineering characterization of bioreactors. Pharm Bioprocess 2:85–99
Megee RD, Kinoshita S, Fredrickson AG, Tsuchiya HM (1970) Differentiation and product formation in molds. Biotechnol Bioeng 12:771–801
Nielsen J (1992) Modelling the growth of filamentous fungi. Adv Biochem Eng Biotechnol 46:187–223
Boswell GP, Davidson FA (2012) Modelling hyphal networks. Fungal Biol Rev 26:30–38
Edelstein L (1982) The propagation of fungal colonies: a model for tissue growth. J Theor Biol 98:679–701
Boswell GP, Jacobs H, Ritz K, Gadd GM, Davidson FA (2007) The development of fungal networks in complex environments. Bull Math Biol 69:605–634
Schnepf A, Roose T, Schweiger P (2008) Impact of growth and uptake patterns of arbuscular mycorrhizal fungi on plant phosphorus uptake—a modelling study. Plant Soil 312:85–99
Meskauskas A, Fricker MD, Moore D (2004) Simulating colonial growth of fungi with the neighbour-sensing model of hyphal growth. Mycol Res 108(11):1241–1256
Yang H, King R, Reichl U, Gilles ED (1992) Mathematical model for apical growth, septation, and branching of mycelial microorganisms. Biotechnol Bioeng 39:49–58
Heaton L, Obara B, Grau V, Jones N, Nakagaki T, Boddy L (2012) Fricker MD: analysis of fungal networks. Fungal Biol Rev 26:12–29
Bartnicki-Garcia S, Hergert F, Gierz G (1989) Computer simulation of fungal morphogenesis and the mathematical basis for hyphal (tip) growth. Protoplasma 153:46–57
Boudaoud A (2003) Growth of walled cells: from shells to vesicles. Phys Rev Lett 91:018104
Goriely A, Tabor M (2003) Biomechanical models of hyphal growth in actinomycetes. J Theor Biol 222:211–218
Tindemans SH, Kern N, Mulder BM (2006) The diffusive vesicle supply center model for tip growth in fungal hyphae. J Theor Biol 238:937–948
Sugden KEP, Evans MR, Poon WCK, Read ND (2007) Model of hyphal tip growth involving microtubule-based transport. Phys Rev E Stat Nonlin Soft Matter Phys 75(3 Pt 1):031909
Heaton LLM, López E, Maini PK, Fricker MD, Jones NS (2010) Growth-induced mass flows in fungal networks. Proc Biol Sci 277:3265–3274
Mabey Gilsenan J, Cooley J, Bowyer P (2012) CADRE the central Aspergillus data REpository 2012. Nucleic Acids Res 40(Database issue):D660–666
Kersey PJ, Allen JE, Christensen M, Davis P, Falin LJ, Grabmueller C, Hughes DST, Humphrey J, Kerhornou A, Khobova J, Langridge N, McDowall MD, Maheswari U, Maslen G, Nuhn M, Ong CK, Paulini M, Pedro H, Toneva I, Tuli MA, Walts B, Williams G, Wilson D, Youens-Clark K, Monaco MK, Stein J, Wei X, Ware D, Bolser DM, Howe KL, et al (2014) Ensembl genomes 2013: scaling up access to genome-wide data. Nucleic Acids Res 42(Database issue):D546–552
Stajich JE, Harris T, Brunk BP, Brestelli J, Fischer S, Harb OS, Kissinger JC, Li W, Nayak V, Pinney DF, Stoeckert CJ, Roos DS (2012) FungiDB: an integrated functional genomics database for fungi. Nucleic Acids Res 40(Database issue):D675–681
Choi J, Cheong K, Jung K, Jeon J, Lee G-W, Kang S, Kim S, Lee Y-W, Lee Y-H (2013) CFGP 2.0: a versatile web-based platform for supporting comparative and evolutionary genomics of fungi and Oomycetes. Nucleic Acids Res 41(Database issue):D714–719
Jung K, Park J, Choi J, Park B, Kim S, Ahn K, Choi J, Choi D, Kang S, Lee Y-H (2008) SNUGB: a versatile genome browser supporting comparative and functional fungal genomics. BMC Genomics 9:586
Pagani I, Liolios K, Jansson J, Chen I-MA, Smirnova T, Nosrat B, Markowitz VM, Kyrpides NC (2012) The Genomes OnLine Database (GOLD) v.4: status of genomic and metagenomic projects and their associated metadata. Nucleic Acids Res 40(Database issue):D571–579
Priebe S, Linde J, Albrecht D, Guthke R, Brakhage AA (2011) FungiFun: a web-based application for functional categorization of fungal genes and proteins. Fungal Genet Biol 48:353–358
Lum G, Min XJ (2011) FunSecKB: the fungal secretome knowledgebase. Database (Oxford) 2011:bar001
Park J, Park J, Jang S, Kim S, Kong S, Choi J, Ahn K, Kim J, Lee S, Kim S, Park B, Jung K, Kim S, Kang S, Lee Y-H (2008) FTFD: an informatics pipeline supporting phylogenomic analysis of fungal transcription factors. Bioinformatics 24:1024–1025
Park J, Lee S, Choi J, Ahn K, Park B, Park J, Kang S, Lee Y-H (2008) Fungal cytochrome P450 database. BMC Genomics 9:402
Garcia-Albornoz M, Thankaswamy-Kosalai S, Nilsson A, Väremo L, Nookaew I, Nielsen J (2014) BioMet Toolbox 2.0: genome-wide analysis of metabolism and omics data. Nucleic Acids Res 42(Web Server issue):W175–181
Pabinger S, Snajder R, Hardiman T, Willi M, Dander A, Trajanoski Z (2014) MEMOSys 2.0: an update of the bioinformatics database for genome-scale models and genomic data. Database (Oxford) 2014:bau004
Boele J, Olivier BG, Teusink B (2012) FAME, the flux analysis and modeling environment. BMC Syst Biol 6:8
Caspi R, Foerster H, Fulcher CA, Kaipa P, Krummenacker M, Latendresse M, Paley S, Rhee SY, Shearer AG, Tissier C, Walk TC, Zhang P, Karp PD (2008) The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases. Nucleic Acids Res 36(Database issue):D623–631
Barrett T, Troup DB, Wilhite SE, Ledoux P, Evangelista C, Kim IF, Tomashevsky M, Marshall KA, Phillippy KH, Sherman PM, Muertter RN, Holko M, Ayanbule O, Yefanov A, Soboleva A (2011) NCBI GEO: archive for functional genomics data sets–10 years on. Nucleic Acids Res 39(Database issue):D1005–1010
Rustici G, Kolesnikov N, Brandizi M, Burdett T, Dylag M, Emam I, Farne A, Hastings E, Ison J, Keays M, Kurbatova N, Malone J, Mani R, Mupo A, Pedro Pereira R, Pilicheva E, Rung J, Sharma A, Tang YA, Ternent T, Tikhonov A, Welter D, Williams E, Brazma A, Parkinson H, Sarkans U (2013) ArrayExpress update–trends in database growth and links to data analysis tools. Nucleic Acids Res 41(Database issue):D987–990
Khaldi N, Seifuddin FT, Turner G, Haft D, Nierman WC, Wolfe KH, Fedorova ND (2010) SMURF: genomic mapping of fungal secondary metabolite clusters. Fungal Genet Biol 47:736–741
Blin K, Medema MH, Kazempour D, Fischbach MA, Breitling R, Takano E, Weber T (2013) antiSMASH 2.0–a versatile platform for genome mining of secondary metabolite producers. Nucleic Acids Res 41(Web Server issue):W204–212
Sanli K, Karlsson FH, Nookaew I, Nielsen J (2013) FANTOM: functional and taxonomic analysis of metagenomes. BMC Bioinform 14:38
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Meyer, V., Fiedler, M., Nitsche, B., King, R. (2015). The Cell Factory Aspergillus Enters the Big Data Era: Opportunities and Challenges for Optimising Product Formation. In: Krull, R., Bley, T. (eds) Filaments in Bioprocesses. Advances in Biochemical Engineering/Biotechnology, vol 149. Springer, Cham. https://doi.org/10.1007/10_2014_297
Download citation
DOI: https://doi.org/10.1007/10_2014_297
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-20510-6
Online ISBN: 978-3-319-20511-3
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)