
Abstract
Thermal and themo-mechanical properties of alkali-silica reaction (ASR) products are poorly studied. The existent property data refers to theoretical considerations and do not account for the fact that ASR products can be crystalline, nanocrystalline and potentially amorphous. Here, the thermal conductivity, heat capacity, and coefficient of thermal expansion of crystalline structures (based on Na- and K-shlykovite), nanocrystalline structure (based on defective K-shlykovite structures), and amorphous ASR product are calculated using molecular simulations. Semi-classical estimates of the thermal conductivity, heat capacity, and standard molar entropy are provided. The anisotropy of thermal conductivity and thermal expansion is quantified. Nanoacoustics parameters (sound velocities, phonon free path and relaxation time) are calculated. These results contribute to completing property data for ASR products.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
The thermal problem is relevant for alkali-silica reaction (ASR) development, as the temperature influences pathology development, stress developement and phase assemblage [1,2,3,4]. A temperature increase on the order of 10 \(^{\circ }\)C is reported to accelerate ASR expansion roughly threefold [5]. Along with an increase in relative humidity and alkalinity, accelerated tests for ASR generally also involve increasing the temperature to enhance reaction kinetics [5]. Higher-temperature protocols include specimen storage at 38\(^{\circ }\)C in the concrete prism test (CPT), as recommended by ASTM [6], CSA [7], and RILEM [8, 9]. Accelerated CPT is usually performed at 60\(^{\circ }\)C [10]. Several authors have adopted temperatures between and above the range of 38–60\(^\circ\)C in the study of ASR [5]. Temperature is also shown to affect remedial measures regarding ASR [11]. Chemo-thermo-mechanical analysis of ASR-affected concrete has been proposed in the literature [12,13,14,15]. In these studies, the use of thermal properties of ASR could help in increasing the precision of predictions. Furthermore, alkali-activated materials (AAM) are susceptible to form ASR products, especially high calcium AAM [16]. The quantification of the thermal and thermo-mechanical properties of ASR products, therefore, helps in predicting the behaviour of these materials.
Recent progress have been made in unveil the details of the atomic structure of ASR products. The main ASR products identified are [17]:
-
K-shlykovite (KCaSi\(_4\)O\(_8\)(OH)\(_3\)·2H\(_2\)O): crystalline products akin to the naturally occurring mineral shlykovite [18].
-
Na-shlykovite (NaCaSi\(_4\)O\(_8\)(OH)\(_3\)·2H\(_2\)O): crystalline products based on the total substitution of potassium in shlykovite structure by sodium [17, 19].
-
ASR-P1 (K\(_{0.52}\)Ca\(_{1.16}\)Si\(_4\)O\(_8\)(OH)\(_{2.84}\)·1.5H\(_2\)O): nanocrystalline products whose atomic structure can be related to defective K-shlykovite with a coherence length of a few nanometers. This product has also been reported as amorphous [20, 21] in the literature, but recent molecular simulations [22] show that defective nanocrystalline K-shlykovite reproduces available experimental X-ray diffraction patterns and pair distribution functions better. The comparison of defective nanocrystalline K-shlykovite structure with available experimental data demonstrate that ASR-P1 can be associated with the disordered/ amorphous products reported in previous studies in the literature [23,24,25]
Na- and K-rich products are the main ASR products in cement systems, as these are the primary alkalis in the pore solution of cement-based materials [26]. However, molecular simulations point out the possibility of having other alkalis (Li and Cs) substituting K counterions with no significant changes in shlykovite structure [27]. The compositional and structural differences among these products signify that variations in thermal properties are expected. The extent of these differences is yet to be quantified. Disordered solids exhibit distinctive vibrational and thermal properties compared to their fully crystalline counterparts (e.g., [28, 29]). In crystals, the vibrational density of states (VDOS) is well-described in low frequencies (up to 1 THz) by the Debye model, which states an evolution of the VDOS \(g(\omega ) \propto \omega ^2\) as the square of the angular frequency \(\omega\) [29, 30]. The absence of long-range periodicity is claimed to be responsible for the presence in non-crystalline materials of an excess contribution to the Debye model, evidenced as a peak in the reduced VDOS \(g(\omega )/\omega ^2\), termed the boson peak [29, 31]. Boson peak was identified for C–S–H [32], which is a nanocrystalline phase [33]. This observation suggests that the boson peak can be relevant for the thermal properties of both nanocrystalline and amorphous ASR products.
To the best of the authors’ knowledge, no experimental measurements have been reported in the literature for the heat capacity of ASR products to date. Estimates based on empirical laws exist for crystalline products [34]. Full classical molecular simulations have been used to calculate the heat capacity of Na- and K-shlykovite [27]. However, full classical molecular dynamics simulations are known to lead to overestimations of the heat capacity of solids (e.g., [35, 36]). Thermal expansion tensors have been computed in the same study for crystalline products with non-reactive force fields [27]. No experimental data, theoretical estimates, or empirical calculations have been proposed for the thermal conductivity and thermal expansion of all ASR products, particularly nanocrystalline and amorphous products. Molecular dynamics (MD) simulations have been successful in providing missing data on thermal properties of cement systems, including C–(A)–S–H [32, 37], portlandite [32, 38], clinker minerals [32, 39, 40], and AF-phases [41, 42]. Thermal conductivity, thermal expansion, and heat capacity have been calculated for these phases. When experimental data is available, the results computed with molecular simulations generally show good agreement with such data, which is the case of thermal conductivity of alite [40], thermal expansion and heat capacity of ettringite and metaettringite [41], and thermal properties of C–(A)–S–H [32, 37, 43]. These results build confidence in using this technique to estimate, even for the first time, thermal properties of various phases of interest in cement systems. Only recently, an atomic structure was proposed for non-crystalline products [22] (based on the ASR-P1 composition provided in [44]). The present study builds on these recent references by providing the thermal properties of ASR products.
In this work, the thermal properties of crystalline, nanocrystalline, and amorphous ASR products are computed using molecular dynamics and reactive force fields. Thermal conductivity and thermal expansion tensors are provided, highlighting anisotropy at the molecular scale. The heat capacities at constant volume and pressure are computed using a semi-classical approach, accounting for quantum corrections. Using the semi-classical simulation for heat capacity computation, we also provide the estimate for the standard entropy. Quantum corrections are also introduced for thermal conductivity. Most of the data are provided for the first time in the literature.
2 Molecular models and methods
2.1 Atomic structure and force fields
The atomic structure of (K)-shlykovite [18] is adopted for crystalline systems. Na-Shlykovite is made by substitution of K counterions by Na, as in [19, 27]. Amorphous and nanocrystalline systems were constructed in a previous work [22] based on defective K-shlykovite structure. For these system the composition reported for ASR-P1 is used as reference [44]. The final composition of these system is K\(_{0.53}\)Ca\(_{1.17}\)Si\(_4\)O\(_8\)(OH)\(_{2.83}\)·1.5H\(_2\)O. To build the ASR-P1 structures, defects were introduced randomly [22]. Four independent atomic structures with different random defects were generated and are considered here. Nanocrystalline and amorphous systems are based on these four defective structures. Amorphicity was obtained using high annealing temperature in well-established simulation protocol [45]. In all cases, unit cell is replicated 3x3x1 fold to obtain a simulation box on the order of 10 nm\(^3\).

Snapshots of the atomic structure depicting Na and K-shlykovite, and defective shlykovite with ASR-P1 composition in nanocrystalline and amorphous forms
All simulations are run with LAMMPS [46]. ReaxFF parameters for Na/K/Si/O/H/Water systems [47], developped to the study of zeolites and already used for potassium ASR products [22], are adopted. Here, Na-shlykovite is for the first time modelled with these ReaxFF parameters. The lattice parameters obtained for the studies system are reported in Table 1. In reference [22], lattice parameters were computed in systems in which unit cell angles were kept the same as the one obtained after Shlykovite relaxation with ReaxFF. Here, the angles are also allowed to change upon NPT relaxation. The results for K-shlykovite can be compared with Zubkova et al. [18] experimental data on lattice parameters \((a, b, c, \alpha , \beta , \gamma )=\) (6.4897 Å, 6.6669 Å, 26.714 Å, 90 \(^{\circ }\), 94.697 \(^{\circ }\), 90 \(^{\circ }\)); and previous data from non-reactive MD simulations [19]: \((a, b, c, \alpha , \beta , \gamma )=\) (6.40 ± 0.04 Å, 6.90 ± 0.04 Å, 27.29 ± 0.30 Å, 90.0 ± 2.4 \(^{\circ }\), 91.6 ± 5.6 \(^{\circ }\), 90.0 ± 0.7 \(^{\circ }\)). The results for Na-shlykovite are consistent with non-reactive MD simulations [19]: \((a, b, c, \alpha , \beta , \gamma )=\) (6.35 ± 0.05 Å, 6.92 ± 0.04 Å, 24.89 ± 0.61 Å, 90.0 ± 3.8 \(^{\circ }\), 89.9 ± 3.6 \(^{\circ }\), 90.0 ± 1.0 \(^{\circ }\)). The results for ASR-P1 obtained here are consistent with the one reported in reference [22] for the systems with fixed angles. Snapshots of the atomic structures are shown in Fig. 1. As observed with simulation ussing ClayFF [19, 27], some of the Na in Na-shlykovite displace to a position closer to the Ca plan in shlykovite.
The system studied here refers to the so-called undrained conditions, i.e., when water is not allowed to exchange with an exterior reservoir (which can be the environment or larger neighboring pores). Undrained conditions are relevant for short timescales, such as fast temperature changes when no significant water content can be evacuated to reequilibrate with the new temperature (and humidity) conditions, and for moderate temperature changes. Drained properties of nanoporous hydrophilic phases like ASR products would correspond to boundary conditions in which water can exchange with the environment (e.g., by drying due to RH or temperature change). It must be noted that the saturating vapor pressure of water increases with temperature, and drained conditions in environments with available liquid water would lead to only a very slight dehydration of the nanoporous solid (water would leave the system due to the thermal expansion of a liquid being generally one order of magnitude larger than that of a solid; in the case of C–S–H, less than one water molecule per Si is lost for a variation of more than 100\(^{\circ }\)C [48]). Drained conditions with significant water loss occur only in environments in which the external vapor reservoir is kept at a partial pressure below saturating vapor pressure. In this case, significant water loss would occur when liquid water changes to vapor above boiling temperature. For the temperatures of interest in ASR accelerated protocols (up to 60 \(^{\circ }\)C, as discussed in the introduction), no significant amount of water is expected to leave the system since all is done below the boiling temperature of water under normal pressure conditions. The undrained conditions considered here are therefore relevant for the conditions observed in accelerated protocols.
In general, pressure effects should not alter significant properties of solids at the molecular scale except if pressures exceeding GPa (i.e., well above the service life conditions of concrete application) are adopted. Thus, pressure effects are not considered hereafter.
2.2 Thermal and thermo-mechanical properties from molecular simulations
Details on computing thermal properties with classical molecular dynamics simulation are recalled in this section.
2.2.1 Coefficient of thermal expansion
The thermal expansion tensor is calculated using the fluctuation-dissipation approach. In this case, it is determined using the cross fluctuations of the "enthalpy" \(H=E+PV\) (calculated using the instantaneous total energy E and instantaneous volume V but the imposed pressure P instead of the instantaneous pressure since this latter fluctuates with the barostat leading, therefore, to erroneously larger fluctuations) and the Lagrangian deformation tensor \(\epsilon _{ij}\) in the N\(\sigma\)T ensemble [49,50,51] using the following equation:
where \(k_B\) is the Boltzmann constant, T is the temperature, \({\textbf{I}}\) is the identity matrix, and the superscript \([\star ]^T\) denotes a matrix transpose; \(\epsilon _{ij} = \frac{1}{2} \left[ (h_{0}^{-1})^T h^T h h_{0}^{-1} - {\textbf{I}} \right]\) is the Lagrangian deformation for a generic (triclinic) system; \(\epsilon _{ij}\) is obtained from the matrices \(h_{0} = [a_0, b_0, c_0]{x, y, z}\) and \(h = [a, b, c]{x, y, z}\) associated with the metric of the reference and deformed supercell, respectively.
Simulations were performed in NPT ensemble with the Nosé-Hoover barostat applied independently along each uniaxial and tangential direction. A timestep of 0.1 fs is adopted, and six trajectories are considered in calculating the average value and standard deviations.
2.2.2 Heat capacity and entropy
Following the standard procedure for solid phases, the isochoric heat capacity is calculated accounting for quantum corrections with [52, 53]:
where \(C_V\) is the molar isochoric heat capacity (respectively, \(C_P\) is the molar isobaric heat capacity), \(3Nk_B\) is the Dulong-Petit relation, m is the total mass, \(\hbar\) is the reduced Planck constant, \(\omega\) is the angular frequency, \(n(\omega ,T) = \left[ \exp \left( \frac{\hbar \omega }{k_b T} \right) -1 \right] ^{-1}\) is Bose-Einstein distribution. In the formula above \(g(\omega )\) is the Vibrational Density of the States (VDOS) [36, 52]:
obtained from the mass-weighted velocity auto-correlation function (VACF) \(\langle m_j {\textbf{v}}_j(t).{\textbf{v}}_j(0) \rangle\). The estimate above is semi-classical since the VDOS is calculated using classical methods, but quantum corrections are introduced. The same approach was successfully used for the heat capacity of various solid phases in cement-based materials [32, 37, 39,40,41].
The isobaric heat capacity is calculated using anisotropic Mayer’s relation (e.g., [54]):
where \(\rho\) is the density, and \(C_{ijkl}\) is the stiffness tensor. Details on the computation of \(C_{ijkl}\) are provided in other work [55].
The entropy S can also be calculated from the VDOS for solids by assuming harmonic oscillators with the expression (e.g., [56, 57]):
This expression is consistent with \(S=\int _0^T C_v(T)/T dT\). As for \(c_V\), this estimate of entropy is semi-classical since the VDOS is computed using classical molecular simulations, but quantum corrections are introduced. Standard molar entropy \(S^0\), a crucial parameter in thermodynamic modeling, is typically determined from \(C_p\) at standard conditions (25\(^{\circ }\) and 1 bar). In this context, we approximate \(S^0\) by integrating \(C_p \approx C_v \left( \frac{C_p(T=273.15K)}{C_v(T=273.15K)}\right)\), where the term \(\left( \frac{C_p(T=273.15K)}{C_v(T=273.15~K)}\right)\) serves as an approximation for the conversion of \(C_v\) into \(C_p\).
The VACFs are calculated in an NVE run following proper equilibration in an NVT run. A timestep of 0.1 fs is adopted, and five trajectories are considered in the calculation of the average value and standard deviations. The VDOS is computed in post-processing the mass-weighted VACF by taking the Fourier transform after linearly interpolating the VACF. Numerical integration is used to get the entropy and heat capacity.
2.2.3 Thermal conductivity
Thermal conductivity tensor is computed using Green-Kubo formalism (e.g., [32, 58]):
where \(\langle J_i(0) \otimes J_j(t) \rangle\) are the components of ensemble-averaged heat flux auto-correlation function (HFACF) HFACF, with \(J_i\) being heat flux; i or j representing the Cartesian directions x, y, and z; \(\otimes\) denotes the dyadic product; and V stands for the system volume. The heat flux vector \(J_i\) is calculated from the viral stress tensor \(\mathbf {\sigma }_p\) and the energy (kinetic and potential) per atom \(E_p\) with \({\textbf{J}}_p = \frac{1}{V} \left[ \sum _p E_p {\textbf{v}}_p - \sum _p \mathbf {\sigma }_p {\textbf{v}}_p \right]\), where \({\textbf{v}}_p\) is the velocity vector.
Quantum corrections are needed for the thermal conductivity for temperatures T below \(T_D\), where \(T_D\) is the Debye temperature. The Debye temperature can be estimated by (e.g., [59, 60]):
in which \(S_v\) is the sound velocity, and N/V is the number density. The volumetric sound velocity reads [61]:
where the longitudinal and transverse sound velocities are, respectively, given by:
both computed from the density \(\rho\), and the diagonal components of the elastic tensor in Voigt notation (The three axial components for \(S_l\), and the three shear components for \(S_t\)).
The quantum-corrected thermal conductivity can be approximated as [62]:
where \(C_v\) is for the quantum-corrected volume heat capacity (corresponding to Eq. 2), and \(\lambda _{GK}\) is classical (Green-Kubo) thermal conductivity.
HFACF are computed in NVE runs for 5 ns; the correlations are calculated using a 10000 timestep correlation window, with input values recorded every timestep, and time window averages computed every 10000 timesteps. Before HFACF calculation, the systems were equilibrated adequately under ambient conditions using Nosé-Hoover barostat and thermostat. The linear and angular moments were zeroed during equilibration to avoid HFACF converging to non-zero values at large timescales. Average values and standard deviations are calculated based on data obtained from 4 independent trajectories.
3 Results
3.1 Coefficient of thermal expansion
The components of the thermal expansion tensor are gathered in Table 2. A larger thermal expansion is obtained in the direction perpendicular to the interlayer, as expected for systems with confined water. Anisotropy in the response decreases with nanocrystallinity and amorphicity. Thermal expansion in the interlayer plane directions increases with nanocrystallinity and amorphicity. Due to the monoclinic symmetry of shlykovite, the response along the layers’ plane directions is not expected to be the same. Still, the expansions along x and y obtained for all products are statistically indistinguishable (considering the variability/ uncertainty of our results). Off-diagonal components are larger in crystalline systems.
The volumetric thermal expansion \(\alpha _{v}=\text {Tr}(\alpha _{ij})\) is larger for Na- and K-shlykovite when compared to ASR-P1. The values for crystalline products can be compared with simulation using ClayFF [27] for which \(\alpha _{v}\) = 15.52 ± 2.96 \(\times\) 10\(^{-5}\)/K for Na-Shlykovite, and \(\alpha _{v}\) = 5.73 ± 0.70 \(\times\) 10\(^{-5}\)/K for K-Shlykovite. The results presented here are obtained with simulations with smaller timesteps and more extensive production lengths, which means they are more reliable. These new results show that thermal expansion is positive for all Cartesian directions, indicating that the previous results for Na-Shlykovite are likely related to simulation imprecision.
These values can be compared with the volumetric thermal expansion coefficients reported for C–(A)–S–H: 1.2\(-\)6.2 \(\times\) 10\(^{-5}\)/K according to various C–(A)–S–H models and Ca/Si ratios [32, 37, 63, 64], since ASR products are also layered calcium silicate hydrates. Siliceous aggregates, clinker minerals and hydrates such as portlandite and AF-phases have volumetric thermal expansion in the range of 2−4 \(\times\) 10\(^{-5}\)/K [65]. Thermal expansion and elastic constant property mismatch between ASR products and the other phases will lead to the rise of thermal stresses in each phase with a temperature variation. These results provided here can be used in future analyses to quantify the contribution of thermal cracking in ASR development under temperature-accelerated conditions.
3.2 Heat capacity and entropy
The VDOS are shown in Fig. 2. Experimental spectra measurements such as infrared adsorption and Raman spectra can be related to the VDOS. The specific per bond frequencies for K-ASR products are already discussed in ref. [22]. The VDOS for Na-shlykovite is computed here for the first time. All VDOS exhibit zero values for frequencies above 120 THz. Identified bands (I-V) in the VDOS correspond to various vibrations: Na–O, K–O, Si–O, and Ca–O vibrations for Band I, Si–O and O–H vibrations for Bands II and III, and intramolecular water vibrations for Bands IV and V. Hydrogen vibrations play a significant role in all bands, primarily contributing to Band V. Between bands II and IV and IV and V, the VDOS vanishes. These findings align with experimental spectra from other silicates, including C–(A)–S–H [17, 32, 37, 66]. In FTIR experiments of ASR products [17], the 28–34 THz (950-1130/cm) band corresponds to asymmetric and symmetric stretching vibrations of Si–O bonds, while the 18-21 THz (600–700/cm) band relates to Si–O–Si bending. The 2400-3800/cm band signifies stretching vibrations of O-H groups in molecular water or hydroxyls, potentially associated with gel water or loosely bound water in the products [17]. ASR-P1 experiments reveal a weak hydroxyl signal, attributed to the absence of well-ordered OH-groups [17, 66]. Raman spectroscopy of ASR products identifies high-frequency symmetrical stretching vibrations of Si–O bonds (24\(-\)34.4 THz), Si–O–Si symmetrical bending vibrations (600–700/cm), and intramolecular Si-O tetrahedra vibrations (12-15 THz) in silicates and ASR products [17, 66]. Specific wavenumbers associated with polymerization levels are noted, but the precision of the VDOS results hinders such identification.

A Total VDOS per ASR product. B VDOS per species
The occurrence of the boson peak is evaluated in the low-frequency VDOS and the reduced VDOS \(g(\omega )/\omega ^2\) in Fig. 3. In the reduced VDOS, a Lorentzian tail was used to describe the quasi-elastic component associated with the fast relaxation process [31, 70]. This contribution was extracted from the reduced VDOS to identify the boson peak. The VDOS at the low-frequency range in Fig. 3 for ASR-P1 shows an excess in the VDOS compared to the Debye model. The reduced VDOS indicates that the boson peak for ASR products appears in the 0.5-2 THz scale. These positions of boson peaks are similar to the ones experimentally identified for vitreous silica in the frequency range \(\nu _{BP}=\omega /2\pi\) of 1–2 THz [71] and 1.5\(-\)2.8 THz [72], and in simulations on C–S–H with varying Ca/Si with frequencies in the range 1\(-\)2.5 THz [32]. The boson peak frequency \(\nu _{BP}\) is reported to increase with temperature, with the intensity of the boson peak decreasing with temperature [71]. This observation helps explain the reduced VDOS of ASR-P1. ASR-P1 amorphous systems were built based on a higher annealing temperature than the nanocrystalline system. Thus, amorphous ASR-P1 would exhibit similar features as higher temperature vitreous silica compared with the lower temperature system (ASR-P1 nanocrystalline). K-shlykovite also shows a boson peak, which can be related to the disorder induced by finite temperature and the intrinsic degree of disorder associated with confined water and counterions. Conversely, Na-shylokoite shows two less pronounced peaks in the reduced VDOS in the 0-2 THz range that can be fairly approximated as negligible effects of the boson peak.

At top, VDOS at low frequency range per ASR product. The Debye model indicating VDOS \(g(\omega ) \propto \omega ^2\) at low frequency range is shown for comparison. At bottom, reduced VDOS per ASR product
The heat capacities of ASR products computed with the semi-classical approach are gathered in Table 3. K-shlykovite exhibits the largest heat capacity; for K products, the heat capacities increase with the disorder, as expected. Values for ASR-P1 are provided for the first time. The values reported here are almost half of the values reported for crystalline products by MD simulations with full classical approach [27] (i.e., without the quantum correction adopted here in the semi-classical approach): 1.860 ± 0.011 J/(g K) for Na-shlykovite, and 1.949 ± 0.024 J/(g K) for K-shlykovite. Full classical simulations are known to lead to overestimations of the heat capacity of solids (e.g., [35, 36]). This observation reaffirms the importance of accounting for quantum effects in the heat capacity estimates for solids.
The isobaric heat capacity can be compared with estimates provided by empirical formula. Various authors provide empirical relations also for the isobaric heat capacity (in J/(mol K)) as a function of the molar volume \(V_m\) [67,68,69].
These formulas rely on the observation that the heat capacity is strongly linearly correlated with formula volume for large sets of minerals [73]. Table 4 gathers the heat capacities obtained with different empirical formulas. Overall, the empirical formula leads to values of \(C_p\) larger than the values obtained by MD. The formula proposed by [68] leads to lower values of \(C_p\), therefore closer to the values computed by MD. Using the expression proposed by [69], the heat capacity of crystalline products was computed and used in thermodynamics calculation by [34].
The standard molar entropy of ASR products computed with the semi-classical approach are also gathered in Table 3. Empirical relations are also provided for the standard molar entropy (in J/(mol\(\cdot\)K)) as a function of the molar volume \(V_m\) [67,68,69].
Again, the empirical formula also lead to values of \(S^0\) larger than the values obtained by MD, and the formula proposed by [68] is closer to the values computed by MD.
Differences in the range of tenths of J/(mol\(\cdot\)K) have been observed for both \(C_p\) and \(S^0\) in the comparison with values calculated from molecular simulations, empirical formulas, and experimental data for other minerals as well [74]. These consistent trends between MD results and empirical formulas underscore the reliability and consistency of the molecular dynamics approach, especially considering the uncertainties in calculations.

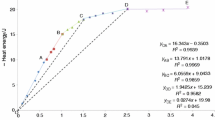
The semi-classical approach enables computing the temperature dependence of \(C_v\) and \(S^0\) from a single VDOS. Figure 4 displays the temperature dependence of \(C_p\) and \(S^0\). The MD results are fitted with the expressions (with T in Kelvins):
where \(a_i\) and \(b_i\) (\(i=0-4\)) are fitting parameters obtained from least squares fitting for each property independently. To avoid the indeterminations as \(T \rightarrow 0\), the fittings are performed from 7 K (this explains why \(a_0\) is a non-zero value). The values of the fitting parameters are listed in Table 5. The coefficient of determination of all fits were \(R^2=\) 1.00. These results can used in to describe the temperature dependence in thermodynamic modelling of ASR development.
3.3 Thermal conductivity
Nanoacoustic parameters including sound velocities, phonon mean free path, and phonon relaxation time are calculated for each ASR products and reported in Table 6. Phonons mean free path \(l_m\) can be estimated in the framework of kinetic theory with [75]:
In the same framework, phonons relaxation time \(\tau _m\) is [61]:
Sound velocities are in line with values previously reported for C–S–H [76]. Phonon mean free path \(l_m\) is on the order of half of the simulation boxes used in MD simulations, suggesting that size effects should not significantly affect the results. The relaxation time is on the order of half of a picosecond.
The Debye temperatures computed for the ASR products (Table 6) are close to the ambient temperature in which the simulations were carried out. Quantum correction might, therefore, need to be included in thermal conductivity estimates. The correction \(\frac{C_v}{3 N k_B}\) used is also reported in Table 6. Table 7 shows the thermal conductivity of ASR products obtained directly from simulation with Green-Kubo formalism (\(\lambda ^{GK}\)) and the quantum-corrected values (\(\lambda ^{corr.}\)). Not accounting for the correction led to an overestimation of the thermal conductivity of approximately 40\(\%\).
Conductivity along the layer plans is larger than in the perpendicular direction for crystalline products. As already discussed in the case of thermal expansion, the monoclinic symmetry of shlykovite means that the behavior along the layers’ plan is not isotropic (therefore, xx and yy components are not expected to be the same). Still, the values obtained here are indistinguishable statistically when the computed uncertainty is accounted for. The results for amorphous products are more isotropic, as expected. Nanocrystalline ASR-P1 also shows an approximately isotropic response despite the layered structure still being present in its atomic structure. The off-diagonal components of the thermal conductivity tensor are closer to zero and can be approximated to zero, considering the standard error reported. These results mean that the frame adopted in the simulation is approximately the principal frame. The volumetric thermal conductivity (the invariant calculated as the trace of the full tensor: \(\lambda _{v}=\text {Tr}\lambda _{ij}/3\)) is close to that reported for other calcium silicate hydrates [32, 76, 77] and amorphous silica [78].
4 Conclusions
Thermal and thermo-mechanical properties of ASR products are provided through molecular simulations. The significance of this work lies in its ability to offer data on thermal properties not yet reported for ASR products, a crucial aspect for future research focusing on the role of temperature in ASR development, including accelerated tests.
The thermal properties of ASR products are comparable to those of C–S–H obtained in previous studies using atomistic simulations, highlighting the similarities in layered silicates’ properties. Anisotropy plays a significant role in the thermal expansion and conductivity of crystalline products. Nanocrystalline products exhibit marked anisotropic thermal expansion, while thermal conductivity is approximated as isotropic.
Quantum corrections are crucial for estimates of thermal properties such as heat capacity and thermal conductivity. Compared to previous simulation results [27], the heat capacity provided here based on the semi-classical approach aligns better with theoretical estimates for heat capacity. Standard molar entropy profiling also indicates that molecular simulations can be a helpful tool in providing data to complete thermodynamic tables.
Future work may focus in upscaling thermal properties to the gel scale [79], as well as simulating temperature effects coupled in both undrained and drained (i.e., coupled with drying) conditions. Even for phases like C–S–H undrained thermal properties remains unknown to date (no experimental data is known, to the best of our knowledge; and no molecular simulation have been done so far considering drained conditions). Simulating drained systems with molecular simulations requires using techniques like Grand Canonical Monte Carlo simulation [80, 81] that are computer intensive and are yet to be applied to all ASR products.
References
Shi Z, Lothenbach B (2020) The combined effect of potassium, sodium and calcium on the formation of alkali-silica reaction products. Cem Concr Res 127:105914. https://doi.org/10.1016/j.cemconres.2019.105914
Shi Z, Lothenbach B (2019) The role of calcium on the formation of alkali-silica reaction products. Cem Concr Res 126:105898. https://doi.org/10.1016/j.cemconres.2019.105898
Maia Neto F.M, Andrade T.W.C.O, Gomes R.M, Leal A.F, Almeida A.N.F, Lima Filho M.R.F, Torres S.M (2021) Considerations on the effect of temperature, cation type and molarity on silica degradation and implications to ASR assessment. Constr Build Mater 299:123848. https://doi.org/10.1016/j.conbuildmat.2021.123848
Bagheri M, Lothenbach B, Scrivener K (2022) The effect of paste composition, aggregate mineralogy and temperature on the pore solution composition and the extent of ASR expansion. Mater Struct 55(7):192. https://doi.org/10.1617/s11527-022-02015-6.
Gautam B.P, Panesar D.K (2017) The effect of elevated conditioning temperature on the ASR expansion, cracking and properties of reactive Spratt aggregate concrete. Constr Build Mater 140:310–320. https://doi.org/10.1016/j.conbuildmat.2017.02.104
International, A. (2015) ASTM C1293 - Standard test method for determination of length change of concrete due to alkali-silica reaction. Technical report, ASTM International, West Conshohocken, PA
Group, C.: CSA A23.2-14A Potential expansivity of aggregates (procedure for length change due to alkali-aggregate reaction in concrete prisms at 38 \(^\circ\)C). Technical report, CSA Group, Mississauga, ON, Canada (2015)
Sims I, Nixon P (2003) RILEM recommended test method AAR-0: detection of alkali-reactivity potential in concrete-outline guide to the use of RILEM methods in assessments of aggregates for potential alkali-reactivity. Mater Struct 36(7):472–479. https://doi.org/10.1007/BF02481527
Rønning TF, Wigum BJ, Lindgård J, Nixon P, Sims I (2021) Recommendation of RILEM TC 258-AAA: RILEM AAR-0 outline guide to the use of RILEM methods in the assessment of the alkali-reactivity potential of concrete. Mater Struct 54(6):206. https://doi.org/10.1617/s11527-021-01687-w
Ideker JH, East BL, Folliard KJ, Thomas MDA, Fournier B (2010) The current state of the accelerated concrete prism test. Cem Concr Res 40(4):550–555. https://doi.org/10.1016/j.cemconres.2009.08.030
Ueda T, Kushida J, Tsukagoshi M, Nanasawa A (2014) Influence of temperature on electrochemical remedial measures and complex deterioration due to chloride attack and ASR. Constr Build Mater 67:81–87. https://doi.org/10.1016/j.conbuildmat.2013.10.020
Ulm F-J, Coussy O, Kefei L, Larive C (2000) Thermo-chemo-mechanics of ASR expansion in concrete structures. J Eng Mech 126(3):233–242. https://doi.org/10.1061/(ASCE)0733-9399(2000)126:3(233)
Comi C, Fedele R, Perego U (2009) A chemo-thermo-damage model for the analysis of concrete dams affected by alkali-silica reaction. Mech Mater 41(3):210–230. https://doi.org/10.1016/j.mechmat.2008.10.010. (Accessed 2023-11-29)
Charpin L, Ehrlacher A (2014) Microporomechanics study of anisotropy of ASR under loading. Cem Concr Res 63:143–157. https://doi.org/10.1016/j.cemconres.2014.05.009. (Accessed 2023-12-24)
Yang L, Pathirage M, Su H, Alnaggar M, Di Luzio G, Cusatis G (2021) Computational modeling of expansion and deterioration due to alkali-silica reaction: effects of size range, size distribution, and content of reactive aggregate. Int J Solids Struct. https://doi.org/10.1016/j.ijsolstr.2021.111220
Wang W, Noguchi T (2020) Alkali-silica reaction (ASR) in the alkali-activated cement (AAC) system: a state-of-the-art review. Constr Build Mater 252:119105. https://doi.org/10.1016/j.conbuildmat.2020.119105
Shi Z, Geng G, Leemann A, Lothenbach B (2019) Synthesis, characterization, and water uptake property of alkali-silica reaction products. Cement Concrete Res 121:58–71. https://doi.org/10.1016/j.cemconres.2019.04.009
Zubkova NV, Filinchuk YE, Pekov IV, Pushcharovsky DY, Gobechiya ER (2010) Crystal structures of shlykovite and cryptophyllite: comparative crystal chemistry of phyllosilicate minerals of the mountainite family. Eur J Mineral 22(4):547–555. https://doi.org/10.1127/0935-1221/2010/0022-2041
Honorio T, Chemgne Tamouya OM, Shi Z, Bourdot A (2020) Intermolecular interactions of nanocrystalline alkali-silica reaction products under sorption. Cem Concr Res 136:106155. https://doi.org/10.1016/j.cemconres.2020.106155
Leemann A, Rezakhani R, Gallyamov E, Lura P, Zboray R, Griffa M, Shakoorioskooie M, Shi Z, Dähn R, Geng G, Boehm-Courjault E, Barbotin S, Scrivener K, Lothenbach B, Bagheri M, Molinari J-F (2021) Alkali-silica reaction - a multidisciplinary approach. RILEM Tech Lett 6:169–187. https://doi.org/10.21809/rilemtechlett.2021.151
Shi Z, Ma B, Lothenbach B (2021) Effect of Al on the formation and structure of alkali-silica reaction products. Cem Concr Res 140:106311. https://doi.org/10.1016/j.cemconres.2020.106311
Honorio T, Wei W (2023) Atomic structure of nanocrystalline and amorphous asr products. Cem Concr Res 181:107521. https://doi.org/10.1016/j.cemconres.2024.107521
Davies G, Oberholster RE (1988) Alkali-silica reaction products and their development. Cem Concr Res 18(4):621–635. https://doi.org/10.1016/0008-8846(88)90055-5
Meral C, Benmore CJ, Monteiro PJM (2011) The study of disorder and nanocrystallinity in C–S–H, supplementary cementitious materials and geopolymers using pair distribution function analysis. Cem Concr Res 41(7):696–710. https://doi.org/10.1016/j.cemconres.2011.03.027
Benmore CJ, Monteiro PJM (2010) The structure of alkali silicate gel by total scattering methods. Cement Concrete Res 40(6):892–897. https://doi.org/10.1016/j.cemconres.2010.02.006
Vollpracht A, Lothenbach B, Snellings R, Haufe J (2016) The pore solution of blended cements: a review. Mater Struct 49(8):3341–3367
Honorio T, Tamouya OMC, Shi Z (2020) Specific ion effects control the thermoelastic behavior of nanolayered materials: the case of crystalline alkali-silica reaction products. Phys Chem Chem Phys 22(47):27800–27810
Phillips WA (ed) (1981) Amorphous Solids: Low-Temperature Properties. Topics in Current Physics, Springer, Berlin, Heidelberg
Hu Y-C, Tanaka H (2022) Origin of the boson peak in amorphous solids. Nat Phys 18(6):669–677. https://doi.org/10.1038/s41567-022-01628-6
Mizuno H, Mossa S, Barrat J-L (2013) Elastic heterogeneity, vibrational states, and thermal conductivity across an amorphisation transition. EPL (Europhysics Letters) 104(5):56001. https://doi.org/10.1209/0295-5075/104/56001
Corezzi S, Caponi S, Rossi F, Fioretto D (2013) Stress-induced modification of the boson peak scaling behavior. J Phys Chem B 117(46):14477–14485. https://doi.org/10.1021/jp4054742
Qomi MJA, Ulm F-J, Pellenq RJ-M (2015) Physical origins of thermal properties of cement paste. Phys Rev Appl 3(6):064010. https://doi.org/10.1103/PhysRevApplied.3.064010
Skinner LB, Chae SR, Benmore CJ, Wenk HR, Monteiro PJM (2010) Nanostructure of calcium silicate hydrates in cements. Phys Rev Lett 104(19):195502. https://doi.org/10.1103/PhysRevLett.104.195502
Jin H, Ghazizadeh S, Provis JL (2023) Assessment of the thermodynamics of Na, K-shlykovite as potential alkali-silica reaction products in the (Na, K)2O-CaO-SiO2-H2O system. Cem Concr Res 172:107253. https://doi.org/10.1016/j.cemconres.2023.107253
Oganov AR, Brodholt JP, David Price G (2000) Comparative study of quasiharmonic lattice dynamics, molecular dynamics and Debye model applied to MgSiO3 perovskite. Phys Earth Planet Inter 122(3):277–288. https://doi.org/10.1016/S0031-9201(00)00197-7
Honorio T, Brochard L (2022) Drained and undrained heat capacity of swelling clays. Phys Chem Chem Phys 24(24):15003–15014. https://doi.org/10.1039/D2CP01419J
Hawchar BM, Honorio T (2022) C-A-S-H thermoelastic properties at the molecular and gel scales. J Adv Concr Technol 20(6):375–388. https://doi.org/10.3151/jact.20.375
Sarkar PK, Goracci G, Dolado JS (2024) Thermal conductivity of Portlandite: molecular dynamics based approach. Cem Concr Res 175:107347. https://doi.org/10.1016/j.cemconres.2023.107347
Claverie J, Kamali-Bernard S, Cordeiro JMM, Bernard F (2021) Assessment of mechanical, thermal properties and crystal shapes of monoclinic tricalcium silicate from atomistic simulations. Cem Concr Res 140:106269. https://doi.org/10.1016/j.cemconres.2020.106269
Barbosa W, Honorio T (2022) Triclinic tricalcium silicate: structure and thermoelastic properties from molecular simulations. Cem Concr Res 158:106810. https://doi.org/10.1016/j.cemconres.2022.106810
Honorio T (2022) Thermal conductivity, heat capacity and thermal expansion of ettringite and metaettringite: effects of the relative humidity and temperature. Cem Concr Res 159:106865. https://doi.org/10.1016/j.cemconres.2022.106865
Honorio T, Carasek H, Cascudo O (2022) Friedel’s salt: temperature dependence of thermoelastic properties. Cem Concr Res 160:106904. https://doi.org/10.1016/j.cemconres.2022.106904
Moshiri A, Morshedifard A, Stefaniuk D, El Awad S, Phatak T, Krzywiński KJ, Rodrigues DF, Qomi MJA, Krakowiak KJ (2023) Organic cross-linking decreases the thermal conductivity of calcium silicate hydrates. Cem Concr Res 174:107324. https://doi.org/10.1016/j.cemconres.2023.107324
Shi Z, Park S, Lothenbach B, Leemann A (2020) Formation of shlykovite and ASR-P1 in concrete under accelerated alkali-silica reaction at 60 and 80 \(^\circ\)C. Cem Concr Res 137:106213. https://doi.org/10.1016/j.cemconres.2020.106213
Bauchy M, Qomi MJA, Ulm F-J, Pellenq RJ-M (2014) Order and disorder in calcium-silicate-hydrate. J Chem Phys 140(21):214503. https://doi.org/10.1063/1.4878656
Thompson AP, Aktulga HM, Berger R, Bolintineanu DS, Brown WM, Crozier PS, Veld PJ, Kohlmeyer A, Moore SG, Nguyen TD, Shan R, Stevens MJ, Tranchida J, Trott C, Plimpton SJ (2022) LAMMPS - a flexible simulation tool for particle-based materials modeling at the atomic, meso, and continuum scales. Comput Phys Commun 271:108171. https://doi.org/10.1016/j.cpc.2021.108171
Psofogiannakis GM, McCleerey JF, Jaramillo E, Duin ACT (2015) ReaxFF reactive molecular dynamics simulation of the hydration of Cu-SSZ-13 Zeolite and the formation of Cu dimers. J Phys Chem C 119(12):6678–6686. https://doi.org/10.1021/acs.jpcc.5b00699
Honorio T, Masara F, Poyet S, Benboudjema F (2023) Surface water in C–S–H: effect of the temperature on (de)sorption. Cem Concr Res 169:107179. https://doi.org/10.1016/j.cemconres.2023.107179.
Cagin T, Karasawa N, Dasgupta S, Goddard WA (1992) Thermodynamic and elastic properties of polyethylene at elevated temperatures. MRS Online Proc Library Arch 278:61
Ray JR (1983) Molecular dynamics equations of motion for systems varying in shape and size. J Chem Phys 79(10):5128–5130. https://doi.org/10.1063/1.445636. (Accessed 2018-02-08)
Honório T, Lemaire T, Tommaso DD, Naili S (2019) Molecular modelling of the heat capacity and anisotropic thermal expansion of nanoporous hydroxyapatite. Materialia 5:100251. https://doi.org/10.1016/j.mtla.2019.100251
Dickey JM, Paskin A (1969) Computer simulation of the lattice dynamics of solids. Phys Rev 188(3):1407–1418. https://doi.org/10.1103/PhysRev.188.1407
Winkler B, Dove MT (1992) Thermodynamic properties of MgSiO3 perovskite derived from large scale molecular dynamics simulations. Phys Chem Miner 18(7):407–415. https://doi.org/10.1007/BF00200963
Wallace DC (1998) Thermodynamics of Crystals. Courier Corporation, n.a
Honorio, T., Razki, S., Bourdot, A., Benboudjema, F.: Mechanical properties of ASR products at the molecular scale. in preparation
Lin S-T, Maiti PK, Goddard William AIII (2010) Two-Phase thermodynamic model for efficient and accurate absolute entropy of water from molecular dynamics simulations. J Phys Chem B 114(24):8191–8198. https://doi.org/10.1021/jp103120q
Pascal TA, Lin S-T, Iii WAG (2011) Thermodynamics of liquids: standard molar entropies and heat capacities of common solvents from 2PT molecular dynamics. Phys Chem Chem Phys 13(1):169–181. https://doi.org/10.1039/C0CP01549K
Allen MP, Tildesley DJ (1989) Computer Simulation of Liquids. Oxford University Press, New York
Gu X, Fan Z, Bao H (2021) Thermal conductivity prediction by atomistic simulation methods: recent advances and detailed comparison. J Appl Phys 130(21):210902. https://doi.org/10.1063/5.0069175
Huang B.L, McGaughey A.J.H, Kaviany M (2007) Thermal conductivity of metal-organic framework 5 (MOF-5): part I. Molecular dynamics simulations. Int J Heat Mass Transf 50(3–4):393–404. https://doi.org/10.1016/j.ijheatmasstransfer.2006.10.002
Bhowmick S, Shenoy VB (2006) Effect of strain on the thermal conductivity of solids. J Chem Phys 125(16):164513. https://doi.org/10.1063/1.2361287
Zheng X, Bourg IC (2023) Nanoscale prediction of the thermal, mechanical, and transport properties of hydrated clay on 106- and 1015-Fold larger length and time scales. ACS Nano 17(19):19211–19223. https://doi.org/10.1021/acsnano.3c05751
Ghabezloo S (2011) Micromechanics analysis of thermal expansion and thermal pressurization of a hardened cement paste. Cem Concr Res 41(5):520–532. https://doi.org/10.1016/j.cemconres.2011.01.023
Krishnan NMA, Wang B, Falzone G, Le Pape Y, Neithalath N, Pilon L, Bauchy M, Sant G (2016) Confined Water in Layered Silicates: The Origin of Anomalous Thermal Expansion Behavior in Calcium-Silicate-Hydrates. ACS Applied Materials & Interfaces 8(51):35621–35627. https://doi.org/10.1021/acsami.6b11587
Johnson, W.H., Parsons, W.H.: Thermal expansion of concrete aggregate materials. US Government Printing Office, ??? ( 1944)
Kirkpatrick RJ, Yarger JL, McMillan PF, Ping Y, Cong X (1997) Raman spectroscopy of C–S–H, tobermorite, and jennite. Adv Cem Based Mater 5(3):93–99. https://doi.org/10.1016/S1065-7355(97)00001-1
Glasser L (2021) The effective volumes of waters of crystallization & the thermodynamics of cementitious materials. Cement 3:100004. https://doi.org/10.1016/j.cement.2021.100004
Glasser L, Brooke Jenkins HD (2016) Predictive thermodynamics for ionic solids and liquids. Phys Chem Chem Phys 18(31):21226–21240. https://doi.org/10.1039/C6CP00235H
Ghazizadeh S, Hanein T, Provis JL, Matschei T (2020) Estimation of standard molar entropy of cement hydrates and clinker minerals. Cem Concr Res 136:106188. https://doi.org/10.1016/j.cemconres.2020.106188
Yannopoulos SN, Kastrissios DT (2002) Spectral features of the quasielastic line in amorphous solids and supercooled liquids: a detailed low-frequency Raman scattering study. Phys Rev E 65(2):021510. https://doi.org/10.1103/PhysRevE.65.021510
Wischnewski A, Buchenau U, Dianoux AJ, Kamitakahara WA, Zarestky JL (1998) Sound-wave scattering in silica. Phys Rev B 57(5):2663–2666. https://doi.org/10.1103/PhysRevB.57.2663
Jund P, Jullien R (2000) Densification effects on the Boson peak in vitreous silica: a molecular-dynamics study. J Chem Phys 113(7):2768–2771. https://doi.org/10.1063/1.1305861
Glasser L, Jenkins HDB (2011) Ambient isobaric heat capacities, Cp, m, for ionic solids and liquids: an application of volume-based thermodynamics (VBT). Inorg Chem 50(17):8565–8569. https://doi.org/10.1021/ic201093p
Lothenbach B, Geiger CA, Dachs E, Winnefeld F, Pisch A (2022) Thermodynamic properties and hydration behavior of ye’elimite. Cement Concrete Res 162:106995. https://doi.org/10.1016/j.cemconres.2022.106995
Ziman JM (2001) Electrons and Phonons: The Theory of Transport Phenomena in Solids Oxford Classic Texts in the Physical Sciences. Oxford University Press, Oxford
Honorio T, Masara F, Huang G, Benboudjema F (2023) Thermal, mechanical, and transport properties of C-S-H at the molecular scale: a force field benchmark. Under revision
Sarkar PK, Mitra N (2021) Thermal conductivity of cement paste: influence of macro-porosity. Cem Concr Res 143:106385. https://doi.org/10.1016/j.cemconres.2021.106385
Cahill DG, Pohl RO (1988) Lattice vibrations and heat transport in crystals and glasses. Annu Rev Phys Chem 39(1):93–121. https://doi.org/10.1146/annurev.pc.39.100188.000521
Ait Hamadouche S, Honorio T (2023) Nanomechanics of ASR gels from coarse-grained simulations. J Eng Mech. https://doi.org/10.1061/JENMDT/EMENG-6926
Masara F, Honorio T, Benboudjema F (2023) Sorption in C–S–H at the molecular level: disjoining pressures, effective interactions, hysteresis, and cavitation. Cement Concrete Res 164:107047. https://doi.org/10.1016/j.cemconres.2022.107047
Honorio T (2019) Monte Carlo molecular modeling of temperature and pressure effects on the interactions between crystalline calcium silicate hydrate layers. Langmuir 35(11):3907–3916. https://doi.org/10.1021/acs.langmuir.8b04156.
Acknowledgements
We thank the financial support of the French National Research Agency (ANR) through the project HEAT COFFEE (ANR-22-CHIN-0007)
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Honorio, T., Razki, S., Bourdot, A. et al. Thermal properties of ASR products. Mater Struct 57, 117 (2024). https://doi.org/10.1617/s11527-024-02388-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1617/s11527-024-02388-w