
Abstract
The present work evaluates chitosan/sodium alginate blended with two nanomaterials of hydroxyapatite (HA) and commercial nanoclay-Cloisite20A for improving bone regeneration. The free nano-HA, nanoclay, and their scaffolds were characterized via TEM, SEM, EDX, FTIR, XRD, and TGA analyses. In vitro bioactivity, degradation, swelling, and cytotoxicity were investigated. For the in vivo studies, two groups of rats were used (n = five) in the study period (37 days). The scaffolds were implanted in the rat’s back. TEM analysis showed the nanorods of HA have a particle length of around 107 nm and a width around 17 nm, which is crystalline in nature, as XRD demonstrated. However, the SEM images showed the formation of honeycomb-like scaffolds. The in vitro bioactivity results through the changes of PBS ions and SEM/EDX analysis after immersion in PBS detected the formation of a calcium phosphate layer which is important for bone regeneration. The cell viability results proved IC50 > 670 µg/mL and IC50: 116 µg/mL for HA and nanoclay, respectively. Furthermore, the histological analysis revealed the highest calcification after 1 month for the nano-HA-containing scaffold supporting them as promising substitute biomaterials for treating bone loss.
Graphical abstract

Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Replacement of bone tissue is necessary to restore function following bone deficiencies, which are usually caused by trauma, surgery, infection, and diseases like osteoporosis and arthritis.[1] Surgery is necessary to promote tissue restoration because the large bone abnormalities cannot be repaired by the body on its own. The advancement of suitable tissue engineering methods may be advantageous over artificial replacement and speed up tissue regeneration.[2]
In the field of tissue engineering for bone loss, a biodegradable scaffold acts as a temporary skeletal frame to help and promote bone tissue regeneration, while it eventually deteriorates and is substituted by new bone tissue that is identical to that of natural bone. In addition to promoting adhesion, proliferation, and differentiation of cells, also the scaffolds should be biocompatible, biodegradable, have a higher surface-to-volume ratio, have a particular mechanical potency, and be cast in esthetic shapes.[3]
Chitosan is a naturally occurring, cationic polysaccharide, semi-crystalline composed of repeating units of d-glucosamine and N-acetyl glucosamine that is derived from partial chitin’s alkaline deacetylation, the primary constituent of crustacean shells.[4] Chitosan could be used for fabricating 3D scaffolds because of its exceptional properties which include non-toxicity, biodegradability, biocompatibility, and bio-functionality. Its biocompatibility is due to the structural analogy to glycosaminoglycans (GAGs), a crucial part of the extracellular matrix of cartilage.[5]
The anionic polymer alginate, known for its biocompatible, hydrophilic, and biodegradable characteristics, is frequently employed in bone tissue engineering. Sodium alginate boosts mechanical strength or structural integrity by forming a strong link with chitosan. In a short time, sodium alginate can create scaffolds, and the amount of porosity can be easily adjusted.[5]
The primary inorganic substance in human bone tissue is hydroxyapatite (HA), which exhibits strong biological activity, bone growth, and biocompatibility.[6,7] Moreover, HA is regarded as one of the most promising materials for bone implants. However, it cannot be employed in therapeutic settings because of its low mechanical strength and excessive brittleness. High-molecular polymers and other macromolecules are commonly mixed with HA to develop composites that can exhibit improved bone-forming capabilities to meet the demands for exceptional bone scaffold components.[8] Both sodium alginate and chitosan can encourage tissue regeneration and has a significant role in bone tissue engineering when combined with hydroxyapatite.[9]
The mineral montmorillonite (MMT) has an exceptional layered structure and strong intercalation capabilities. It can also be chemically altered to improve its compatibility with a polymer, making it an excellent choice for polymer nanocomposites, such as Cloisite20A. Cloisite20A is also known as nanoclays and is made of layered mineral silicate nanoparticles. A novel class of tissue engineering materials could be developed by mixing nanoclay with chitosan scaffolds.[10]
The current study aims to assess the bone-regenerative activity of the synthesized, characterized biocomposite scaffolds chitosan/alginate/nano-HA and chitosan/alginate/nanoclay. The characteristics of the scaffold, in vitro bioactivity in PBS, and cell viability are all covered in the current work, along with the impact of adding nano-HA and nanoclay. Furthermore, the calcification efficiency of scaffolds on rats as models for applications involving bone tissue engineering.
Materials and method
Chemicals and reagents
Chitosan, medium molecular weight from 50 to 190 kDa, with a deacetylation degree of 90.2%, was supplied from Fluka, Germany. Acetic acid (glacial, 100%) was obtained from ADWIC, Egypt. Sodium hydroxide (NaOH) extra pure was supplied from (Loba Chemie, India). Montmorillonite clay (MMT) modified with dimethyl and di-hydrogenated quaternary ammonium (Cloisite 20A) were procured from BYK-Chemie, GmbH, Germany. Twofold distilled water was used for preparing all the aqueous solutions. Sodium alginate was procured from Fisher Chemical, USA. Calcium Nitrate [Ca (NO3)2], Ammonium Phosphate [(NH4)3PO4], and Ammonium Hydroxide (NH4OH) were purchased from Sigma-Aldrich. Disodium hydrogen phosphate (MWt = 141.96; MERCK, Germany), potassium dihydrogen phosphate (99%, MWt = 136.09; Alfa Aesar, Germany), sodium chloride (MWt = 58.44; El-Nasr pharmaceutical chemicals, Egypt), and hydrochloric acid (MWt = 36; El-Nasr pharmaceutical chemicals, Egypt) were provided. All the reagents were used as received.
The fabrication of hydroxyapatite nanorods
A wet chemical precipitation method was used to synthesize the hydroxyapatite (HA) nanorods.[11]
In the present work, the Ca (NO3)2 and (NH4)3PO4 were utilized as precursors to produce HA nanorods, while the precipitating agent was NH4OH. Firstly, a 500-mL flask with three necks was filled with a freshly made Ca (NO3)2 aqueous solution (0.042 M). Secondly, two separate solutions of (NH4)3PO4 (0.025 M) and NH4OH were prepared in two flasks. These solutions were poured into the Ca (NO3)2 solution drop by drop, keeping the pH of the resultant solution at 10. The white precipitate formed through the addition process confirms the HA formation. Then, the solution was stirred for 2 h, the distilled water was used for washing of the resultant precipitate until neutralization, filtered, and dried, and then the obtained white powder was calcined at 300°C for 2 h.
Preparation of chitosan/alginate/nano-HA and chitosan/alginate/nanoclay scaffolds
The bio-nanocomposite scaffolds of nano-HA and nanoclay contain chitosan as a building unit and were prepared via the freeze-dry method. Briefly, 2% (w/v) chitosan was dissolved in 1% acetic acid and using a magnetic stirrer it stirred for 12 h at room temperature. Then, the alginate was added to chitosan solution and agitated for 2 h to obtain a homogeneous solution. Then, nano-HA (1% by weight) was added to the previously prepared chitosan/alginate solution and agitated again for another 2 h. The obtained gel-like solution was transferred to 24-well culture plates to produce a dried scaffold and pre-frozen at − 20°C for 48 h before being freeze-dried (Freeze dryer, Model-SCANAC, Cool Safe, Denmark), which was adjusted at − 90°C for 48 h. Finally, the prepared scaffolds were neutralized by submerging them in a 0.2 M NaOH solution, washing twice with deionized water, and further lyophilization. The same protocol was followed for fabricating chitosan/alginate/nanoclay scaffolds, while the nanoclay was added instead of nano-HA. Another control sample containing only chitosan/alginate was prepared via the same process.
Characterization techniques
The morphological features, including size, shape, and uniformity, were examined using a high-resolution transmission electron microscope (Model JEM 2010, Japan) that operated at 200 kV of accelerating voltage and was coupled to a Gatan camera (Model Erlangshen ES500). The surface morphology and porous structure of the prepared scaffolds were imaged by a Scanning Electron Microscope (SEM; JEOL model JSM-IT100). The SEM was attached to Energy-Dispersive x-ray spectroscopy (EDX), which was employed to detect the elemental composition. The x-ray diffraction (XRD) technique was used to determine the crystallinity/amorphous nature of the synthesized HA nanoparticles and clay, using an x-ray diffractometer (XRD-6000 Shimadzu) at 40 kV with current adjusted at 30 mA with CuKa1 radiation λ = 1.54056 Å, and the angle range was 10–80°. The crystallite size of the nanoparticles was evaluated from the peaks broadening in the XRD pattern measured following Scherrer’s equation
where D is the crystallite size (nm), λ is the x-ray beam wavelength (λ = 01.54056 Å), β = FWHM is the full-width at half-maximum, and θ is the diffraction angle (deg). The UV–Vis absorption spectra were obtained using a Shimadzu UV-2450 spectrophotometer.
Measurements of produced nanoparticles and scaffolds using FTIR analysis provided structural information within the range of 4000 to 400 cm−1. Zeta potential measurements were made in a disposable cell using a Zetasizer Nano ZS (Model Malvern) at 25°C. The scaffold formulation was freeze-dried using a Freeze dryer, SCANAC, Cool Safe, Denmark and was adjusted at − 90°C. To learn more about the heat degradation of synthesized materials, thermogravimetric analysis (TGA), performed in a DSC-60 Plus Series SHIMADZU, was used. Under a nitrogen atmosphere, samples weighing 10 to 15 mg were heated from 25 to 800°C at a flow rate of 60 mL/min. All the experiments were carried out within 10°C/min. Ca and P element concentrations were measured using a chemical kit and a UV/visible spectrometer (model SP-2000UV) at 585 and 640 nm, respectively, was used to determine the concentrations.
The cell viability test
Fresh stock solutions of the nano-HA and nanoclay were prepared in media (DMEM) to determine the half-maximal cytotoxic concentration (CC50). The Vero (green monkey kidney cells) cell line was used to test the cytotoxic potential of tested materials using the 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT) technique as previously reported.[12] In a typical experiment, the Vero cells were seeded in a 96-well plate (100 µL/well and a density of 3×105 cells/mL) and set for incubation for 24 h at 37°C in 5% CO2.
Then, various concentrations of the investigated nanoparticles were applied to the cells in triplicates. After 72 h, the supernatant was removed and sterile 1× PBS was used to wash the cell monolayers three times before adding the MTT solution (20 L of a 5-mg/mL stock solution). For 4 h, the cells were incubated at 37°C, followed by medium aspiration. The crystals of formazan that were produced were dissolved in 200 µL of acidified isopropanol (0.073 mL of HCl in 50 mL of isopropanol is equal to 0.04 M of HCl in 100% isopropanol) in each well.
The absorbance of formazan solutions was measured at λmax 540 and 620 nm, serving as a reference wavelength using a multi-well plate reader (BMGLABTECH ®FLUOstar Omega, Germany). To calculate the proportion of cytotoxicity in comparison to untreated cells using the following equation
The plot of % cytotoxicity versus sample concentration was used to measure the concentration, which displayed 50% cytotoxicity (TC50).
In vitro studies
Preparation of phosphate buffer saline (PBS)
After dissolving 1.38 g of disodium hydrogen phosphate in 700 mL of distilled water, 0.19 g of potassium dihydrogen phosphate was added to the initial mixture. The mixture was mixed with 8 g of sodium chloride and then the pH was adjusted to 7.4 with a few drops of hydrochloric acid. Use distilled water to finish the aforesaid solution up to 1000 mL.[13]
Bioactivity evaluation
The in vitro bioactivity of the synthesized scaffolds was tested at 37°C in PBS at one, 4, 7, 14, and 28 days, followed by immersion of the scaffolds in 100 mL of PBS in plastic containers. Five mL was removed and replaced with fresh PBS after each period to measure the various concentrations of PBS ions using a UV spectrophotometer. The prepared scaffolds were taken out of the PBS, cleaned, and allowed to dry before being examined using a SEM and EDX to verify the development of a calcium phosphate layer on the surface of the scaffold.
Degradation
The degradation behavior of the scaffold was studied in PBS medium at 37°C at one, 4, 7, 14, and 28 days. The scaffolds were immersed in PBS and incubated for 28 days. After each period, the scaffolds were dried and weighed. The scaffold’s initial weight was noted as W0 and the dry weight after immersion time as Wt, and the degradation of the scaffold was calculated using Equation (1) [14]:
Swelling
The swelling study of the scaffolds was operated in PBS medium at 37°C at one, 4, 7, 14, and 28 days. The scaffolds were immersed in PBS and incubated for 28 days. After each period, the scaffolds were gently put on filter paper to eliminate the adsorbed PBS, weighed, and recorded as Ww. Wd was referred to the scaffold’s initial weight. The ratio of swelling is calculated by Equation (2) [15]:
Experimental animals and design
The protocol of the in vivo study followed the guidelines of the ethical committee of the National Research Center, Egypt (Approval No. 19/248). Ten rats were selected for this study, weighing 150 to 200 g and aged between 3 and 4 months. The intraperitoneal administration of sodium pentobarbital (0.025 g/kg body weight) was used for anesthetization of rats. The hair on rat’s back was then shaved and disinfected using 5% iodine mixed with alcohol. Four incisions were surgically created in the dorsum, and subcutaneous pockets were carefully prepared to a depth of 15 mm. Two incisions were filled with the tested biocomposite scaffolds, containing Nanoclay and Nano-HA. The third incision was filled with the control material, while the fourth incision was left empty. The incisions were closed using black silk sutures. The tested rats were divided randomly into two groups of five each according to the 37-day evaluation period.
After 1 week, five rats were sacrificed by inhalation of ether. The tissues around the incision lines were excised and immersed in 10% buffered formalin and fixed for 48 h and then processed for paraffin embedding. Followed by preparation of 5-µm cut sections by a microtome, stained using hematoxylin and eosin (H&E). Prepared histological sections were examined blindly to examine response of tissue to the implanted materials. Tissue reactions were scored using the following scoring system:
Capsule | |
1 | Thin, less than 150 µm |
2 | Thick, more than 150 µm |
Inflammation | |
0 | No reaction with non or few inflammatory cells |
1 | Mild reaction with fewer than 25 inflammatory cells |
2 | Moderate reactions with inflammatory cells between 25 and 125 |
3 | Severe reaction with 125 and more cells |
Dystrophic calcification | |
0 | No dystrophic calcification |
1 | Dystrophic calcification |
The statistical analysis
The Statistical Package for Social Sciences (SPSS), version 20, was used to collect data and conduct statistical analysis. The results were expressed using frequency (number) and percentage. The Chi-square test with Yates’ adjustment was utilized to compare groups. Microsoft Excel (Microsoft Corporation, Redmond, Washington, USA) was utilized to produce representative figures. P values ≤ 0.05 were regarded as significant.
Results and discussion
The nano-HA and nanoclay characterization
The FTIR studies
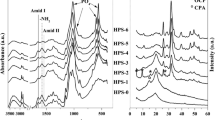
Firstly, the FTIR spectra of nano-HA ([Fig. 1(a)], Supplemental Information) showed several absorption peaks corresponding to carbonate (CO32−), phosphate (PO43−), hydroxyl (OH−), and water molecule, which are characteristic of the HA. Regarding the phosphate group, vibrational modes are observed at 2001 and 2074 cm−1, attributed to asymmetric stretching modes ν1 and ν3 of phosphate ions. Additionally, three peaks at 963, 1036, and 1098 cm−1 correspond to the ν1 symmetric and ν3 asymmetric stretching modes of P–O in phosphate ions. The group of bands at 471 cm−1, 564 cm−1, and 601 cm−1 are identified for ν2 and ν4 symmetric bending modes for O–P–O exist in phosphate ions. The appearance of those peaks in a narrow and sharp form confirms the formation of crystalline nano-HA.
For the hydroxyl groups, 632 and 3430 cm−1 are characterized for the OH–symmetric stretching mode of the HA or the water molecule. While –OH ν2 bending mode peak appeared at 1635 cm−1. For the carbonate groups, the peaks observed at 873 cm−1, 1321 cm−1, and 1417 cm−1 in the IR spectrum are attributed to components of ν2, ν1, and ν3 symmetric modes of carbonate ions.
Secondly, the stretching vibration of the –OH in water molecules is responsible for the twin peaks in the nanoclay FTIR spectra ([Fig. 1(b)], Supplemental Information), located at 1540–1640 and 3423 cm−1. The peaks that appeared at the region from 466 to 529 cm−1 are due to Si–O–Al and Si–O–Mg vibrational modes, while the peak centered at 1042 cm−1 corresponds to Si–O–Si vibrations.[16] The C–H bond, which is characteristic of the organic modifier of clay, dimethyl di-hydrogenated quaternary ammonium, is verified by the emergence of two sharp peaks presented at 2925 and 2857 cm−1. Furthermore, the peak that appeared at 1450 cm−1 may be due to the –CH2 bending vibration in the organic modifier.[17]
The zeta potential
As previously mentioned, the zeta-potential measurement directly relates to the stability of the nanoparticle dispersion.[11] The zeta potentials of the produced nano-HA and nanoclay ([Fig. 2(a and b)], Supplemental Information) were 15.59 and − 13.2 mV, respectively; these values reflect the particle’s resistance to aggregation.
The EDX measurements
The chemical composition of the prepared nano-HA and nanoclay was justified by the EDX results ([Fig. 3(a and b)], Supplemental Information). The obtained EDX pattern of nano-HA showed that it exhibited typical peaks with mass percentages of carbon, oxygen, phosphorous, and calcium at 8.33%, 36.68%, 17.18%, and 37.82%, respectively. As measured, the Ca/P ratio is 2.20, indicating a slightly high Ca content for the sample prepared, which closely aligns with what has been detected in the literature.[18] While the EDX pattern of the nanoclay provided the presence of the elements of carbon, oxygen, magnesium, aluminum, silicon, potassium, and calcium with mass percentages of 38.08%, 41.04%, 0.87%, 5.23%, 14.25%, 0.20%, and 0.33%, respectively.
The TEM imaging
The morphological analysis detected by the high-resolution transmission electron microscope (HR-TEM) for the synthesized nano-HA and the nanoclay is represented in [Fig. 1(b and c)]. The TEM images of nano-HA showed that the particles had a mostly rod-shaped morphology[19] with a particle length size around 107 nm and a width of 15–20 nm, according to the histogram calculated Fig. 4 Supplemental Information.[19] Furthermore, the Selective Area Diffraction (SAED) image (inset [Fig. 1(b)] showed the presence of ring patterns with well-defined dots conform crystalline nature of the synthesized HA. The particles appeared in definite shape, indicating their well capping from aggregation.[20] Additionally, the HR-TEM image ([Fig. 4(c)] Supplemental Information) for lattice fringes that represent the lattice crystal space showed d-spacing of 0.258 nm, 0.298 nm, and 0.348 nm, corresponding to 310, 211, and 002, respectively, that fit the XRD results discussed below. However, the TEM images of nanoclay appeared as lamellae thin sheets with many folds due to the organic modifier in the clay.

(a) FTIR spectra of the synthesized CH/Alg, CH/Alg/nano-HA, and CH/Alg/nanoclay scaffolds, (b) the HR-TEM images of synthesized nano-HA (inset its SAED, (c) the HR-TEM images of the nanoclay, and (d) the XRD pattern for nano-HA and the nanoclay.
The XRD analysis
Figure 1(c) displays the XRD patterns of nano-HA and nanoclay. The XRD graph of nano-HA showed the characteristic peaks of HA, as reported previously.[21] The main peaks appeared at 2θ of 25.54°, 31.59°, 32.52°, 33.84°, 39.43°, 46.44°, 49.28°, and 53.06°, which are indexed to (022), (211), (112), (300), (310), (312) (213), and (004), (JCPDS #09-0432) for the crystalline nature of HA.[22] The crystal size calculated using Scherrer’s equation revealed formation of particles with size about 17.5 nm that are comparable to the TEM results.[23,24,25,26,27,28] The appearance of sharp peaks in the XRD pattern implies the sample crystallinity, which may be the result of the fact that the prepared HA is very rich in Ca; this result is matched with the obtaining from the TEM results. Furthermore, it was previously[29] reported that the degree of crystallinity increased as the ratio of Ca/P increased. However, the XRD pattern of the nanoclay showed peaks identical to those detected in the literature.[30]
The TGA analysis
(Figure 5(a) Supplemental Information) displays the whole thermal TGA curve of the anno-HA, which displayed three thermal curves. The weight loss below 100°C corresponds to the vaporization of the trapped moisture water molecules. Around 261.2°C, the 2nd weight loss was due to the loss of structural water. At 334.6°C, the third curve represents the reaction between two molecules of calcium monohydrogen phosphate that produce pyrophosphate and water. The last mass loss at about 638°C is anticipated to be dehydroxylation, indicating that HA had been produced in its purest state.[31] The TGA curve for the nanoclay ([Fig. 5(b)], Supplemental Information) was somewhat resemble to the corresponding TGA findings reported earlier.[32] The curve revealed a slight weight loss peak at 100°C that is correlated to water molecule loss. The broad peak for weight loss from 165.22 to 407.6°C may correspond to the decomposition of the organic modifier. The last weight loss detected from 407.6 to 698.8°C was due to the thermal decomposition of the inorganic residue in nanoclay.
The chitosan/alginate/nano-HA and chitosan/alginate/nanoclay characterization
The FTIR studies
Figure 1(a) represents the FTIR spectra of CH/Alg, CH/Alg/nano-HA, and CH/Alg/nanoclay. The FTIR spectra of the three scaffolds are matched in general peaks. First, the peak at 3404–3424 cm−1 is assigned to the OH stretching vibrations or to the interaction between the amide group and the OH group of chitosan. Liu et al.[9] suggested that the changes in the OH group of chitosan after loading with HA may be due to the formation of hydrogen bonding between C=O groups in chitosan and OH– groups in HA. The peak seen within the range from 1637 to 1643 cm−1 is due to the C=O in the alginate or the amide group in the chitosan. In addition, the FTIR spectrum of CH/Alg/nano-HA showed major peaks similar to those of the free nano-HA, such as the phosphate group, indicating the presence of the HA phase. Furthermore, the FTIR spectrum of CH/Alg/nanoclay showed a stretching vibration peak of the Si–O network at 1037 cm−1.[33]
The sharp peak at 1559 cm−1 in the spectra of nano-HA and nanoclay scaffolds due to the amide II could be attributed to the NH bending vibrations linked to CN stretching vibrations. The section of sharp peaks in the nano-HA and nanoclay scaffolds FTIR spectra within the range from 650 to 400 cm−1 are because the phosphate groups and other metal–O groups, which provided another confirmation that linkage occurred between the building unit of the scaffold (chitosan), and the doping nanomaterials.
The SEM and EDX measurements
SEM and EDS analyses were utilized on their lyophilized sponges to ensure the real presence of nano-HA or nanoclay in the fabricated scaffold [Fig. 6(a–c)] Supplemental Information. As presented in SEM images in [Fig. 2(a)], crystalline aggregates of Ca and P, as measured by EDX, appeared on the scaffold walls, indicating the deposition of nano-HA on the surface of the CH/Alg/HP scaffold; however, the determined Ca/P ratio was close to the obtained for nano-HA only. Although the scaffold SEM images showed several spheroids of various diameters with pore sizes around 52.5 µm, as calculated from its histogram,[25] the SEM result of the Nanoclay-doped scaffolds revealed noticeable porosity microstructure with variable pore sizes and shapes, but a minimal decrease in CS/Alg/nanoclay pore sizes was detected in comparison with the CS/Alg/nano-HA scaffold. The clay particles were heavily deposited on the mouth of the scaffold, reducing the diameter of the pores 56.5 µm rather than control sample. Furthermore, the EDX analysis detected the same elements that appeared in the free nanoclay, which emphasizes the creation of a clay-doped CH/Alg scaffold. On the other hand, the control sample (CH/Alg) chemical composition detected by EDX showed only the elements (C, N, O, and Na) that make up the control scaffold. Also, the SEM images of the control sample showed a few sets of pores (pore numbers less than those of scaffolds containing HA or nanoclay) with larger pore diameters around 92 µm.

(a) Representative SEM images for the control (CH/Alg), the CH/Alg/nano-HA, and for the CH/Alg/nanoclay; (b) ions concentrations of P ion, (c) Ca ion, and (d) corresponding pH values recorded at the same intervals of immersion in PBS.
Brennanetal et al.[34] stated that pores existing in scaffolds were advantageous for the regeneration of bone tissue and could be chosen according to their pore diameter. Additionally, Brennanetal et al. proved that pores of scaffolds between 100 and 200 µm exhibit blatant osteogenic effects in vitro. Furthermore, Roosa et al.[35] recorded that scaffolds with pore diameters between 350 and 800 µm can exhibit bone tissue protruding outside the scaffold, whereas those with pore diameters under 350 µm exhibit pronounced in-stent bone growth.
The XRD analysis
The x-ray diffraction patterns for the fabricated three scaffolds ([Fig. 7(a–c)] Supplemental Information) demonstrated amorphous phases with only a few crystalline peaks. The peaks observed at 2θ range of 21° to 23.5° could be caused by the semi-crystallinity of chitosan that is generally found within this range.[36] Alginate is known for its amorphous nature. The other peaks observed are due to the incorporation of the crystalline nano-HA and nanoclay, which became evidence for their successful dispersion inside the polymer matrix. To sum up, the findings of FTIR and XRD analyses indicated that the integration of nano-HA and nanoclay into chitosan scaffolds did not cause any damage to their crystal structure.
TGA analysis
The thermogram curve for the control sample (CH/Alg) ([Fig. 8(a)] Supplemental Information) showed decomposition for mass loss at about 145.9°C anticipated for the removal of the loosely bound water in the CH/Alg, and the second degradation and terminated peak at 408°C with weight lost 44.80% may be due to the pyrolysis reaction of chitosan.
Rizwan et al. found that chitosan thermal breakdown occurred between 217 and 300°C,[37] whereas sodium alginate thermal decomposition occurred between 200 and 248°C. The thermograms of scaffolds containing nano-HA nano nanoclay ([Fig. 8(b and c)], Supplemental Information) showed their decomposition at lower temperature peaks with respect to the TGA curves of the free nanomaterials. This late decomposition may be due to the fusing of their crystalline structure by the polymer matrix.
In vitro bioactivity evaluation
Changes in PBS ions’ concentrations
Using a UV spectrophotometer, the concentrations of calcium and phosphorus in the PBS were measured at various immersion times. Figure 2 shows the P and Ca release from the prepared scaffolds, whereas [Fig. 2(b)] illustrates that the P concentration was progressively raised from the 1st day of immersion until it decreased at the point of 14 days, which is due to the precipitation of P ion on the scaffold surface. Figure 2(c) shows the calcium ion, where at the beginning, the concentration of Ca ion is the highest because of the leakage of ions from the prepared scaffolds and then the Ca concentrations decreased sharply over time until the point of 14 days that correspond to the maximum precipitation of Ca ion on the scaffold surface. These findings indicated the calcium phosphate layer was formed on the surface of the scaffold. For confirmation after this period, the scaffolds were eliminated down so that SEM and EDX investigations could be used to see the precipitations on their surfaces. Figure 2(d) shows the differences in pH value that correspond to the P and Ca release, where the increase and decrease of these ions affect the pH value. The pH increase for the two samples (CH/Alg/HA and CH/Alg/Clay) was due to the high release of Ca ion in the beginning, which is considered an alkaline metal. With time, the concentration of Ca ion decreased and P ion increased, decreasing the pH value because of the acidic nature of P.
Scaffold surface changes after soaking in PBS
The formation of a calcium phosphate layer by SEM coupled with EDX (Fig. 3) was used for analysis of the prepared scaffold. Figure 3(a) shows the surface morphology of the CH/Alg scaffold after immersion in PBS for 14 days. EDX analysis showed precipitations of Na and Cl ions on the scaffold surface. Figure 3(b) displays the image of the CH/Alg/HA surface, and EDX analysis indicated the formation of the calcium phosphate layer. The SEM and the EDX analyses shown in Figure 3(c) for the CH/Alg/clay scaffold does not show any precipitation of Ca ions. So, the best sample is CH/Alg/HA, which enhances bone regeneration.

SEM/EDX images of (a) CH/Alg, (b) CH/Alg/HA, and (c) CH/Alg/clay, after 14 days of immersion in PBS. (d) Swelling (%) of CH/Alg, CH/Alg/HA, and CH/Alg/clay.
In vitro studies
For investigation of the mass loss % of the prepared scaffolds, degradation studies were applied in PBS solution after 28 days of incubation. As shown in Fig. 9 Supplemental Information, HA and clay affected the degradation behavior, especially the HA composite scaffold showed a lower degradation. In detail, incorporating the inorganic filler in the polymer matrix decreased the porosity, decreasing the exposed area to the PBS solution, and the scaffold became more compact, subsequently decreasing the degradation rate.[38] As known, the formation and replacement of newly formed bone tissue required a low degradation rate to assess the new cells to attach and proliferate.[38] Thus, adding HA and clay to the polymer matrix enhances the proliferation and cell attachment.
Swelling studies
Swelling studies were used to detect the interconnected pores of the studied porous scaffolds by water absorption. The significance of these interconnected pores stems from their function in gaseous exchange, nutrition transportation, and cell migration.[15] Figure 3(d) reveals that the control scaffold had the highest swelling ratio at the beginning due to the porosity, and the swelling behavior of the HA and clay composite scaffolds happened slower than the control sample, consistent with the degradation results. The swelling rate would be slowed down if an inorganic filler that reduces porosity was present. It is noticed that the swelling of the HA composite scaffold happened more slowly, and this also agrees with the degradation results. With time, saturation occurred; therefore, the swelling results are partly stable.
The cytotoxicity studies
The MTT assay is an analytical method that measures the metabolic activity of cells and serves as a gauge for their cytotoxicity, viability, and proliferation. For this colorimetric assay to work, metabolically active cells must reduce a yellow tetrazolium salt (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide or MTT) to purple formazan crystals. The purple formazan was dissolved in DMSO, and the color was measured at 570 nm. In the current study, the MTT assay was used to evaluate the anti-proliferation effects of serial concentrations (3.9, 7.8, 15.6, 31.25, 62.5, 125, 250, 500, and 1000 µg/mL) from hydroxyapatite and nanoclay in Vero Normal cells. Based on cellular proliferation, Vero Normal Cells gradually proliferated after 72-h incubation with 90%, 85.9%, 77.8%, 69.5%, 65.8%, 63.8 %, 61.8%, 58.7%, and 56.2% from hydroxyapatite, respectively, and 83.3, 78.1%, 73.2%, 67.2%, 59%, 55.4%, 53.5%, 21.6%, and 12.2% from nanoclay, respectively. Moreover, the potential inhibitory effect was detected after their incubation to concentration (1000 µg), exhibiting cytotoxic activity (56.2%) in the case of hydroxyapatite and (12.2%) in the case nanoclay, respectively. Our findings confirmed that the cytotoxic activity of hydroxyapatite and nanoclay was examined at IC50 > 670 µg/mL and IC50: 116 µg/mL, respectively (Fig. 10 Supplemental Information).
Histological findings
I-Capsule
Figure 4(a–d) shows the histological findings of the capsule scoring. After 1 week, 80% of the cases of the control and nano-HA groups had a score (1), compared to 40% of the nanoclay group and 100% of the negative control group. Score (2) was present in 20% of the cases of the control and nano-HA groups, 60% of the nanoclay group cases, and absent in the negative control group. The groups’ differences were not statistically significant (p = 0.493). After a month, Fig. 11. Supplemental Information, score (1) was recorded in 100% of the cases of the control group and negative control group compared to 60% of the cases of the Nano-HA group and 40% of the nanoclay group. Score (2) was recorded in 40% of the cases of the nano-HA group compared to 60% of the cases of the Nanoclay group and was absent in the control and negative control groups. The groups’ differences were not statistically significant (p = 0.402).

(a) Showing no/few inflammatory cells in Nano-HA group after 1 month (score 0). The capsule is thin (score1, the arrow). X100, H&E; (b) showing thick capsule (the black arrow) and dense inflammatory cells (the blue arrow, more than 125, score 3) in the Nanoclay group after 1 month. H&EX400; (c) showing some edema, with few inflammatory cells (fewer than 25 cell) score 1 in the Nano-HA group after 1 week; (d) showing positive calcification stained black, VK in the Nano-HA group after 1 month, X200.
II-Inflammation
(Figure 12 Supplemental Information) shows the histological findings of the inflammation scoring. After 1 week, 60% of the cases of the control and negative control groups had a score (1) compared to 40% of the cases of the Nano-HA group. 60% of the cases of the Nano-HA and Nanoclay groups had a score (2), while only 40% of the control and negative control groups had a score (2). 40% of the cases of the nanoclay group had a Score (3). The groups’ differences were not statistically significant (p = 0.941). After 1 month, 40% of the cases of the control group and Nano-HA group had a score (0) compared to 100% in the negative control group. 60% of the cases of the control group, Nano-HA group, and nanoclay group had a score (1). 20% of the cases of the Nanoclay group had Scores (2) and (3). The groups’ differences were not statistically significant (p = 0.626).
III-Calcification
(Figure 13 Supplemental Information) shows the histological findings of the calcification scoring. After 1 week, the capsule was observed in 20% of the Nano-HA group and in 40% of the Nanoclay group but was absent in the control and negative control groups. The groups’ differences were not statistically significant (p = 0.759). After 1 month, the capsule was observed in 80% of the Nano-HA group and in 20% of the Nanoclay group but was absent in the control and negative control groups. The groups’ differences were not statistically significant (p = 0.083).
Conclusion
Biocomposite scaffolds with nano-HA and nanoclay based on chitosan/alginate have a macroporous nature, with pore sizes varying between 100 and 200 µm. The presence of nanomaterials in the polymer matrix did not affect nanomaterials’ crystallinity. The cytotoxicity effect of nano-HA is much less than that of nanoclay, and incorporating nano-HA has improved the calcium deposition on the scaffold surfaces more than nanoclay-doped scaffolds. Furthermore, tested scaffolds showed significant calcification. Adding the nano-HA to chitosan/alginate scaffolds provided a new strategy for promising scaffolds for therapeutic applications, especially bone tissue engineering.
Data availability
The data presented in this study are available upon request from the corresponding author.
References
S. Wei, J.-X. Ma, L. Xu, X.-S. Gu, X.-L. Ma, Biodegradable materials for bone defect repair. Mil. Med. Res. 7, 1–25 (2020)
G. Montalbano, G. Borciani, G. Cerqueni, C. Licini, F. Banche-Niclot, D. Janner, S. Sola, S. Fiorilli, M. Mattioli-Belmonte, G. Ciapetti, Collagen hybrid formulations for the 3d printing of nanostructured bone scaffolds: an optimized genipin-crosslinking strategy. Nanomaterials 10, 1681 (2020)
G. Chen, N. Kawazoe, T. Tateishi, Collagen-based scaffolds. Nat. Polym. Biomed. Appl. 1, 396–415 (2008)
P.J. VandeVord, H.W.T. Matthew, S.P. DeSilva, L. Mayton, B. Wu, P.H. Wooley, Evaluation of the biocompatibility of a chitosan scaffold in mice. J. Biomed. Mater. Res. Off. J. Soc. Biomater. Jpn. Soc. Biomater. Aust. Soc. Biomater. Korean Soc. Biomater. 59, 585–590 (2002)
Z. Li, H.R. Ramay, K.D. Hauch, D. Xiao, M. Zhang, Chitosan–alginate hybrid scaffolds for bone tissue engineering. Biomaterials 26, 3919–3928 (2005)
W. Nie, C. Peng, X. Zhou, L. Chen, W. Wang, Y. Zhang, P.X. Ma, C. He, Three-dimensional porous scaffold by self-assembly of reduced graphene oxide and nano-hydroxyapatite composites for bone tissue engineering. Carbon N. Y. 116, 325–337 (2017)
P. Melo, R. Naseem, I. Corvaglia, G. Montalbano, C. Pontremoli, A. Azevedo, P. Quadros, P. Gentile, A.M. Ferreira, K. Dalgarno, Processing of Sr2+ containing poly l-lactic acid-based hybrid composites for additive manufacturing of bone scaffolds. Front. Mater. 7, 601645 (2020)
M. Saleem, S. Rasheed, C. Yougen, Silk fibroin/hydroxyapatite scaffold: a highly compatible material for bone regeneration. Sci. Technol. Adv. Mater. 21, 242–266 (2020)
D. Liu, Z. Liu, J. Zou, L. Li, X. Sui, B. Wang, N. Yang, B. Wang, Synthesis and characterization of a hydroxyapatite-sodium alginate-chitosan scaffold for bone regeneration. Front. Mater. 8, 648980 (2021)
J.-H. Lin, C.-W. Lin, C.-H. Huang, C.-L. Huang, C.-W. Lou, Manufacturing technique and mechanical properties of plastic nanocomposite. Compos. Part B Eng. 44, 34–39 (2013)
A. Abdelnabi, M.K. Hamza, O.M. El-Borady, T.M. Hamdy, Effect of different formulations and application methods of coral calcium on its remineralization ability on carious enamel. J. Med. Sci. 8, 94–99 (2020)
A. Mostafa, A. Kandeil, Y.A.M.M. Elshaier, O. Kutkat, Y. Moatasim, A.A. Rashad, M. Shehata, M.R. Gomaa, N. Mahrous, S.H. Mahmoud, FDA-approved drugs with potent in vitro antiviral activity against severe acute respiratory syndrome coronavirus 2. Pharmaceuticals 13, 443 (2020)
S.K. Motwani, S. Chopra, S. Talegaonkar, K. Kohli, F.J. Ahmad, R.K. Khar, Chitosan–sodium alginate nanoparticles as submicroscopic reservoirs for ocular delivery: formulation, optimisation and in vitro characterisation. Eur. J. Pharm. Biopharm. 68, 513–525 (2008)
E. El-Meliegy, M. Mabrouk, S.A.M. El-Sayed, B.M. Abd El-Hady, M.R. Shehata, W.M. Hosny, Novel Fe2O3-doped glass/chitosan scaffolds for bone tissue replacement. Ceram. Int. 44, 9140–9151 (2018)
K. Kapat, A.P. Rameshbabu, P.P. Maity, A. Mandal, K. Bankoti, J. Dutta, D.K. Das, G. Dey, M. Mandal, S. Dhara, Osteochondral defects healing using extracellular matrix mimetic phosphate/sulfate decorated GAGs-agarose gel and quantitative micro-CT evaluation. ACS Biomater. Sci. Eng. 5, 149–164 (2018)
F.H. Elmergawy, M.S. Nassif, O.M. El-Borady, M. Mabrouk, D.I. El-Korashy, Physical and mechanical evaluation of dental resin composite after modification with two different types of Montmorillonite nanoclay. J. Dent. 112, 103731 (2021)
A.K. Behera, C. Mohanty, Fabrication and characterization of nano-biocomposites using natural fiber and soy resin. Mater. Today Proc. 47, 1183–1188 (2021)
J.K. Abifarin, D.O. Obada, E.T. Dauda, D. Dodoo-Arhin, Experimental data on the characterization of hydroxyapatite synthesized from biowastes. Data Br. 26, 104485 (2019)
T.S. Roy, S. Akter, M.R. Fahim, M.A. Gafur, T. Ferdous, Incorporation of Ag-doped ZnO nanorod through graphite hybridization: effective approach for degradation of ciprofloxacin. Heliyon 9, e13130 (2023). https://doi.org/10.1016/j.heliyon.2023.e13130
M. Bagum, S. Islam, E. Khan, J. Khandaker, F. Ahmed, Degradation of metronidazole from aqueous environment using hydrothermally synthesized ZnO, N-doped ZnO, and ZnO/AC nanoparticles. Adv. Condens. Matter Phys. (2023). https://doi.org/10.1155/2023/8706698
A. Chandrasekar, S. Sagadevan, A. Dakshnamoorthy, Synthesis and characterization of nano-hydroxyapatite (n-HAP) using the wet chemical technique. Int. J. Phys. Sci. 8, 1639–1645 (2013)
J.-K. Han, H.-Y. Song, F. Saito, B.-T. Lee, Synthesis of high purity nano-sized hydroxyapatite powder by microwave-hydrothermal method. Mater. Chem. Phys. 99, 235–239 (2006)
M. Ahmed, S. Islam, M. Bashar, M.A. Hossain, F. Ahmed, Synthesis and characterizations of CH3NH3PbI3: ZnS microrods for optoelectronic applications. Adv. Mater. Sci. Eng. 2022, 1–14 (2022). https://doi.org/10.1155/2022/7606339
M. Ahmed, S. Islam, M. Bashar, F. Ahmed, One-step synthesis of (CH3NH3)2CuCl4:ZnS thin film for optoelectronic applications. Cryst. Res. Technol. 57, 2100235 (2022). https://doi.org/10.1002/crat.202100235
M.T. Ahmed, S. Islam, F. Ahmed, Synthesis of (CH3NH3)2CuCl4 nanoparticles by antisolvent engineering. J. Cryst. Growth 587, 126637 (2022). https://doi.org/10.1016/j.jcrysgro.2022.126637
M.T. Ahmed, S. Islam, F. Ahmed, Optical and electrical properties of perovskite microrods. Materwiss. Werksttech. 53, 790–797 (2022)
M. Chand, S. Islam, M. Ahmed, M. Bashar, F. Ahmed, Synthesis and characterization of lithium-doped copper zinc tin sulfide (CZTS) thin films. AIP Adv. 13, 125225 (2023). https://doi.org/10.1063/5.0173862
M. Ahmed, S. Islam, F. Ahmed, Structural, vibrational, and electronic properties of γ-graphyne, γ-graphyne oxide, and boron nitride doped γ-graphyne: a DFT study. AIP Adv. 12, 125110 (2022). https://doi.org/10.1063/5.0129855
G.M. Poralan, J.E. Gambe, E.M. Alcantara, R.M. Vequizo, X-ray diffraction and infrared spectroscopy analyses on the crystallinity of engineered biological hydroxyapatite for medical application. IOP Conf. Ser. Mater. Sci. Eng. 79, 12028 (2015)
P. Kumar, K.P. Sandeep, S. Alavi, V.D. Truong, R.E. Gorga, Effect of type and content of modified montmorillonite on the structure and properties of bio-nanocomposite films based on soy protein isolate and montmorillonite. J. Food Sci. 75, N46–N56 (2010)
M.R. Senra, R.B. de Lima, D.H.S. de Souza, M.F.V. de Marques, S.N. Monteiro, Thermal characterization of hydroxyapatite or carbonated hydroxyapatite hybrid composites with distinguished collagens for bone graft. J. Mater. Res. Technol. 9, 7190–7200 (2020)
K.E. Strawhecker, E. Manias, Structure and properties of poly (vinyl alcohol)/Na+ montmorillonite nanocomposites. Chem. Mater. 12, 2943–2949 (2000)
E.A. Naumenko, I.D. Guryanov, R. Yendluri, Y.M. Lvov, R.F. Fakhrullin, Clay nanotube–biopolymer composite scaffolds for tissue engineering. Nanoscale 8, 7257–7271 (2016)
C.M. Brennan, K.F. Eichholz, D.A. Hoey, The effect of pore size within fibrous scaffolds fabricated using melt electrowriting on human bone marrow stem cell osteogenesis. Biomed. Mater. 14, 65016 (2019)
S.M.M. Roosa, J.M. Kemppainen, E.N. Moffitt, P.H. Krebsbach, S.J. Hollister, The pore size of polycaprolactone scaffolds has limited influence on bone regeneration in an in vivo model. J. Biomed. Mater. Res. Part A Off. J. Soc. Biomater. Jpn. Soc. Biomater. Aust. Soc. Biomater. Korean Soc. Biomater. 92, 359–368 (2010)
J.A. Sowjanya, J. Singh, T. Mohita, S. Sarvanan, A. Moorthi, N. Srinivasan, N. Selvamurugan, Biocomposite scaffolds containing chitosan/alginate/nano-silica for bone tissue engineering. Colloids Surf. B Biointerfaces 109, 294–300 (2013)
M. Rizwan, K.M. Zia, M.A. Javaid, M. Zuber, W. Aftab, S. Rehman, Synthesis and molecular characterization of chitosan/alginate blends based polyurethanes biocomposites. Int. J. Biol. Macromol. 180, 324–331 (2021)
M. Peter, N.S. Binulal, S.V. Nair, N. Selvamurugan, H. Tamura, R. Jayakumar, Novel biodegradable chitosan–gelatin/nano-bioactive glass ceramic composite scaffolds for alveolar bone tissue engineering. Chem. Eng. J. 158, 353–361 (2010)
Acknowledgments
The authors Acknowledge Ass. Prof. Dr. Nemany A.N.Hanafy and Miss. Hanaa El Borolsy, Nanomedicine department, Nanoscience and Nanotechnology, Kafrelsheikh University for his help in the in vivo part.
Funding
The author also acknowledges the Science Technology & Development Fund (STDF), Egypt, Grant No: 43450 for funding this manuscript.
Author information
Authors and Affiliations
Contributions
Conception: G.A., S. A.M. El-S., O.M. El-B., H.S.A. E., M.E. K, and M. El-K.; Data curation; G.A., S. A.M. El-S., O.M. El-B., H.S.A. E., and M.E. K.; Formal analysis: A.M. El-S., and O. M. El-B.; Investigation: G.A., S. A.M. El-S., and O.M. El-B.; experimental design: G.A., S. A.M. El-S., O.M. El-B., H.S.A. E., M.E. K, and M. El-K; Supervision: O.M. El-B., H.S.A. E., M.E. K, and M.; Visualization: G.A., S. A.M. El-S., O.M. El-B., H.S.A. E., M.E. K, and M. El-K.; Writing: G.A., S. A.M. El-S., O.M. El-B., H.S.A. E., M.E. K, and M. El-K.
Corresponding author
Ethics declarations
Conflicts of interest
The authors declare that there is no conflict of interest.
Ethical approval
The protocol of the in vivo study was followed the guidelines of the ethical committee of the National Research Center, Egypt (Approval no. 19/248).
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Ali, G., El-Sayed, S.A.M., El-Borady, O.M. et al. Nanoclay and nano-hydroxyapatite chitosan-based scaffold for bone regeneration: Morphological, spectral, structural, thermal, in vitro, and in vivo studies. MRS Communications 14, 699–710 (2024). https://doi.org/10.1557/s43579-024-00607-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1557/s43579-024-00607-7