
Abstract
Pulse electrodeposition is utilized to prepare Co–Fe–P metallic nano-glasses (NGs) containing glassy grains with an average size D < 100 nm. The effects of glass–glass interfaces (GGIs) on thermodynamic and mechanical properties of NGs are systematically studied. The glass transitions in NGs as characterized by differential scanning calorimetry and dynamic mechanical analysis show that they are strongly affected by D or the volume fraction of GGIs. In comparison with those for conventional metallic glasses, a predominant internal-friction peak corresponding to the glass transition is observed for NGs with reduced D, suggesting that the GGIs are in a glassy state much different with that of glassy grains in NGs. Nanoindentation measurements on the hardness and volume of shear transformation zones reveal that the GGIs could accommodate plastic strains in NGs, where homogeneous plastic deformation occurs.
Graphical abstract

Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Metallic glasses (MGs) are classified as one of the disordered alloys and have attracted much attention over decades. Due to the disordered atomic structure, their mechanical properties, such as high yield strength [1, 2] and large elastic limit [3], can be easily reproduced. Up to now, numerous kinds of MGs are available and most of them are synthesized through rapid quenching from their molten state. However, due to the lack of microstructural defects, such as grain boundaries, their properties are mainly tuned by varying the chemical compositions. Thus, in comparison with the wide-spread structural applications of crystalline alloys, the practical application of MGs is extremely limited.
Recently, two-dimensional defects, specifically defined as glass–glass interfaces (GGIs) that separate glassy nano-sized grains or MG blocks, have been introduced into MGs [4,5,6,7], resulting in a different kind of nanomaterials called metallic nano-glasses (NGs) [8], and a new approach in modifying the properties of MGs. The GGIs can enable NGs with electronic [9], mechanical [10,11,12,13], and magnetic [14,15,16,17,18] properties much different with those of conventional MGs. Based on Mossbauer spectroscopy measurements, Ghafari et al. [9] found that the contribution of itinerant electrons at GGIs to magnetic moments is higher than those in conventional MGs. Wang et al. [19] suggested that the shear transformation zones prefer to initiate and propagate inside the soft GGIs with excess free volumes, avoiding the localized plastic deformation and thus enhancing the plasticity of Sc75Fe25 NGs. Witte et al. [20] reported that Fe90Sc10 NGs are ferromagnetic at room temperature although Fe10Sc10 MGs are paramagnetic because of the unique electronic structure at GGIs. In other words, a glassy region [21] different with the interiors of glassy grains, which could be in an energetically stable state, exists in NGs [22,23,24]. Nevertheless, the complete knowledge of the glassy phases of GGIs is still lacking, and needs further investigation.
Since the GGIs can provide NGs with extraordinary properties that generally do not exist in MGs, it would be meaningful to investigate the effects of GGIs on thermodynamic properties of NGs comprehensively. To date, there are only a few studies that reveal the thermodynamic properties of NGs. For examples, Mohri et al. [25] reported that the glass transition temperature (Tg) in Ti34Zr14Cu22Pd30 NGs is lower than those of its bulk MG counterparts; Nandam et al. [26] found that with the existence of GGIs the Tg is increased in Zr50Cu50 NGs, while it is decreased in Pd80Si20 NGs [27], as compared to those of conventional MGs. It is worth noting that the sample size effects on Tg in glassy polymers are well established [28,29,30,31]. In contrast, a comprehensive study on the effects of sizes of glassy grains (and correspondingly the volume fractions of GGIs) on Tg of NGs, is currently unavailable.
Molecular dynamics simulations have played an important role in investigating the mechanical properties of NGs. In comparison, experimental approach is mostly achieved through compression tests on a few NGs. For instance, Franke et al. [32] found that the propagation of shear bands could be inhibited in Sc75Fe25 NGs, which may offer extra plasticity. Wang et al. [19] suggested that formation of multiple shear bands could be the dominant deformation mechanisms in Sc75Fe25 NGs under compression, rather than localized shear banding. Guo et al. [12] reported that the creep plasticity in Ni78P22 NGs is more pronounced through nanoindentation measurements. Although the obtained experimental results are consistent with those simulated by molecular dynamics [33,34,35,36,37,38,39], unlike the sample size effects on the plasticity of MGs [1, 2, 40, 41], the experimental results that reveal the effects of sizes of glassy grains on mechanical behaviors of NGs, are yet incomplete.
Most of NGs are successfully fabricated through inert gas condensation [42, 43], magnetron sputtering [44, 45] and severe plastic deformation [46, 47]. However, the main technological barrier to the practical application of NGs still exists due to the fact that the above-mentioned method cannot economically produce NGs with dimensions larger than centimeters. Recently, electrodeposition as a facile manufacturing route has been utilized to prepare large-size NGs [48], which is well known as an alternative method that economically produces metallic materials with various sizes and shapes for different applications. So far, only very few studies have been reported in the preparation of NGs by the electrodeposition method, and the development of other NG systems through this technique becomes critical for their technological application.
In this work, a new Co–Fe–P NG system with the sizes of glassy grains smaller than 100 nm, prepared through pulse electrodeposition, is reported. The nanostructure of NGs is subsequently adjusted by heat treatment, and the effects of sizes of glassy grains on thermodynamic and mechanical properties are studied by differential scanning calorimetry (DSC), dynamic mechanical analysis (DMA) and nanoindentation. The results suggest that GGIs are in a glassy state different with that of the interiors of glassy grains. Compared with Co–Fe–P MGs, the nanostructured Co–Fe–P sample is found to exhibit different thermodynamic and mechanical behaviors due to the existences of GGIs.
Results and discussion
The schematic of fabrication of Co–Fe–P thick-film sample is illustrated in Fig. 1a. A specific Teflon mold is designed for depositing specimens with a rectangular shape over a titanium substrate. For the micro-structured specimens, they are synthesized by direct-current (DC) electrodeposition with a working potential of 0.95 V, whereas the nano-structured specimens are synthesized by pulse electrodeposition with various pulse parameters. The nano- and micro-structured specimens are denoted as x-NG and x-MG, respectively, where x refers to the averaged size D of glassy grains as estimated by calculating the mean size of at least 20 grains. The calculation of D is based on the scanning electron microscopy (SEM) images on the cross-sections or surface morphology of x-NG or x-MG, respectively [25]. The huge difference in D of x-NG and x-MG is attributed to different deposition mechanisms in the electroplating process. During the electrodeposition of Co–Fe–P films, Fe2+, Co2+ and H2PO2− ions initially gather on the titanium substrate. The redox reactions of H2PO2− with lower energy barrier first occur and P clusters are thereafter formed, in which the Co and Fe atoms are most likely to deposit; then, Co2+ and Fe2+ cations agglomerate at different sites of P clusters, forming the primitive nano-sized Co–Fe–P glassy clusters [3]. Compared with DC electrodeposition, the pulse electrodeposition can efficiently suppress the growth of nano-sized glassy clusters. By lowering the duty cycle, the ion source from the electrolyte for the growth of Co–Fe–P glassy clusters is reduced. Meanwhile, the increase in pulse frequency can help in replenishing ions near the substrate more often, which is beneficial to the nucleation of glassy clusters under large current density or high working potential. Therefore, D of x-NG prepared by pulse electrodeposition is generally much smaller than that of x-MG prepared by DC electrodeposition.

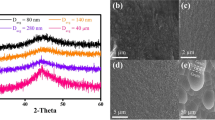
(a) Schematics of preparation of Co–Fe–P samples. (b) XRD patterns and FWHM (degree) of XRD peak for Co–Fe–P samples with different averaged sizes of glassy grains D.
The X-ray diffraction (XRD) patterns of exfoliated Co–Fe–P thick films are shown in Fig. 1b. In the XRD patterns, a typical broad peak can be observed at \(2\theta\) ~ 45° without any sharp crystalline peak, which demonstrates the fully glassy structure of the Co–Fe–P samples. Furthermore, it can be found that the peak at 45° is gradually flattened when the average size of glassy grains is reduced to below 1.0 micron. After subtracting the background, the full width at half maximum (FWHM) of the corresponding peak increases from 5° to 7°. Therefore, it is evident that the Co–Fe–P samples could be in different structures characterized by the averaged size of glassy grains D, as shown in Fig. 2a and b. Unlike nano-structured samples, the micro-structured samples do not exhibit any sub-structures inside the micron-sized grains. The uneven and rough surfaces of glassy grains shown in Fig. 2b could be caused by the low overpotential applied in DC electrodeposition. During the fabrication processes, electric fields around the surfaces of grains may be heterogeneous, where some deposited in contact with the electrolyte may be reversely dissolved into the electrolyte. Based on the energy dispersive spectroscopy (EDS) analysis, the chemical compositions of Co–Fe–P samples are determined to be Co65Fe14P21. The EDS elemental mapping shows that all three elements are homogeneously distributed in the films, suggesting that the Co65Fe14P21 film is dense and has fully disordered atomic structure. In addition, transmission electron microscopy (TEM) images of 75 nm-NGs are illustrated in Fig. 3. It could be seen that glassy grains with sizes less than 10 nm are formed. The selected area electron diffraction (SAED) patterns with halo rings further confirm the amorphous state of nano-structured samples.

SEM images of as-prepared Co–Fe–P samples (a) 75 nm-NGs in the cross sections, and (b) 50 \({\upmu \mathrm{m}}\)-MGs at the surfaces, prepared by pulse and DC electrodepositions, respectively.

TEM images and SAED patterns of 75 nm-NGs.
In addition, the effects of heat treatment and working potential of electrodeposition on the microstructure of Co–Fe–P samples are studied. Different annealing time (at ~ 473 K) and working potentials are chosen in this work. As shown in Fig. 4a–c, D of NG samples gradually increases with increasing annealing time. The mechanisms of growth of glassy grains may be similar with those of polycrystalline materials. It could be caused by the increased kinetic energy of GGI atoms at elevated temperatures that leads to their diffusions into adjacent glassy grains. As a result, the excess free volumes at GGIs will be reduced. During the heat treatment, glassy grains with small sizes may be joined together and new glassy grains with larger sizes will eventually reduce the total areas of GGI and the free energy of the sample.

SEM images of cross sections (a)–(e) or surface morphology (f) of Co–Fe–P samples under different heat treatment conditions and pulse electrodeposition parameters. (a) The 75 nm-NGs prepared with a working potential of 3.8 V; (b) The 75 nm-NGs annealed for 5 h; (c) the 75 nm-NGs annealed for 8 h. The Co–Fe–P films prepared by pulse electrodeposition with a working potential of 3.8 V (d), 1.9 V (e), and 1.0 V (f).
Similarly, by altering the working potential which consists of a constant voltage (between 1.0 and 3.8 V) and zero voltage in a cycle (a period of 0.0001 s), D can be also varied, as shown in Fig. 4d–f. To be specific, glassy grains with lower D are more likely to be formed under high working potentials. If the working potential is fixed at above 1.9 V, glassy grains with D < 100 nm would be formed in Co–Fe–P samples. During the pulse electrodeposition, an increase in working potential can induce a larger current density, which provides more embryonic sites for the formation of glassy clusters in a short time. Meanwhile, the growth of each site would be greatly constrained if a large quantity of glassy clusters exist. Therefore, the working potentials is maintained between 1.9 to 3.8 V for the fabrication of Co–Fe–P NGs during pulse electrodeposition.
The relation between D and annealing time is presented in Fig. 5a, and the volume fraction of GGIs is also shown, assuming that the Co–Fe–P samples contain spherical glassy grains and GGIs with a width as high as 1.0 nm [6,7,8]. It can be seen that D will increase rapidly from 75 to 125 nm if the films are annealed for 1 h. When D reaches 300 nm, additional annealing time becomes no longer effective in increasing D. The average size of glassy grains does not change much upon further heat treatment. The relation between D and working potential is depicted in Fig. 5b. At a working potential of 1.0 V, D is as large as 50 \(\upmu{\mathrm{m}}\) and the volume fraction of GGIs is about 0.03%, suggesting that the microstructure of Co–Fe–P samples prepared by pulse electrodeposition is similar with those fabricated through DC electrodeposition. In contrast, 75 nm-, 250 nm-, 400 nm- and 500 nm-NGs with estimated GGI volume fractions of 13.8%, 4.58%, 2.91% and 2.34% can be obtained at working potentials of 3.8 V, 3.0 V, 2.6 V and 1.9 V, respectively. Thus, with a combination of different annealing time and working potentials, it is evident that Co–Fe–P samples with designated D can be fabricated for the investigation on the effects of sizes of glassy grains D on thermodynamic and mechanical properties of NGs.

GGIs volume fraction and D of Co–Fe–P samples obtained under (a) different annealing time, and (b) working potentials.
Thermodynamic properties of Co–Fe–P samples are firstly investigated by DSC. Typical heat-flow curves at a heating rate of 5 K/min for 75 nm-NGs and 50 \(\upmu{\mathrm{m}}\)-MGs are shown in Fig. 6a. Tg is defined as the onset temperature of endothermic heat flow, and the crystallization temperature (Tx) is located with the sharp exothermic peak in the curve. The apparent activation energies (Ea) for the grain growth (above the temperature Tgrowth) and crystallization processes are determined using the Kissinger equation written as:

(a) DSC heat-flow curves for Co–Fe–P samples; those close to Tg are magnified in the inset. (b) Plots of Kissinger equation for the crystallizations in 75 nm-NGs and 50 \(\upmu{\mathrm{m}}\)-MGs at a heating rate of 5 K/min. (c) The effects of D on Tg and Tc of Co–Fe–P samples at 5 K/min. (d) The enthalpy change in glass transition of Co–Fe–P samples with different D at 10 K/min. The inset is the plots for Eq. (2) that determines \(\Delta {\mathrm{H}}_{I}\) (the slope of fitting straight line).
where \(\beta\) is the heating rate, T is Tgrowth or Tx, R is the universal gas constant and C is a constant.
Figure 6b presents the illustration plots of Eq. (1) for the calculation on Ea for the crystallization transitions in 75 nm-NGs and 50 \(\upmu{\mathrm{m}}\)-MGs. The enthalpy change in glass transition (\(\Delta {H}_{g}\)= Hsc − Hg) and that in crystallization transition (\(\Delta {H}_{x}\)= Hsc − Hc) for Co–Fe–P films are determined by taking the integrations of heat flow throughout the endothermic process, and the exothermic peak for crystallization, respectively, where Hg, Hsc and Hc are the enthalpies of glass, supercooling liquid and crystalline states, respectively. Table 1 lists the results of thermodynamic properties for 75 nm-NGs and 50 \(\upmu{\mathrm{m}}\)-MGs under different heating rates.
As it can be seen in Fig. 6c, Tx of Co–Fe–P NGs generally decreases with decreasing D. To be specific, Tx are 646 K, 656 K, 658 K and 660 K for Co–Fe–P samples with D = 75 ± 9 nm, 300 ± 32 nm, 400 ± 32 nm and 50 ± 7 \(\upmu{\mathrm{m}}\), respectively. As listed in Table 1, although the 75 nm-NG sample has an activation energy of crystallization (Ea) very close to that of 50 \(\upmu{\mathrm{m}}\)-MGs, it has a much smaller \(\Delta {\mathrm{H}}_{x}\) than the 50 \(\upmu{\mathrm{m}}\)-MG sample. The lower Tx and smaller \(\Delta {H}_{x}\) of Co–Fe–P sample with reduced D could be attributed to its increased volume fraction of GGIs (Fig. 5a). In the GGIs of 75 nm-NG sample, there are more excess free volumes which may become nucleation sites at elevated temperatures, hence, the crystallization is more likely to occur in the supercooling-liquid region of GGIs at a lower Tx, and the supercooling-liquid state with a lower enthalpy Hsc leads to a smaller \(\Delta {\mathrm{H}}_{x}\) for the crystallization.
Similarly, Tg of Co–Fe–P NGs decreases with decreasing D, which is 505 K, 508 K, 510 K and 512 K for Co–Fe–P sample with D = 75 ± 9 nm, 300 ± 32 nm, 400 ± 32 nm and 50 ± 7\(\upmu{\mathrm{m}}\), respectively. In previous work on glassy polymers [28,29,30,31], Tg is found to decrease with decreasing sample size, resulting from the sample surfaces with additional free volumes favorable for a lower Tg. In the Co–Fe–P NGs, excess free volumes provided by GGIs can also reduce Tg as compared with conventional MGs. Figure 6d shows the strong size effect on \(\Delta {\mathrm{H}}_{g}\) of Co–Fe–P samples with the reduced D < 1 μm, attributing to the increased volume fraction of GGIs that have a lower enthalpy Hg than conventional MGs. Assume the GGI is in a glass state that has an enthalpy change in glass transition of\(\Delta {H}_{I}\), which is different with that (\(\Delta {H}_{0 })\) of adjacent nano-sized glassy grains, \(\Delta {\mathrm{H}}_{g}\) of Co–Fe–P samples can be expressed as
where x is the volume fraction of GGIs. Since x = 0.03% for the Co–Fe–P sample with D = 50 \(\upmu{\mathrm{m}}\) is close to zero, \(\Delta {H}_{0}\) could be approximated as \(\Delta {\mathrm{H}}_{g}\)=20.4 J/g of the sample. The inset in Fig. 5d shows the plots of \({\Delta H}_{g}-(1-\mathrm{x})\Delta {\mathrm{H}}_{0}\) versus x, demonstrating that \(\Delta {\mathrm{H}}_{I}\) could be determined by using Eq. (2) and the value is 120.8 J/g. Since \(\Delta {\mathrm{H}}_{I}\) of glass state in the GGI region is much higher than \(\Delta {H}_{0}\), it is concluded that the glass phase of GGI is more thermodynamically stable as compared with that of glassy grains.
The glass transition in Co–Fe–P NGs is furthered studied by DMA. The internal friction (IF) or mechanical loss (Q−1) and storage modulus (E’) of Co–Fe–P samples are shown in Fig. 7. The results well characterize the evolution of microstructures and structural transitions in the samples at elevated temperatures, i.e., the growth of glassy grains, glass transition and crystallization that occur at T’growth, T’g and T’x, marked as the peak temperatures of typical IF peaks, respectively. The values of those peak temperatures for 75 nm-NGs and 50 m\(\upmu\)-MGs are listed in Table 2, in consistent with those determined by DSC. Remarkably, although T’g = 524 K for 75 nm-NGs is close to that of 521 K for 50 \(\upmu\)m-MGs, their internal friction and storage modulus during the glass transition are much different. The storage modulus of 50 \(\upmu\)m-MG is significantly reduced or softened from 50 to 20 GPa when its glass state transforms into a supercooling liquid state, while that of 75 nm-NG is reduced from 22 to 18 GPa. The results reveal two important features of GGIs, as follows: First, the GGIs as glassy structures could have much lower shear modulus as compared with that of conventional MGs. That is the reason why 75 nm-NG with a large volume fraction of GGIs would have a much smaller storage modulus than 50 \(\upmu\)m-MGs whose volume fraction (x = 0.03%) of GGIs can be neglected. Second, the glass state of GGIs is more disordered in atomic structures than that of conventional MGs, reflecting by the much less significantly elastic softening during the glass transition in 75 nm-NG. This structural characteristic of GGIs was also proved by previously reported work of Mossbauer spectroscopy [9] and positron annihilation [49] analyses.

The mechanical loss (Q−1) and storage modulus (E’) of Co–Fe–P samples (top: 50 \(\upmu\)m-MGs; bottom: 75 nm-NGs) at elevated temperatures under 0.8 Hz testing frequency.
By subtracting the background of internal friction, the intensity of IF peak for glass transition (Q−1 = 0.032) of 75 nm-NGs can be found to be much larger than that (Q−1 = 0.025) of 50 \(\upmu\)m-MGs, suggesting that the transition from the glass state to supercooling liquid state in the GGI region contributes significantly to the internal frictions of NGs during the glass transition processes. The predominant IF peak of glass transition in 75 nm-NGs can be explained by the large \(\Delta {\mathrm{H}}_{I}\) of glass state in the GGI region as determined from the aforementioned DSC results, as well as the large volume fraction of GGIs in 75 nm-NGs. Such strong IF peak of glass transition in NGs could clearly indicate that GGIs could be in a glass state much different with that of glassy grains in NGs.
In the measurement on elastic modulus and hardness of NGs at room temperature, different peak loads from 5 to 10 mN are applied to avoid experimental errors. The bottom surfaces of samples are used for nanoindentation tests, as exfoliated from the Ti substrate which had been well polished by 0.25 μm-diamond polishing paste to have a mirror surface before electrodeposition. The thickness of specimens is larger than 50 μm and the contact areas of each individual indent are ~ 4.23 µm2, ~ 2.69 µm2, ~ 2.51 µm2 and ~ 2.22 µm2 for samples with D = 75 nm, 300 nm, 400 nm and 50 μm, respectively. As it can be seen in the inset of Fig. 8a, the obtained reduced modulus (Er) and hardness (H) of sample with D = 75 nm are found to be dependent on the peak load once it is lower than 7000 μN. Similarly, Er and H of samples with different D are found to be dependent on the peak load lower than about 7000 μN. It is suggested the microstructures of as-deposited samples might not be in their equilibrium states after heat treated at 473 K for 2 h. Therefore, to avoid the effects of thermally driven relaxation of microstructures on the measurements on Er and H, a peak load of 10,000 μN is applied, and the relations between Er and H and D of Co–Fe–P samples are shown in Fig. 8a. As expected, both Er and H are significantly reduced in Co–Fe–P samples with D < 500 nm, mainly resulting from the increased volume fraction of GGIs where there are excess free volumes. It is noted that the relation between H and D does not follow the Hall–Petch relation that well describes the grain-size effects on mechanical strength in polycrystalline materials. It could be also caused by the increased volume fraction of GGIs in NGs with reduced D, and GGIs have much less mechanical strength than conventional MGs. The results are consistent with those reported from molecular dynamics simulation [37, 39, 50,51,52,53,54,55].

(a) Size effect on elastic modulus Er and hardness H. The inset shows data under different peak loads (Pmax) for 75 nm-NGs. (b) Load vs depth curves for Co–Fe–P samples with different D. The inset shows the magnified pop-in curve. (c) Typical plots of creep curves and fits; the inset shows the calculation of strain rate sensitive (SRS) factor m. (d) The effects of D on STZ volumes and SRS factors.
The curves of indentation force versus depth (h) are presented for Co–Fe–P samples, as shown in Fig. 8b. The first pop-in can be found at a contact depth hc close to 200 nm at the curve for 50 \(\upmu{\mathrm{m}}\)-MGs, indicating the formation of shear bands. By reducing D to 400 nm, the first pop-in shifts to around hc = 350 nm for 400 nm-NGs. Furthermore, no pop-in can be found in the curve for 75 nm-NGs with h < 500 nm. In general, the appearance of pop-ins can be attributed to the formation and propagation of a localized shear band in MGs. The increase in hc with decreasing D indicates the mechanical strength is decreased in NGs with reduced D, in consistent with the results on hardness as described above. Thus, it is indicative that shear banding might be restricted to occur in softer NGs with D < 100 nm [27, 37].
The effects of D on the deformation behaviors of Co–Fe–P samples are further investigated by creep behavior as measured by the time dependent indentation depth h(t). Typical results and fitting curve for 75 nm-NGs are shown in Fig. 8c. For a Berkovich tip, h(t) can be fitted using the following empirical equation:
where ho is the initial contact depth, to is initial time and a, b or k are the fitting constants. The relation between hardness H and strain rate \(\dot{\upepsilon }\) can be determined when the parameters in Eq. (3) are obtained from the fittings. Subsequently, the strain rate sensitivity (SRS) factor m is expressed as:
Moreover, the volume (\(\Omega )\) of shear transformation zone (STZ) can be estimated according to Johnson’s [56] and Pan’s [57] models, as follows:
where kB is Boltzmann constant, T is temperature, \({C}^{\mathrm{^{\prime}}}=2{\mathrm{G}}_{0}\mathrm{R\zeta }{\upgamma }_{\mathrm{c}}^{2}\sqrt{1-{\uptau }_{\mathrm{CT}}/{\uptau }_{\mathrm{c}}}/\sqrt{3}{\uptau }_{\mathrm{c}}, R\approx 0.25,\upzeta \approx 3, {\upgamma }_{\mathrm{c}}=0.027, {\mathrm{G}}_{0}/{\uptau }_{\mathrm{c}}\approx 27.78\mathrm{ , }{\uptau }_{\mathrm{CT}}/{\uptau }_{\mathrm{c}}=1-(\frac{0.016}{0.036}){(T/{T}_{\mathrm{g}})}^{2/3}\). Figure 8d shows the SRS factors and \(\Omega\) for Co–Fe–P samples with different D. As it can be seen, larger STZ volume can be initiated in Co–Fe–P samples with smaller D. For 75 nm-NGs, the STZ volume can be as high as 7.81 nm3, as compared to 4.40 nm3 and 3.64 nm3 for 300 nm- and 400 nm-NGs. At the same time, the STZ volume for 50 \(\upmu{\mathrm{m}}\)-MGs is as low as 2.86 nm3. By assuming a spherical STZ, its radius would be around 1.23 nm in 75 nm-NGs, which is close to the width of GGI regions. An increase of STZ volume can be well explained by the previous molecular dynamics studies on NGs [37]. It has been demonstrated that a much higher amount of STZ embryos can be initiated with a lower energy barrier in NGs, due to the softness of GGIs. Those embryos are also found to be uniformly distributed throughout the specimen without localization in a specific zone which occurs in MGs. Due to the large quantity and uniform distribution of STZ embryos without the formation of a localized shear band after passing the yield point, STZ volumes in NGs are thus much higher than those in MGs, which would eventually cause an ultra-high STZ volume of 7.81nm3 and the absence of pop-ins in 75 nm-NGs. Therefore, compared with 50 \(\upmu{\mathrm{m}}\)-MGs, it is evidential that the GGIs with high volume fractions in Co–Fe–P NGs may act as nucleation sites for the formation of STZs and accommodate plastic strains across the nanostructures, resulting in homogenous deformation in Co–Fe–P NGs.
Conclusions
In summary, Co–Fe–P NGs with different averaged sizes D of glassy grains have been successfully prepared by pulse electrodeposition. The effects of GGIs on the thermodynamic and mechanical properties of NGs are investigated by DSC, DMA and nanoindentation. It is found that the GGIs have a larger enthalpy change in glass transition and more predominant internal-friction peak for glass transition, while they have lower elastic modulus and hardness, than MGs containing microstructures with D > 50 μm. The results indicate that GGIs could be in a glass state much different with that of glassy grains or conventional MGs. Based on the STZ volume as estimated from nanoindentation measurements, it is suggested that the GGIs with high volume fractions in Co–Fe–P NGs may act as nucleation sites for the formation of STZs and accommodate plastic strains across the nanostructures, resulting in homogenous deformation in the Co–Fe–P samples.
Methodology
The Co–Fe–P samples were prepared by electrodeposition on the working electrode, i.e., titanium, via a conventional three-electrode cell system. Graphite rod was used as the counter electrode. Saturated calomel electrode was utilized to measure and adjusted the working potential during electrodeposition. Analytical grade chemical reagents were used for the preparation of electrolytic solution and was kept at 333 K during the electroplating process. The composition of the electrolyte was summarized as follows: FeSO4·7H2O 0.06 mol/L, CoSO4·7H2O 0.04 mol/L, C6H5Na3O7·2H2O 0.2 mol/L, H3BO3 0.5 mol/L and NaH2PO2·H2O 0.2 mol/L. Boric acid was used as PH buffer and concentrated sulfuric acid was added to adjust the solution PH down to 3–4. For the fabrication of Co–Fe–P films containing microstructures, direct current electrodeposition was used. The working potential was kept at 0.95 V and the deposition time was varied from 3 to 7 h. Pulse electrodeposition was employed to prepare Co–Fe–P NGs. The duty cycle of the pulse was kept at 15% and the pulse frequency of 100 kHz was applied. The working potential could be changed from 1.9 to 3.8 V and the fabrication process could take as long as 48 h. Teflon mold was designed to deposit Co–Fe–P films with a rectangular shape (35 mm × 3.5 mm × 0.1 mm). The as-prepared Co–Fe–P films were peeled off mechanically. Heat treatments of the as-prepared Co–Fe–P samples were performed in a tube furnace at 473 K in vacuum for 2 h to avoid possible oxidation.
The crystal structure of Co–Fe–P samples were characterized by X-ray diffraction (XRD, Rigaku Smartlab) using a 9 kW X-ray source (λ = 0.154 nm). Scanning electron microscopy (SEM, TESCAN VEGA3) equipped with energy dispersive spectroscopy (EDS) was used to identify the microstructure and composition of the films. High resolution images of glassy grains with sizes less than 50 nm were obtained by transmission electron microscopy (TEM, JOEL JEM-2011). Their thermodynamic properties were measured by differential scanning calorimetry (DSC, TA Instruments Q200) at heating rates of 3, 5, 10, 20, 40 K/min. Nanoindentation (Hysitron TI 900 TriboIndenter) with a typical Berkovich indenter was used to study their mechanical properties. Before measurements, tip-area function calibration with a load profile consisting of the processes of loading (for 5 s), holding (for 2 s) and unloading (for 5 s) was done on the standard sample (quartz) where 100 indents could be generated with forces varying from 100 μN (with hc = 11 nm) to 10 mN (with hc = 338 nm). The force was varied by approximately 4.762% (decreasing from maximum to minimum) when a subsequent indentation was carried out, and the resulting indent was 20-μm away from its nearest neighbors. It should be noted that the tip-area function calibration was performed after the indentation axis calibration and optic-probe tip offset calibration had been completed. The elastic modulus and hardness of the films were obtained by performing at least 100 indentation events for each film with applied loads changing from 0 to 10 mN at a constant loading rate of 0.2 mN/s. For the creep experiment, the peak load was held for 200 s and at least 10 indentation events were performed for each specimen. The thermodynamic behavior was further investigated by dynamic mechanical analysis (DMA, TA Instruments Q800). Rectangular Co–Fe–P samples were mounted on a 20-mm dual cantilever clamp. During the dynamic experiment, a constant oscillating amplitude of 20 \(\upmu{\mathrm{m}}\), at a single frequency of 0.8 Hz, was applied on the specimen from 303 to 673 K under a heating rate of 1 K/min. A 5-min soak time was applied at 303 K before the measurement.
Data availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Y. Wang, J. Zhang, K. Wu, G. Liu, D. Kiener, J. Sun, Nanoindentation creep behavior of Cu–Zr metallic glass films. Mater. Res. Lett. 6, 22 (2018)
A. Dubach, R. Raghavan, J.F. Löffler, J. Michler, U. Ramamurty, Micropillar compression studies on a bulk metallic glass in different structural states. Scr. Mater. 60, 567 (2009)
Y. Shen, X.C. Zheng, G.P. Zheng, Mechanical properties and crystallization behaviors of microstructured Co-Fe-P amorphous alloys. Metall. Mater. Trans. A 42, 211 (2011)
J. Jing, A. Krämer, R. Birringer, H. Gleiter, U. Gonser, Modified atomic structure in a Pd-Fe-Si nanoglass: a Mössbauer study. J. Non. Cryst. Solids 113, 167 (1989)
H. Gleiter, T. Schimmel, H. Hahn, Nanostructured solids—from nano-glasses to quantum transistors. Nano Today 9, 17 (2014)
H. Gleiter, Nanoglasses: a new kind of noncrystalline materials. Beilstein J. Nanotechnol. 4, 517 (2013)
H. Gleiter, Nanoglasses: a new kind of noncrystalline material and the way to an age of new technologies? Small 12, 2225 (2016)
N. Chen, D.V. Louzguine-Luzgin, K. Yao, A new class of non-crystalline materials: Nanogranular metallic glasses. J. Alloys Compd. 707, 371 (2017)
M. Ghafari, H. Hahn, H. Gleiter, Y. Sakurai, M. Itou, S. Kamali, Evidence of itinerant magnetism in a metallic nanoglass. Appl. Phys. Lett. 101, 243104 (2012)
X. Wang, F. Jiang, H. Hahn, J. Li, H. Gleiter, J. Sun, J. Fang, Sample size effects on strength and deformation mechanism of Sc75Fe25 nanoglass and metallic glass. Scr. Mater. 116, 95 (2016)
C. Pei, R. Zhao, Y. Fang, S. Wu, Z. Cui, B. Sun, S. Lan, P. Luo, W. Wang, T. Feng, The structural and dynamic heterogeneities of Ni-P nanoglass characterized by stress-relaxation. J. Alloys Compd. 836, 155506 (2020)
C. Guo, Y. Fang, F. Chen, T. Feng, Nanoindentation creep behavior of electrodeposited Ni-P nanoglass films. Intermetallics 110, 106480 (2019)
S.P. Singh, M.R. Chellali, L. Velasco, Y. Ivanisenko, E. Boltynjuk, H. Gleiter, H. Hahn, Deformation-induced atomic rearrangements and crystallization in the shear bands of a Tb75Fe25 nanoglass. J. Alloys Compd. 821, 153486 (2020)
M. Ghafari, X. Mu, J. Bednarcik, W.D. Hutchison, H. Gleiter, S.J. Campbell, Magnetic properties of iron clusters in Sc75Fe25 nanoglass. J. Magn. Magn. Mater. 494, 165819 (2020)
A. Baksi, S.H. Nandam, D. Wang, R. Kruk, M.R. Chellali, J. Ivanisenko, I. Gallino, H. Hahn, S. Bag, Ni60Nb40 nanoglass for tunable magnetism and methanol oxidation. ACS Appl. Nano Mater. 3, 7252 (2020)
C. Wang, X. Mu, M.R. Chellali, A. Kilmametov, Y. Ivanisenko, H. Gleiter, H. Hahn, Tuning the Curie temperature of Fe90Sc10 nanoglasses by varying the volume fraction and the composition of the interfaces. Scr. Mater. 159, 109 (2019)
A. Stoesser, M. Ghafari, A. Kilmametov, H. Gleiter, Y. Sakurai, M. Itou, S. Kohara, H. Hahn, S. Kamali, Influence of interface on structure and magnetic properties of Fe50B50 nanoglass. J. Appl. Phys. 116, 134305 (2014)
J. Cheng, T. Li, S. Ullah, F. Luo, H. Wang, M. Yan, G. Zheng, Giant magnetocaloric effect in nanostructured Fe-Co-P amorphous alloys enabled through pulse electrodeposition. Nanotechnology 31, 385704 (2020)
X.L. Wang, F. Jiang, H. Hahn, J. Li, H. Gleiter, J. Sun, J.X. Fang, Plasticity of a scandium-based nanoglass. Scr. Mater. 98, 40 (2015)
R. Witte, T. Feng, J.X. Fang, A. Fischer, M. Ghafari, R. Kruk, R.A. Brand, D. Wang, H. Hahn, H. Gleiter, Evidence for enhanced ferromagnetism in an iron-based nanoglass. Appl. Phys. Lett. 103, 073106 (2013)
O. Adjaoud, K. Albe, Microstructure formation of metallic nanoglasses: insights from molecular dynamics simulations. Acta Mater. 145, 322 (2018)
Y. Ritter, D. Opu, H. Gleiter, K. Albe, Structure, stability and mechanical properties of internal interfaces in Cu64Zr36 nanoglasses studied by MD simulations. Acta Mater. 59, 6588 (2011)
O. Adjaoud, K. Albe, Interfaces and interphases in nanoglasses: surface segregation effects and their implications on structural properties. Acta Mater. 113, 284 (2016)
D. Danilov, H. Hahn, H. Gleiter, W. Wenzel, Mechanisms of nanoglass ultrastability. ACS Nano 10, 3241 (2016)
M. Mohri, D. Wang, J. Ivanisenko, H. Gleiter, H. Hahn, Thermal stability of the Ti-Zr-Cu-Pd nano-glassy thin films. J. Alloys Compd. 735, 2197 (2018)
S.H. Nandam, Y. Ivanisenko, R. Schwaiger, Z. Śniadecki, X. Mu, D. Wang, R. Chellali, T. Boll, A. Kilmametov, T. Bergfeldt, H. Gleiter, H. Hahn, Cu-Zr nanoglasses: atomic structure, thermal stability and indentation properties. Acta Mater. 136, 181 (2017)
S.H. Nandam, O. Adjaoud, R. Schwaiger, Y. Ivanisenko, M.R. Chellali, D. Wang, K. Albe, H. Hahn, Influence of topological structure and chemical segregation on the thermal and mechanical properties of Pd-Si nanoglasses. Acta Mater. 193, 252 (2020)
D. Morineau, Y. Xia, C. Alba-Simionesco, Finite-size and surface effects on the glass transition of liquid toluene confined in cylindrical mesopores. J. Chem. Phys. 117, 8966 (2002)
J.L. Keddie, R.A.L. Jones, R.A. Cory, Size-dependent depression of the glass transition temperature in polymer films. Europhys. Lett. 27, 59 (1994)
Q. Jiang, H.X. Shi, J.C. Li, Finite size effect on glass transition temperatures. Thin Solid Films 354, 283 (1999)
Q. Jiang, X.Y. Lang, Glass transition of low-dimensional polystyrene. Macromol. Rapid Commun. 25, 825 (2004)
O. Franke, D. Leisen, H. Gleiter, H. Hahn, Thermal and plastic behavior of nanoglasses. J. Mater. Res. 29, 1210 (2014)
B. Cheng, J.R. Trelewicz, Controlling interface structure in nanoglasses produced through hydrostatic compression of amorphous nanoparticles. Phys. Rev. Mater. 3, 035602 (2019)
S. Yuan, P.S. Branicio, Tuning the mechanical properties of nanoglass-metallic glass composites with brick and mortar designs. Scr. Mater. 194, 113639 (2021)
Z.D. Sha, P.S. Branicio, H.P. Lee, T.E. Tay, Strong and ductile nanolaminate composites combining metallic glasses and nanoglasses. Int. J. Plast. 90, 231 (2017)
S. Adibi, P.S. Branicio, R. Ballarini, Compromising high strength and ductility in nanoglass-metallic glass nanolaminates. RSC Adv. 6, 13548 (2016)
S. Adibi, P.S. Branicio, S.P. Joshi, Suppression of shear banding and transition to necking and homogeneous flow in nanoglass nanopillars. Sci. Rep. 5, 1 (2015)
Z.D. Sha, P.S. Branicio, Q.X. Pei, Z.S. Liu, H.P. Lee, T.E. Tay, T.J. Wang, Strong and superplastic nanoglass. Nanoscale 7, 17404 (2015)
S. Adibi, P.S. Branicio, Y.W. Zhang, S.P. Joshi, Composition and grain size effects on the structural and mechanical properties of CuZr nanoglasses. J. Appl. Phys. 116, 043522 (2014)
S. Xie, E.P. George, Size-dependent plasticity and fracture of a metallic glass in compression. Intermetallics 16, 485 (2008)
R.T. Qu, S.G. Wang, X.D. Wang, S.J. Wu, Z.F. Zhang, Shear band fracture in metallic glass: sample size effect. Mater. Sci. Eng. A 739, 377 (2019)
Q. Hu, J. Wu, B. Zhang, Synthesis and nanoindentation behaviors of binary CuTi nanoglass films. Physica B 521, 28 (2017)
K. Zheng, P. Branicio, Synthesis of metallic glass nanoparticles by inert gas condensation. Phys. Rev. Mater. 4, 1 (2020)
Z. Śniadecki, D. Wang, Y. Ivanisenko, V.S.K. Chakravadhanula, C. Kübel, H. Hahn, H. Gleiter, Nanoscale morphology of Ni50Ti45Cu5 nanoglass. Mater. Charact. 113, 26 (2016)
M. Mohri, D. Wang, J. Ivanisenko, H. Gleiter, H. Hahn, Investigation of the deposition conditions on the microstructure of TiZrCuPd nano-glass thin films. Mater. Charact. 131, 140 (2017)
X.D. Wang, Q.P. Cao, J.Z. Jiang, H. Franz, J. Schroers, R.Z. Valiev, Y. Ivanisenko, H. Gleiter, H.J. Fecht, Atomic-level structural modifications induced by severe plastic shear deformation in bulk metallic glasses. Scr. Mater. 64, 81 (2011)
H. Shao, Y. Xu, B. Shi, C. Yu, H. Hahn, H. Gleiter, J. Li, High density of shear bands and enhanced free volume induced in Zr70Cu20Ni10 metallic glass by high-energy ball milling. J. Alloys Compd. 548, 77 (2013)
C. Guo, Y. Fang, B. Wu, S. Lan, G. Peng, X.L. Wang, H. Hahn, H. Gleiter, T. Feng, Ni-P nanoglass prepared by multi-phase pulsed electrodeposition. Mater. Res. Lett. 5, 293 (2017)
J.X. Fang, U. Vainio, W. Puff, R. Würschum, X.L. Wang, D. Wang, M. Ghafari, F. Jiang, J. Sun, H. Hahn, H. Gleiter, Atomic structure and structural stability of Sc75Fe25 nanoglasses. Nano Lett. 12, 458 (2012)
D. Şopu, Y. Ritter, H. Gleiter, K. Albe, Deformation behavior of bulk and nanostructured metallic glasses studied via molecular dynamics simulations. Phys. Rev. B 83, 1 (2011)
K. Albe, Y. Ritter, D. Şopu, Enhancing the plasticity of metallic glasses: shear band formation, nanocomposites and nanoglasses investigated by molecular dynamics simulations. Mech. Mater. 67, 94 (2013)
D. Şopu, K. Albe, Influence of grain size and composition, topology and excess free volume on the deformation behavior of Cu-Zr nanoglasses. Beilstein J. Nanotechnol. 6, 537 (2015)
Y. Zhao, X. Peng, C. Huang, B. Yang, N. Hu, M. Wang, Super ductility of nanoglass aluminium nitride. Nanomaterials 9, 1 (2019)
B. Cheng, J.R. Trelewicz, Interfacial plasticity governs strain delocalization in metallic nanoglasses. J. Mater. Res. 34, 2325 (2019)
O. Adjaoud, K. Albe, Influence of microstructural features on the plastic deformation behavior of metallic nanoglasses. Acta Mater. 168, 393 (2019)
W.L. Johnson, K. Samwer, A universal criterion for plastic yielding of metallic glasses with a (T/Tg)2/3 temperature dependence. Phys. Rev. Lett. 95, 2 (2005)
D. Pan, A. Inoue, T. Sakurai, M.W. Chen, Experimental characterization of shear transformation zones for plastic flow of bulk metallic glasses. Proc. Natl. Acad. Sci. USA 105, 14769 (2008)
Acknowledgments
This work was supported by a grant from the Research Grants Council of the Hong Kong Special Administrative Region, China (PolyU152607/16E).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors have no conflicts of interest to declare that are relevant to the content of this article.
Rights and permissions
About this article

Cite this article
Li, T., Ma, K. & Zheng, G. The effects of glass–glass interfaces on thermodynamic and mechanical properties of Co–Fe–P metallic nano-glasses. Journal of Materials Research 36, 4951–4962 (2021). https://doi.org/10.1557/s43578-021-00429-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1557/s43578-021-00429-6