
Abstract
Graphite nitride carbon nanosheets have received more and more attention toward the photocatalytic research and applications. Ultrathin g-CN nanosheets with porous structure were synthesized successfully by thermal calcination of melamine supramolecular complexes, which was obtained by pre-treating melamine in nitric acid solution at different concentrations (0.5–2 mol/L). Effects of HNO3 pre-treatment on the microstructure of supramolecular complexes were studied. The characteristics of g-CN nanosheets were investigated by X-ray diffractometry, X-ray photoelectron spectroscopy, transmission electron microscopy and Fourier transform infrared spectroscopy. The degradation performance for RhB and water splitting hydrogen production performance were used to evaluate the photocatalytic performances of g-CN nanosheets. The morphology and microstructure of HNO3/melamine supramolecular complexes are different from those of melamine precursor due to the better arrangement of the melamine units. Ultrathin porous g-CN nanosheets which possess a thickness of less than 2 nm were successfully prepared by calcination of melamine pre-treated with 1.0 mol/L nitric acid. The g-CN(1.0) nanosheets possess the highest photocatalytic degradation performance and water splitting hydrogen production performance due to the effective separation of photogenerated carriers and high specific surface area providing a large number of active sites.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Environmental damage and energy crisis are becoming more and more serious with the rapid development of economy. Since Fujishima and Honda discovered water photocatalysis on a TiO2 electrode in 1972 [1], solar photocatalytic technology has become an ideal way for solving environmental damage and energy crisis, including organic pollutant degradation, CO2 reduction and water splitting hydrogen generation [2, 3, 4]. In the fields of photocatalytic science and technology, graphitic carbon nitride (g-CN) on numerous visible light-responsive semiconductor materials has attracted more and more attention due to its suitable energy band gap, proper energy band position, unique electronic structure and stability [5, 6, 7]. However, in most cases, bulk g-CN synthesized by direct thermal calcination displayed a low photocatalytic property because of the limited specific surface area, relatively narrow optical utilization region and higher recombination probability of photogenerated carriers [8].
Many strategies for enhancing the photocatalytic performance of g-CN semiconductors have been developed, mainly including two categories. One is to optimize the electron and energy band structure, including heteroatoms doping [9], defects controlling [10] and construction on heterojunction [11]. The other is to develop morphology regulation by constructing nanosheets [12], nanotubes [13], nanoparticles [14] and porous structures [15]. Among them, g-CN nanostructures, especially the g-CN nanosheets, have some unique physical properties such as effective photogenerated carrier separation and a large number of active sites, which is beneficial for improving the photocatalytic performance. Up to now, several routes have been realized for the preparation of nanosheets from the bulk g-CN, mainly including ultrasonic liquid exfoliation [12], chemical liquid exfoliation [16] and thermal oxidation etching [17]. It is noteworthy that the ultrasonic liquid exfoliation reaction process is time-consuming, chemical liquid exfoliation involves some other functional groups, and thermal-oxidized nanosheets offer a rather low yield.
Another effective strategy for the improvement of photocatalytic performance is to introduce porous structure in the materials, which can effectively increase the specific surface area, thus significantly promoting high charge separation efficiency and providing more active centers of photocatalytic performance. Up to now, hard-templates [18] and soft-templates [19] are the most common methods to prepare porous g-CN. However, hard-templates require sacrificial materials and hazardous chemicals to remove templates, which are costly and time-consuming. Soft-templates usually retain carbon residues of some template polymers in the catalyst, which leads to the deterioration in photocatalytic activity. In recent years, researchers have developed methods to prepare the g-CN with porous structure by calcining melamine-based supramolecular complexes for significantly increasing the specific surface area [20, 21]. The formation of hydrogen bonded supramolecular assembly plays key roles in separating photogenerated charges, promoting mass adsorption of reactants and providing more reaction active sites.
Herein, ultrathin graphitic carbon nitride nanosheets with porous structure were synthesized by direct thermal calcination of HNO3/melamine supramolecular complexes as precursors obtained by pretreating melamine in nitric acid. The prepared ultrathin g-CN nanosheets with porous structure displayed admirable visible photocatalytic performance for the degradation of organics and hydrogen production.
Results and discussion
Characterizations
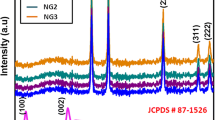
The X-ray diffractometry (XRD) patterns of g-CN nanosheets obtained from pre-treated melamine with different concentrations of nitric acid solutions and its bulk counterpart are displayed in Fig. 1(a). All samples exhibited two typical diffraction peaks of graphite carbon nitride, which indicates that all samples maintain the basic g-CN materials’ crystal structure. The diffraction peaks located around 12.9° and 27.7° for bulk g-CN refer to the (100) and (002) lattice planes, corresponding to the in-plane structural packing motif of tri-s-triazine units and the interlayer stacking of aromatic segments, respectively [22, 23]. What is more, the (002) peak of g-CN nanosheets, especially g-CN(1.0), become weaker and broader in varying degrees in comparison with that of bulk g-CN, indicating that the crystallinity and interlaminar periodic correlation length of tri-s-triazine building blocks decrease [24, 25]. It is noted that the (100) peak of 1.0-g-CN becomes weaker, which can be ascribed to the broken hydrogen bond between the interlaminar framework during the calcination process of HNO3/melamine [26]. Figure 1(b) shows the XRD patterns of melamine and melamine treated with a 1.0 mol/L nitric acid solution. Compared with that of melamine, new and well-resolved peaks at 9.86°, 17.10°, 29.24°, and 30.98° appear in the XRD pattern of 1.0-HNO3/melamine, indicating that hydrogen bonded supramolecular complexes formed through the interaction between melamine and nitric acid [27, 28].

(a) XRD patterns of g-CN. (b) XRD patterns of melamine and 1.0-HNO3/melamine.
Figure 2 shows the scanning electron microscopic (SEM) morphologies of melamine and melamine pre-treated with different concentrations of nitric acid solutions. Figure 2(a) is the morphology of melamine, exhibiting the sphere-like particles of an average size of about 20 µm. As shown in Fig. 2(b), 0.5-HNO3/melamine displays rod-like particles of about 0.2 µm in diameter and 2.5 µm in length. The morphology of 1.0-HNO3/melamine, as shown in Fig. 2(c), also exhibits a rod-like morphology, and the particle size is bigger than that of 0.5-HNO3/melamine. With the increase in nitric acid concentration, the morphology of 2.0-HNO3/melamine is composed of many small particles with an average size of 0.2 µm. The morphologies of HNO3/melamine are much different from those of un-treated melamine.

SEM morphologies of (a) melamine, (b) 0.5-HNO3/melamine, (c) 1.0-HNO3/melamine, and (d) 2.0-HNO3/melamine.
Morphologies of g-CN products obtained with different precursors are shown in Fig. 3. The bulk g-CN shows solid agglomerates of several micrometers, as shown in Fig. 3(a). The g-CN nanosheets obtained from melamine precursors pre-treated with different nitric acid solutions possess typical 2D nanosheet architectures with rough and uneven surfaces in comparison with those of bulk g-CN, as shown in Figs. 3(b)–3(d). The morphology of g-CN(1.0) nanosheets shows many irregular overlapped nanosheets with delaminated structure. Abundant irregular pores were also clearly seen in the magnified inset image in Fig. 3(c).

SEM morphologies of (a) bulk g-CN, (b) g-CN(0.5), (c) g-CN(1.0), and (d) g-CN(2.0).
Bulk g-CN and g-CN nanosheets were further studied by transmission electron microscopy (TEM), as presented in Fig. 4. Agglomerates with several micrometers of bulk g-CN can be observed in Fig. 4(a). All g-CN(0.5), g-CN(1.0), and g-CN(2.0) samples show a typical layered stacking structure of several hundred nanometers in size, as shown in Figs. 4(b)–4(d). The edge of the layered structure is curled due to the spontaneous reduction of surface energy of the nanosheets. As the width of crimping edges can reflect the thickness of the nanosheets to some extent, g-CN(1.0) nanosheets present the smallest thickness among all samples. For reflecting the thickness more directly, Figs. 4(e) and 4(f) show the atomic force microscopy (AFM) image of g-CN(1.0) nanosheets and corresponding thickness fluctuation along the line throughout the nanosheet in AFM. The nanosheet possesses a rough surface with a sheet thickness of approximately 1.5 nm, suggesting that the ultrathin nanosheets can be achieved by direct calcination process. Large amounts of irregular pores exist in the g-CN(1.0) nanosheets, as shown in the inset of Fig. 4(c). The results of TEM and AFM analyses confirm that the ultrathin g-CN nanosheets with porous structure are successfully prepared by direct calcination of HNO3/melamine supramolecular complexes in our experiment. The preparation process of ultrathin g-CN nanosheets with a porous structure is shown in Scheme 1. The possible formation process is depicted as follows. During the self-assembly process of supramolecular complexes, NH⋯O(N) hydrogen bond forms, leading to the rod-like or small particle-like morphology of the precursor [27]. The HNO3/melamine supramolecular complexes were calcined, and the NO3− functional groups gradually decompose and finally form the ultrathin and porous structures of g-CN nanosheets.

TEM morphologies of (a) bulk g-CN, (b) g-CN(0.5), (c) g-CN(1.0), (d) g-CN(2.0), (e) AFM image of g-CN(1.0), and (f) height fluctuation along the line in the AFM throughout g-CN(1.0) nanosheets.

Schematic illustration for preparation process of ultrathin porous g-CN nanosheets.
FT-IR spectra were used to investigate the chemical bonding of all g-CN samples. Infrared absorption patterns of g-CN nanosheets are similar to those of bulk g-CN, as shown in Fig. 5(a). The peak around 805 cm−1 originates from the characteristic band breathing mode of triazine units [29]. The strong wide peak ranging from 900 to 1800 cm−1 refers to the characteristic stretching vibration of aromatic CN heterocyclic units [30]. The strong absorption band in the range 3000 to 3600 cm−1 can be ascribed to –NH3 and –OH stretches of bulk g-CN, originating from water adsorbed on the surface or uncondensed amino groups [31]. The above FT-IR characteristic spectra illustrate that the g-CN can be synthesized by calcining the HNO3/melamine supramolecular complexes.

(a) FT-IR spectra, (b) XPS survey spectra, (c) high resolution XPS spectra of N 1s, and (d) high resolution XPS spectra of C 1s.
The chemical state and elemental composition of g-CN(1.0) and the bulk g-CN are shown in Figs. 5(b)–5(d). The results of survey X-ray photoelectron spectroscopy (XPS) spectra show that there are C, N and O elements in both samples. A weak O 1s peak forms the absorbed CO2 and H2O. Taking g-CN(1.0) nanosheets as an example, high resolution XPS spectra of N 1s were deconvoluted into four peaks at 398.6, 399.6, 401.0, and 404.4 eV, respectively. The dominant peak at 398.6 eV is ascribed to sp2-hybridized nitrogen in the C-containing aromatic ring (C–N–C) [32]. The peaks at 399.6 and 401.0 eV are indexed to the tertiary nitrogen in the N(–C)3 group and the amino function group (C–N–H) [33]. And the weakest peak at 404.4 eV refers to the positive charge localization in heterocycles or charging effects [34]. The high resolution spectra of C 1s are resolved into three peaks with bonding energies at 288.1, 286.1, and 284.8 eV, respectively. The strongest peak at 288.1 eV and the weakest peak at 286.1 eV are identified as sp2-hybridized carbon in the tri-s-triazine ring (N–C–N) [35]. And the peak at 284.8 eV is relevant to the surface adventitious carbon, such as amorphous carbon or graphite [35]. The main binding energies of N 1s and C 1s in g-CN(1.0) exhibit higher values than those in bulk g-CN, which may be related to the breakdown of long-distance atomic arrangement between layers and the formation of nanosheet structures.
Photocatalytic performance
Figure 6 shows the photocatalytic performances of different g-CN nanosheets, including photocatalytic degradation of RhB dyes (a, b) and photocatalytic hydrogen production (c, d). RhB degradation efficiencies of different samples are shown in Fig. 6(a). Prior to irradiation, the equilibrium of adsorption–desorption was first carried out with a continuous magnetic stirring for 30 min in dark. Clearly, all samples can reach the adsorption–desorption equilibrium in 10 min. The RhB adsorption of g-CN(0.5), g-CN(1.0), and g-CN(2.0) are approximately 16.8%, 18.8%, and 17.3%, respectively, while the RhB adsorption of bulk g-CN is only 4.9%. The degradation rate of bulk g-CN is approximately 46.2% under visible light irradiation for 16 min. All g-CN nanosheets possess much higher degradation efficiency than that of bulk g-CN. Among them, g-CN(1.0) displays the highest degradation efficiency at 99.6% at the same condition as bulk g-CN. Figure 6(b) shows the photodegradation kinetics of RhB, which is linearly fitted using degradation efficiency data for a better illustrating photodegradation rate. The reaction rate constant k of all samples is shown in the inset of Fig. 6(b), assuming that the reaction follows first-order kinetics [36]. The g-CN nanosheets displayed greatly enhanced k values in comparison with bulk g-CN. The highest k value of g-CN(1.0) nanosheets is 0.352 min−1, which is 9.72 times as high as that of bulk g-CN, indicating that the excellent photodegradation activity of g-CN nanosheets can be obtained by optimizing HNO3 pretreating process.

Photocatalytic performances of different g-CN nanosheets. (a) C/C0 of RhB degradation, (b) RhB degradation kinetics plots, (c) hydrogen production versus time, and (d) hydrogen production rate. The inset shows the recycling tests of g-CN(1.0) nanosheets.
The plots of hydrogen amount versus irradiation time are given in Fig. 6(c). Bulk g-CN totally produces 33.06 µmol hydrogen with 4 h of visible light irradiation, while g-CN(0.5) and g-CN(2.0) nanosheets increase the hydrogen production to 63.42 and 91.72 µmol respectively. The g-CN(1.0) nanosheets possess the highest photocatalytic hydrogen production of 154.95 µmol. The hydrogen production rates of all samples are calculated from their production data, as displayed in Fig. 6(d). It is clearly seen that the photocatalytic hydrogen production rate of bulk g-CN is only 826.43 µmol/(h g). However, g-CN nanosheets exhibit increased photocatalytic hydrogen production rates. In particular, g-CN(1.0) nanosheets achieve the highest rate of 3860.53 µmol/(h g), which is 4.7 times that of bulk g-CN. The apparent quantum yield (AQY) of g-CN(1.0) is calculated to be 4.3% at 420 nm.
Free-standing g-C3N4 nanosheets of 2 nm thickness prepared by Yang et al. showed a hydrogen production rate of 1860 µmol/(h g) [37]. Liu and co-workers synthesized mesoporous g-C3N4 nanosheets by using the hydrothermal process to transfer dicyandiamide into the MCA-like complex, achieving a hydrogen production rate of 2738 µmol/(h g) [38]. Compared with above studies, the as-prepared ultrathin porous g-CN nanosheets possess a greatly enhanced photocatalytic hydrogen activity.
Stability is the main factor for the potential application of photocatalysis. Therefor the stability of g-CN(1.0) was also evaluated by recycling tests of photocatalytic hydrogen production, as shown in the inset of Fig. 6(d). The photocatalytic hydrogen production of g-CN nanosheets was sustained without noticeable degradation after five cycles, confirming the excellent photocatalytic stability of the g-CN(1.0) nanosheets.
Mechanism discussion
The mechanism of photocatalytic process of g-CN nanosheets is shown in Fig. 7. Generally speaking, the photocatalytic RhB degradation and hydrogen production process of g-CN can be divided into several steps, including surface adsorption, optical absorption (including photogenerated carriers), carrier transfer and surface reaction. When the photocatalyst is irradiated by photons whose energy is higher than or equal to that of the bandgap, the electrons occupied in the valence band (VB) can be excited and then rapidly migrate to the empty conduction band (CB), generating electron–hole pairs. Some separated electrons and holes move to the surface of the photocatalyst. The holes in VB can oxidize electron donors including organic molecules and OH− groups. In addition, electron receptors such as oxygen molecules and hydrogen ion can be reduced by the electrons on CB. The surface photocatalytic reactions are complex and can be described as follows:

Schematic diagram of photocatalytic process of g-CN nanosheets.
Figure 8(a) displays the optical absorption abilities of bulk g-CN and g-CN nanosheets. All samples show a powerful absorption ability in the visible light region. Compared with bulk g-CN, the g-CN nanosheets display low absorption intensities and blue shift of absorption edges. The band gap energies of bulk g-CN and g-CN(1.0) are calculated to be 2.65 and 2.69 respectively from the Tauc plots inserted in Fig. 8(a). The blue shift and the increased band gap energy are attributed to the quantum confinement effect induced by the existence of ultrathin nanosheets and pores in photocatalysts, resulting in the shift of the conduction and VB edges [32, 39]. It is clear to find that the optical absorption capacity is not the factor improving the photocatalytic performance.

(a) UV-vis DRS spectra, (b) PL emission spectra, (c) EIS Nyquist plots, and (d) effects of capture agents on the photocatalytic RhB degradation.
The separation and transport abilities of photogenerated charge carriers for as-prepared samples recorded by photoluminescence (PL) spectra are illustrated in Fig. 8(b). Compared with bulk g-CN, the PL emission peaks of g-CN nanosheets display a slight blue shift, further confirming the above-mentioned UV-vis DRS results. Generally, the low PL intensity indicates low charges’ recombination rate [40]. It is worth noting that g-CN nanosheets exhibit a remarkable quenching of the PL intensities in comparison with that of bulk g-CN, especially in the g-CN(1.0) nanosheets, suggesting that optimized ultrathin nanosheets with a porous structure is favorable for the effective transfer and separation of photogenerated carrier. Effective separation and transfer of photogenerated carriers play key roles in enhancing the photocatalytic performance of g-CN nanosheets. For a better understanding of the interface charge transfer rate of the as-prepared samples, electrochemical impedance spectroscopy (EIS) was conducted, as shown in Fig. 8(c). Compared with bulk g-CN, the g-CN nanosheets have a smaller arc radius on the EIS plots, especially the g-CN(1.0) nanosheets, suggesting a much lower electron-transfer resistance and faster interface charge transfer efficiency [41].
In order to investigate the active oxidation mechanism, three capture agents (10 mM) were added into the solutions during the photocatalytic degradation process for 16 min, where BQ, IPA, and NaI are used for capturing •O2−, •OH, and h+, respectively. As shown in Fig. 8(d), in bulk g-CN system, the degradation rate decreases from 46% to 39%, 43% and 13% with the addition of BQ, IPA, and NaI, respectively, indicating that h+ radicals play a key role in the photocatalytic process in a direct oxidation route. However, for g-CN(1.0) nanosheets, it is a little different from bulk g-CN. The degradation rate decreases from 99% to 63%, 99%, and 32% with the addition of BQ, IPA, and NaI, respectively, suggesting that •O2− and h+ play the key roles in the photocatalytic degradation process, and almost no •OH radicals are produced in the photocatalytic degradation process.
The N2 adsorption–desorption isotherms curves were recorded with the aim to further study the porous structure and specific surface area of g-CN. As shown in Fig. 9, The N2 adsorption–desorption isotherms exhibit typical IV isotherms characteristic of H3 hysteresis loops, suggesting the existence of mesoporous and macroporous structures in g-CN nanosheets. A faint hysteresis loop of the bulk g-CN sample is observed, suggesting a poor pore structure. It can be seen from pore-size distribution curves inserted in Fig. 9 that g-CN(1.0) samples possess a wide distribution region from 10 to 100 nm centered at 70 nm, further confirming the mesoporous/macroporous structures. This result is in accordance with that of TEM morphology observation. The Brunauer–Emmett–Teller (BET) surface areas of the as-prepared products are calculated according to the N2 adsorption–desorption isotherms. The BET surface areas of g-CN(0.5), g-CN(1.0), and g-CN(2.0) samples are 49.4, 59.2, and 28.7 cm2/g, respectively, which are higher than those of bulk g-CN (15.6 cm2/g) obviously. The enlarged specific surface areas of optimized ultrathin g-CN nanosheets can be ascribed to the reduced thickness and the existence of abundance of pores [32]. The photocatalyst with a larger BET surface area can provide more catalytic active sites for surface catalytic reaction, thus improving the photocatalytic performance.

N2 adsorption–desorption isotherms and pore-size distribution.
Conclusions
In summary, the ultrathin g-CN nanosheets with porous structure were successfully synthesized by the direct calcination of melamine/HNO3 supramolecular complexes. The photocatalytic degradation performances and water splitting hydrogen production were performed under visible light irradiation. All g-CN nanosheets prepared from melamine pre-treated with different HNO3 concentrations display a higher photocatalytic degradation rate and water splitting hydrogen production rate than those of bulk g-CN. In particular, the g-CN(1.0) sample possesses the highest photocatalytic performance which can degrade 96.6% of 10 mg/L RhB in 16 min with a reaction rate constant k of 0.352 min−1, and enhance the photocatalytic hydrogen production rate to 3860.5 µmol/(h g). The effective separation rate and a larger number of catalytic active centers contribute to the excellent photocatalytic performances of as-prepared g-CN nanosheets.
Experimental section
Preparation of g-CN samples
Firstly, 10 g melamine was added into 200 mL ethanol with magnetic stirring, and then 100 mL nitric acid solutions with different concentrations of 0.5, 1.0 and 2.0 mol/L were added into the melamine/ethanol mixture. The obtained solutions were evaporated in an 80 °C water bath with magnetic stirring for 12 h, and then dried at 60 °C for 12 h in a vacuum oven. The obtained supramolecular complexes were denoted as 0.5-HNO3/melamine, 1.0-HNO3/melamine and 2.0-HNO3/melamine, respectively.
The HNO3/melamine supramolecular complexes were placed into a covered crucible which was wrapped by an aluminum foil for reduction of sublimation. Then the covered crucible was calcined at 530 °C for 4 h in a muffle furnace at a heating rate of 2 °C/min. According to the different HNO3 concentrations, the obtained final products are denoted as g-CN(0.5), g-CN(1.0), and g-CN(2.0), respectively. For comparison, the melamine without any pretreatment was calcined directly to prepare the bulk graphitic carbon nitride (denoted as bulk g-CN) in the same process.
Characterization
The crystal phases of all samples were performed using an X-ray diffractometer (XRD; Rigaku D/MAX2500, Tokyo, Japan) with Cu Kα radiation. All morphologies and microstructures were acquired on a field emission scanning electron microscope (FESEM; HitachiSU8020, Tokyo, Japan) and a transmission electron microscope (TEM; JEM-2100F, Tokyo, Japan). The electronic states of N and C were obtained by X-ray photoelectron spectroscopy (XPS; ESCALAB 250, Thermo-VG Scientific Corporate, Waltham, Massachusetts) using the monochromatic Al Kα linear excitation source. Fourier transform infrared spectroscopy (FTIR; Nicolet-6700, Thermo-VG Scientific Corporate, Waltham, Massachusetts) was used to collect infrared spectra. UV-vis absorption spectra were examined by a UV3600 diffuse reflectance spectrometer. PL spectra were evaluated by a spectrometer (PerkinElmer LS55, PerkinElmer Life and Analytical Sciences, Waltham, Massachusetts) with excitation wavelength at 310 nm. The N2 adsorption/desorption isotherms were recorded on a Quadrasorb-EVO (Quantachrome Corporation, Boynton Beach, Florida) porosimeter after samples being vacuum-dried at 130 °C overnight. Atomic force microscopic images were recorded on an atomic force microscope (AFM; Dimension 3100, Bruker Nano/Veeco Inc., Santa Barbara, California).
Electrochemical impedance spectroscopy (EIS) was carried out on an electrochemical system (CHI 660E, Shanghai, China) in the dark condition via a standard three-electrode system with a working electrode, a platinum wire counter electrode, and a standard calomel reference electrode (SCE). Sodium sulfate (0.5 M) was used as the electrolyte solution. 2 µL g-CN aqueous suspension (1 mg/mL) was drop-cast on the surface of a glassy carbon electrode to form the working electrode.
Photocatalytic performance
Photocatalytic performances of the samples were estimated by means of organics degradation and water splitting hydrogen production. RhB solution was utilized as the organics for the measurement of degradation activities. And the degradation was carried out by a photochemical reactor (XPA-7, Nanjing, China) containing a 250 W metal halide lamp with a cut-off filter (λ > 420 nm) which could realize visible light irradiation. 10 mg photocatalyst was added into 10 mL RhB solution with 10 mg/L concentration. Subsequently, the suspensions were placed in a photochemical reactor and the equilibrium of adsorption–desorption was achieved by continuous stirring under the dark condition for 30 min. Then mixed solutions were illuminated for 4, 8, 12, and 16 min, respectively. Then the illuminated solutions at every given time interval were centrifuged to remove the catalyst. Absorbency after irradiation was evaluated by a UV1800 spectrometer, Kyoto, Japan. According to the Lambert–Beer law, the standard linear relationship between absorbance and RhB concentration can be calculated by the following equation:
where D is the decolored rate reflecting the degradation degree, C is the remnant RhB concentration, C0 is the initial RhB concentration, A is absorbency after irradiation, and A0 is original absorbency.
The photocatalytic hydrogen production was investigated by connecting a glass gas circulation system with a top-irradiation vessel. 10 mg photocatalyst and 10 mL triethanolamine were mixed into a 100 mL aqueous solution as the holes’ receptor. 3 mass% Pt was used as co-catalyst by dissolving H2PtCl6·6H2O in the aqueous solution. All solutions were evacuated carefully to remove air completely. The hydrogen production process was carried out under visible light (≥400 nm) irradiation using a 300 W Xeon-lamp (Beijing, China) with a glass filter at the room temperature. In addition, the component and content of produced gases were analyzed using a gas chromatograph. The AQY for H2 production was determined by using a similar procedure with 50 mg photocatalyst and monochromatic light (λ = 420 nm). The light was obtained by locating a band pass filter and a neutral density filter below a 300 W Xeon-lamp. The AQY was calculated using the following equation:
References
A. Fujishima and K. Honda: Electrochemical photolysis of water at a semiconductor electrode. Nature 238, 37 (1972).
J. Wen, J. Xie, X. Chen, and X. Li: A review on g-C3N4-based photocatalysts. Appl. Surf. Sci. 391, 72 (2017).
X. Li, J. Yu, M. Jaroniec, and X. Chen: Cocatalysts for selective photoreduction of CO2 into solar fuels. Chem. Rev. 119, 3962 (2019).
R. Shen, W. Liu, D. Ren, J. Xie, and X. Li: Co1.4Ni0.6P cocatalysts modified metallic carbon black/g-C3N4 nanosheet Schottky heterojunctions for active and durable photocatalytic H2 production. Appl. Surf. Sci. 466, 393 (2019).
Y. Ren, D. Zeng, and W. Ong: Interfacial engineering of graphitic carbon nitride (g-C3N4)-based metal sulfide heterojunction photocatalysts for energy conversion: A review. Chin. J. Catal. 40, 289 (2019).
X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J.M. Carlsson, K. Domen, and M. Antonietti: A metal-free polymeric photocatalyst for hydrogen production from water under visible light. Nat. Mater. 8, 76 (2009).
J. Fu, J. Yu, C. Jiang, and B. Cheng: g-C3N4-Based heterostructured photocatalysts. Adv. Energy Mater. 8, 1701503 (2018).
X. Wang, K. Maeda, X. Chen, K. Takanabe, K. Domen, Y. Hou, X. Fu, and M. Antonietti: Polymer semiconductors for artificial photosynthesis: Hydrogen evolution by mesoporous graphitic carbon nitride with visible light. J. Am. Chem. Soc. 131, 1680 (2009).
Y. Luo, J. Wang, S. Yu, Y. Cao, K. Ma, Y. Pu, W. Zou, and C. Tang: Nonmetal element doped g-C3N4 with enhanced H2 evolution under visible light irradiation. J. Mater. Res. 33, 1268 (2017).
Y.P. Yuan, L.S. Yin, S.W. Cao, L.N. Gu, G.S. Xu, P. Du, H. Chai, and C. Xue: Microwave-assisted heating synthesis: A general and rapid strategy for large-scale production of highly crystalline g-C3N4 with enhanced photocatalytic H2 production. Green Chem. 16, 4663 (2014).
R. Shen, J. Xie, Q. Xiang, X. Chen, J. Jiang, and X. Li: Ni-based photocatalytic H2-production cocatalysts. Chin. J. Catal. 40, 240 (2019).
S. Fu, Y. He, Q. Wu, and Y. Wu: Visible-light responsive plasmonic Ag2O/Ag/g-C3N4 nanosheets with enhanced photocatalytic degradation of Rhodamine B. J. Mater. Res. 31, 2252 (2016).
B. Liu, L. Ye, R. Wang, J. Yang, Y. Zhang, R. Guan, L. Tian, and X. Chen: Phosphorus-doped graphitic carbon nitride nanotubes with amino-rich surface for efficient CO2 capture, enhanced photocatalytic activity, and product selectivity. ACS Appl. Mater. Interfaces 10, 4001 (2018).
X. Jin, V.V. Balasubramanian, S.T. Selvan, D.P. Sawant, M.A. Chari, G.Q. Lu, and A. Vinu: Highly ordered mesoporous carbon nitride nanoparticles with high nitrogen content: A metal-free basic catalyst. Angew. Chem., Int. Ed. 48, 7884 (2009).
Q. Liang, Z. Li, X. Yu, Z-H. Huang, F. Kang, and Q-H. Yang: Macroscopic 3D porous graphitic carbon nitride monolith for enhanced photocatalytic hydrogen evolution. Adv. Mater. 27, 4634 (2015).
J. Tong, L. Zhang, F. Li, K. Wang, L. Han, and S. Cao: Rapid and high-yield production of g-C3N4 nanosheets via chemical exfoliation for photocatalytic H2 evolution. RSC Adv. 5, 88149 (2015).
P. Niu, L. Zhang, G. Liu, and H-M. Cheng: Graphene-like carbon nitride nanosheets for improved photocatalytic activities. Adv. Funct. Mater. 22, 4763 (2012).
Y. Zheng, Y. Jiao, J. Chen, J. Liu, J. Liang, A. Du, W. Zhang, Z. Zhu, S.C. Smith, M. Jaroniec, G.Q. Lu, and S.Z. Qiao: Nanoporous graphitic-C3N4@carbon metal-free electrocatalysts for highly efficient oxygen reduction. J. Am. Chem. Soc. 133, 20116 (2011).
Y-S. Jun, J. Park, S.U. Lee, A. Thomas, W.H. Hong, and G.D. Stucky: Three-dimensional macroscopic assemblies of low-dimensional carbon nitrides for enhanced hydrogen evolution. Angew. Chem., Int. Ed. 52, 11083 (2013).
T. Jordan, N. Fechler, J. Xu, T.J.K. Brenner, M. Antonietti, and M. Shalom: “Caffeine doping” of carbon/nitrogen-based organic catalysts: Caffeine as a supramolecular edge modifier for the synthesis of photoactive carbon nitride tubes. ChemCatChem 7, 2826 (2015).
C. Liu, X. Dong, Y. Hao, X. Wang, H. Ma, and X. Zhang: A novel supramolecular preorganization route for improving g-C3N4/g-C3N4 metal-free homojunction photocatalysis. New J. Chem. 41, 11872 (2017).
N. Xiao, S. Li, S. Liu, B. Xu, Y. Li, Y. Gao, L. Ge, and G. Lu: Novel PtPd alloy nanoparticle-decorated g-C3N4 nanosheets with enhanced photocatalytic activity for H2 evolution under visible light irradiation. Chin. J. Catal. 40, 352 (2019).
Y. Li, Z. Jin, L. Zhang, and K. Fan: Controllable design of Zn–Ni–P on g-C3N4 for efficient photocatalytic hydrogen production. Chin. J. Catal. 40, 390 (2019).
K. He, J. Xie, Z. Liu, N. Li, X. Chen, J. Hu, and X. Li: Multi-functional Ni3C Cocatalyst/g-C3N4 nanoheterojunctions for robust photocatalytic H2 evolution under visible light. J. Mater. Chem. A 6, 13110 (2018).
C. Fan, Q. Feng, G. Xu, J. Lv, Y. Zhang, J. Liu, Y. Qin, and Y. Wu: Enhanced photocatalytic performances of ultrafine g-C3N4 nanosheets obtained by gaseous stripping with wet nitrogen. Appl. Surf. Sci. 427, 730 (2018).
Y. Kang, Y. Yang, L-C. Yin, X. Kang, L. Wang, G. Liu, and H-M. Cheng: Selective breaking of hydrogen bonds of layered carbon nitride for visible light photocatalysis. Adv. Mater. 28, 6471 (2016).
L. Shi, F. Wang, L. Liang, K. Chen, M. Liu, R. Zhu, and J. Sun: In site acid template induced facile synthesis of porous graphitic carbon nitride with enhanced visible-light photocatalytic activity. Catal. Commun. 89, 129 (2017).
J-S. Shen, Q-G. Cai, Y-B. Jiang, and H-W. Zhang: Anion-triggered melamine based self-assembly and hydrogel. Chem. Commun. 46, 6786 (2010).
Z. Li, Y. Ma, X. Hu, E. Liu, and J. Fan: Enhanced photocatalytic H2 production over dual-cocatalyst-modified g-C3N4 heterojunctions. Chin. J. Catal. 40, 434 (2019).
Q. Liang, Z. Li, Z-H. Huang, F. Kang, and Q-H. Yang: Holey graphitic carbon nitride nanosheets with carbon vacancies for highly improved photocatalytic hydrogen production. Adv. Funct. Mater. 25, 6885 (2015).
S.C. Yan, Z.S. Li, and Z.G. Zou: Photodegradation performance of g-C3N4 fabricated by directly heating melamine. Langmuir 25, 10397 (2009).
Q. Han, B. Wang, J. Gao, Z. Cheng, Y. Zhao, Z. Zhang, and L. Qu: Atomically thin mesoporous nanomesh of graphitic C3N4 for high-efficiency photocatalytic hydrogen evolution. ACS Nano 10, 2745 (2016).
Q. Gu, Y. Liao, L. Yin, J. Long, X. Wang, and C. Xue: Template-free synthesis of porous graphitic carbon nitride microspheres for enhanced photocatalytic hydrogen generation with high stability. Appl. Catal., B 165, 503 (2015).
R.C. Dante, P. Martín-Ramos, A. Correa-Guimaraes, and J. Martín-Gil: Synthesis of graphitic carbon nitride by reaction of melamine and uric acid. Mater. Chem. Phys. 130, 1094 (2011).
M. Wang, M. Fang, C. Tang, L. Zhang, Z. Huang, Y.G. Liu, and X. Wu: A C3N4/Bi2WO6 organic–inorganic hybrid photocatalyst with a high visible-light-driven photocatalytic activity. J. Mater. Res. 31, 713 (2016).
Z. Feng, L. Zeng, Y. Chen, Y. Ma, C. Zhao, R. Jin, Y. Lu, Y. Wu, and Y. He: In situ preparation of Z-scheme MoO3/g-C3N4 composite with high performance in photocatalytic CO2 reduction and RhB degradation. J. Mater. Res. 32, 3660 (2017).
S. Yang, Y. Gong, J. Zhang, L. Zhan, L. Ma, Z. Fang, R. Vajtai, X. Wang, and P.M. Ajayan: Exfoliated graphitic carbon nitride nanosheets as efficient catalysts for hydrogen evolution under visible light. Adv. Mater. 25, 2452 (2013).
Q. Liu, X. Wang, Q. Yang, Z. Zhang, and X. Fang: Mesoporous g-C3N4 nanosheets prepared by calcining a novel supramolecular precursor for high-efficiency photocatalytic hydrogen evolution. Appl. Surf. Sci. 450, 46 (2018).
G. Zhang, M. Zhang, X. Ye, X. Qiu, S. Lin, and X. Wang: Iodine modified carbon nitride semiconductors as visible light photocatalysts for hydrogen evolution. Adv. Mater. 26, 805 (2013).
J. Fang, H. Fan, Z. Zhu, L.B. Kong, and L. Ma: “Dyed” graphitic carbon nitride with greatly extended visible-light-responsive range for hydrogen evolution. J. Catal. 339, 93 (2016).
M.Z. Rahman, J. Ran, Y. Tang, M. Jaroniec, and S.Z. Qiao: Surface activated carbon nitride nanosheets with optimized electro-optical properties for highly efficient photocatalytic hydrogen production. J. Mater. Chem. A 4, 2445 (2016).
Acknowledgments
This work was financially supported by the 111 Project “New Materials and Technology for Clean Energy” (B18018) and the Fundamental Research Funds for the Central Universities (JZ2019HGBZ0142).
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Zhu, B., Xu, G., Li, X. et al. Ultrathin porous g-CN nanosheets fabricated by direct calcination of pre-treated melamine for enhanced photocatalytic performance. Journal of Materials Research 34, 3462–3473 (2019). https://doi.org/10.1557/jmr.2019.294
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1557/jmr.2019.294