
Abstract
Conventional dissolution tests only assess the aqueous release of drugs to ensure quality and performance, without indicating whether absorption occurs through the portal or the lymphatic circulation. To address this issue, this study aimed to develop novel first-generation dissolution models that could investigate the release and uptake of oral lymphotropic drugs and examine relevant formulation issues. Dissolution of three commercial lymphotropic drug products (Terbinafina, Apo-terbinafine, and Lamisil) was done using modified versions of USP Apparatus II and IV. The developed models contained a lymphatic compartment filled with artificial chylomicrons to account for absorption through intestinal lymphatic pathway. The various products exhibited different release profiles into the aqueous media and the lymphatic media across the two tested models. The modified USP IV apparatus demonstrated greater distinction in aqueous release patterns. However, the release pattern into the lymphatic media remained similar in both models. This work represents a progress in meeting the challenges posed by the increasing complexity of pharmaceutical products containing lipophilic drugs or formulations, and has the potential to contribute towards the development of in-vitro bioequivalence standards for formulations targeting intestinal lymphatics.
Graphical Abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Dissolution testing, as defined by the United States Pharmacopeia (USP), measures the rate and extent of drug solution formation from a dosage form [1]. Its fundamental importance was recognized in 1957, when Nelson discovered a correlation between in-vivo blood concentrations of oral theophylline salts and their in-vitro dissolution [2]. Contemporary dosage form dissolution testing has been further developed and refined to aid in the drug development process. It now reflects the suitability of developed formulations in the early stages of product development and allows for the selection of formulations that will advance into in-vivo studies. It also serves as a quality control measure and a means of comparison between different commercial products containing the same active pharmaceutical ingredient (bioequivalence assessment) [3]. Furthermore, in-vitro in-vivo correlations (IVIVC) utilize in-vitro data to predict in-vivo performance in humans, bridging the gap between pre-clinical and clinical studies [4,5,6].
There are several types of dissolution apparatus commonly used in pharmaceutical research and development, and the choice of apparatus depends on various factors, including the type of dosage form, pharmacopeial and regulatory requirements, and drug properties [5]. The general performance tests monographed in the United States Pharmacopeia (USP) chapters on Dissolution < 711> [7], and Drug Release < 724> [8] details guidelines and standards for testing pharmaceutical dosage forms, and for solid oral dosage forms, USP type Apparatus 1 and 2 are most frequently used [1]. However, these standard dissolution equipment only estimate the aqueous release of drugs from formulations, without reflecting the pathways through which they are absorbed.
Most oral drugs when absorbed intracellularly (active or passive), they pass through the portal vein to the liver before entering the general circulation [9, 10]. However, certain drugs can enter the general circulation via the intestinal lymphatics (lymphotropic drugs) instead of simply conventional enteric absorption. These xenobiotics are packaged into triglyceride-rich lipoproteins called chylomicrons and then exocytosized out of the enterocytes to be taken up by the intestinal lymphatics [11, 12]. This method of drug absorption and delivery offers several pharmacokinetic and pharmacodynamics advantages, including shunting away from first-pass enteric and hepatic metabolism, potentially higher bioavailability, and increased efficacy of various treatment modalities [13,14,15]. Yet, quantifying this pathway directly is not possible without measurements in the lymphatic fluid, which requires invasive procedures [16].
In a previous study, we presented an in-vitro model crafted to predict, inhibit, and enhance lymphatic uptake. The foundation of the model was based on the interaction of drugs with chylomicrons, a process well-documented for its predictive abilities in assessing intestinal lymphatic uptake [17].
Here we report the first lymph-focused dissolution models. This study aimed to develop innovative first-generation dissolution models that provide a deeper understanding of the release and uptake of oral lymphotropic drugs, thereby enhancing formulation design strategies. In light of the growing number of formulations and delivery systems designed for lymphatic transport, these models also have the potential to eventually contribute to the establishment of bioequivalence guidelines specifically for lymphotropic formulations.
The proposed models consider both pathways through which released drugs may enter the general systemic circulation and incorporate an artificial chylomicron-containing compartment within the dissolution vessel. This compartment adds a lipid dissolution sink for lymphotropic drugs to the setup. By analyzing the media in both the dissolution vessel and lymphatic compartments, the drug content in each compartment can be assessed, providing valuable insights into drug behavior.
Materials and Methods
Materials
Sodium chloride (NaCl, CAS: 7647-15-5), potassium phosphate monobasic (KH2PO4, CAS: 7778-77-0), potassium phosphate dibasic (K2HPO4, CAS: 7758-11-4), sodium hydroxide (NaOH, CAS: 1310-73-2) and triethylamine (CAS: 121-44-8) were all obtained from Caledon Laboratories (Ontario, Canada). Hydrochloric acid (36.5–38%) was from BDH Inc. (Ontario, Canada) whereas artificial chylomicrons media (Intralipid® [16]) was purchased from Fresenius Kabi Ltd (Toronto, Ontario, Canada). HPLC-grade acetonitrile and o-phosphoric acid (85%) were both products of Fisher Chemicals (Ontario, Canada).
Equipment
The tools and equipment used in the different experiments included dialysis bags with molecular weight cut-off, MWCO: 12–14 kDa and 45 mm-width (Spectra/Por molecularporous membrane tubing SP4) from Fisher Scientific (Ontario, Canada), Amicon Ultra-0.5 30 KDa centrifugal filtering units (Millipore, Sigma-Aldrich, Darmstadt, Germany), accumet® XL20 pH/conductivity meter (Fischer Scientific, Massachusetts, USA) and density kit (XPR/XSR-Ana) from Mettler Toledo (Ohio, USA). For HPLC analysis, Shimadzu HPLC (LC-10AD, Shimadzu Corporation, Kyoto, Japan) equipped with SIL-10 A (Shimadzu Auto Injector) and UV-VIS detector (SPD-10AV) was used. Analysis was performed using Kinetex™ C18 column (250 mm ×4.6 mm×5 μm) from Phenomenex (California, USA), and resultant peak areas were integrated using LabSolutions software (Shimadzu Corporation, Kyoto, Japan).
Preparation of the Dissolution Media
Standard Simulated Gastric Fluid (pH = 1.2) and Phosphate Buffer (pH = 6.8)
USP41-NF36 protocol was employed to prepare the simulated gastric fluid (without enzymes, pH 1.2) and the phosphate buffer (pH 6.8) (p 5754 and 5748, respectively) [1].
Modified Simulated Gastric Fluid
An adapted gastric fluid was prepared by adding 0.9 g of sodium chloride, 3.27 mL of hydrochloric acid and 3.2 g of potassium dihydrogen phosphate to make 1000 mL solution using water. The pH of this solution was attuned to 1.9.
Measurement of Density of the Prepared Dissolution Media
The density of 80 mL-samples of each fluid was measured at a temperature of 25 ± 0.2 °C. The designated amount of fluid was added to the beaker and the sinker was fully submerged. Any air bubbles that adhered to the sinker were removed and the draft shield was closed. Once the balance was stabilized, the readings were recorded.
Measuring the Dissolution of Terbinafine Products Using the Developed Models
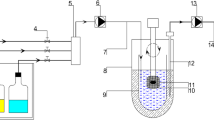
Dissolution of three commercial products of the lymphotropic drug terbinafine was tested using modified USP Apparatus II and IV (Fig. 1). The used products were Terbinafina (Laboratorio Chile, Chile), Apo-terbinafine (Apotex, Toronto, Canada) and Lamisil (Manufactured by Novartis Pharma Produktions GmbH, Germay for Novartis Pharma AG, Switzerland). Each product was tested between 4 and 6 times using each developed methodology. For terbinafine hydrochloride tablets, FDA Dissolution Methods Database calls for 500 mL media, maintained at 37 ± 0.5 °C and stirred at 50 rpm [18]. The same conditions were applied in these experiments.

Illustration of the a Modified USP Apparatus II (Top) and b Modified USP Apparatus IV (Bottom) used to study the dissolution of various commercial products of the lymphotropic drug, terbinafine. To each model a dialysis bag (molecular weight cut-off, MWCO:12-14kD and 45 mm-width) containing 5mLof artificial chylomicrons – was added to mimic lymphatic uptake. Part of this illustration was created with www.BioRender.com
Dissolution Testing via Modified USP Apparatus II
Freshly prepared and degassed media was used (250 mL of modified simulated gastric fluid (pH = 1.9)). Dialysis bag containing 5mL of artificial chylomicrons mediawas added to mimic lymphatic uptake. Initially, the dialysis bag was attached either to the bottom of the paddle or the paddle shaft. However, a systematic assessment led to determining that the most effective configuration involved placing the dialysis bag within the dissolution media in the vessel of the modified USP Apparatus II. This positioning ensured continuous contact with the media throughout the experiment, enabling valuable comparisons between the Apparatus II and Apparatus IV setups as further illustrated in the following section. Samples were taken from the vessel media at different time points (5, 15, 30, 45, and 60 min). From the dialysis bags, 60-minute samples were taken after the end of the experiment. These samples of 0.2 mL were then diluted with 0.8 mL acetonitrile before being filtered into the HPLC vial for analysis. Also from the bag, 0.5 mL was added to the centrifugal filtering unit and centrifuged at 10 K g for 10 min. The filtrate was collected and analysed using HPLC. The calculated concentrations for the samples were plotted to determine the cumulative percent dissolved over time using DDSolver [19].
Dissolution Testing via Modified USP Apparatus IV
This apparatus included a pump that moved the media from a container through a flow cell of 22.6 mm internal diameter. The cell held the dosage form that was placed over 25 glass beads of 1 mm size positioned in the bottom of the cell. The media used for the test were placed in a constant temperature water bath, maintained at 37 ± 0.5 ° C and pumped through the cell. The dissolved drug along with any other substances that pass through the flow cell were collected in a vessel comparable to the USP Apparatus II vessel. First, simulated gastric fluid (SGF) was pumped through the cells at a flow rate of 8 mL/min for 15 min. This was followed by phosphate buffer (pH 6.8) at a flow rate of 16 mL/min for 45 min. Upon switching to the second media, dialysis bag containing 5 mL of artificial chylomicrons media was added to the collection vessel. Here, the collection vessel would start filling as the experiment progressed, therefore, attaching the bag to a fixed point (paddle bottom or shaft) would not have provided the consistent media contact that the chosen position did, making it a critical choice for this study. During the dissolution test, 5 mL samples were collected at 5, 15, 30, 45, and 60 min. The media volume was weighed and exact volume was calculated using the previously determined density of the media. Samples from the dialysis bags containing the artificial chylomicrons were collected at the end and processed as described with the apparatus II.
HPLC Instrumentation and Chromatographic Conditions
The mobile phase was composed of a mixture of acetonitrile and water containing 0.02 M ortho-phosphoric acid and 0.01 M triethylamine (40:60 v/v) and was eluted isocratically at 25 ºC and a flow rate of 1 mL/min. All samples were injected at 20 µl, and detection was set at 224 and 283 nm.
Statistical Analysis
All dissolution groups were established with 4–6 independent replicates, and the results are presented as mean ± standard error of the mean (SE). Statistical differences were assessed using an ANOVA test with a significance level of α = 0.05, where p-values below 0.05 were deemed statistically significant in all instances.
Results
As depicted in Fig. 2, results of performance of the different products in the modified USP Apparatus II demonstrated that terbinafine release into the dissolution media was highest with Lamisil followed by Terbinafina, then Apo-terbinafine (87.90%, 83.17% and 71.68%, respectively). For the lymphatic uptake aspect of the model system, Terbinafina and Lamisil accumulated more drug in the lymphatic compartment than Apo-terbinafine. However, Terbinafina had higher lymphatic uptake than Lamisil (2.58%, 2.32%, respectively). Apo-terbinafine had 1.71% in the lymphatic compartment of this model.

Cumulative percentage of dissolved terbinafine from the commercial products into modified USP apparatus II. The Line graph represents the dissolution vessel profile and the columns represent the % of the drug with respect to the total dose in the lymphatic vicinity (dialysis membrane containing the artificial chylomicrons) after 60 min. Data represent the mean values and bars represent the stansard error (n = 4–6). * Denotes statistically significant from other groups p < 0.05
The results obtained from the modified USP Apparatus IV revealed notable differences among the tested products (Fig. 3). Specifically, Terbinafina demonstrated superior release as documented in the collection vessel compared to Lamisil, with both exhibiting significantly higher release than Apo-terbinafine (101.93%, 83.93%, and 11.48%, respectively). A similar phenomenon was observed in the lymphatic compartment, with Terbinafina exhibiting the highest accumulation in the lymphatic compartment (2.14%), followed by Lamisil (1.16%) and Apo-terbinafine (0.21%).

Cumulative percentage of dissolved terbinafine from the commercial products into modified USP apparatus IV. The Line graph represents the dissolution vessel profile and the columns represent the % of the drug with respect to the total dose in the lymphatic vicinity (dialysis membrane containing the artificial chylomicrons) after 60 min. Data represent the mean values and bars represent the stansard error (n = 4–6). * Denotes statistically significant from other groups p < 0.05
Discussion
Following oral administration, immediate-release dosage forms have to disintegrate to liberate drugs that dissolve in the physiological fluid before moving across the GIT [20, 21]. While most drugs travel from the GIT into the systemic circulation via portal blood, others might take a different route to the systemic circulation through intestinal lymphatic voyage via chylomicrons [13, 16]. The latter mainly have log P > 5 and solubility in long chain triglycerides > 50 mg/g, however other molecular descriptors may also play a role [17]. These drugs get packaged into chylomicrons which are taken up by the lymphatics rather than blood capillaries once they get exocytosized from the enterocytes [22, 23].
Dissolution testing of oral dosage forms quantifies the release of the active pharmaceutical ingredient (API) in a specified dissolution media that might mimic the physiological environment in which the same process would happen in-vivo [24]. For drugs not subjected to first-pass effect or other physiological phenomenon and whose permeability is not limited, dissolution testing performance can be an indicative method of the in-vivo drug bioavailability in plasma [25]. However, not all drugs reach the systemic circulation via the portal vein. For those xenobiotics a more representative dissolution model should be considered; one that considers both blood and lymphatic pathways through which candidate APIs may reach the general circulation.
In the proposed models for the measurement of dissolution of lymphotropic drugs, artificial chylomicrons [16, 17] were added to the dissolution media in the vessel of modified USP Apparatus II and to the media in the collection vessel of modified USP Apparatus IV to facilitate the drug uptake into a lymphatic-like environment and hence account for the proportion of the lymphatic uptake of the dissolved drug.
Terbinafine was the selected model drug in this study. It has been documented to go through intestinal lymphatics [26]. The molecular descriptors of terbinafine are in Table 1. It comes in various oral solid dosage forms including tablets and granules [27]. Three commercial product of 250 mg terbinafine hydrochloride tablets were sampled and utilized in this work (Terbinafina, Apo-terbinafine and Lamisil).
Terbinafine hydrochloride is listed officially in the United States Pharmacopoeia, British Pharmacopoeia, and European Pharmacopoeia [28]. For collecting preliminary data, a standard USP Apparatus II was employed with different media, namely standard simulated gastric fluid (SGF, pH = 1.2), modified simulated gastric fluid (modified SGF, pH = 1.9), and phosphate buffer (pH = 6.8). Standard simulated gastric fluid has been reported as a dissolution medium for terbinafine tablets and other dosage forms in the literature [20]. Preliminary findings (data not shown) indicated that the cumulative percent dissolved of terbinafine from the different products in SGF was quite low. The highest amount of the drug detected in the media was about 15%. Nevertheless, the performance of the tablets in terms of dissolution rate was increased in the modified SGF, and was ultimately utilized instead of the standard SGF. Results in phosphate buffer results showed close to zero drug release over time.
Being a basic drug whose solubility is pH-dependent with maximum solubility at acidic pH, modified SGF yielded understandably better results than the basic phosphate buffer. As seen from the solubility graph of terbinafine (Fig. 4), the drug solubility in both pHs of the standard and modified SGFs (1.2 and 1.9, respectively) remains the same. However, the superior performance of the modified SGF than the standard SGF might be attributed to the excipients utilized in the different products as will be discussed later.

Solubility graph of terbinafine hydrochloride in different pHs. Data was predicted by ADMET predictor (version 10.4 (Simulations Plus Inc., Lancaster, CA, USA)
Results obtained from modified Apparatus II could be justified by the excipients used in the different products. While all three products are pharmaceutical equivalent according to the FDA definitions, differences in the formulation, specifically in the excipients used are evident (Table 2). Terbinafina and Lamisil have identical formulations, while the percent ratio of the excipients in the formulation and processing variables are not known. In contrast, excipients utilized in Apo-terbinafine are different. Sodium starch glycolate was used as the disintegrant in Terbinafina and Lamisil, while croscarmellose sodium was used in Apo-terbinafine. Sodium starch glycolate is known to be highly effective in promoting a more rapid tablet disintegration and dissolution, while croscarmellose sodium may be less effective in this regard, likely due to functional differences between the two disintegrants. Specifically, sodium starch glycolate has a higher swelling and wicking capacity, meaning it can absorb additional water and distribute it more evenly throughout the tablet, leading to more uniform disintegration. It also has greater compressibility, allowing it to be used in higher concentrations without compromising tablet hardness or friability, ultimately enhancing tablet disintegration and dissolution [29, 30].
Although the hydrophobicity of hydroxypropyl cellulose and hydroxypropylmethyl cellulose is different [31]. Yet, that did not affect the performance of their formulations (Terbinafina and Lamisil, respectively). However, for Apo-terbinafine, the usage of methylcellulose in Apo-terbinafine could have impacted the drug release. Methylcellulose is a water-soluble polymer that can form a gel-like matrix in the presence of water [32]. The presence of this gel can decrease tablet porosity, water diffusivity, and increase the time it takes for the tablet to disintegrate, all of which can result in a slower drug release rate from the tablet. This property is utilized in controlled release formulations [33, 34]. The hydration of methyl cellulose is lower compared to both hydroxypropyl methyl cellulose [35] and hydroxypropyl cellulose [31]. The adhesive property of methyl cellulose, which could potentially block tablet pores, may explain the prolonged disintegration time observed in Apo-terbinafine tablets compared to Lamisil and Terbinafina.
Moreover, the manufacturing process of these products could also have affected their dissolution performance [36]. Terbinafina and Lamisil may have been manufactured under conditions that result in a more porous or more rapidly dissolving tablet. For example, the less compression force used during production could impact the tablets and impart superior dissolution performance compared with Apo-terbinafine. Other factors could be granulation or direct compression, however, this information is proprietary [37].
In an attempt to simulate the changing conditions inside the gastrointestinal tract, modified USP Apparatus IV was utilized [38]. A dynamic dissolution protocol with two different media was applied: one that mimicked the environment of the stomach (modified SGF), followed by one that simulated the environment of the intestines (phosphate buffer with a pH of 6.8). In experiments, when switching to the intestinal-like media, a dialysis bag containing artificial chylomicrons media was added to the collection vessel to account for the intestinal lymphatic uptake of the dissolved drug.
The observed differences in release when the modified USP Apparatus IV was used were attributed to the rate of disintegration and dissolution of the different products. For Terbinafina and Lamisil, a faster disintegration and dissolution resulted in higher drug release, whereas Apo-terbinafine exhibited low release due to its slow disintegration and subsequent dissolution, which may be attributed to its excipients or tableting processes as previously discussed.
It is noteworthy to mention that the solution in the collection vessel turned turbid at completion of the experiments. The solubility of the drug and the pH change between the stomach and intestine may explain this displayed phenomenon of precipitation. A dissolved basic drug entering a basic intestinal environment may cause first a supersaturated solution followed by precipitation out of solution [39]. However, due to sink conditions and drug absorption from the intestinal lumen, the drug redissolves and is subsequently absorbed [40, 41]. In this experiment set-up, the presence of phosphate buffer in the collection vessel, combined with simulated gastric fluid, altered the pH of the local media. As the solubility of terbinafine is pH-dependent, and when the pH of the media is suboptimal, the drug can precipitate out of the solution, as observed in this case.
To ensure the ability of the model to distinguish lymphotropic drugs accurately, a comparative analysis with the biphasic method [21] was conducted. Interestingly, the accumulation in the octanol phase in the biphasic model showed almost identical behaviour for both a lymphotropic drug (rifampicin) and a non-lymphotropic drug (Ibuprofen [21]). However, in the model proposed in this paper, the uptake into lymphatic compartment varied among different lymphotropic drugs (terbinafine and rifampicin), despite the fact that the aqueous solubility of rifampicin was comparable to one of the terbinafine products (data not shown).
Nonetheless, refinements in the proposed experimental models can be tailored according to the specific goals of the study. For the second model, it is important to consider the more alkaline pH conditions in the collection vessel, which can present solubility challenges, especially for drugs with basic properties, as observed in our tested compounds. To address this concern, one potential strategy is to introduce surfactants into the dissolution media, which can enhance drug solubility under alkaline conditions. Additionally, continuous sampling from the lymphatic compartment could be explored as a refinement. This approach would allow for the simultaneous monitoring of drug uptake profiles into the lymphatic vicinity along with the aqueous dissolution profile. That can help gain deeper insights into the dynamics of drug absorption and distribution, particularly in the context of lymphatic uptake.
Overall, the evolution of dissolution testing and USP apparatus represents an on-going commitment to enhancing the quality assessment of pharmaceutical products. The innovative models introduced herein have the potential to expand this evolution by focusing on the often-underestimated lymphatic absorption pathway facilitated by chylomicrons and the increasingly recognized use of hydrophobic xenobiotics in formulations. These models offer greater discriminatory power in dissolution testing, allowing for precise evaluations in complex contexts. Additionally, they open doors to post-approval changes, enabling pharmaceutical companies to adapt to evolving regulatory requirements and emerging scientific insights with precision and efficacy.
Conclusion
Lymphotropic candidate formulations are specifically designed to facilitate uptake via the intestinal lymphatic system as an alternative absorption route to the enteric-portal pathway. However, conventional dissolution tests assess drug release in aqueous media solely for quality and performance assurance, without contemplating the absorption pathway, be it portal or lymphatic. This study showed that it may be possible to develop lymphatic-focused dissolution models to assess formulations and factors potentially affecting chylomicron uptake. With the challenges encountered in solubilizing hydrophobic drugs and the increased focus on lipid based formulations of xenobiotics, it is prudent that dissolution testing and development are also developed and refined in an attempt to more accurately assess increase possible lymphatic uptake, and variables such as excipients and manufacturing that may impact formulation performance. Given the rising complexity of pharmaceutical products and further refining and developing performance testing methods must also be reconceptualised and refined accordingly where possible.
References
United States Pharmacopeia and National Formulary USP 41- NF 36. The United States Pharmacopeial Convention, Rockville. 2018.
Nelson E. Solution rate of theophylline salts and effects from oral administration. J Am Pharm Assoc. 1957;46(10):607–14. https://doi.org/10.1002/jps.3030461012.
US Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research. Office of Generic Drugs/Office of Generic Drugs Policy. Approved Drug Products With Therapeutic Equivalence Evaluations. https://www.fda.gov/media/71474/download?attachment. 2024. Accessed 24 Mar 2024.
Grady H, Elder D, Webster GK, Mao Y, Lin Y, Flanagan T, et al. Industry’s view on using quality control, biorelevant, and clinically relevant dissolution tests for pharmaceutical development, registration, and commercialization. J Pharm Sci. 2018;107(1):34–41. https://doi.org/10.1016/j.xphs.2017.10.019.
Uddin R, Saffoon N, Sutradhar KB. Dissolution and dissolution apparatus: a review. Int J Cur Biomed Phar Res. 2011;1(4):201–7.
Azarmi S, Roa W, Löbenberg R. Current perspectives in dissolution testing of conventional and novel dosage forms. Int J Pharm. 2007;328(1):12–21. https://doi.org/10.1016/j.ijpharm.2006.10.001.
United States Pharmacopeia. <711 > Dissolution. United States Pharmacopeia and National Formulary USP 43- NF 38. The United States Pharmacopeial Convention, Rockville. 2023. https://doi.org/10.31003/USPNF_M99470_03_01.
United States Pharmacopeia. <724 > Drug Release. United States Pharmacopeia and National Formulary USP 43- NF 38. The United States Pharmacopeial Convention, Rockville. 2020. https://doi.org/10.31003/USPNF_M99490_05_01.
Vimalson DC. Techniques to enhance solubility of hydrophobic drugs: an overview. Asian J Pharm. 2016;10(2):S67–75.
Vishwakarma N, Jain A, Sharma R, Mody N, Vyas S, Vyas SP. Lipid-based nanocarriers for lymphatic transportation. AAPS PharmSciTech. 2019;20(2):83. https://doi.org/10.1208/s12249-019-1293-3.
Managuli RS, Raut SY, Reddy MS, Mutalik S. Targeting the intestinal lymphatic system: a versatile path for enhanced oral bioavailability of drugs. Expert Opin Drug Deliv. 2018;15(8):787–804. https://doi.org/10.1080/17425247.2018.1503249.
Cifarelli V, Eichmann A. The intestinal lymphatic system: functions and metabolic implications. Cell Mol Gastroenterol Hepatol. 2019;7(3):503–. https://doi.org/10.1016/j.jcmgh.2018.12.002. 13.
Yousef M, Silva D, Chacra NB, Davies NM, Löbenberg R. The lymphatic system: a sometimes-forgotten compartment in pharmaceutical sciences. J Pharm Pharm Sci. 2021;24:533–47. https://doi.org/10.18433/jpps32222.
Khan AA, Mudassir J, Mohtar N, Darwis Y. Advanced drug delivery to the lymphatic system: lipid-based nanoformulations. Int J Nanomed. 2013;8(1):2733–44. https://doi.org/10.2147/IJN.S41521.
Zhang Z, Lu Y, Qi J, Wu W. An update on oral drug delivery via intestinal lymphatic transport. Acta Pharm Sin B. 2021;11(8):2449–68. https://doi.org/10.1016/j.apsb.2020.12.022.
Yousef M, Park C, Le TS, Chacra NB, Davies NM, Löbenberg R. Simulated lymphatic fluid for in-vitro assessment in pharmaceutical development. Dissolution Technol. 2022. https://doi.org/10.14227/DT290222P86.
Yousef M, Park C, Henostroza M, Chacra NB, Davies NM, Löbenberg R. Development of a novel in-vitro model to study lymphatic uptake of drugs via artificial chylomicrons. Pharmaceutics. 2023;15(11):2532. https://doi.org/10.3390/pharmaceutics15112532.
US Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research. Office of Pharmaceutical Quality/Office of New Drug Products Division of Biopharmaceutics. Dissolution Methods Database. 2022. https://www.accessdata.fda.gov/scripts/cder/dissolution/dsp_SearchResults.cfm. Accessed 20 Apr 2024.
Zhang Y, Huo M, Zhou J, Zou A, Li W, Yao C, et al. DDSolver: an add-in program for modeling and comparison of drug dissolution profiles. AAPS PharmSciTech. 2010;12:263–71. https://doi.org/10.1208/s12248-010-9185-1.
Devadasu VR, Deb PK, Maheshwari R, Sharma P, Tekade RK. Physicochemical, pharmaceutical, and biological considerations in GIT absorption of drugs. In: Tekade RK, editor. Dosage Form Design considerations. Vol I. Massachusetts: Academic Press, Elsevier Inc.; 2018. pp. 149–78. https://doi.org/10.1016/B978-0-12-814423-7.00005-8.
Silva DA, Melo KJC, Davies NM, Chacra NB, Ferraz HG, Löbenberg R. A BCS-based bowaiver approach using biphasic dissolution test. Dissolution Technol. 2021;28(4):40–8. https://doi.org/10.14227/DT280421P40.
Porter CJH, Charman WN. Intestinal lymphatic drug transport: an update. Adv Drug Del Rev. 2001;50(1–2):61–80. https://doi.org/10.1016/S0169-409X(01)00151-X.
Trevaskis NL, Kaminskas LM, Porter CJH. From sewer to saviour-targeting the lymphatic system to promote drug exposure and activity. Nat Rev Drug Discov. 2015;14(11):781–803. https://doi.org/10.1038/nrd4608.
Dressman JB, Amidon GL, Reppas C, Shah VP. Dissolution testing as a prognostic tool for oral drug absorption: immediate release dosage forms. Pharm Res. 1998;15(1):11–22. https://doi.org/10.1023/A:1011984216775.
Li S, He H, Parthiban LJ, Yin H, Serajuddin AT. IVIVC considerations in the development of immediate-release oral dosage form. J Pharm Sci. 2005;94(7):1396–417. https://doi.org/10.1002/jps.20378.
Baheti A, Srivastava S, Sahoo D, Lowalekar R, Prasad Panda B, Padhi BK, et al. Development and pharmacokinetic evaluation of industrially viable self-microemulsifying drug delivery systems (SMEDDS) for terbinafine. Curr Drug Deliv. 2016;13(1):65–75. https://doi.org/10.2174/1567201812666150120153357.
Kuminek G, Rauber GS, Riekes MK, De Campos CEM, Monti GA, Bortoluzzi AJ, et al. Single crystal structure, solid state characterization and dissolution rate of terbinafine hydrochloride. J Pharm Biomed Anal. 2013;78:105–11. https://doi.org/10.1016/j.jpba.2013.02.001.
Kanakapura B, Penmatsa VK. Analytical methods for determination of terbinafine hydrochloride in pharmaceuticals and biological materials. J Pharm Anal. 2016;6(3):137–49. https://doi.org/10.1016/j.jpha.2016.01.003.
Shafiq-un-Nabi S, Shakeel F, Talegaonkar S, Ali J, Baboota S, Ahuja A, et al. Formulation development and optimization using nanoemulsion technique: a technical note. AAPS PharmSciTech. 2007;8(2):E12–7. https://doi.org/10.1208/pt0802028.
Berardi A, Janssen PHM, Dickhoff BHJ. Technical insight into potential functional-related characteristics (FRCs) of sodium starch glycolate, croscarmellose sodium and crospovidone. J Drug Deliv Sci Technol. 2022;70:103261. https://doi.org/10.1016/j.jddst.2022.103261.
Brady J, Dürig T, Lee PI, Li J-X. (2017) Polymer Properties and Characterization. In: Qiu Y, Chen Y, Zhang GGZ, Yu LU, Mantri R, editors. Developing Solid Oral Dosage Forms. Massachusetts: Academic Press, Massachusetts, 2017. pp. 181–223.
Hu Z, Patten T, Pelton R, Cranston ED. Ynergistic stabilization of emulsions and emulsion gels with water-soluble polymers and cellulose nanocrystals. ACS Sustain Chem Eng. 2015;3(5):1023–31. https://doi.org/10.1021/acssuschemeng.5b00194.
Aulton ME, Taylor KMG. Aulton’s Pharmaceutics E-Book: the design and manufacture of medicines. 6th ed. Amsterdam: Elsevier Health Sciences; 2021.
Cheng Y, Qin H, Acevedo NC, Shi X. Development of methylcellulose-based sustained‐release dosage by semisolid extrusion additive manufacturing in drug delivery system. J Biomed Mater Res Part B Appl Biomater. 2021;109(2):257–68. https://doi.org/10.1002/jbm.b.34697.
Ford JL. Thermal analysis of hydroxypropylmethylcellulose and methylcellulose: powders, gels and matrix tablets. Int J Pharm. 1999;179(2):209–. https://doi.org/10.1016/S0378-5173(98)00339-1.
Arshad MS, Zafar S, Yousef B, Alyassin Y, Ali R, AlAsiri A, et al. A review of emerging technologies enabling improved solid oral dosage form manufacturing and processing. Adv Drug Deliv Rev. 2021;178:113840. https://doi.org/10.1016/j.addr.2021.113840.
Silva DA, Al-Gousous J, Davies NM, Chacra NB, Webster G, Lipka E, et al. Simulated, biorelevant, clinically relevant or physiologically relevant dissolution media: the hidden role of bicarbonate buffer. Eur J Pharm Biopharm. 2019;142:8–19. https://doi.org/10.1016/j.ejpb.2019.06.006.
Singh I, Aboul-Enein HY. Advantages of USP apparatus IV (flow-through cell apparatus) in dissolution studies. J Iran Chem Soc. 2006;3:220–22. https://doi.org/10.1007/BF03247211.
Okumu A, DiMaso M, Löbenberg R. Computer simulations using GastroPlus™ to justify a biowaiver for etoricoxib solid oral drug products. Eur J Pharm Biopharm. 2009;72(1):91–8. https://doi.org/10.1016/j.ejpb.2008.10.019.
De la Cruz-Moreno MP, Montejo C, Aguilar-Ros A, Dewe W, Beck B, Stappaerts J, et al. Exploring drug solubility in fasted human intestinal fluid aspirates: impact of inter-individual variability, sampling site and dilution. Int J Pharm. 2017;528(1–2):471–84. https://doi.org/10.1016/j.ijpharm.2017.05.072.
Sugita K, Takata N, Yonemochi E. Dose-dependent solubility–permeability interplay for poorly soluble drugs under non-sink conditions. Pharmaceutics. 2021;13(3):323. https://doi.org/10.3390/pharmaceutics13030323.
Funding
This work was supported by MITACS Accelerate Internship (IT24899). It is important to note that the opinions, interpretations, and conclusions presented in this study are solely those of the authors and do not necessarily reflect the views of the funding agency.
Author information
Authors and Affiliations
Contributions
M.Y.: Conceptualization, Methodology, Investigation, Formal analysis, Writing – original draft, Writing – review & editing.; C.P.: Methodology, Formal analysis, Writing – review & editing; N.B.C.: Investigation, Supervision, Writing – review & editing; N.M.D.: Conceptualization, Methodology, Supervision, Writing – review & editing, Resources, Project administration, Funding acquisition; R.L.: Conceptualization, Methodology, Supervision, Writing – review & editing, Resources, Project administration.
Corresponding authors
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Yousef, M., Park, C., Chacra, N.B. et al. Novel First-Generation Dissolution Models to Investigate the Release and Uptake of Oral Lymphotropic Drug Products. AAPS PharmSciTech 25, 187 (2024). https://doi.org/10.1208/s12249-024-02866-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1208/s12249-024-02866-y