
Abstract
The classically used nontargeted chemotherapeutic approach to pancreatic cancer has a dual drawback of suboptimal drug delivery at the target site and the systemic side effects produced by the unfettered exposure of the drug to healthy tissue. This study has the objective of developing novel poly(2-ethyl-2-oxazoline) (PETOX)–based long circulating liposomes loaded with gemcitabine and irinotecan for the treatment of pancreatic ductal adenocarcinoma, with a juxtaposition to PEGylated and uncoated liposomes. A PETOX−cholesteryl chloroformate lipopolymer conjugate (PETOX–ChC) with a carbonate linkage was prepared and characterized by 1H NMR, FTIR, and DSC. Liposomes were prepared using the thin film hydration technique followed by freeze–thaw and membrane extrusion methods. Liposome characterization includes particle size determination, zeta potential determination using a zetameter, and structural elucidation using 31P NMR and cryo-TEM. The PETOXylated liposomes showed a particle size of 180.1 ± 2.2 nm and a zeta potential of − 33.63 ± 1.23 mV. The liposomal combination therapy of gemcitabine and irinotecan was found to have an IC50 value 39 times lower in comparison to the drug combination in solution, while the PEGylated and PETOXylated liposomes showed IC50 values 1.6 times lower and 2 times lower than that of uncoated liposomes, respectively, against Mia PaCa II pancreatic cancer cell line. The PEGylated and PETOXylated liposomes showed 4.1 and 5.4 times slower macrophagial uptake in vitro in comparison to the uncoated liposomes respectively. The PEGylated liposomes showed 11% higher in vitro macrophagial uptake in comparison to PETOXylated liposomes.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
Pancreatic cancer is a collective term used for the various types of neoplasms originating from the exocrine and endocrine fractions of the pancreas. The National Cancer Institute estimates that 57,600 new cases of pancreatic cancer will be reported in 2020, along with 47,050 deaths associated with this morbid condition (1). Further, as the disease is characterized by early micrometastatic spread, long-term survival following surgery is poor with 80% of patients experiencing a local or distant recurrence within 2 years (2). Of these cancers, more than 90% are pancreatic ductal adenocarcinoma (PDAC) which is of exocrine origin (3). Treatment options remain limited and chemotherapy remains the first-line treatment for advanced PDAC, even after decades of research.
Burris et al. (4) established gemcitabine monotherapy as the gold standard for first-line treatment of pancreatic cancer in 1997. Gemcitabine (2′,2′-difluoro-2′-deoxycytidine) has a dual mechanism of action of being both a ribonucleotide reductase inhibitor and a deoxycytidine analog; after triphosphorylation, it is incorporated into newly formed DNA and causes DNA chain termination (5). The first combination chemotherapy regimen to demonstrate a statistically significant survival benefit versus gemcitabine monotherapy in a phase III clinical trial was reported by Conroy et al. (6) in 2011. It consisted of 5-flurouracil, irinotecan, and oxaliplatin. Gillam et al. (7) conducted a global, randomized, open-label, phase 3 trial with liposomal irinotecan in metastatic pancreatic cancer after previous gemcitabine-based therapy and found that treatment extends survival with a manageable safety profile in these patients. Irinotecan is a synthetic derivative of camptothecin which acts by inhibiting topoisomerase I, and as a prodrug termed the CPT-11 form, is converted predominantly in the liver by carboxylesterases to the active metabolite SN-38 (8). Multiple clinical trials have assessed the viability of a combination of gemcitabine and irinotecan for the treatment of pancreatic cancer (9,10,11). However, no clinical trials have been conducted for the combination of gemcitabine with liposomal irinotecan for pancreatic cancer at the time of writing this article.
A liposome is a spherical concentric vesicle composed of a phospholipid bilayer employed as a drug delivery system. By virtue of their significant drug-carrying capacity of both hydrophilic and hydrophobic drugs and as a biocompatible lipid exterior, liposomes offer a viable means of local delivery of therapeutic agents to the site of interest by active or passive targeting while reducing systemic toxicity. Liposomes are the most common lipid-based formulation for drug delivery and the most successful nanodelivery system to date (12).
The uncoated liposome surface is strongly affected by physical interactions with specific circulating proteins (complement proteins, laminin, fibronectin, C-reactive protein, type I collagen, and immunoglobulins) in a phenomenon commonly referred to as opsonization, which tags the nanoparticles for uptake by the mononuclear phagocyte system and subsequent clearance (13,14). Depending on the size and composition of the liposome, macrophagial uptake can occur within minutes after administration and remove the liposomes from the circulation, leading to loss of systemically available drugs as well as immunosuppression (15). Extensive research was undertaken to overcome these problems by surface modification of the liposomes with inert polymers to form a barrier to opsonization and produce long circulating liposomes, also known as stealth liposomes. These long circulating liposomes achieve passive targeting of solid tumors by harnessing enhanced permeability and retention (EPR), demonstrated by Matsumura and Maeda (16) in 1986 using copolymer of styrene and maleic acid conjugated to neocarzinostatin.
A major development in the last decade of the twentieth century was the synthesis of PEGylated liposomes with a prolonged circulation time in the blood. PEG–liposomes containing polyethylene glycol derivatives of phosphatidyl ethanolamine (PEG–lipid) are not readily taken up by macrophages in the RES and, hence, remain in the circulation for a relatively long period (17). However, there are multiple issues associated with PEGylation of liposomes. Klibanov et al. (18) were the first to address the uptake of intravenously injected PEGylated nanoparticles by the MPS being the main rate-limiting step in targeted delivery to tumors. Dams et al. (19) managed to show that the enhanced clearance of a second dose of PEGylated liposomes in mice is mediated by unidentified soluble protein produced reactionary to the first injection and introduced the term ‘accelerated blood clearance effect’ (ABC effect) of PEGylated liposomes. Ishida et al. (20,21) traced back the established ABC effect to PEG-specific immunoglobulin M (IgM) in rats by showing that transfusion of serum collected from rats pretreated 5 days before with PEGylated liposomes into treatment naive rats could elicit enhanced clearance of PEGylated liposome in these rats. Ishida et al. (22) further went on to show that the liposomes elicit an anti-PEG IgM response in a T cell–independent manner and appear to be a TI-2 antigen; splenic MZ B cells may be essential for the immune response against PEGylated liposomes. While previous studies found treatment-induced anti-PEG immunological elements in patients, Armstrong et al. (23) found preexisting anti-PEG in 80% of the patients enrolled in a trial with a sample size of 15 patients. The group also demonstrated that the anti-PEG elements had no effect on the unmodified drug in the plasma. Yang et al. (24) found detectable levels of anti-PEG Ab in ∼ 72% of the contemporary specimens (18% IgG, 25% IgM, 30% both IgG and IgM). The worrying fact about the findings is that the same group reported 56% of serum samples collected during 1970–1999 (20% IgG, 19% IgM, and 16% both IgG and IgM), pointing out a marked and rapid increase in the prevalence of anti-PEG in the general populace. Due to the rising issues with the use of PEG discussed above, the search for a viable replacement for PEG remains imperative.
The first report of a ring-opening polymerization reaction of 2-substituted 2-oxazolines (PETOX) was made in 1966 by parallel study groups of Tomalia et al. (25) Seeliger et al. (26), Kagiya et al. (27), and Bassiri et al. (28). The utility of PETOX conjugates in extending the circulation time of drugs was explored by Verlander et al. (29) in 1992, when they attempted direct conjugation of unterminated PETOX to human protein C. A novel attempt to impart stealth properties to liposomes using PETOX was undertaken by Woodle et al. (30). Zalipsky et al. (31) performed a similar study in mice where they found that the PETOX-conjugated liposomes exhibited long plasma half-life and low hepatosplenic uptake. Xu et al. (32) developed long circulating and pH-sensitive PETOXylated liposomes in their study.
The first PETOX-based therapeutic regimen to enter human clinical studies was put forth by Moreadith et al. (32) in 2017. The study involved SER-214, a PETOX conjugate of rotigotine for treatment in Parkinson’s syndrome used as a once-weekly IV injectable delivered in a standard insulin syringe. The study showed all the subjects completed dosing without any significant adverse events. A single IV injection of SER-214 of 20 mg was deemed safe, well-tolerated, and demonstrated predictable pharmacokinetics, and the clinical study has now entered the multidose phase of the trial (33).
The primary objective of this study was to develop a stealth liposomal delivery system based on PETOX for passive targeted delivery of gemcitabine and irinotecan for the treatment of PDAC. A lipopolymer conjugate of PETOX and cholesteryl chloroformate was prepared, and the process was verified using analytical techniques such as proton NMR, differential scanning calorimetry (DSC), and Fourier transform infrared spectroscopy (FTIR) spectroscopy. The study also employed techniques such as dynamic light scattering (DLS), cryo-TEM imaging, MTT cytotoxicity assay, and live confocal imaging to determine the characteristics of blank, gemcitabine- and irinotecan-loaded uncoated, and stealth liposome formulations. DLS provided the particle size analysis of the uncoated and the surface-modified liposomes, while cryo-TEM imaging of the liposomes provided an understanding of their lamellarity. A comparison of the cytotoxicity of the blank, drug-loaded uncoated, and surface-modified liposomes in Mia PaCa II pancreatic cancer cell line was performed using the MTT cytotoxicity assay. Lastly, live confocal imaging was employed to assess the uptake of the liposomes by human macrophages in KG1 cell line.
EXPERIMENTAL SECTION
Materials
Poly(2-ethy-2-oxazoline) (PETOX) MW 5000 g/mol (I24066-50) was purchased from Polysciences, Inc. (Warrington, PA). Gemcitabine hydrochloride, cholesteryl chloroformate (ChC), trisodium citrate dihydrate, citric acid monohydrate, sodium dodecyl sulfate (SDS), HEPES buffer, d-(+)-trehalose dihydrate, and potassium carbonate were purchased from Sigma Aldrich (St. Louis, MO). Irinotecan hydrochloride was purchased from MedChem Express. MTT reagent, 4-dimethylaminopyridine (DMAP), and N,N′-dicyclohexylcarbodiimide (DCC) were purchased from Acros Organics (NJ). Cholesterol, Tween 80, dimethyl formamide (DMF), acetonitrile, and dichloromethane (DCM) were purchased from Fisher Scientific (Hampton, NH). Cellulose ester dialysis membranes (12–14 kDa) were purchased from Spectrum Laboratories (CA). RPMI 1640 medium and IMDM medium were purchased from Cellgro Mediatech Inc. (Herndorn, VA). NucBlue® Live reagent and LysoTracker Deep Red were purchased from Invitrogen Inc. (Carlsbad, CA). Soy phosphatidylcholine (J61675) was purchased from Alfa Aesar (Tewksbury, MA). 1,2-Distearoyl-sn-glycero-3-phosphoethanolamine–conjugated polyethylene glycol (DSPE-mPEG 2000) was purchased from Avanti Polar Lipids. Inc. (Alabaster, AL).
Cells
Mia PaCa II pancreatic cancer cells were obtained from Dr. Surinder Batra and Dr. Rakesh Singh of the University of Nebraska Medical Center, Omaha, NE. KG-1 human macrophage cells (ATCC® CCL-246™) were purchased from the American Type Culture Collection (ATCC) (Manassas, VA).
Methods
Conjugation of PETOX with Cholesteryl chloroformate
A lipopolymer conjugate of PETOX and ChC with a carbonate linkage was prepared by esterification of the hydroxy end group and the chloroformate group, respectively, based on the protocol discussed by Xu et al. (33). The scheme of conjugation is shown in Fig. 1a. A solution of PETOX, ChC, DCC, DMAP, and potassium carbonate was made in 15 mL of DCM. The mixture was mechanically agitated for 24 h at room temperature in a nitrogen environment. The mixture was concentrated under vacuum using a rotary evaporator and further subjected to vacuum for 5 h. The obtained product was washed with cold 1 M HCl and n-hexane to remove the side products and unreacted reactants.

a Conjugation of PETOX and cholesteryl chloroformate. b Proton NMR spectra of PETOX. c Proton NMR spectra of cholesteryl chloroformate. d Proton NMR spectra of PETOX-cholesterol conjugate
Determination of Conjugation with Proton Nuclear Magnetic Resonance
The conjugation between PETOX and ChC was verified using proton nuclear magnetic resonance (1H NMR) spectra which were obtained on an Ascend TM 400-MHz Bruker spectrometer. The sample was dissolved in deuterated chloroform by vortexing for 2–3 min and analyzed. A total of 50 scans were accumulated with a 90° pulse width, pulse length of 16 μs, and 1 s relaxation delay. The obtained spectra were analyzed using TopSpin® software 4.0. The study was performed using pure PETOX, pure ChC, blank, and the PETOX–ChC.
Determination of Conjugation with Differential Scanning Calorimetry
The conjugation between PETOX and ChC was verified using DSC (Shimadzu DSC-60, Kyoto, Japan) with a thermal analysis operating system (Shimadzu TA-60WS, Kyoto, Japan). About 8 mg of sample was weighed in an aluminum pan and crimped which was used as the sample pan. An empty crimped aluminum pan was used as the reference pan. The pans were heated from room temperature to 300°C at the rate of 10°C/min in a nitrogen environment (flow rate 20 mL/min). The DSC study was performed using pure PETOX, pure ChC, blank, and PETOX–ChC.
Determination of Conjugation with Fourier Transform Infrared Spectroscopy
A sample size of 20 mg was used to scan the lipopolymer conjugate. FTIR was carried out using IR-Prestige 21 (Shimadzu, Columbia, MD). All the spectra were recorded as a mean of 15 scans, with a resolution of 4 cm−1 and in the range of 800–4000 cm−1. The obtained spectra were analyzed specifically in the range of 1000–3000 cm−1. FTIR was carried out to observe for new bonds being formed or broken between PETOX and ChC during the conjugation process.
Preparation of Liposomes
Multilamellar liposomes were prepared using the thin film hydration technique, where a film of soy PC and cholesterol (molar ratio 3:2) was hydrated with 3 mL of the aqueous hydrating medium comprised of HEPES, Tween 80, and sodium chloride dissolved in filtered water. The hydrating medium for the drug-loaded liposomes had gemcitabine hydrochloride and irinotecan hydrochloride in a molar ratio of 3:4 dissolved in the above medium. The mixture was vortexed and bath sonicated till completely dispersed. These multilamellar liposomes were subjected to 10 cycles of freeze thawing using liquid nitrogen and water bath at 40°C. These freeze-thawed liposomes were subjected to membrane extrusion with 11 passes through 200 nm pore size polycarbonate membranes using an Avanti Mini-Extruder (Avanti Polar Lipids, Alabaster, AL, SKU: 610000-1EA). The unilamellar liposomes were dialyzed (12–14 kDa cutoff range) against 5% glucose to remove free drugs, unassociated lipids, and micelles. Surface modification of the liposomes was achieved by the post-insertion method, which involved adding the surface modification agent to preformed liposomes. PEG-DSPE (3 mol%) and PETOX–ChC (3 mol%) were added to the liposomes and incubated at 40oC for 12 h. The liposomes were dialyzed against 5% glucose with a membrane (12–14 kDa cutoff range) for 12 h.
Characterization of Liposomes
Particle Size and Zeta Potential
Measurements were carried out using a Brookhaven Zetameter (ZetaPlus, Brookhaven Instruments Corporation, Holtsville, NY). One hundred microliters of the liposomal dispersion was diluted to 8 mL with 0.45 μm filtered DI water, and the particle size and zeta potential were measured. Each sample was measured five times. All measurements were performed in triplicate.
Determination of Encapsulation Efficiency
The drug encapsulation efficiency of the liposomes was calculated as the percent ratio of the amount of drug entrapped in the nanoparticles to the amount of drug initially added to the liposomes. The encapsulation efficiency of the liposomes was assessed with HPLC (Shimadzu LC 2030C) which utilized an Agilent Zorbax SB C18 column as the stationary phase and a mobile phase comprising of acetonitrile and citrate buffer (0.1 M at pH 3.2) at a ratio of 7:3 with isocratic elution. The oven temperature was 40°C and the flow rate was 1.0 mL/min. A PDA UV/VIS detector was used at 255 nm and 272 nm wavelengths to detect irinotecan and gemcitabine, respectively. Gemcitabine and irinotecan had retention times of 1.387 min and 2.563 min, respectively. The liposomes were lysed with the mobile phase and the drugs were extracted by vortexing and bath sonication for a total of 5 min. The resulting stock was subjected to serial dilution and filtered using 0.22-μm filter before analyzing by HPLC.
Structural Elucidation with 31P NMR
Proton-decoupled 162 MHz 31P NMR liposome spectra were obtained on a Bruker Avance III HD 400 NMR. The sample comprised of a total of 50 mM lipids and 10% (v/v) deuterium oxide (D2O) in water. A total of 10,000 scans were accumulated using a 90° pulse width of 14 μs and 1 s relaxation delay. The obtained spectra were analyzed using Bruker TopSpin 3.5 software.
Cryo-TEM Imaging
A 3-μL aliquot of the purified and concentrated liposome sample preparation was applied to glow-discharged Quantifoil TEM grids (Hatfield, PA) and used for plunge freezing into liquid ethane. The frozen grids were then transferred to a FEI TF30 field emission gun transmission electron microscope at liquid nitrogen temperature. Images were recorded using a Gatan 4k × 4k CCD camera (Gatan, Inc., Pleasanton, CA).
In Vitro Drug Release
The in vitro release of gemcitabine and irinotecan from the nanoparticulate formulation was studied using a dialysis membrane. The release medium used was 0.1 M phosphate buffer pH 7.4 at 37°C. A 2-mL suspension of nanoparticles was prepared in the release medium and sealed into the dialysis bag. This bag was then inserted into a falcon tube containing 13 mL of release medium (receiver compartment). The tubes were placed in the incubator-shaker at 150 rpm at 37°C. Samples were collected at 24, 48, and 72 h time intervals by removing 200 μL from the receiver compartment and by replacing it with 200 μL fresh release medium. The aliquots were filtered through 0.2 μm syringe filter and analyzed for drug content using the HPLC method discussed before.
MTT Cytotoxicity Assay
The cytotoxicity of the drug solution, blank and drug-loaded uncoated liposomes, and surface-modified liposomes was studied using MTT cytotoxicity assay. Mia PaCa II pancreatic cancer cells were grown in RPMI medium supplemented with 10% fetal bovine serum (FBS) and plated in a 96-well plate at a seeding density of 3500 cells per well. After plating the cells, the plates were incubated overnight in a humidified chamber at 37°C with 5% carbon dioxide. On the second day of the experiment, eight different concentrations of the treatments comprised of uncoated and surface-modified (PEGylated and PETOXylated) liposomes, with blank and drug-loaded subsets, were added to the cells. A blank media treatment was also carried out as a control. A 100-μL treatment was added to each well and the plates were returned to incubation. After an incubation period of 24, 48, and 72 h, 30 μL of MTT solution (5 mg/mL) prepared in sterile phosphate buffer (pH 7.4) was added. The plate was then incubated for a period of 4 h. After 4 h, the treatment was removed, and the cells were lysed using a 1:1 solution of 20% (w/v) SDS and DMF. The plates were kept on the incubator-shaker (MaxQ 4450, Thermo Scientific) for 1 h at 37°C. The analysis was carried out on the microplate reader (Multiskan MCC) at 540 nm.
Determination of Macrophagial Uptake of Liposomes by Confocal Microscopy
The in vitro macrophagial uptake of fluorescently labeled uncoated, PEGylated, and PETOXylated liposomes was assessed with a Nikon Ti-E inverted microscope with a Yokagawa spinning disc for confocal imaging and a Sutter DG4 high-speed wavelength switcher for widefield imaging. The liposomes used for the determination of macrophagial uptake were loaded with FD&C Yellow #5 dye for green fluorescence (488 nm) and were PEGylated and PETOXylated as per the procedure mentioned before. The concentration of the fluorescent dye was quantified on a microplate reader (Multiskan MCC). KG1 human macrophage cells were used to assess the uptake of the liposomes and were cultured under recommended conditions. MatTek glass bottom plates were prepared by adding a thin film of 2% gelatin on the cover slip for the adherence of the cells. KG1 macrophage cells were spun down in the centrifuge at 1500 rpm for 5 min to form a pellet of cells. The cells were resuspended in IMDM culture medium with LysoTracker Red, and 1 mL cell of suspension was added to each gelatin-coated plate. The plate was kept at room temperature for 10 min and spun down using a centrifuge at 750 rpm for 3 min. The supernatant medium from the plate was removed and 1 mL of fresh IMDM medium was added with 2 drops of NucBlue stain. An aliquot of 20 μL of liposomes was added to the plate and the plate was assessed using confocal microscopy.
Statistical Analyses
The data was obtained from measurements done in triplicates. The statistical analysis of the experimental data was performed using single-factor ANOVA test. A statistically significant difference was termed if the p value was determined to be less than 0.05.
RESULTS
Conjugation of PETOX with Cholesteryl Chloroformate
The lipopolymer conjugate obtained from the conjugation of PETOX with cholesteryl chloroformate was a white powder with a yield of 73%. This conjugation of PETOX with ChC was confirmed using 1H NMR, DSC, and FTIR spectroscopy.
Determination of Conjugation with Proton Nuclear Magnetic Resonance
The 1H NMR spectra for PETOX, ChC, and the conjugate are shown in Fig. 1b, c, and d. The peak at the chemical shift of 7.29 was contributed by the solvent deuterated chloroform (CDCl3) and was observed in the NMR scans of the starting materials, PETOX (Fig. 1b) and ChC (Fig. 1c). The peaks observed at 3.46, 2.39, 2.3, and 1.13 were contributed by the PETOX polymer, while the peaks with chemical shifts of 0.71, 0.884, 0.9, 0.927, 0.947, and 1.05 were provided by ChC along with multiple resonances in the range of 1.07–2.08. The NMR spectrum of the lipopolymer conjugate in Fig. 1d shows the indicative peaks of PETOX and ChC. However, the peaks from the individual components were not superimposable and showed a slight difference in chemical shifts, indicating a chemical reaction between the individual components PETOX and ChC. Since the conjugate shows the indicative peaks of both the components with a slight change in chemical shift, the conjugation of PETOX and ChC can be confirmed with 1H NMR.
Determination of Conjugation with Differential Scanning Calorimetry
An overlay of the DSC thermograms of the PETOX cholesterol conjugate (red), cholesteryl chloroformate (green), and PETOX (black) is shown in Fig. 2. The conjugate shows different thermal behavior than either of the individual components. Cholesteryl chloroformate shows an endothermic peak at 120°C near its reported melting point of 115–117°C. PETOX shows a degradation pattern after 220°C. The conjugate shows neither of these events at the specific temperatures. The conjugate shows an exothermic peak around 125°C and a subsequent degradation pattern. These results further suggest that the conjugation of PETOX and cholesteryl chloroformate was successful.

Overlay of DSC thermogram of PETOX–cholesterol conjugate and the individual chemicals
Determination of Conjugation with Fourier Transform Infrared Spectroscopy
The FTIR spectrum of PETOX–ChC is shown in Fig. 3 with the characteristic peaks highlighted. The major functional groups of PETOX–cholesterol conjugate that could be detected are the carbonyl groups (large sharp peak at 1635 cm−1), alkane-based C–H stretching (two small peaks just under 3000 cm−1), alkene-based C–H stretching (small peak just above 3000 cm−1), and the alkane-based CH2 stretching (peak at 1470 cm−1). Carbon–carbon double bond stretching is commonly shown at 1680–1640 cm−1 and is buried under the carbonyl peak due to much poorer IR absorbing capabilities. The data obtained from FTIR spectroscopy thus confirms the conjugation of PETOX and ChC.

FTIR spectrum of PETOX–cholesterol conjugate
Characterization of Liposomes
Particle Size and Zeta Potential
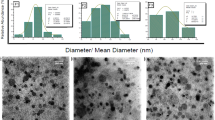
The particle sizes and zeta potential of the uncoated and surface-modified liposomes are shown in Table I. Both PEGylated liposomes and PETOXylated liposomes showed an increase in their hydrodynamic diameter and polydispersity. PETOXylated liposomes showed a higher increase in particle size due to the higher molecular weight and Flory length of PETOX compared to PEG, but both surface-modified liposomes had a particle size in the range of 150–200 nm. Both surface-modified liposomes showed a statistically significant (p < 0.05) reduction in the anionic zeta potential of the liposome.
Structural Elucidation with 31P NMR
It was observed from Fig. 4 that the processes of freeze thawing and extrusion provide liposomes with decreased signal at − 13.7 ppm. It may be inferred that the freeze thawing results in a thermodynamically stable equilibrium between untreated and extruded liposomes, which brings forth a hypothesis that a reduction in the peak at − 13.7 ppm with a corresponding increase in the − 0.21 peak intensity was indicative of a trend toward unilamellarity in liposomes. Extruded liposomes showed the highest signal at − 0.21 ppm with the lowest signal at − 13.7 ppm, which suggests that this sample had the highest proportion of unilamellar liposomes.

31P NMR spectra of untreated, freeze-thawed, and extruded liposomes
Cryo-TEM Imaging
The photomicrographs of cryo-TEM of uncoated liposomes without surface modification are shown in Fig. 5a and revealed that there are multiple bilayers in the vesicles. It should also be noted that a small fraction of unilamellar liposomes is observed in a population with a majority of multilamellar liposomes. The cryo-TEM of the extruded liposomes in Fig. 5b shows liposomes with a single bilayer and lower particle size.

a Cryo-TEM image of multilamellar liposomes. b Cryo-TEM image of unilamellar liposomes
Determination of Drug Encapsulation in Liposomes
The encapsulation efficiencies and loading capacities of the various ample sets of liposomes for gemcitabine and irinotecan are shown in Table II. From the data presented in Table II, it is evident that encapsulation efficiency was significantly less than that of irinotecan, and the drug ratios were adjusted so as to attain similar loading capacities. However, due to the high potency of the drugs gemcitabine and irinotecan, it was observed that the low encapsulation efficiency did not hamper the efficacy of the formulation, as seen from the data presented in further sections. It should also be considered that the combination of two drugs colocalized in the liposome may mutually influence the loading capacity.
Drug Release In Vitro
The objective of the study was to assess the effect of surface modifications by PEG-DSPE and PETOX–ChC on the release characteristics of gemcitabine and irinotecan from these formulations. The in vitro drug release patterns of gemcitabine and irinotecan from the uncoated and surface-modified liposomes are presented in Table III. The release of gemcitabine was significantly higher (p < 0.05) than PEGylated and PETOXylated at all three time points. PETOXylated liposomes showed a significantly higher (p < 0.05) release of gemcitabine compared to PEGylated liposomes. It is therefore evident that surface modification of liposomes reduces the release of gemcitabine from the liposomes. Irinotecan release was more or less similar in the three sample sets across the three time points and a statistically significant (p < 0.05) difference was not encountered. Thus, irinotecan release from the liposomes does not seem to be affected by the surface modification of the liposomes.
MTT Cytotoxicity Assay
The data provided pertaining to the MTT cytotoxicity of the drugs and formulations is from the 24-, 48-, and 72-h data sets. Comparison of the cytotoxicity of the combination of gemcitabine and irinotecan and individual drug solutions at 24, 48, and 72 h is shown in Fig. 6 in panels a, b, and c, respectively, while Table IV shows the IC50 values for the sample sets across various time points. The treatment included the individual drug solutions of gemcitabine and irinotecan and a combination of both these drugs with gemcitabine and irinotecan in a molar ratio of 3:4 in solution. The combination of gemcitabine and irinotecan exhibited a significantly greater toxicity (p < 0.05) as compared to either individual drugs at all three-exposed time points of 24, 48, and 72 h. The combination of gemcitabine and irinotecan in solution exhibited an IC50 value of 10.65 ± 3.19 nM at 72 h treatment. However, the individual drugs gemcitabine and irinotecan never reached IC50 under the experimental conditions tested for these cells.

a Percent survival of Mia PaCa II cells after treatment with individual drugs gemcitabine and irinotecan and a combination of both these drugs at 24 h of incubation post-treatment. b Percent survival of Mia PaCa II cells after treatment with individual drugs gemcitabine and irinotecan and a combination of both these drugs at 48 h of incubation post-treatment. c Percent survival of Mia PaCa II cells after treatment with individual drugs gemcitabine and irinotecan and a combination of both these drugs at 72 h of incubation post-treatment
The drug-loaded liposomes had the drugs in the same molar ratio and overall concentration as the drug solution. The data pertaining to these sets of experiments from the 24-, 48-, and 72-h time points is illustrated in Fig. 7 in panels a, b, and c, respectively, while the IC50 values are provided in Table IV. The drug-loaded liposomes exhibited an IC50 value of 0.27 ± 0.09 nM with 72 h treatment, which was significantly lower (p < 0.05) than that of the drug solution. The drug-loaded liposomes displayed a statistically significant (p < 0.05) decrease in IC50 from 24 to 72 h. The IC50 for the blank liposomes could not be determined with the highest concentration at 72 h as the overall survival of the cells remained higher than 50% at all tested concentrations. The percent survival of the Mia PaCa II pancreatic cancer cells after 72 h of post-treatment incubation is represented in Fig. 8b. The treatment included the free drugs gemcitabine and irinotecan in combination, blank liposomes, and drug-loaded liposomes.

a Percent survival of Mia PaCa II cells after treatment with the free drugs gemcitabine and irinotecan in combination, blank liposomes, and drug-loaded liposomes at 24 h of incubation post-treatment. b Percent survival of Mia PaCa II cells after treatment with the free drugs gemcitabine and irinotecan in combination, blank liposomes, and drug-loaded liposomes at 48 h of incubation post-treatment. c Percent survival of Mia PaCa II cells after treatment with the free drugs gemcitabine and irinotecan in combination, blank liposomes, and drug-loaded liposomes at 72 h of incubation post-treatment

a Percent survival of Mia PaCa II cells after treatment with uncoated liposomes, PEGylated liposomes, and PETOXylated liposomes at 24 h of incubation post-treatment. b Percent survival of Mia PaCa II cells after treatment with uncoated liposomes, PEGylated liposomes, and PETOXylated liposomes at 48 h of incubation post-treatment. c Percent survival of Mia PaCa II cells after treatment with uncoated liposomes, PEGylated liposomes, and PETOXylated liposomes at 72 h of incubation post-treatment
The treatment included the uncoated liposomes, PEGylated liposomes, and PETOXylated liposomes. A higher volume of surface-modified liposomes was added to the medium due to overall lower drug encapsulation in the surface-modified liposomes as compared to uncoated liposomes. The data pertaining to these sets of experiments from the 24-, 48-, and 72-h time points is illustrated in Fig. 8 in panels a, b, and c, respectively, while the IC50 values are provided in Table IV. The uncoated liposomes showed an IC50 value of 0.27 ± 0.09 nM of the gemcitabine and irinotecan combination with 72 h treatment. The PEGylated liposomes showed an IC50 value of 0.16 ± 0.02 nM, while that of PETOXylated liposomes, it was 0.12 ± 0.03 nM at 72 h of treatment.
Determination of Macrophagial Uptake of Liposomes by Confocal Microscopy
Macrophagial uptake of liposomes was studied using a confocal microscope. Macrophages were exposed to liposomes loaded with green fluorescent dye and visualized under the confocal microscope. The localization of green fluorescence (488 nm) and increasing intensity of the green fluorescence were used to quantify the liposomal internalization by the macrophages. The data obtained from the experiment is represented in Fig. 9a and summarized in Table V. The uncoated liposomes show a rapid uptake characterized by the single sharp peak at 141 s and later show rapidly and consistently decreasing levels as the internalized liposomes were disintegrated and the dye was metabolized. The PEGylated liposomes show two distinct peaks, which may be an indication of heterogeneity in the extent of surface modification of the liposomes where liposomes with higher surface density of PEGylation will be internalized later than those with lesser surface density of PEGylation. The PEGylated liposomes show an initial spike at 231 s, while a second spike in the uptake was seen at the 682-s time point. This may be an indication of slower disintegration of the internalized PEGylated liposomes compared to the uncoated liposomes, thus leading to lowered intracellular degradation of the dye. This might explain the overall higher AUC values for liposomal internalization of PEGylated liposomes. The PETOXylated liposomes show very little uptake compared to both other samples but follows the general trend of PEGylated liposomes with a small initial spike at 321 s and a second spike at 862 s.

a Macrophagial internalization of liposomes. b Lysosomal activity in macrophages
The lysosomal activity of macrophages was observed by staining the cells with LysoTracker Red and assessment of red fluorescence at 561 nm. Lysosomal activity in the macrophages data is shown in Fig. 9b and summarized in Table VI. Lysosomal activity is indicative of the general trend of active uptake in the cell and may not translate into a quantification of lysosomal activity caused by uptake of liposomes exclusively. The uncoated liposomes showed the earliest spike in fluorescence at 141 s, while the highest levels of fluorescence and the highest AUC were shown by the PEGylated liposomes. This might be an indication of the difference in the way the liposomes are disintegrated after internalization by the macrophages. Both PEGylated and PETOXylated liposomes showed an initial spike at 412 s and 231 s, respectively, while a second spike was seen at 682 s and 592 s, respectively.
DISCUSSION
The study aimed to develop PETOXylated liposomes as a replacement to the PEGylated liposomes with three prime factors in consideration: development of liposomes that can utilize the EPR effect, cytotoxicity in Mia PaCa II cell lines, and evasion of macrophagial uptake in the KG1 cell line. The study addressed these factors categorically with the experiments discussed above.
The passage of the liposomes through the leaky vasculature of the tumor blood vessels is dependent on the factor including but not limited to particle size and plasma circulation time. The size of the gaps between the endothelial cells lining the tumor capillaries ranges from 100 to 780 nm depending on the cancer type, as opposed to that in a typical normal endothelium of 5–10 nm (34,35). While it was known that liposomes should usually be smaller than 400 nm in size in order to utilize the EPR effect, more effective extravasation was shown to occur with particles smaller than 200 nm (36,37). It was therefore the objective of this study to develop liposomes smaller than 200 nm.
The structural elucidation of the liposomes was achieved by the use of orthogonal techniques likes DLS, 31P NMR, and cryo-TEM imaging. It is evident from Table I that all the categories of liposomes developed in this study were below 200 nm in particle size and thus can achieve passive targeting of the tumor by harnessing the EPR effect. The 31P NMR spectra shown in Fig. 4 provided insight into the lamellarity of the liposomes and their state as they are subjected to various formulation techniques. It may be inferred that freeze thawing results in a thermodynamically stable equilibrium between untreated and extruded liposomes, which brings forth a hypothesis that a reduction in the peak at − 13.7 ppm with a corresponding increase in the − 0.21 peak intensity was indicative of a trend toward unilamellarity in liposomes. Extruded liposomes showed the highest signal at − 0.21 ppm with the lowest signal at − 13.7 ppm, which suggests that this sample had the highest proportion of unilamellar liposomes. Indeed, the hypothesis drawn was confirmed by the cryo-TEM images of untreated and extruded liposomes shown in Fig. 5a and b, respectively. The cryo-TEM of the untreated liposomes shows that there are multiple bilayers in most of the vesicles present. These liposomes also have a larger particle size and higher polydispersity as verified with DLS and are shown in Table I. It should also be noted that there is a certain small fraction of unilamellar liposomes in a population with a majority of multilamellar liposomes. It is therefore evident that the untreated liposomes are predominantly multilamellar. The cryo-TEM image of extruded liposomes shown in Fig. 5b shows liposomes with a single bilayer. These liposomes have a lower particle size and polydispersity, as verified from the data obtained using DLS. Cryo-TEM further proves that the liposomes obtained by freeze thawing and subsequent extrusion yield unilamellar liposomes.
It is evident from the 31P NMR spectra of the liposomes that the signal peak at − 0.21 ppm is representative of the unshielded phospholipids comprised of solubilized phospholipids, micelles, and the outermost layer of the unilamellar liposomes. This finding has a precedent in the literature and Moreau et al. (38) have reported that the appearance of an additional central spectral component around the isotropic chemical shift is attributable to the presence of small vesicles or micellar structures. Fröhlich et al. (39) reported that dispersions of multilamellar vesicles give rise to broad 31P NMR spectra due to the restricted anisotropic motion, whereas small unilamellar vesicles are characterized by a narrow line spectrum. The results discussed above show that clearly there is more to this matter than the reported literature, with multiple peaks in the spectra indicative of the state of phospholipids in the sample. Other studies (40,41) have reported that the 31P NMR spectra of multilamellar liposomes present distinct peaks in the 30–40-ppm range, indicative of bilayer or hexagonal phases within them. However, this study did not find any such characteristic peak in that range; instead, the peak characteristic of shielded phospholipids at −13.7 ppm was found.
There are three techniques used for surface modification of liposomes: pre-insertion method, post-insertion method, and post modification by chemical reaction. The pre-insertion method involves integrating the lipopolymer conjugate for surface modification in the lipid film itself used for thin film hydration and has a dual drawback of requiring more lipopolymer to achieve enough surface density to promote brush conformation of the outward-oriented lipopolymer and inward-oriented lipopolymer which jeopardizes the drug loading and does not contribute to stealth property (42,43). Attachment of stealth lipopolymers to liposome by chemical reaction has not been utilized to a significant extent due to multiple factors such as the inherent fragile nature of the liposome structure, solvent and reaction conditions, the difficulties in the separation of free reactants and modified product, the limited availability of aqueous reagents, aggregation and phase segregation, and the possibilities of side reactions (43). The post-insertion method involves inserting the lipopolymer conjugate into preformed liposomes and mitigates the drawbacks in the pre-insertion and post modification by chemical reaction methods. The post insertion of lipopolymers onto the outward surface of the liposomes is driven by the hydrophobic interaction of membrane lipids and the hydrophobic part of lipopolymers, thus keeping the internal space in the vesicle intact and drastically reduces the required amount of lipopolymer (43). Hence, this study utilized the post-insertion method for surface modification of the liposomes with PETOX lipopolymer conjugate.
The data from the MTT assay provided insight into the cytotoxicity of the drugs and the formulations. The cytotoxicity of the combination of gemcitabine and irinotecan in solution was significantly (p < 0.05) higher than either of the individual drugs in solution, and the cytotoxicity of the drug-loaded uncoated liposomes was significantly (p < 0.05) higher than that of the drug solution, as shown by Figs. 6 and 7, respectively. While there was no significant difference (p > 0.05) between the cytotoxicity of uncoated, PEGylated, and PETOXylated liposomes at 72 h, there was a significant difference between their cytotoxicity at 24- and 48-h data sets as shown in Fig. 8. This addresses a crucial point of long circulating liposome-based delivery, which is the interaction of the liposomes with the cells and the release of drug at the target site.
The findings from the confocal experiments conducted in this study provide a unique insight into the behavior of nanoparticles on short-term exposure to macrophages, which remained unaddressed in previous reports of a similar nature as far as any quantitative analysis is concerned. The basis for these set of experiments was the correlation reported by Allen et al. (44) between the in vitro evasion of macrophagial uptake by nanoparticles and their behavior in vivo. The PEGylated liposomes show two distinct peaks, which may be an indication of heterogeneity in the extent of surface modification of the liposomes where liposomes with higher surface density of PEGylation will be internalized later than those with lesser surface density of PEGylation. This may also be an indication of slower disintegration of the internalized PEGylated liposomes compared to the conventional liposomes and a difference in intracellular fate, thus leading to lowered intracellular degradation of the dye. This might explain the overall higher AUC values for liposomal internalization of PEGylated liposomes. The reader should note that the experiment which assessed LysoTracker Red–stained lysosomes provided an idea of the overall activity in the cellular apparatus concerned with phagocytosis and not just the lysosomal activity associated with liposomal uptake, which may explain the apparent lack of superimposability in Fig. 9a and b obtained from quantifying the macrophagial internalization of liposomes and the lysosomal activity in macrophages, as well as the higher standard deviation in the data obtained for the lysosomal activity in macrophages in Fig. 9b. While it may provide a rudimentary insight into the intracellular fate of the uptaken liposomes, it is difficult to draw crystallized conclusions from data concerning the lysosomal activity in macrophages.
The targeting strategy employed by stealth nanoparticles discussed in this article relies on clinically significant degree of EPR effect, the applicability of which has been debated for years. While there have been reports that have demonstrated significant EPR effect in carcinomas, these have not extrapolated to all tumors like sarcomas (45). It is therefore evident from this premise that while the findings from this article hold relevance in the treatment of tumors that show significant EPR effect, the same cannot be employed across the spectrum of all tumors. While it is known that the latter stages of carcinomas display EPR effect, there is also an apparent lapse in understanding of the stage at which a tumor will be clinically relevant, and this poses a problem with patient stratification for treatment. An integration of these findings leads to a conclusion that the stealth liposomes developed in this study cannot be employed as a magic bullet solution but rather a part of a regimen that addresses the condition in its entirety.
PEG-based long circulating nanoparticles are still the gold standard in the clinical setting, not because of the lack of complications associated with the use of PEG but because of the lack of exploration of viable alternatives. The search for better stealth polymers should be extended to the linkers and the anchor of the stealth conjugate as well. Some of these alternatives for PEG have reached a stage of viability that warrants clinical trials, but the replacement of PEG and the subsequent clinically ubiquity of these stealth polymers is yet to be achieved.
CONCLUSIONS
The study successfully developed surface-modified unilamellar anionic liposomes of the particle size range of 150–200 nm with PEG and PETOX separately to provide drug-loaded uncoated and surface-modified liposomes. A PETOX–cholesterol lipopolymer conjugate with carbonate linkage was prepared, and the conjugation was verified with orthogonal analytical techniques comprised of DSC, NMR, and FTIR. A solution of gemcitabine and irinotecan exhibited significantly greater cytotoxicity (p < 0.05) as compared to either individual drug at all time points in a cytotoxicity assay as shown in Fig. 6 using the Mia PaCa II cell line. The drug-loaded liposomes showed a significantly (p < 0.05) higher cytotoxicity in three intermediate concentrations as compared to drug solution as shown in Fig. 7. PETOXylated drug-loaded liposomes and PEGylated drug-loaded liposomes did not show statistically significant (p < 0.05) differences in cytotoxicity compared to the uncoated drug-loaded liposomes at 72 h as shown in Fig. 8. While both surface-modified liposomes showed significant (p < 0.05) lower uptake by human macrophages (KG1 cell line) in vitro than the uncoated liposomes, PETOXylated liposomes show a higher degree of evasion of macrophagial uptake in vitro in comparison to PEGylated and uncoated liposomes. The uncoated liposomes showed the fastest uptake by the macrophages, and while both surface-modified liposomes showed significant (p < 0.05) lower uptake than the uncoated liposomes, PETOXylated liposomes showed the least uptake as shown in Fig. 9a.
Abbreviations
- ABC:
-
Accelerated blood clearance
- ChC:
-
Cholesteryl chloroformate
- Cryo-TEM:
-
Transmission electron cryomicroscopy
- DCC:
-
N,N′-Dicyclohexylcarbodiimide
- DCM:
-
Dichloromethane
- DMAP:
-
4-Dimethylaminopyridine
- DSC:
-
Differential scanning calorimetry
- DSPE:
-
1,2-Distearoyl-sn-glycero-3-phosphoethanolamine
- FTIR:
-
Fourier transform infrared spectroscopy
- HPLC:
-
High-performance liquid chromatography
- MTT:
-
(3-(4,5-Dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide)
- NMR:
-
Nuclear magnetic resonance
- PDAC:
-
Pancreatic ductal adenocarcinoma
- PE:
-
Phosphatidyl ethanolamine
- PEG:
-
Polyethylene glycol
- PETOX:
-
Poly(2-ethyl-2-oxazoline)
- RES:
-
Reticuloendothelial system
References
Howlader N, Noone AM, Krapcho M, Miller D, Brest A, Yu M, et al., editors. SEER cancer statistics review, 1975-2017. Bethesda: National Cancer Institute. https://seer.cancer.gov/csr/1975_2017/, based on November 2019 SEER data submission, posted to the SEER web site, April 2020
Sohal D, Mangu P, Khorana A, Shah M, Philip P, O’Reilly E, et al. Metastatic pancreatic cancer: American Society of Clinical Oncology clinical practice guideline. J Clin Oncol. 2016;34:2784–96. https://doi.org/10.1200/JCO.2016.67.1412.
National Cancer Institute. Pancreatic cancer: an agenda for action. Report of the Pancreatic Cancer Progress Review Group. NIH Publication No. 01-4940. Bethesda (MD): NCI, 2001.
Burris H III, Moore M, Andersen J, Green M, Rothenberg M, Modiano M, et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. J Clin Oncol. 1997;15:2403–13. https://doi.org/10.1200/JCO.1997.15.6.2403.
Heinemann V, et al. Comparison of the cellular pharmacokinetics and toxicity of 2′, 2-difluoro deoxycytidine and 1-beta-D-arabinofuranosylcytosine. Cancer Res. 1988;48(14):4024–31.
Conroy T, Desseigne F, Ychou M, Bouche O, Guimbaud R, Becouarn Y, et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med. 2011;364:1817–25. https://doi.org/10.1056/NEJMoa1011923.
Wang-Gillam A, Li CP, Bodoky G, Dean A, Shan YS, Jameson G, et al. Nanoliposomal irinotecan with fluorouracil and folinic acid in metastatic pancreatic cancer after previous gemcitabine-based therapy (NAPOLI-1): a global, randomized, open-label, phase 3 trial. Lancet. 2016;387(10018):545–57. https://doi.org/10.1016/S0140-6736(15)00986-1.
Swami U, Goel S, Mani S. Therapeutic targeting of CPT-11 induced diarrhea: a case for prophylaxis. Curr Drug Targets. 2013;14(7):777–97. https://doi.org/10.2174/1389450111314070007.
Rocha Lima CM, Savarese D, Bruckner H, et al. Irinotecan plus gemcitabine induces both radiographic and CA 19-9 tumor marker responses in patients with previously untreated advanced pancreatic cancer. J Clin Oncol. 2002;20(5):1182–91. https://doi.org/10.1200/JCO.2002.20.5.1182.
Stathopoulos GP, Rigatos SK, Dimopoulos MA, Giannakakis T, Foutzilas G, Kouroussis C, et al. Treatment of pancreatic cancer with a combination of irinotecan (CPT-11) and gemcitabine: a multicenter phase II study by the Greek Cooperative Group for Pancreatic Cancer. Ann Oncol. 2003;14(3):388–94. https://doi.org/10.1093/annonc/mdg109.
Stathopoulos GP, Syrigos K, Polyzos A, Fountzilas G, Rigatos SK, Ziras N, et al. Front-line treatment of inoperable or metastatic pancreatic cancer with gemcitabine and capecitabine: an intergroup, multicenter, phase II study. Ann Oncol. 2004;15(2):224–9. https://doi.org/10.1093/annonc/mdh065.
Felice B, Prabhakaran MP, Rodríguez AP, Ramakrishna S. Drug delivery vehicles on a nano-engineering perspective. Mater Sci Eng C Mater Biol Appl. 2014;41:178–95. https://doi.org/10.1016/j.msec.2014.04.049.
Salmaso S, Caliceti P. Stealth properties to improve therapeutic efficacy of drug nanocarriers. J Drug Deliv. 2013. Article ID 374252, 19 page. https://doi.org/10.1155/2013/374252.
Nilsson B, Ekdahl KN, Mollnes TE, Lambris JD. The role of complement in biomaterial-induced inflammation. Mol Immunol. 2007;44:82–94. https://doi.org/10.1016/j.molimm.2006.06.020.
Maruyama K, Yuda T, Okamoto A, Kojima S, Suginaka A, Iwatsuru M. Prolonged circulation time in vivo of large unilamellar liposomes composed of distearoyl phosphatidylcholine and cholesterol containing amphipathic poly (ethylene glycol). Biochim Biophys Acta. 1992;1128:4–9. https://doi.org/10.1016/0005-2760(92)90255-T.
Matsumura Y, Maeda H. A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986;46(12,1):6387–92.
Allen TM, Hansen C, Martin F, Redemann C, Yau-young A. Liposomes containing synthetic lipid derivatives of poly (ethylene glycol) show prolonged circulation half-lives in vivo. Biochim Biophys Acta. 1991;1066(1):29–36. https://doi.org/10.1016/0005-2736(91)90246-5.
Klibanov AL, Maruyama K, Torchilin VP, Huang L. Amphipathic polyethylene glycols effectively prolong the circulation time of liposomes. FEBS Lett. 1990;268(1):235–7. https://doi.org/10.1016/0014-5793(90)81016-h.
Dams ETM, Laverman P, Oyen WJG, Storm G, Scherphof GL, van der Meer JWM, et al. Accelerated blood clearance and altered biodistribution of repeated injections of sterically stabilized liposomes. J Pharmacol Exp Ther. 2000;292(3):1071–9.
Ishida T, Maeda R, Ichihara M, Irimura K, Kiwada M. Accelerated clearance of PEGylated liposomes in rats after repeated injections. J Control Release. 2003;88:35–42. https://doi.org/10.1016/s0168-3659(02)00462-5.
Ishida T, Ichihara M, Wang X, Yamamoto K, Kimura J, Majima E, et al. Injection of PEGylated liposomes in rats elicits PEG-specific IgM, which is responsible for rapid elimination of a second dose of PEGylated liposomes. J Control Release. 2006;112:15–25. https://doi.org/10.1016/j.jconrel.2006.01.005.
Ishida T, Wang X, Shimizu T, Nawata K, Kiwada H. PEGylated liposomes elicit an anti-PEG IgM response in a T cell-independent manner. J Control Release. 2007;122(3):349–55. https://doi.org/10.1016/j.jconrel.2007.05.015.
Armstrong JK, Hempel G, Koling S, Chan LS, Fisher T, Meiselman HJ, et al. Antibody against poly (ethylene glycol) adversely affects PEG-asparaginase therapy in acute lymphoblastic leukemia patients. Cancer. 2007;110:103–11. https://doi.org/10.1002/cncr.22739.
Yang Q, Jacobs TM, McCallen JD, Moore DT, Huckaby JT, Edelstein JN, et al. Analysis of pre-existing IgG and IgM antibodies against polyethylene glycol (PEG) in the general population. Anal Chem. 2016;88(23):11804–12. https://doi.org/10.1021/acs.analchem.6b03437.
Tomalia DA, Sheetz DP. Homopolymerization of 2-alkyl- and 2-aryl-2-oxazolines. J Polym Sci A Polym Chem. 1966;4:2253–65. https://doi.org/10.1002/pol.1966.150040919.
Seeliger W, Aufderhaar E, Diepers W, Feinauer R, Nehring R, Thier W, et al. Recent syntheses and reactions of cyclic imidic esters. Angew Chem Int Ed. 1966;5:875–88. https://doi.org/10.1002/anie.196608751.
Kagiya T, Narisawa S, Maeda T, Fukui K. Ring-opening polymerization of 2-substituted 2-oxazolines. J Polym Sci B Polym Lett. 1966;4:441–5. https://doi.org/10.1002/pol.1966.110040701.
Bassiri TG, Levy A, Litt M. Polymerization of cyclic imino ethers. I. Oxazolines. J Polym Sci B Polym Lett. 1967;5:871–9. https://doi.org/10.1002/pol.1967.110050927.
Verlander WH, Madurawe RD, Subramanian A, Kumar G, Sinai-Zingde G, Riffle J. Polyoxazoline-peptide adducts that retain antibody activity. Biotechnol Bioeng. 1992;39:1024–30.
Woodle M, Engbers C, Zalipsky S. New amphipathic polymer-lipid conjugates forming long-circulating reticuloendothelial system-evading liposomes. Bioconjug Chem. 1994;5:493–6. https://doi.org/10.1021/bc00030a001.
Zalipsky S, Hansen CB, Oaks JM, Allen TM. Evaluation of blood clearance rates and biodistribution of poly(2-oxazoline)-grafted liposomes. J Pharm Sci. 1996;85:133–7. https://doi.org/10.1021/js9504043.
Xu H, Zhang W, Li Y, Ye FF, Yin PP, Yu X, et al. The bifunctional liposomes constructed by poly(2-ethyl-oxazoline)-cholesteryl methyl carbonate: an effectual approach to enhance liposomal circulation time, pH-sensitivity and endosomal escape. Pharm Res. 2014;31(11):3038–50. https://doi.org/10.1007/s11095-014-1397-0.
Moreadith R, et al. Clinical development of a poly(2-oxazoline) (POZ) polymer therapeutic for the treatment of Parkinson’s disease – proof of concept of POZ as a versatile polymer platform for drug development in multiple therapeutic indications. Eur Polym J. 2017;88:524–52. https://doi.org/10.1016/j.eurpolymj.2016.09.052.
Haley B, Frenkel E. Nanoparticles for drug delivery in cancer treatment. Urol Oncol. 2008;26(1):57–64. https://doi.org/10.1016/j.urolonc.2007.03.015.
Deshpande PP, Biswas S, Torchilin VP. Current trends in the use of liposomes for tumor targeting. Nanomedicine (London). 2013;8(9):1509–28. https://doi.org/10.2217/nnm.13.118.
Danhier F, Feron O, Préat V. To exploit the tumor microenvironment: passive and active tumor targeting of nanocarriers for anticancer drug delivery. J Control Release. 2010;148(2):135–46. https://doi.org/10.1016/j.jconrel.2010.08.027.
Sawant RR, Torchilin VP. Challenges in development of targeted liposomal therapeutics. AAPS J. 2012;14(2):303–15. https://doi.org/10.1208/s12248-012-9330-0.
Moreau C, Floch ML, Segalen J, Leray G, Metzinger L, Certaines JD, et al. Static and magic angle spinning 31P NMR spectroscopy of two natural plasma membranes. FEBS Lett. 1999;461(3):258–62. https://doi.org/10.1016/s0014-5793(99)01461-1.
Fröhlich M, Brecht V, Peschka-Süss R. Parameters influencing the determination of liposome lamellarity by 31P-NMR. Chem Phys Lipids. 2001;109(1):103–12. https://doi.org/10.1016/S0009-3084(00)00220-6.
Yeagle PL. Phosphorus NMR of membranes. In: Berliner LJ, Reuben J, editors. Biological magnetic resonance, vol. 9. Boston: Springer; 1990. https://doi.org/10.1007/978-1-4615-6549-9_1.
Seelig J. 31P nuclear magnetic resonance and the head group structure of phospholipids in membranes. Biochim Biophys Acta Rev Biomembr. 1978;515(2):105–40. https://doi.org/10.1016/0304-4157(78)90001-1.
Awasthi VD, Garcia D, Klipper R, Goins BA, Phillips WT. Neutral and anionic liposome-encapsulated hemoglobin: effect of post-inserted poly (ethylene glycol)-distearoylphosphatidylethanolamine on distribution and circulation kinetics. J Pharmacol Exp Ther. 2004;309:241–8. https://doi.org/10.1124/jpet.103.060228.
Nag OK, Awasthi V. Surface engineering of liposomes for stealth behavior. Pharmaceutics. 2013;5(4):542–69. Published 2013 Oct 25. https://doi.org/10.3390/pharmaceutics5040542.
Allen TM, Austin GA, Chonn A, Lin L, Lee KC. Uptake of liposomes by cultured mouse bone marrow macrophages: influence of liposome composition and size. Biochim Biophys Acta. 1991;1061(1):56–64. https://doi.org/10.1016/0005-2736(91)90268-d.
Hansen AE, Petersen AL, Henriksen JR, Boerresen B, Rasmussen P, Elema DR, et al. Positron emission tomography based elucidation of the enhanced permeability and retention effect in dogs with cancer using copper-64 liposomes. ACS Nano. 2015;9(7):6985–95. https://doi.org/10.1021/acsnano.5b01324.
Acknowledgments
The cryo-TEM images were obtained with cooperation from Dr. Wei Zhang at the University of Minnesota, MN, USA. The pancreatic cancer cell lines were obtained from Dr. Surinder Batra and Dr. Rakesh Singh of the University of Nebraska Medical Center, Omaha, NE.
Funding
This work was supported by the Department of Pharmacy Sciences and the Department of Chemistry at Creighton University, Omaha, NE, USA. The confocal imaging research was conducted at the Integrated Biomedical Imaging Facility at Creighton University, Omaha, NE. This facility is supported by the Creighton University School of Medicine and grants GM103427 and GM110768 from the National Institute of General Medical Science (NIGMS), a component of the National Institutes of Health (NIH). The facility was constructed with support from grants from the National Center for Research Resources (RROI 64 69) and the NIGMS (GM103427).
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Deodhar, S., Dash, A.K., North, E.J. et al. Development and In Vitro Evaluation of Long Circulating Liposomes for Targeted Delivery of Gemcitabine and Irinotecan in Pancreatic Ductal Adenocarcinoma. AAPS PharmSciTech 21, 231 (2020). https://doi.org/10.1208/s12249-020-01745-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1208/s12249-020-01745-6