
Abstract
The oral route is the most widely accepted and commonly used route for administration. However, this route may not be suitable for certain drug candidates which suffer from the problem of low aqueous solubility and gastrointestinal absorption and extensive first-pass effect. Nanotechnology-based approaches can be taken up as remedies to overcome the disadvantages associated with the oral route. Among the various nanocarriers, lipidic nanocarriers are widely used for oral delivery of bioactive molecules owing to their several advantages. Active targeting of bioactive molecules via lipidic nanocarriers has also been widely attempted to improve oral bioavailability and to avoid first-pass effect. This active targeting approach involves the use of ligands grafted or conjugated onto a nanocarrier that is specific to the receptors. Active targeting increases the therapeutic efficacy as well as reduces the toxic side effects of the drug or bioactive molecules. This review mainly focuses on the challenges involved in the oral delivery of drugs and its approaches to overcome the challenges using nanotechnology, specifically focusing on lipidic nanocarriers like liposomes, solid lipid nanoparticles, and nanostructured lipid carriers and active targeting of drug molecules by making use of ligand-conjugated lipidic nanocarriers.
Similar content being viewed by others
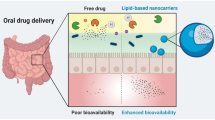
Avoid common mistakes on your manuscript.
INTRODUCTION
Oral Delivery of Drugs
The oral route is the most commonly used and accepted route for the administration of a number of drugs to treat or cure many diseases. The oral route is also the most preferred by patients, owing to its advantages including self-administration, noninvasiveness, ease of use, and reliability of oral dosage forms. With respect to the drug delivery aspects, the oral route is the most preferred as it offers different pH ranges throughout the entire gastrointestinal tract (GIT) facilitating the drug absorption from different sites of the GIT (1). The human intestinal epithelium is composed of villi, which provide a highly absorptive surface in the GIT for the absorption of drugs (2). The gut-associated lymphatic system which is a part of Peyer’s patches contains follicle-associated epithelium (FAE), that consists of enterocytes (absorptive cells), goblet cells (mucus-secreting cells), and microfold cells (M cells). These M cells play an active role in the delivery of drugs as they have a high transcytotic capacity and are less protected by the mucus (3,4).
Challenges in the Oral Delivery of Drugs and Bioactive Molecules
Despite of the advantages offered by the oral route, delivering drugs through the oral route is challenging as the human GIT forms a formidable barrier for most of the active pharmaceutical ingredients (API) (5). The continuous secretion of mucus which protects the GIT and the acidic gastric environment are other hurdles for the effective oral delivery of the drugs. The drug molecule, in order to reach the systemic circulation intact and to provide the required therapeutic effect, should pass through various physical and chemical barriers of the oral route (1). Following oral administration, the drug molecule passes through the following barriers:
-
1.
The oral, gastric, and intestinal fluid environment: The fluid environment of the GI system like (i) various enzymes in the stomach fluid, intestinal fluid, and in the saliva; (ii) different pH range in the stomach and intestine; (iii) gut motility; and (iv) specific transport mechanism changes rapidly (6). The GI membrane also acts as a physical barrier, restricting the absorption of the drug that in turn additionally influences the efficacy and toxicity of the orally administered drugs (1).
-
2.
The mucosal layer of the stomach and intestine: The mucus layer of the GI tract acts as an important barrier for the permeation of the drug molecules. The mucosal layer protects the exposed epithelial surface by trapping and clearing the pathogens and foreign particles (2). It also lubricates the epithelium as the drug molecule passes through thereby decreasing the residence time of the drug that fail to penetrate through the loosely adhered GI mucus layer (7). The thickness of the mucus layer throughout the GIT varies to a larger extent (8). The mucus layer is thickest in the stomach and colon, thereby protecting the stomach from acid and from bacteria in the colon (9). Besides this, the pH of the mucus is also different in different regions of the GIT affecting the absorption of the drug molecules.
-
3.
Intestinal epithelial cells: The intestinal epithelium is the first line of defense acting as a barrier which allows the selective absorption of the molecules. It also protects the host from the pathogens that enter via the GIT (10). The physical integrity of the epithelium is mainly due to the presence of tight junction proteins which form a connection between two adjacent cells through the intercellular spaces (11). These tight junctions along with acting as a barrier for the diffusion of solutes through the intercellular spaces also act as an inhibiting barrier for the paracellular transport of macromolecules (12).
Apart from the barriers of the GIT, poor solubility, permeability, and stability of the drug molecules in the GI environment also make the oral delivery of the therapeutic agents a challenging task (5). These hurdles and challenges can be overcome by utilizing nanotechnology approaches for the oral delivery of the drug molecules. Nanotechnology exclusively offers the possibility of developing unique drug delivery systems and pharmaceutical dosage forms (13).
Nanocarriers for the Oral Delivery of Drug Molecules
Nanocarrier systems are generally classified as substrates having a nanosize range that can encapsulate active pharmaceutical ingredients within a conjugated, synthesized, or complexed nanodimensions (13). Nanocarriers by virtue of their small size provide a larger surface area for drug encapsulation, thereby aiding in achieving a higher therapeutic concentration of the drug at the target site (14). These nanocarriers are aimed at decreasing the systemic side effects, increasing the solubility of the drug and blood residence time, controlling drug release and targeting the drug to the disease site, or crossing the barriers (15). There are various nanocarrier systems used for the delivery of drugs, such as polymeric nanoparticles, dendrimers, lipidic nanocarriers (like liposomes, ethosomes, phytosomes, solid lipid nanoparticles, nanostructured lipid carriers, etc), and several others. In this review, focus is given on the active targeting of the drugs and bioactive molecules by making use of lipidic nanocarriers via the oral route of administration, in which area there is dearth of reviews.
Lipidic Nanocarriers in Oral Drug Delivery
Lipid nanocarriers comprise a wide range of nanotechnology-based drug delivery systems. Both lipophilic and hydrophilic agents can be incorporated into these lipidic vesicles. Liposomes are self-assembled enclosed concentric vesicular systems made up of phospholipid bilayers. Ethosomes and transfersomes are the lipidic vesicles with high deformability and flexibility which can be obtained by varying their composition; however, they mostly have been found useful in the delivery of drugs across the skin. Solid lipid nanoparticles (SLN) and nanostructured lipid nanocarriers (NLC) are designed to provide higher stability to the vesicle and also to increase the encapsulation rate (16). These lipidic nanocarriers are mainly designed to improve the absorption of the drugs, to target the drug to a specific site, and to overcome the toxicological issues (17). Their use as drug delivery systems and their route of administration largely depend on the architecture and particle size of the vesicles (18).
DRUG DELIVERY BY ACTIVE TARGETING USING LIPIDIC NANOCARRIERS
Active targeting is a drug delivery approach in which the ligand-bound nanocarriers will be specifically retained and taken up by the targeted cells. Active targeting is also called as ligand-mediated targeting. The ligands used for targeting are specific either to the surface substance or to receptors that are overexpressed in the particular sites. The targeted drug delivery system mainly involves modifying or conjugating the nanocarrier surface with the targeting ligand, which is supposed to have affinity to the receptors overexpressed on the cells. The interaction between the ligand and the receptor will be either through physical adsorption or by forming covalent bonds. The fundamentals of active targeting include mainly the strategies for ligand conjugation, particle size and shape, ligand density of the actively targeted nanocarrier, surface hydrophobicity, and charge on the surface of the nanocarrier as well as on the ligand. Conjugating the nanocarriers with the targeting ligands greatly enhances the therapeutic efficacy of the active moiety and also reduces the toxic side effects (14). The nanocarriers conjugated with the ligands are taken up by receptor-mediated endocytosis, thereby achieving the intracellular drug delivery. Some examples of ligands include antibodies and their fragments (IgG, IgA, and IgM), proteins (transferrin), peptides (RGD and integrins), nucleic acid–based ligands (aptamers), and small molecules (folic acid and lectins) (19). Nanocarriers like SLN, NLC, and liposomes are widely used for the active targeting of the therapeutic agents.
Solid Lipid Nanoparticles
These are colloidal nanocarrier systems which are prepared from lipids that remain solid at both room and body temperature (20). SLNs are considered to be first-generation lipid nanocarriers. They are designed to carry and deliver hydrophobic and hydrophilic drugs and macromolecules (proteins, peptides, genes, and antigens). The drugs are either dispersed within the lipid matrix (solid solution model) or dispersed inside the core or on the shell (core–shell model). Lipids like glycerol palmitosterate, tricaprin, and glyceryl behenate are used in the preparation of SLNs. The stability of the SLNs is attributed to the use of emulsifiers like Tween-80, sodium dodecyl sulfate, and poloxamers (21).
SLNs can be classified into the following:
-
1.
Type 1: It is a homogeneous matrix model, wherein the drug is either molecularly dispersed in the lipid core or present in the form of an amorphous cluster. This type of SLNs provides controlled release of the drug.
-
2.
Type 2: It is a drug-enriched shell model wherein the melted lipid has less concentration of the drug. This type of SLNs provides a burst release of the drug.
-
3.
Type 3: It is a drug-enriched core model. This type of SLNs provides prolonged release of the drug.
SLNs are prepared by various methods using solid lipid, emulsifier, and water/solvent. SLNs are prepared by adopting the following methods: high-pressure homogenization, ultrasonication, solvent evaporation method, solvent emulsification–diffusion method, supercritical fluid method, microemulsion-based method, spray drying method, etc. (22).
Nanostructured Lipid Carriers
NLCs are composed of spatially different lipid molecules. A mixture of solid lipid and liquid lipids forms the lipid matrix of NLCs (16). A melting point depression is caused by the addition of liquid lipids; however, despite of this depression of melting point, the NLCs remain solid at body temperature. A blend of solid and liquid lipids is used in NLCs. The structural differences between solid and liquid lipids make them not to fit in together to form a crystal. This creates a lot of imperfection which results in the accumulation of more drug in amorphous clusters and in molecular form (23).
The types of NLC are as follows:
-
1.
Imperfect NLCs: The solid matrix of the NLCs is imperfectly arranged. It forms an imperfect crystal model containing a matrix with many void spaces wherein the drug molecules can be incorporated.
-
2.
Amorphous NLCs: The matrix is solid amorphous in nature. It is obtained by mixing the lipids that do not recrystallize after homogenization. This type of NLCs reduces the expulsion of the drug upon storage (24).
-
3.
Multiple NLCs: The matrix is composed of multiple oils and fat in water. These NLCs are prepared based on the principle that solubility of the lipophilic drug in liquid lipids is higher than solid lipids. The solid lipids are blended in a higher amount of oils. This type of NLCs is used to provide controlled release of the drugs and the lipid matrix prevents drug leakage (25).
NLCs are prepared by various techniques such as high-pressure homogenization, hot homogenization technique, cold homogenization technique, microemulsion technique, high shear homogenization/ultrasonication, melting dispersion method, solvent emulsification (evaporation and diffusion techniques), solvent injection, double emulsion technique, etc. (24,26).
Liposomes
Liposomes are extensively used carriers for the delivery of a wide variety of therapeutic agents. Liposomes are mainly composed of phospholipids like phosphatidylcholine and cholesterol which form a self-assembled amphiphilic system upon addition of aqueous phase (27). Based on the use of lipids, structure of the lipid bilayers, and size, liposomes are generally classified as small unilamellar vesicles (SUV), large unilamellar vesicles (LUV), multillamelar vesicles (MLV), and multivesicular vesicles (MVV). These vesicular carriers have great potential for drug delivery by the oral route (28).
Liposomes are prepared by various methods and some of them are sonication, French pressure cell, extrusion method, microemulsification, lipid film hydration, membrane extrusion, freeze–thaw method, reverse phase evaporation method, ether/ethanol injection method, etc. (29).
In liposomes, the polar heads of phospholipids face toward the aqueous phase whereas nonpolar groups are arranged inside the bilayers. Because of these attractive structural features, liposomes can encapsulate both hydrophilic as well as hydrophobic therapeutic agents. The hydrophilic drugs are entrapped inside the inner aqueous core, whereas the hydrophobic agents get entrapped within lipid bilayers. The liposomal vesicular diameter lies within a wide range, i.e., 20 to 50 μm. Since the 1970s, liposomes are used as drug delivery carriers in a variety of diseases but still remained as a carrier of choice due to remarkable stability, control on size, safety, and biocompatibility because of its bioresemblance and biomimetic properties. Also, liposomes can be easily prepared and there is virtuous possibility of surface modification for conjugation of variety of ligands for targeted delivery of drugs (30).
Even though all these lipidic nanocarriers exhibit the great potential for oral delivery, still their use for drug delivery is limited due to certain challenges.
CHALLENGES FOR ORAL DELIVERY OF LIPIDIC NANOCARRIERS
The most important challenge for oral delivery of conventional lipidic nanocarriers is that when administered orally, they will be exposed to the harsh environment of the GI tract. The lipidic components, especially those of liposomes, are highly susceptible to certain GI environment like gastric acid, GI enzymes like lipases and proteases, bile salts, and GI movements, which may result in the loss of structural integrity of the liposomes (31). Drug leakage due to bile salt emulsification may reduce the liposomal payload. Intestinal lipases hydrolyze the liposomal phospholipids, which may result in vesicular disruption. Another challenge faced by lipidic nanocarriers, specifically liposomes, is poor permeability across the GI membrane. There are hardly any lines of evidence available for the uptake of intact liposomes across the GI epithelium (32,33). These barriers for the absorption of nanocarriers upon oral administration make them suitable candidates for essential surface modification. Because of these challenges, researchers are continuously focusing on active transport by identifying certain gateways in the form of specific cell surface receptors.
In order to circumvent the above problems, ligand-conjugated lipidic nanocarriers for active targeting of the GI cell surface receptor as well as targeting certain transporters have been given more attention nowadays. The GI epithelia are mainly composed of enterocytes, M cells, and immune cells. A large number of receptors are expressed on the surface of enterocytes, M cells, and immune cells. These include lectin receptors, sugar receptors, receptors for amino acids and vitamins, integrins, transferrins, mannose receptors, Fc receptors, and claudin 4 receptors (34,35).
Surface conjugation of liposomes with specific ligands for GI epithelial cell surface epitopes might boost the specific cellular uptake and transepithelial transport. Due to continuous moving of the GI environment, there may be chances of rapid clearance of lipidic nanocarriers. This can be avoided through strong receptor–ligand interactions. Thus, it results in the accumulation of nanocarriers at the absorption sites. Since the last few decades, varieties of GI receptors have been investigated as potential targets for drug delivery. In this context, ligand-conjugated lipidic nanocarriers for active targeting have emerged as a promising drug delivery approach (36).
INTERACTION/UPTAKE OF CONVENTIONAL AND LIGAND-CONJUGATED LIPIDIC NANOCARRIERS AT THE INTESTINAL EPITHELIUM
For the active targeting of nanocarriers, there are many target sites available on the GI epithelium for each type of ligands. For example, lectins can be used as ligands for M cells as well as for enterocytes. Possible pathways by which the actively targeted (ligand-conjugated) and conventional lipidic nanocarriers interact with different sites of intestinal epithelium are shown in Fig. 1.

Challenges in the oral delivery of drugs and other active agent drugs and the use of ligand-conjugated lipidic nanocarriers to overcome the challenges. The different motifs shown on the surface of lipidic nanocarriers in the above figure indicate the “ligands” used for active targeting
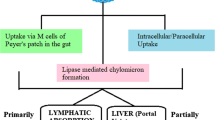
Based on the anatomical as well as physiological architectures of the small intestine, there may be many possibilities of interactions of lipidic nanocarriers based on their composition. The conventional lipidic noncarriers like liposomes, SLNs, NLCs, etc., which are mostly composed of dietary lipids, undergo the general pathways of lipid absorption. The same fact can be possible even with ligand-conjugated lipidic nanocarriers, too. In order to directly cross the intestinal barrier, the nanocarriers have to follow either the paracellular or transcellular pathways (34). Paracellular permeation is a size-dependent pathway, which generally permits the particles with the size less than 200 nm. Based on structural intactness of lipidic nanocarriers, the presence of other GI contents like food and enzymes as well as the peristaltic movement of the intestine, some of the lipidic nanocarriers undergo disruption and may release the contents at the site of the intestine. These contents may fail to cross the intestinal epithelium and get degraded due to enzymatic action of lipases, pancreatin, trypsin, chymotrypsin, or pepsin (37). In case if these lipidic nanocarriers withstand the intestinal environment, bile salts attack these nanocarriers and emulsify them to form small lipid droplets. The triglyceride portion of these lipid droplets get converted to monoglycerides and fatty acids by pancreatic lipases. Again, in association of bile salts and intestinal phospholipids, these monoglycerides form micelles which diffuse into enterocytes. Inside the enterocytes, these monoglycerides combine with the apolipoproteins (Apo B) and cholesterol at the endoplasmic reticulum as well as Golgi body to form the chylomicron particle, which exocytosized into the lymphatic circulation (38,39) (Fig. 2).

Interaction of conventional and ligand-conjugated lipidic nanocarriers on intestinal membrane
M-cell–mediated transport is another pathway for the active transport of lipidic nanocarriers. As discussed earlier, the M-cell surface expresses certain receptors like lectin receptors, mannose receptors, β1 integrins, Fc receptors, claudin 4 receptors, etc. The use of their specific ligand to conjugate with lipidic nanocarriers may be an attractive strategy for active targeting. In active targeting, the drug-loaded lipidic nanocarriers, which possess a specific ligand, undergo receptor-mediated endocytosis and deliver the contents in the lymphatic circulation. At the base of M cells, depending on the nature of the carrier as well as the drug, macrophages or lymphocytes may digest nanocarriers and allow the bioactive molecules to reach the lymphatic circulation.
Furthermore, there are certain transporter pathways for the entry of various nutrients as well as minerals through the enterocytes. These transporters include vitamin B12, folic acid, biotin, transferrin, niacin, etc. Surface anchoring of these transporter molecules or vitamins to the lipidic nanocarriers enables them to undergo transcytosis process by enterocytes into the systemic circulation. Drugs with poor permeability can be effectively carried into the systemic circulation by this mechanism if encapsulated in the lipidic nanocarriers anchored with the above transporters (40). Another approach where we can focus on the clearance of lipidic nanocarriers includes active targeting of immune cells like dendritic cells. Mannose receptors which are specifically expressed on dendritic cells can be easily targeted if lipidic nanocarriers are conjugated with mannose. Also, cationic lipidic nanocarriers are generally recognized by dendritic cells and endocytosized to deliver the drug into lymphatic circulation. There are several pathways for the transport of bioactive molecules via lipidic nanocarriers across/into the intestine, and hence, multiple pathways may be involved in this process. Thus, the exact mechanism for the transport of a particular lipidic nanocarrier is very difficult to predict (41).
LIPIDIC NANOCARRIERS USED FOR ACTIVE TARGETING OF DRUGS UPON ORAL ADMINISTRATION
Various reports on the application of different lipidic nanocarriers such as SLNs, NLCs, and liposomes for active targeting of the bioactive molecules upon oral administration are discussed below. The concise form of all these reports along with additional reports is given in Table I.
SLNs for Active Targeting of Bioactive Molecules via Oral Administration
Only a few reports are available on the active targeting of bioactive molecules via ligand-conjugated SLNs upon oral administration. Zhang et al. (42) developed insulin-encapsulated SLNs, which were further conjugated with lectin (wheat germ agglutinin) to study the efficiency of lectin-decorated lipidic nanocarriers as a carrier system for the oral delivery of proteins and peptides. They observed that the stability of wheat germ agglutinin (WGA)-conjugated SLNs was greater than that of the bare SLNs (without WGA) in protecting insulin against the degradative effect of digestive enzymes in vitro. Furthermore, they also observed an increase in the oral bioavailability of insulin when administered in the form of SLNs which were conjugated with WGA in comparison with the insulin-loaded bare SLNs. The results showed a receptor-mediated endocytosis for the WGA-conjugated SLNs containing insulin. Thus, it can be concluded that the lectins can be used effectively for increasing the oral bioavailability and also to protect the insulin from the derogative effect of gastric enzymes.
Fan et al. (44) developed SLNs loaded with salmon calcitonin (sCT) which were modified using two kinds of peptides and were evaluated for their ability to transport the protein drugs across intestinal barriers. CSK (CSKSSDYQC peptide ligand; a peptide that has affinity to goblet cells) and IRQ (IRQRRRR, a cell-penetrating peptide) were used for this study. CSK-modified sCT SLN and IRQ-conjugated sCT SLN were prepared. These modified SLNs were compared with the unmodified SLNs (without peptide ligands) and it was observed that the modified SLNs provided much better protection to the sCT. The modified SLNs facilitated the drug permeation in excised rat duodenum mucosa and also internalization on a Caco-2/HT29-MTX co-cultured cells. The absolute bioavailability of salmon calcitonin was enhanced with the modified SLNs, and thus, it was concluded that both the peptides CSK and IRQ could be used efficiently for the transport of protein drugs to the intestine.
Pooja et al. (45) developed SLNs conjugated with WGA to improve the oral bioavailability of paclitaxel (PTX). The anticancer activity against A549 lung cancer cells after internalization through N-acetyl-d-glucosamine receptors was enhanced by PTX-loaded SLN conjugated with WGA (LPSN) in comparison with free PTX. Biodistribution studies in rats showed that the lung targeting ability and oral bioavailability of PTX was significantly improved by LPSN. The study concluded that the increase in bioavailability and targetability could be due to the cumulative effect of bioadhesive property of the nanocarriers and the presence of targeting ligand (WGA).
Octaarginine (SA-R8) is a cell-penetrating peptide, which is used as a carrier for the delivery of bioactive. Zhang et al. (43) designed SLNs, modified with stearic acid–octaarginine, as carriers in order to enhance the oral absorption of insulin (SAR8-Ins-SLNs). A dramatic change in the zeta potential of the SLNs from − 32.13 to 29.87 was observed, which was due to the binding of SLNs to positively charged SA-R8. The SLNs and SA-R8 partially protected insulin from proteolysis which was confirmed by in vitro degradation experiment. The SA-R8-Ins-SLNs increased the Caco-2 cell internalization in comparison with insulin solution. A significant hypoglycemic effect in diabetic rats was observed in vivo with SA-R8-Ins-SLNs when compared with Ins-SLNs without SA-R8.
The transport of drugs and bioactive molecules across the intestinal epithelium is often restricted by lyso-endosomal degradation in epithelial cells which acts as a physiological barrier to the transport of drugs. Xu et al. (46) developed SLNs having a unique feature of endosomal escape for the oral delivery of proteins. The objective of the study was to protect the model protein drug, insulin (INS) from lyso-endosomal degradation. The research group developed two types of SLNs using double emulsion method containing an endosomal escape agent, hemagglutinin-2 (GLFEAIEGFIENGWEGMIDGWYG; HA2) in its solid lipid shell and an aqueous core loaded with insulin. INS-HA2-W-SLNs (HA2 peptide loaded in the aqueous phase of SLN) and INS-HA2-O-SLNs (HA2 peptide loaded in the oil phase of SLN) were prepared and studied for their release kinetics. The release studies showed that INS-HA2-O-SLNs possess a faster release in comparison with the INS-HA2-W-SLNs. INS-HA2-O-SLNs also facilitated the escape of the loaded insulin from the acidic endosomes and thereby the biological activity of the insulin was preserved. INS-HA2-O-SLNs also showed less distribution in endosomes and lysosomes. The study concluded that SLNs with an endosomal escaping agent (HA2) could significantly protect the protein/peptide drugs from lyso-endosomal degradation, thereby enhancing their therapeutic efficacy.
NLCs for Active Targeting of Bioactive Molecules by the Oral Route
With NLCs also, there are few reports available on the active targeting of drugs using ligand-conjugated NLCs via the oral route. Zhou et al. (49) developed biotin-conjugated oridonin NLCs (Bio-ori-NLCs) to enhance the oral bioavailability of oridonin. Biotin receptors are largely present in the jejunum of the intestine, and thus, the Bio-ori-NLCs possessed greater intestinal permeation in the jejunum. The in vivo pharmacokinetics studies showed a significant increase in the oral bioavailability of Bio-ori-NLCs when compared with the nonconjugated oridonin NLCs. Thus, the study concluded that the enhancement of bioavailability is mainly due to biotin modification, involving a ligand-based active transportation of the drug. The study also elucidated the possible mechanisms of drug absorption: (1) During the compete lipolysis of NLCs, a part of Bio-ori-NLCs survives in the harsh GI environment and is transported directly by biotin receptor–mediated endocytosis and (2) the small particle size of the NLCs and the interaction of the ligand receptor may have prolonged the GI residence time thereby improving the contact of the carriers with the epithelial membrane which in turn might have promoted the absorption of the drug.
Zhao et al. (48) studied the targeting efficacy of a pentapeptide (Thr-Lys-Pro-Pro-Arg) to macrophages. They prepared pentapeptide-grafted-NLCs (Pen-NLC) and studied its anti-inflammatory effect in rats. An increased internalization of Pen-NLCs by macrophages was observed, both in vitro and in vivo, in comparison with the bare-NLCs (without conjugation of pentapeptide) and pure pentapeptide. The Pen-NLCs also showed a considerable increase in anti-inflammatory effect in vivo.
To overcome the low oral bioavailability of docetaxel (DTX), Fang et al. (50) developed cysteine-modified NLCs. Cysteine which was linked to NLCs using PEG2000-monosterate (cNLCs) was used as a targeting ligand to achieve the interaction with mucin of the intestinal mucus layer. cNLCs showed a greater intestinal absorption in the total intestinal segments. In vivo imaging of cNLCs showed an improved accumulation in the blood when compared with unmodified NLCs and DTX solution.
In another study, an attempt was made to improve the oral bioavailability of tripterine by developing cell-penetrating peptide (CPP)-coated tripterine NLCs (CT-NLCs) (47). Ste-R6L2, a new kind of CPP which is mainly composed of arginine and leucine, was used in this study. The in vitro drug release studies by the dialysis bag diffusion method showed a controlled release of the drug from CT-NLCs. The MTT assay in Caco-2 cells showed a lower intestinal cytotoxicity for CT-NLCs in comparison with the tripterine solution. CT-NLCs showed an increased absorption of tripterine in rat duodenum and jejunum than the tripterine-loaded NLCs without CPP and tripterine solution. The researchers also speculated that the CPPs of Ste-R6L2 interact with the cell membrane, destabilize the bilayer, and thus transport the NLCs across by endocytotic entry or direct cell membrane penetration. Pharmacokinetics studies showed that CT-NLCs exhibited a maximum oral bioavailability in comparison with T-NLCs and tripterine solution.
Tian et al. (51) developed NLCs containing curcumin (Cur) using taurocholic acid (TCA) as a ligand for the uptake of NLCs mediated by a bile acid transporter system in order to increase the oral absorption of Cur. TCA-modified Cur-loaded NLCs (Cur-TCA NLCs) exhibited a sustained release in vitro. In situ intestinal perfusion studies showed an increased absorption and permeation of Cur-TCA NLCs than unmodified Cur NLCs. The pharmacokinetic studies showed that Cur-TCA NLCs exhibited good oral bioavailability of curcumin when compared with the unmodified Cur NLCs.
Liposomes for Active Targeting of Drug Molecules via Oral Administration
Compared to NLCs and SLNs, more literature is available on the active targeting of liposomes upon oral administration. Liposomes are proven as versatile drug delivery systems for the delivery of peptide/protein drugs and for vaccines as well. Extensive reports are available on the application of liposomes as drug delivery carriers for many diseases via almost all the routes. Many liposomal products are available in the market for the treatment of cancer, skin diseases, pulmonary diseases, etc. (63,64). In the last few decades, scientists are focusing their attention on the liposomal delivery system for oral administration of several various drugs and bioactive molecules including proteins, peptides, and vaccines. This may be due to the protective encapsulation of susceptible therapeutic agents within the liposomes, excellent permeation through the gut membrane by transcellular or paracellular pathway as well as the potential sites for the absorption within the GI tract (65). The various receptors expressed on the GI epithelium and their active targeting by specific ligand-conjugated liposomes are discussed below.
Lectin-Based Targeting
Enterocytes are the major populations of the GI epithelium, which play a major role in the transport of guest molecules into systemic circulation through various receptors as well as transporters. Also, another population of cells which contributes in the transport of a variety of therapeutic agents is M cells which are located on Peyer’s patches of the intestine. M cells count 1% of intestinal epithelial cells. Other than these, there are certain immune cells like dendritic cells which are also useful for the transport of certain agents across the GI membranes (40,66).
Lectins are a naturally occurring group of glycoproteins which can specifically bind to the intestinal glycoprotein receptors known as lectin receptors or glycoprotein receptors. Lectin receptors are located on enterocytes as well as on M-cell surfaces. There are many naturally occurring lectins from plants which possess active targeting potential to the enterocytes as well as M cells. Lectins also have been found to be useful ligands for the specific recognition of cell surface glycoconjugates on the intestinal membrane. Varieties of lectins are available as ligands for the liposomes for specific delivery of therapeutic agents via active targeting through receptor-mediated endocytosis. These include Ulex europaeus agglutinin I (UEA I), WGA, and tomato lectin (TL) or tilex europaeus agglutinin I (IJEAI) (67).
A recent study explored the active targeting potential of UEA I to the mouse intestinal Peyer’s patches where it exclusively bound to the M-cell surface lectin receptor. This advocates the existence of exclusive fucosylated glycoconjugates on the surface of mouse intestinal M cell (68).
Chen et al. (69) developed lectin-specific liposomes such as UEA I liposomes and WGA liposomes and investigated their Peyer’s patch targeting potential. In this study, lectins were modified with N-glutaryl-phosphotidylethanolamine (NGPE) and incorporated within the liposome bilayers; further, the liposomes were stabilized by adopting the polymerization technique. The lectin-conjugated liposomes showed specific recognition toward the mice Peyer’s patches when compared to the lectin-free liposomes (69).
Li et al. (56) demonstrated the efficiency of lectin like UEA I to anchor the BSA antigen–loaded liposomes for targeted delivery to intestinal M cells of mice. UEA I is composed of lectin-specific α-L-fucose residues, which most exclusively bind to the M cells of mouse Peyer’s patches and thus help in crossing the intestinal epithelial membrane. The developed UEA I–modified liposomes induced strong mucosal as well as systemic response as compared to unmodified liposomes. Thus, the study strongly supported the use of lectin-modified liposomes for the oral delivery of vaccines (56).
Gupta and Vyas (57) developed liposomes encapsulated with hepatitis B surface antigen (HBsAg) coupled with UEA I in order to enhance the transmucosal uptake by M cells. The results showed that UEA I–conjugated liposomes specifically targeted the M cells of Peyer’s patches. The targeting potential of UEA I was due to the specific interaction of UEA I with α-L-fucose residues at the M-cell surface. To determine the in vitro activity and specificity of lectin, bovine submaxillary mucin (BSM) was used as a biological model. The lectinized liposomes showed an increased activity with BSM in comparison with the nonlectinized liposomes, and the specificity to L-fucose was also maintained with the lectinized liposomes. The study concluded that the lectinized liposomes mainly targeted the M cells and could be used as a potential module to develop effective mucosal vaccines.
The specific binding potential of WGA to N-acetyl-d-glucosamine and sialic acid which are present on the glycocalyx of intestinal enterocytes has been reported. WGA was effectively bound to the human enterocytes and taken up via receptor-mediated endocytosis through the epidermal growth factor receptor present on the surface of enterocytes. Thus, the use of WGA-conjugated liposomes could be used for active targeting of intestinal epidermal growth factor receptor for the delivery of drugs (66,70).
Zhang et al. (53) developed and evaluated the potential of lectin-conjugated liposomes loaded with insulin by oral administration. They coupled terminal amino groups of WGA as well as TL through carbodiimide chemistry to N-glutaryl-phosphatidylethanolamine which was incorporated with other lipids. The hypoglycemic activity and bioavailability of orally administered conventional liposomes and modified liposomes were compared with subcutaneous injection of insulin. The results suggested that lectin-conjugated liposomes enhanced the insulin absorption orally through active targeting mechanism.
WGA liposomes as well as WGA-SLNs were prepared to examine the transport potential across the rat intestinal membrane by using insulin as a model drug. They investigated the best part of the intestine for the absorption of the developed formulations. They also compared the stability of the formulations in pepsin as well as in trypsin solutions. WGA liposomes showed better binding ability to the glycoprotein receptors on the M cells as compared to WGA-SLNs (42). It was reported that receptor-mediated endocytosis was the absorption mechanism for WGA-modified carriers.
Mannose-Based Targeting
Mannose receptors are widespread throughout the GIT including the small as well as large intestines, but most predominantly expressed on the surface of immune cells like dendritic cells and macrophages. Mannose receptors are transmembrane receptors which recognize the specific bacterial, viral, fungal, or other parasite surface carbohydrate groups and stimulate the activation of macrophageal activation to clear them from the body. Thus, mannose receptor is considered as a marker for the reticuloendothelial system (71,72). Mannose-based targeting motif specifically binds to the mannose receptors. These strategies were found to be useful for active targeted delivery of a variety of drugs as well as vaccine (73).
Witoonsaridsilp et al. (74) developed mannosylated liposomes, which exhibited bioadhesive potential to mannose receptors. They investigated the GI permeability of the protein such as lysozyme by using liposomes as a carrier which was conjugated with mannose receptor-specific binding ligand N-octadecyl-D-mannopyranosylamine. They proved that mannosylated liposomes successfully permeated across the Caco-2 cell monolayers as compared to the conventional liposomes and a free drug. In their findings, there was almost 2.5-fold more permeability of mannosylated liposomes as compared to the conventional liposomes and 7-fold more compared to the drug solution. Thus, mannose-based targeting approach established the suitable active targeting strategy for the drugs using liposomes (74).
In another study, Pukanud et al. (55) designed bioadhesive mannosylated liposomes for oral delivery of drug. They prepared acyclovir loaded with two different kinds of mannosylated liposomes. In one formulation, liposome was conjugated with mannosamine HCl, and in another type of liposomes, p-aminophenyl-α-D-mannopyranoside was used. They found that size was increased in case of p-aminophenyl-α-D-mannopyranoside compared to mannosamine HCl. This may be due to the grafting of a larger group on the liposomes. The absorption of acyclovir through the everted sac of mouse ileum was significantly higher in both types of mannosylated liposomes as compared to the conventional liposomes and pure drug (55).
The application of the mannosylated proliposomes which were conjugated with mannose receptor–specific ligand N-octadecyl-D-mannopyranosylamine (SAMAN) was studied. Busrelin acetate was used as a model drug. The mannosylated busrelin acetate–loaded proliposomes were prepared by the co-precipitation method. It was found that there was about 1.2-fold higher permeability in Caco-2 monolayers with conjugated liposomes in comparison with conventional liposomes and 2.2-fold higher permeability than the drug solution. This significant increase in permeability might be due to active targeting of mannosylated liposomes to the mannose receptors on intestinal enterocytes (61).
Vitamin-Based Targeting
Absorption of vitamins and minerals across the intestine is a well-established natural mechanism. Enterocytes have a versatile system for active absorption of the essential moieties through the specific transporters. This is a carrier-mediated transport system which is different and specific for each and every xenobiotics. Understanding these transporter systems can facilitate the effective drug delivery strategies via the oral route (35,40). Vitamins such as vitamin B9, vitamin B1, vitamin B3, and vitamin B7 and iron are actively taken up by the intestinal epithelial cells (75). As there is a wide distribution of receptors for vitamins and minerals, the effective uptake of various drug-loaded carriers can be possible by enterocytes. Various reports have explored the use of these ligands for active targeted delivery of drugs via the oral route by focusing on the various transport mechanisms (76).
Vitamins like biotin, which cannot be synthesized endogenously but supplied through the diet, is taken up by enterocytes via biotin receptor from receptor-mediated endocytosis. Biotin receptors are distributed throughout the small intestine, but these are nonspecific receptors. It was found that biotin coupling with glucagon-like peptide-1 increased the hypoglycemic efficacy upon oral administration. This occurred due to enhanced intestinal permeability due to receptor-mediated endocytosis by biotin receptors (77).
Zhang et al. (59) explored the use of biotinylated liposomes as an efficient carrier for the oral delivery of insulin. Biotin-conjugated liposomes were prepared by incorporating the specific phospholipids conjugated with biotin. There was a significant increase in liposomal uptake by biotin receptor through receptor-mediated endocytosis in rat intestine. This finally enhanced the hypoglycemic effect of insulin due to effective absorption in the systemic circulation. The oral bioavailability of biotinylated liposomes was almost double than that of the conventional liposomes. Also, the study revealed the advantage of biotinylated liposomes to increase GI stability. Thus, these reports can provide a strong platform for the oral delivery of other proteins and peptide by using biotinylated liposomes as potential carriers.
Similarly, scientists gave attention on folate receptors by designing folate-conjugated liposomes for active delivery of various drugs. Folic acid is effectively transported across the intestine in the most suitable form as folate via folate receptors which are present throughout the GIT. Thus, folic acid or folate-conjugated liposomal systems may improve the specific uptake of drugs as well as other bioactive molecules (6,78).
Anderson et al. (79) used poly(ethylene oxide)–folic acid (PEO-FA) derivative and incorporated in liposomes to improve the transport across the Caco-2 cells. They first prepared FA–PEO–cholesterol conjugate which was later adsorbed on the liposome surface. In this study, Texas Red-Dextran 3000 (TR-dex) was used as a specific marker. Normally, it is poorly absorbed due to higher molecular weight and neutral hydrophilic nature. There was a significant increase in intracellular accumulation of TR-dex in case of FA-coated TR-dex liposomes than uncoated TR-dex liposomes, which was confirmed by fluorescence microscopy (79).
In a similar study, Agrawal et al. (60) prepared insulin-loaded stable liposomes functionalized with folic acid to improve oral bioavailability. Further, to improve the intestinal stability, the liposomes were coated with positively charged poly(allyl amine) hydrochloride (PAH) as well as negatively charged poly(acrylic acid) (PAA). In order to anchor the folic acid as a targeting ligand to the liposomes, folic acid−poly(allyl amine) hydrochloride conjugation was done. The results confirmed the conformational as well as the biological stability of insulin within the liposomes in simulated biological fluids. Successful delivery of liposomes was observed in Caco-2 cells as well as in an ex vivo intestinal uptake study. Also, the results of the in vivo hypoglycemic study supported the increased oral bioavailability of conjugated liposomes in comparison with conventional liposomes. It was concluded that the expected outcomes of the developed formulation were due to folic acid conjugation to liposomal surface as well as stabilization with polyelectrolytes (6).
Transferrin Receptor–Based Targeting
Transferrin (Tf) receptors are high molecular weight proteins (MW 70,000–80,000 Da), expressed on intestinal epithelial cells especially at the villous region of epithelial cells and the crypt region of the duodenum (80). Other than that, macrophages as well as lymphocytes also express the Tf receptors at Peyer’s patches. Upon binding with transferrin receptors, the ligand (transferrin) undergoes transcytosis on epithelial cells. The biological half-life of transferrin is very high (8 days). It can show the partial antagonistic effect on the degradation of trypsin and chymotrypsin, and thus, its GI stability is very high. The use of Tf as a targeting ligand for Tf receptors can improve the GI stability of the carriers and may help in the effective transport of drugs (81,82). Shah and Shen (83) studied Caco-2 cell monolayer transport of insulin–Tf conjugate prepared through disulfide linkage. It was observed that there were 5- to 15-fold increases in the permeability of insulin. Xia et al. (52) investigated the Tf receptor–mediated transcytosis of insulin–transferrin conjugate using Caco-2 cells. Additionally, the oral delivery of insulin–Tf conjugate was investigated in streptozotocin-induced diabetic rats. The insulin–Tf conjugate exhibited a sustained hypoglycemic effect after oral administration in fasted diabetic rats.
Tf-mediated targeted oral delivery of proteins like insulin, several genes, and anticancer drugs has been reported extensively by using various nanocarriers. Thus, the use of Tf in active targeting approach along with liposomes as carriers may be an effective strategy to enhance the intestinal transport of the drugs (52,84). However, hardly few reports are available on the Tf-conjugated liposomes for active targeted delivery of drugs via the oral route.
M-Cell–Based Targeted Delivery
M cells are specialized intestinal epithelial cells, mainly located at the FAE lying over the Peyer’s patches and other lymphoid clusters. M cells can be easily differentiated from enterocytes due to the absence of microvilli on the surface. Topographically, M cells exhibit a microfold on the apical side; hence, they are called M cells or microfold cells. M cells have underneath infiltrating lymphocytes for antigen clearance. This makes an attractive targeting site for drugs as well as vaccine delivery. M cells possess high transcytosis potential than enterocytes and dendritic cells. The M-cell surface expresses many receptors like β1-integrin, claudin 4, and neonatal Fc receptor, whose receptors provide an opportunity for targeted delivery of many therapeutic agents as well as vaccines (85,86).
β1-Integrin–Based Targeted Delivery
Integrins belong to a family of cell adhesion molecules involved in various ligand-to-cell communications. There are many types of integrins throughout the body. Intestinal M cells represent β1-integrin on the apical side which shows specific binding affinity toward arginine-glycine-aspartic acid (RGD) motif of many biological molecules. After binding with β1-integrin, M cell undergoes receptor-mediated endocytosis of the entire binding assembly. Many reports suggested that the use of the β1-integrin targeting strategy may improve the intestinal delivery of many drugs (34,87,88).
Scientists also have investigated the binding potential of the RGD peptide to M-cell surface β1-integrin receptor of the human intestine. They used the human FAE model derived from Caco-2 cells and Raji B cells. They adopted the cDNA array to identify the targeting motif and immunofluorescence to study the expression pattern. The study revealed the existence of RGD binding motif in adequate density (89).
Ding et al. (90) formulated RGD-modified PEGylated liposomes loaded with indocyanine green, which is a near-infrared dye. Covalent conjugation of the RGD peptide was achieved by using amide linkage with the distal end of DSPE-PEG2000-NH2 lipid. The study indicated the effective targeting of RGD-modified liposomes to the integrins; additionally, PEGylation improved the stability of the liposomes in vivo. This study explored the use of RGD-conjugated liposomes for the active targeted delivery of drugs to the intestine.
Claudin 4-Based Targeted Delivery
Claudin 4 is a member of the claudin receptor family. It is a transmembrane receptor located on the M-cell surface and it contributes to the formation of the tight junction. It has control over the influx of nutrients, biofactors, and ions (91). Expression of claudin 4 is reported in mouse as well as human M cells. Claudin 4 receptor is involved in endocytic mechanism of transport (58).
The specific ligand for claudin is Clostridium perfringens enterotoxin (CPE), which is a high molecular weight (35 kDa) single polypeptide. In humans, it is responsible for illness associated with food poisoning (92). CPE is composed of two different domains. One is the N-terminal domain (22 kDa) which is cytotoxic and another is the C-terminal domain (13 kDa) which is the binding motif. It has been reported that CPE polypeptide is used as a targeting ligand for claudin receptors in different types of cancers (93,94).
Lo et al. (58) formulated the CPE–peptide conjugate with recombinant influenza hemagglutinin (CPE–HA conjugate) by chemical modification. The conjugate was delivered along with a cholera toxin as an adjuvant. The results revealed the increased mucosal M-cell delivery of recombinant influenza hemagglutinin that was mediated through claudin 4, which further induced the IgA response more effectively. Thus, the use of CPE peptide as a ligand for the conjugation with liposomes effectively targeted the M cells (58).
Fc Receptor–Based Targeted Delivery
Fc receptors or neonatal Fc receptors have been reported to be present on M cells to mediate the immunoglobulin transport across the intestinal barrier. Fc receptors naturally appear in neonates to transport IgG from mother’s breast milk to neonatal intestine. The expression of Fc receptors generally diminishes with age but still appears at certain levels. The use of IgG antibody fragments or whole IgG antibody as a ligand to target the Fc receptors has been found to be an effective strategy for active delivery of drugs or proteins across the mucosal barrier. Conjugation of the IgG antibody or fragments to the liposomal surface may provide an additional benefit of protection to the drugs or proteins (95,96).
CONCLUSION
Delivering the drugs via the oral route is a challenging task because of involvement of issues and hurdles such as low permeability and low solubility of the drugs. Nanocarriers have been considered as one of the best options to overcome these problems. Among the nanotechnology-based drug delivery systems, lipidic nanocarriers are more suitable for oral administration. Active targeting approach involves the use of ligands that are specific to receptors thereby increasing the targetability and therapeutic efficacy of the drugs. There is a scarcity of literature in the area of active targeting of the drugs and bioactive molecules using lipidic nanocarriers via the oral route. Hence, this review opens an avenue for the researchers to explore more in this research area.
References
Moss DM, Curley P, Kinvig H, Hoskins C, Owen A. The biological challenges and pharmacological opportunities of orally administered nanomedicine delivery. Expert Rev Gastroenterol Hepatol. 2018;12(3):223–36.
Ensign LM, Cone R, Hanes J. Oral drug delivery with polymeric nanoparticles: the gastrointestinal mucus barriers. Adv Drug Deliv Rev. 2012;64(6):557–70.
Ghosh S, Roy T. Nanoparticulate drug-delivery systems: lymphatic uptake and its gastrointestinal applications. J Appl Pharm Sci. 2014;4(6):123–30.
Plapied L, Duhem N, des Rieux A, Préat V. Fate of polymeric nanocarriers for oral drug delivery. Curr Opin Colloid Interface Sci. 2011;16(3):228–37.
Bernkop-Schnürch A. Reprint of: Nanocarrier systems for oral drug delivery: do we really need them? Eur J Pharm Sci. 2013;50(1):2–7.
Agrawal AK, Harde H, Thanki K, Jain S. Improved stability and antidiabetic potential of insulin containing folic acid functionalized polymer stabilized multilayered liposomes following oral administration. Biomacromolecules. 2013;15(1):350–60.
Lai SK, Wang YY, Hanes J. Mucus-penetrating nanoparticles for drug and gene delivery to mucosal tissues. Adv Drug Deliv Rev. 2009;61(2):158–71.
Sandzen B, Blom H, Dahlgren S. Gastric mucus gel layer thickness mearured by direct light microscopy. An experimental study in the rat. Scand J Gastroenterol. 1988;23(10):1160–4.
Hollingsworth MA, Swanson BJ. Mucins in cancer: protection and control of the cell surface. Nat Rev Cancer. 2004;4(1):45–60.
Shahbazi M-A, Santos A, Improving Oral H. Absorption via drug-loaded nanocarriers: absorption mechanisms, intestinal models and rational fabrication. Curr Drug Metab. 2013;14(1):28–56.
Vllasaliu D, Fowler R, Garnett M, Eaton M, Stolnik S. Barrier characteristics of epithelial cultures modelling the airway and intestinal mucosa: a comparison. Biochem Biophys Res Commun. 2011;415(4):579–85.
Sonaje K, Chuang EY, Lin KJ, Yen TC, Su FY, Tseng MT, et al. Opening of epithelial tight junctions and enhancement of paracellular permeation by chitosan: microscopic, ultrastructural, and computed-tomographic observations. Mol Pharm. 2012;9(5):1271–9.
Tyagi P, Subramony JA. Nanotherapeutics in oral and parenteral drug delivery: key learnings and future outlooks as we think small. J Control Release. 2018;272(January):159–68.
Öztürk-Atar K, Eroğlu H, Çalış S. Novel advances in targeted drug delivery. J Drug Target. 2018;26(8):633–42.
Elbayoumi TA, Torchilin VP. Tumor-specific antibody-mediated targeted delivery of Doxil reduces the manifestation of auricular erythema side effect in mice. Int J Pharm. 2008;357(1–2):272–9.
Sala M, Diab R, Elaissari A, Fessi H. Lipid nanocarriers as skin drug delivery systems: properties, mechanisms of skin interactions and medical applications. Int J Pharm. 2018;535(1–2):1–17.
Haghiralsadat F, Amoabediny G, Naderinezhad S. Overview of preparation methods of polymeric and lipid-based (noisome, solid lipid, liposome) nanoparticles: a comprehensive review. 2018;6(4):383–400.
Attama AA, Momoh MA, Builders PF. Lipid nanoparticulate drug delivery systems : a revolution in dosage form design and development. 2012;
Bertrand N, Wu J, Xu X, Kamaly N, Farokhzad OC. Cancer nanotechnology: the impact of passive and active targeting in the era of modern cancer biology. Adv Drug Deliv Rev. 2014;66:2–25.
Schwarz C, Mehnert W, Lucks JS, Miiller RH. Solid lipid nanoparticles (SLN) for controlled drug delivery. I. Production, characterization and sterilization. J Control Release. 1994;30(93):83–96.
Buse J. Properties, engineering and applications of lipid-based nanoparticle drug-delivery systems: current research and advances. Nanomedicine. 2010;5:1237–60.
Mäder K, Mehnert W. Solid lipid nanoparticles: production, characterization and applications. Adv Drug Deliv Rev. 2001;47(2–3):165–96.
Domingo C, Saurina J. An overview of the analytical characterization of nanostructured drug delivery systems: towards green and sustainable pharmaceuticals: a review. Anal Chim Acta. 2012;744:8–22.
Gaba B, Fazil M, Ali A, Baboota S, Sahni JK, Ali J. Nanostructured lipid (NLCs) carriers as a bioavailability enhancement tool for oral administration. Drug Deliv. 2015;22(6):691–700.
Jaiswal P, Gidwani B, Vyas A. Nanostructured lipid carriers and their current application in targeted drug delivery. Artif Cells Nanomed Biotechnol. 2016;44(1):27–40.
Pardeike J, Hommoss A, Müller RH. Lipid nanoparticles (SLN, NLC) in cosmetic and pharmaceutical dermal products. Int J Pharm. 2009;366(1–2):170–84.
He H, Lu Y, Qi J, Zhu Q, Chen Z, Wu W. Adapting liposomes for oral drug delivery. Acta Pharm Sin B. 2018;20.
Filatova LY, Klyachko NL, Kudryashova EV. Targeted delivery of anti-tuberculosis drugs to macrophages: targeting mannose receptors. Russ Chem Rev. 2018;87(4):374–91.
Akbarzadeh A, Rezaei-Sadabady R, Davaran S, Joo SW, Zarghami N, Hanifehpour Y, et al. Liposome: classification, preparation, and applications. Nanoscale Res Lett. 2013;8(1):102.
Chono S, Kaneko K, Yamamoto E, Togami K, Morimoto K. Effect of surface-mannose modification on aerosolized liposomal delivery to alveolar macrophages. Drug Dev Ind Pharm. 2010;36(1):102–7.
Martins S, Sarmento B, Ferreira DC, Souto EB. Lipid-based colloidal carriers for peptide and protein delivery–liposomes versus lipid nanoparticles. Int J Nanomedicine. 2007;2(4):595–607.
Torchilin VP. Recent advances with liposomes as pharmaceutical carriers. Nat Rev Drug Discov. 2005;4(2):145–60.
Wu W, Lu Y, Qi J. Oral delivery of liposomes. Ther Deliv. 2015;6(11):1239–41.
des Rieux A, Pourcelle V, Cani PD, Marchand-Brynaert J, Préat V. Targeted nanoparticles with novel non-peptidic ligands for oral delivery. Adv Drug Deliv Rev. 2013;65(6):833–44.
Hamman JH, Demana PH, Olivier EI. Targeting receptors, transporters and site of absorption to improve oral drug delivery. Drug Target Insights. 2007;2:71–81.
Roger E, Lagarce F, Garcion E, Benoit JP. Biopharmaceutical parameters to consider in order to alter the fate of nanocarriers after oral delivery. Nanomedicine. 2010;5(2):287–306.
Khan AA, Mudassir J, Mohtar N, Darwis Y. Advanced drug delivery to the lymphatic system: lipid-based nanoformulations. Int J Nanomedicine. 2013;8:2733–44.
Harde H, Das M, Jain S. Solid lipid nanoparticles: an oral bioavailability enhancer vehicle. Expert Opin Drug Deliv. 2011;8(11):1407–24.
Managuli RS, Raut SY, Reddy MS, Mutalik S. Targeting the intestinal lymphatic system: a versatile path for enhanced oral bioavailability of drugs. Expert Opin Drug Deliv. 2018;15(8):787–804.
Tsuji A, Tamai I. Carrier-mediated intestinal transport of drugs. Pharm Res. 1996;13(7):963–77.
Li X, Yu M, Fan W, Gan Y, Hovgaard L, Yang M. Orally active-targeted drug delivery systems for proteins and peptides. Expert Opin Drug Deliv. 2014;11(9):1435–47.
Zhang N, Ping QN, Huang GH, Han X, Cheng Y, Xu W. Transport characteristics of wheat germ agglutinin-modified insulin-liposomes and solid lipid nanoparticles in a perfused rat intestinal model. J Nanosci Nanotechnol. 2006;6(9–10):2959–66.
Zhang ZH, Zhang YL, Zhou JP, Lv HX. Solid lipid nanoparticles modified with stearic acid–octaarginine for oral administration of insulin. Int J Nanomedicine. 2012;7:3333.
Fan T, Chen C, Guo H, Xu J, Zhang J, Zhu X, et al. Design and evaluation of solid lipid nanoparticles modified with peptide ligand for oral delivery of protein drugs. Eur J Pharm Biopharm. 2014;88(2):518–28.
Pooja D, Kulhari H, Kuncha M, Rachamalla SS, Adams DJ, Bansal V, et al. Improving efficacy, oral bioavailability, and delivery of paclitaxel using protein-grafted solid lipid nanoparticles. Mol Pharm. 2016;13(11):3903–12.
Xu Y, Zheng Y, Wu L, Zhu X, Zhang Z, Huang Y. Novel solid lipid nanoparticle with endosomal escape function for oral delivery of insulin. ACS Appl Mater Interfaces. 2018;10(11):9315–24.
Chen Y, Yuan L, Zhou L, Zhang ZH, Cao W, Wu Q. Effect of cell-penetrating peptide-coated nanostructured lipid carriers on the oral absorption of tripterine. Int J Nanomedicine. 2012;7:4581.
Zhao C, Fan T, Yang Y, Wu M, Li L, Zhou Z, et al. Preparation, macrophages targeting delivery and anti-inflammatory study of pentapeptide grafted nanostructured lipid carriers. Int J Pharm. 2013;450(1–2):11–20.
Zhou X, Zhang X, Ye Y, Zhang T, Wang H, Ma Z, et al. Nanostructured lipid carriers used for oral delivery of oridonin: an effect of ligand modification on absorption. Int J Pharm. 2015;479(2):391–8.
Fang G, Tang B, Chao Y, Xu H, Gou J, Zhang Y, et al. Cysteine-functionalized nanostructured lipid carriers for oral delivery of docetaxel: a permeability and pharmacokinetic study. Mol Pharm. 2015;12(7):2384–95.
Tian C, Asghar S, Wu Y, Chen Z, Jin X, Yin L, et al. Improving intestinal absorption and oral bioavailability of curcumin via taurocholic acid-modified nanostructured lipid carriers. Int J Nanomedicine. 2017;12:7897–911.
Xia CQ, Wang J, Shen WC. Hypoglycemic effect of insulin-transferrin conjugate in streptozotocin induced diabetic rats. J Pharmacol Exp Ther. 2000;295:594–600.
Zhang N, Ping QN, Huang GH, Xu WF. Investigation of lectin-modified insulin liposomes as carriers for oral administration. Int J Pharm. 2005;294(1–2):247–59.
Ling SS, Yuen KH, Magosso E, Barker SA. Oral bioavailability enhancement of a hydrophilic drug delivered via folic acid-coupled liposomes in rats. J Pharm Pharmacol. 2009;61(4):445–9.
Pukanud P, Peungvicha P, Sarisuta N. Development of mannosylated liposomes for bioadhesive oral drug delivery via M cells of Peyer’s patches. Drug Deliv. 2009;16(5):289–94.
Li K, Zhao X, Xu S, Pang D, Yang C, Chen D. Application of Ulex europaeus agglutinin I-modified liposomes for oral vaccine: ex vivo bioadhesion and in vivo immunity. Chem Pharm Bull. 2011;59(5):618–23.
Gupta PN, Vyas SP. Investigation of lectinized liposomes as M-cell targeted carrier-adjuvant for mucosal immunization. Colloids Surf B: Biointerfaces. 2011;82(1):118–25.
Lo DD, Ling J, Eckelhoefer AH. M cell targeting by a Claudin 4 targeting peptide can enhance mucosal IgA responses. BMC Biotechnol. 2012;12(1):7.
Zhang X, Qi J, Lu Y, He W, Li X, Wu W. Biotinylated liposomes as potential carriers for the oral delivery of insulin. Nanomedicine. 2014;10(1):167–76.
Agrawal U, Sharma R, Gupta M, Vyas SP. Is nanotechnology a boon for oral drug delivery? Drug Discov Today. 2014;0(10):1530–46.
Yingsukwattana K, Puttipipatkhachorn S, Ruktanonchai U, Sarisuta N. Enhanced permeability across Caco-2 cell monolayers by specific mannosylating ligand of buserelin acetate proliposomes. J Liposome Res. 2016;26(1):69–79.
Zhang X, Qi J, Lu Y, He W, Li X, Wu W. Biotinylated liposomes as potential carriers for the oral delivery of insulin. Nanomedicine: nanotechnology, biology and medicine. 2014;10(1):167–76.
Fricker G, Kromp T, Wendel A, Blume A, Zirkel J, Rebmann H, et al. Phospholipids and lipid-based formulations in oral drug delivery. Pharm Res. 2010;27(8):1469–86.
Rogers JA, Anderson KE. The potential of liposomes in oral drug delivery. Crit Rev Ther Drug Carrier Syst. 1998;15(5):60.
Morishita M, Peppas NA. Is the oral route possible for peptide and protein drug delivery? Drug Discov Today. 2006;11(19–20):905–10.
Gabor F, Bogner E, Weissenboeck A, Wirth M. The lectin–cell interaction and its implications to intestinal lectin-mediated drug delivery. Adv Drug Deliv Rev. 2004;56(4):459–80.
Clark MA, Hirst BH, Jepson MA. Lectin-mediated mucosal delivery of drugs and microparticles. Adv Drug Deliv Rev. 2000;43(2–3):207–23.
Clark MA, Jepson MA, Simmons NL, Hirst BH. Differential surface characteristics of M cells from mouse intestinal Peyer’s and caecal patches. Histochem J. 1994;26:271–80.
Chen H, Torchilin V, Langer R. Lectin-bearing polymerized liposomes as potential oral vaccine carriers. Pharm Res. 1996;13(9):1378–83.
Wirth M, Kneuer C, Lehr CM, Gabor F. Lectin-mediated drug delivery: discrimination between cytoadhesion and cytoinvasion and evidence for lysosomal accumulation of wheat germ agglutinin in the Caco-2 model. J Drug Target. 2002;10(6):439–48.
Irache JM, Salman HH, Gamazo C, Espuelas S. Mannose-targeted systems for the delivery of therapeutics. Expert Opin Drug Deliv. 2008;5(6):703–24.
Takahashi K, Donovan MJ, Rogers RA, Ezekowitz RA. Distribution of murine mannose receptor expression from early embryogenesis through to adulthood. Cell Tissue Res. 1998;292(2):311–23.
Fievez V, Plapied L, des Rieux A, Pourcelle V, Freichels H, Wascotte V, et al. Targeting nanoparticles to M cells with non-peptidic ligands for oral vaccination. Eur J Pharm Biopharm. 2009;73(1):16–24.
Witoonsaridsilp W, Paeratakul O, Panyarachun B, Sarisuta N. Development of mannosylated liposomes using synthesized N-octadecyl-D-mannopyranosylamine to enhance gastrointestinal permeability for protein delivery. AAPS PharmSciTech. 2012;13(2):699–706.
Roger E, Kalscheuer S, Kirtane A, Guru BR, Grill AE, Whittum-Hudson J, et al. Folic acid functionalized nanoparticles for enhanced oral drug delivery. Mol Pharm. 2012;9(7):2103–10.
Chatterjee NS, Kumar CK, Ortiz A, Rubin SA, Said HM. Molecular mechanism of the intestinal biotin transport process. Am J Phys Cell Phys. 1999;277:C605–13.
Youn YS, Chae SY, Lee S, Kwon MJ, Shin HJ, Lee KC. Improved peroral delivery of glucagon-like peptide-1 by site-specific biotin modification: design, preparation, and biological evaluation. Eur J Pharm Biopharm. 2008;68(3):667–75.
Ashokkumar B, Mohammed ZM, Vaziri ND, Said HM. Effect of folate over supplementation on folate uptake by human intestinal and renal epithelial cells. Am J Clin Nutr. 2007;86:159–66.
Anderson KE, Stevenson BR, Rogers JA. Folic acid–PEO-labeled liposomes to improve gastrointestinal absorption of encapsulated agents. J Control Release. 1999;60(2–3):189–98.
Banerjee D, Flanagan PR, Cluett J, Valberg LS. Transferrin receptors in the human gastrointestinal tract. Relationship to body iron stores. Gastroenterology. 1986;91:861–9.
Qing X, Yang X, Yang X, Qian Z, Kui W. Drug delivery via the transferrin receptor-mediated endocytosis pathway. J Chin Pharm Sci. 2009;18:7–13.
Zhang L, Shi Y, Song Y, Duan D, Zhang X, Sun K, et al. Tf ligand-receptor-mediated exenatide-Zn2+ complex oral-delivery system for penetration enhancement of exenatide. J Drug Target. 2018:1–0.
Shah D, Shen WC. Transcellular delivery of an insulin-transferrin conjugate in enterocyte-like Caco-2 cells. J Pharm Sci. 1996;85(12):1306–11.
Li H, Qian ZM. Transferrin/transferrin receptor-mediated drug delivery. Med Res Rev. 2002;22:225–50.
Garinot M, Fiévez V, Pourcelle V, Stoffelbach F, des Rieux A, Plapied L, et al. PEGylated PLGA-based nanoparticles targeting M cells for oral vaccination. J Control Release. 2007;120(3):195–204.
KuoLee R, Chen W. M cell-targeted delivery of vaccines and therapeutics. Expert Opin Drug Deliv. 2008;5(6):693–702.
des Rieux A, Ragnarsson EG, Gullberg E, Préat V, Schneider YJ, Artursson P. Transport of nanoparticles across an in vitro model of the human intestinal follicle associated epithelium. Eur J Pharm Sci. 2005;25(4–5):455–65.
Yun Y, Cho YW, Park K. Nanoparticles for oral delivery: targeted nanoparticles with peptidic ligands for oral protein delivery. Adv Drug Deliv Rev. 2013;65(6):822–32.
Gullberg E, Keita ÅV, Sa'ad YS, Andersson M, Caldwell KD, Söderholm JD, et al. Identification of cell adhesion molecules in the human follicle-associated epithelium that improve nanoparticle uptake into the Peyer’s patches. J Pharmacol Exp Ther. 2006;319(2):632–9.
Ding J, Feng M, Wang F, Wang H, Guan W. Targeting effect of PEGylated liposomes modified with the Arg-Gly-Asp sequence on gastric cancer. Oncol Rep. 2015;34(4):1825–34.
Morin PJ. Claudin proteins in human cancer: promising new targets for diagnosis and therapy. Cancer Res. 2005;65(21):9603–6.
McClane BA, Chakrabarti G. New insights into the cytotoxic mechanisms of Clostridium perfringens enterotoxin. Anaerobe. 2004;10(2):107–14.
Ebihara C, Kondoh M, Hasuike N, Harada M, Mizuguchi H, Horiguchi Y, et al. Preparation of a claudin-targeting molecule using a C-terminal fragment of Clostridium perfringens enterotoxin. J Pharmacol Exp Ther. 2006 Jan 1;316(1):255–60.
Gao Z, McClane BA. Use of Clostridium perfringens enterotoxin and the enterotoxin receptor-binding domain (C-CPE) for cancer treatment: opportunities and challenges. J Toxicol. 2012;2012:1–9.
Yoshida M, Claypool SM, Wagner JS, Mizoguchi E, Mizoguchi A, Roopenian DC, et al. Human neonatal Fc receptor mediates transport of IgG into luminal secretions for delivery of antigens to mucosal dendritic cells. Immunity. 2004;20:769–83.
Pridgen EM, Alexis F, Kuo TT, Levy-Nissenbaum E, Karnik R, Blumberg RS, et al. Transepithelial transport of Fc-targeted nanoparticles by the neonatal fc receptor for oral delivery. Sci Transl Med. 2013;5(213):213ra167.
Author information
Authors and Affiliations
Corresponding author
Additional information
Guest Editor: Sanyog Jain
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Shreya, A.B., Raut, S.Y., Managuli, R.S. et al. Active Targeting of Drugs and Bioactive Molecules via Oral Administration by Ligand-Conjugated Lipidic Nanocarriers: Recent Advances. AAPS PharmSciTech 20, 15 (2019). https://doi.org/10.1208/s12249-018-1262-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1208/s12249-018-1262-2