
Abstract
Partition coefficient (log P) is a key physicochemical characteristic of lipophilic drugs which plays a significant role in formulation development for oral administration. Lipid-based formulation strategies can increase lymphatic transport of these drugs and can enhance bioavailability many folds. The number of lipophilic drugs in pharmacopoeias and under discovery are continuously increasing and making the job of the formulation scientist difficult to develop suitable formulation of these drugs due to potent nature and water insolubility of these drugs. Recently, many natural and synthetic lipids are appearing in the market which are helpful in the development of lipid-based formulations of these types of drugs having enhanced solubility and bioavailability. One such reason for this enhanced bioavailability is the accessibility of the lymphatic transport as well as avoidance of first-pass effect. This review discusses the impact of lipophilicity in enhancing the intestinal lymphatic drug transport thereby reducing first-pass metabolism. The most appropriate strategy for developing a lipid-based formulation depending upon the degree of lipophilicity has been critically discussed and provides information on how to develop optimum formulation. Various formulation strategies are discussed in-depth by classifying lipid-based oral drug delivery systems with case studies of few marketed formulations with challenges and opportunities for the future of the formulations.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
INTRODUCTION
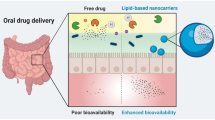
The need for a preferred delivery of therapeutics via the oral routes arises from the fact that this route is patient friendly and the easiest one for self-administration. However, the delivery of therapeutics orally is constantly challenged by the physicochemical properties of drug molecules that display poor solubility, extreme first-pass metabolism, and instability in the gastrointestinal tract (GIT). Also, the chances of dose dumping and inter- and intrasubject variability challenge their quaint success (1). Of the new molecules discovered or being invented through drug discovery and screening pathways that have therapeutic effect, most of them are potent and are lipophilic in nature (2,3). This hydrophobic nature limits the dissolution and absorption of the drug and limits the bioavailability for achieving therapeutic benefits by the oral route. Research in improving the solubility for drug molecules has been addressed by using many techniques such as physical modifications (micronization/nanonization, nanocrystal, solid dispersion), chemical modifications (salt formation or prodrug formation), solubilization (use of wetting agents, co-solvents or pH adjustment), carrier system approach (complexation, microemulsion, liposomes), etc. Such techniques have paved the way for increasing the oral product portfolio for lipophilic molecules (4,5).
In the past decade, much of the attention is gained in the research related to the use of lipids and lipophilic excipients in delivery by the oral route (6,7). This is based on the empirical findings of their advantageous effects on the absorption profile of lipophiles following concurrent administration with lipids and postprandial changes in physicochemical and physiological state in the GIT (8,9). This approach has also been extended to include hydrophilic drugs for tweaking the solubilization effect in the GIT for achieving pharmaceutical benefits (10,11,12,13).
Lipids play a diverse role in the cellular and developmental functions of the body as well as a physiological role in the functioning of cells. They are ubiquitously present in the entire body and form a part of normal diet in human population (14). Lipids have also been known to play a diverse role in the absorption of medications from the GIT. As it is known, in most of the cases, there are wide alterations in postprandial absorption of medications following ingestion of a lipid-rich diet which significantly alters the bioavailability of the formulations (15,16). Marking a correlation as to why these lipids are so important in improving the bioavailability of the molecules is based on the fact that “like dissolves like.” The use of lipids in the formulation of dosage form leads to the involvement of the lymphatic system and activation of enterocyte transport mechanisms at the absorption site. Lipids also make the intestinal environment favorable for solubilization of lipophiles in the media for their transport through the cellular linings for systemic availability. Factors that are known to govern this increased solubilization power due to lipid moieties and superior dissolution character are related to enhancement in the capacity of emulsification and rate of dispersion. Various lipid-based drug delivery systems used include oil emulsions (17), suspensions, self-micro, nano-emulsifying systems (18,19), and lipid complexes and may also include liposomes to some extent. The use of liquid lipids such as triglycerides (long or medium chain), mixed surfactant systems (mixed mono- and diglycerides) or hydrophilic solvents (20), solid lipids (such as glyceryl monostearate), etc. are employed in formulation. A detailed discussion on the physiological, biopharmaceutical, and formulation aspects for these systems is covered in this manuscript which gives important directions for maximizing therapeutic benefits of orally administered drugs.
GASTROINTESTINAL LIPID PROCESSING AND DRUG SOLUBILIZATION
The importance of a lipid-based system for oral delivery lies in the fact that the formulation will have access to the lymphatics that has extensive drainage throughout the body, as an absorption pathway, and will lead to avoidance of first-pass effect of the portal system and subsequent increase in bioavailability of formulation. Designing a lipid-based system requires important considerations in choosing the right combination of lipids, surfactant/solubilizers, and form of the drug (18). An ideal drug candidate for a lipid-based system will be the one with high lipophilicity (log P > 5) and having adequate solubility in triglycerides (21). Once a pharmaceutical formulation with the requisite characteristic is designed, it can be presented in either solid or liquid dosage form for administration orally. The foremost step after the administration of a drug orally for it to be absorbed systemically is their disintegration and dissolution in the GI milieu. For a poorly water-soluble drug candidate, the dissolution rate is a rate-limiting step. An important aspect is thus the rate and extent of absorption of drug molecules across the GIT that is determined by a balance between the solubility of the drug and its permeation through the epithelial linings in the GIT. The lipid-based system will exert an added effect of presenting the drug at the molecular level, with enhanced dissolution rate, decreased metabolism, decreased efflux by transporters, prolongation of residence time in the GIT, increased permeation across the intestinal wall, and possibility of lymphatic transport for systemic absorption. A brief description of the fate of lipids is given herein. Following oral ingestion of a triglyceride-rich lipid formulation along with exogenously administered dietary triglycerides, it gets acted upon by gastric lipase enzyme in the GIT, and along with churning action in the stomach, it gets converted to an emulsion form. This emulsion form consists of molecular dispersion of fatty acid with the drug in dissolved state in its core. Upon reaching the small intestine, the conversion of triglycerides occurs to the end product monoglycerides and free fatty acid through a series of conversion processes under the effect of pancreatic lipase and co-lipase enzymes (22). The presence of this digested form of lipids stimulates secretion from the gallbladder which leads to the formation of micellar structures and vesicles of formulation and lipid fractions such as chylomicrons (23). The absorption of lipid fractions and solubilized drug is then governed by the equilibrium between the dissociated micelles in the intermicellar phase and micellar phase (24,25,26). Thus, the use of exogenous lipids enhances the solubilization power of formulation in the intestine which is beneficial for its passage to the lymphatics and avoidance of first-pass metabolism.
Understanding how the lipids in formulation and those present in food leads to changes in the nature of GI fluids and subsequent solubilization capacity of the drug in milieu is important. The solubilization of the drug that occurs is dependent on the intermolecular forces that are present among solute molecules when in solid state and is also determined by the extent of solute-solvent interaction when present in solution state in media. The three factors that govern this solubilization power are intrinsic solubility of the drug in aqueous media, presence of endogenous solubilizing component (present in food), and amount of exogenous solubilizing component (present in formulation, e.g., surfactant, co-solvents, etc.). The presence of this solubilizing components, i.e., lipids, in itself leads to the stimulation of physiological feedback process of the release of bile salts in the media which further aids in the solubilization process. Thus, it is of importance to also understand the role of lipids as excipients in the formulation for favoring these physiological changes of enhancing the solubilization potential of the drug in GI fluid and enhancing the bioavailability of formulation after postprandial administration. Further, an important consideration herein is the selection of a solubilizing species that impacts the drug solubilization capacity in the media. Generally, long chain fatty acids increase the solubilization capacity to a higher extent than short or medium chain ones. A detailed explanation of this relationship is provided elsewhere in the manuscript.
PHYSIOLOGICAL ASPECTS
The influence of the alimentary canal and related digestive processes on lipid formulations projects opportunities and auxiliary challenges that will thrust our research to decipher the process of the gastrointestinal tract for lipid excipients. For example, certain lipids are well known for restraining drug metabolism, efflux through p-glycoprotein inhibition, and intensifying transportation through lymphatic systems by reducing drug clearance for the hydrophobic drug (27). There are several physiological factors that affect the absorption of lipid-based systems from the intestine which finally alters the bioavailability of the active pharmaceutical ingredients.
Intestinal Lymphatic Drug Transport and Its Importance
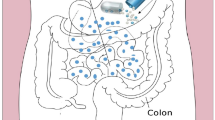
Extensive drainage network of the lymphatic system plays a key role to transport drugs to the systemic circulation. Specific disease-targeted delivery and avoidance of the first-pass metabolism are key benefits of lymphatic systems (28). Majority of the drug that has been administered orally goes into systemic circulation via the portal vein (29). However, the role of lipoproteins for the transport of exogenous compounds through the intestinal lymph can suggest necessities of lipid administration for better transport and lipoprotein formation (19). Accordingly, lymphatic transport can be utilized for drug delivery to the systemic circulation.
Ways to Deliver Drug to Lymphatic Systems Through the Intestinal Route
Lymphatic delivery of drugs primarily occurs in three ways (Fig. 1). First, the porous wall gets formed due to single layers and nonfenestrated endothelial cell. Highly gapped and overlapped structure of these cells allows the macromolecule to transport through the porous wall by opening their paracellular route (30,31). Second, Peyer’s patch contributes to the transport of the drug to lymphatic vessels. Aggregated or isolated lymphoid follicles incorporated in the structure of Payer’s patch provide an entry point to the drugs for lymphatic delivery (32,33). Third, the intestinal walls act as a primary route for lymphatic transport. Intestinal wall transport occurs mainly through paracellular gap, cytochrome P450 reticence, P-glycoprotein gap, and transcellular absorption. Chylomicrons play a crucial role for the transport of drugs through the intestinal walls (8).

Lymphatic delivery of drugs
The Barrier for Transport of Lipids from the Intestine Through the Lymphatic Systems
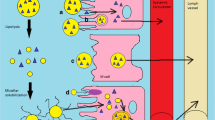
Lipid transport through the intestine is limited to unstirred water layer and brush border membranes. Generally, the transport of medium and short chain fatty acids containing lipids is limited to brush border membranes, whereas the unstirred water layer before brush border plays an important role in the transport of long chain fatty acid (34). The transport of lipids from the intestinal route can be elaborated by unstirred water layer model (Fig. 2).

Transport barriers for oral lipid formulation across GIT
Unstirred Water Layer Model
An unstirred lipid-water layer acts as a barrier for the lipid to be absorbed, as the solubility of the lipid in aqueous media is very low. Unstirred water present just before the apical membrane separates the enterocyte brush border and bulk fluid phase of the lumen (35). Higher Km value for active transport and lower value of permeability for passive transport through the intestine largely depend on the unstirred water layer (36). An acidic microclimate abutting to brush border of the lumen forms due to Na+/H+ and Cl−/HCO3 exchange near the brush border (37). To gain access through the brush border of the lumen, the solute material must cross the unstirred water layer.
Enhancing Lymphatic Drug Transport
The transport of drug through the intestines to lymphatic systems relies on digestion and absorption of lipid (8). Chylomicron production should be encouraged for lipid digestion. There are two specific ways to increase the uptake of the drug to lymphatic systems, namely, formulation approach and prodrug approach (38). In the prodrug approach, the physicochemical properties of the drug are altered with the attachment of the functional group so that it can be metabolized easily via lymphatic systems (39). Here, we focus only on formulation approaches to bypass the intestinal barrier. A small group of the lipid-based formulation can be put to use for favorable uptake by lymphatic systems.
Role of P-Glycoprotein and Multidrug-Resistant Protein and CYP3A
P-glycoprotein (P-gp) was first characterized as an ATP-dependent pump which is responsible for the efflux of anticancer agents and is the product of MDR1 (multidrug resistance gene) found in humans (40). It is a polarized pump which is located near the brush border of the intestines, is energy dependent, and thus works against concentration gradient. The numbers of tissues including the liver, rectum, kidney, bladder, lungs, spleen, pancreas, blood-brain barrier cells, adrenals with colon, esophagus, stomach, and jejunum are the sites where P-gp is found and gets involved in orally delivered drug (41,42). Overlap in specificities for particular substrates demonstrated the role of CYP3A along with P-gp. CYP3A is an enzyme responsible for phase 1 drug metabolizing activity. Fifty to 70% of currently administered drugs are likely to get metabolized through CYP3A enzymes (43,44). P-gp’s estimated role to alter the characteristic of the plasma membrane and the role of the metabolizing activity of CYP3A in the endoplasmic reticulum suggest a spatial relationship between P-gp and CYP3A. Moreover, P-gp can act as an inhibitor or inducer for CYP3A, as the concentration of the administered drug increases inside the endoplasmic reticulum and it may activate CYP3A for metabolism.
Nonionic surfactants like Nonidet P-40, Tween 80, and Triton X-100 reverse the effect of P-gp by inhibiting drug binding to P-gp at a very low concentrations (45). An increase in permeability to basolateral from apical is shown by Pluronic P85 which inhibits P-gp efflux systems (46). Benzyl alcohol anesthetics also decrease activity of P-gp by membrane fluidization (47). Some common P-gp inhibitors which show pharmacological effects as well are amiodarone, felodipine, omeprazole, diltiazem, lansoprazole, cyclosporine, erythromycin, clarithromycin, colchicines, etc. Dietary lipid in feed could modulate levels of cytochrome P450s. Levels of pulmonary cytochrome P450s were reduced significantly in Sprague-Dawley rats that were fed 20% of corn oil (48). Grapefruit juice is a potential inhibitor of CYP3A and also acts as an activator of P-gp-mediated drug efflux transporter (49). These results clearly show the impact of P-gp and CYP3A on the bioavailability of different drug molecules. Inhibition or activation of one of them can be useful for rational formulation designing and increasing bioavailability by higher drug accumulation to selected organs or tissues.
Biopharmaceutical Aspects
Bioavailability enhancement of newer drug substances is often accomplished by the incorporation of lipids in drug delivery systems (50). While the empirical solubility-permeability-based biopharmaceutics classification system (BCS) is often utilized in the selection of appropriate drug substances for the development of such lipidic carrier systems, the use of developability classification system (DCS) in conjunction with BCS provides a more strategic approach for the development of such drug candidates into suitable drug products (51). Recent trends in newer chemical entities (NCE) indicate favoring compounds belonging to BCS class II (good permeability and poor solubility) and class IV (poor solubility and permeability). These newer molecules exhibit superior potencies and selectivity with regard to drug target sites than their predecessors but present more challenges to oral delivery. Their inherent solubility and permeability properties translate to their poor bioavailability and variable pharmacokinetics leading to suboptimal efficacies in humans with subsequent failures in phase III clinical trials (51,52).
Strategies for bioavailability improvement of such compounds would traditionally include solid amorphous dispersions, drug substance salt formation, and particle size reduction or use of excipients like lipids for improvement of dissolution rates with increase in apparent solubility (53,54). Reports suggest the use of lipid excipients with surfactants as suitable formulation strategy for addressing solubility and permeability issues with the advantage of sustaining the drug in solution state both prior to and after administration (55,56). In practice, lipid formulations arise from the mixture of five classes of excipients involving pure triglyceride oils, mixed glycerides, lipophilic surfactants (HLB < 12), hydrophilic surfactants (HLB > 12), and water-soluble co-solvents (54,55). Pouton and subsequently Savla et al. described the use of these mixture blends into the development of suitable lipid-based formulations with the variations in excipient content and hydrophilicity along with relative abilities of keeping the drug in solubilized state during the transit through the gastrointestinal tract (18,51). The transit in solubilized form is considered essential based on the fact that poor dissolution of the precipitated drug in the GI tract would result in variable absorption, bioavailability, and pharmacokinetics leading to suboptimal efficacy (6,55,57). The various biopharmaceutical considerations for designing suitable lipidic drug carriers for the optimal oral delivery of the newer drugs are mentioned below.
Designing Lipid Formulations
The lipid-based drug delivery systems (LBDDS) include various oil- and lipid-based preparations like oil solutions, emulsions, dispersions, micelles, self-emulsifying drug delivery systems (SEDDS), and self-microemulsifying drug delivery systems (SMEDDS) (56). The LBDDS present various challenges to the formulation scientist with respect to their in vivo drug absorption after the successful oral delivery of the therapeutic agents (6,18). Important considerations while designing such formulations to ensure the continued solubility of the drug in the carrier system would include the physicochemical properties of the drug, the thermodynamic parameters of the formulation along with the gastrointestinal physiological parameters which such LBDDS shall encounter during its transit through the GI tract (58,59). Further, the solubilized drug encapsulated in such formulation matrices is subjected to various drug release moderations including the change in the solubility from the solution state in matrix to the emulsion state when coming in contact with the GI fluids and subsequent formation of mixed micellar systems after the digestion of the lipid skeleton (18,51). The lipid formulation classification system based on the BCS and DCS classification systems classifies lipidic formulation into four types, with each having distinct formulation characteristics in terms of the use of excipients, the compounds which may be incorporated into them, and their fate on the GI fluid interactions (6,18,51). The biocompatible type I lipid formulations typically include the usage of oils to generate mixed micellar forms in contact with the digestive fluids and would typically be used for compounds such as vitamin D analogs and retinoids whose drug load can be entirely solubilized in the oil matrix. The type II formulations which include the usage of lipid mixtures with water-insoluble surfactants tend to form formulations having self-emulsifying properties (51). Water-soluble surfactants and co-solvent usage are the highlights of type III lipid formulations, which also possess aqueous environment-initiated self-emulsifying properties (59,60). Based on droplet size of the dispersion formed, they are classified into SEDDS (droplet size > 200 nm) or SMEDDS (droplet size < 200 nm) (57). The last type of the lipid-based formulation includes the usage of water-soluble surfactants and co-solvents which are intended to form fine dispersions in contact with the aqueous environment and transcending to rapid drug release with absorption (59). However, care should be taken while considering the precipitation potential of the poorly water-soluble compounds into the GI tract on initial contact with GI fluid (40,61).
Correlation of the Physicochemical Properties of the LBDDS with the Choice of Excipients and Lipolysis
The choice of suitable excipients for the formulation of the drug molecule into the appropriate LBDDS would encompass various physical, chemical, and biological considerations in conjunction with their safety-associated toxicological evaluations (62,63). The physical considerations would include solubilization potential (both in the matrix and on exposure to external fluids), solvent capacity, droplet size, dispersion characteristics, and hydrophobicity of the active ingredient (61,64). The chemical characteristics would include the stability of the formulation on exposure to various external environmental factors, the excipient drug compatibility, and the purity and the level of the degradants on exposure to the GI fluids (20,65,66). The biological parameters would involve digestibility of the lipidic excipients, interactions with the GI digestion and absorption modulators, and interactions with efflux transporters and metabolism regulators such as P-glycoprotein and cytochrome among others (60). Further, the usage of excipients also necessitates the usage of excipients at levels which have low toxic potential with major focus on the safety and efficacy in the patients (63).
The use of oils including the long chain triglycerides (LCT) and medium chain triglycerides (MCT) can improve the solubilization and dissolution profile of the lipophilic drugs. This is afforded by the generation of the micellar components encompassing the active ingredient after lipid digestion by the GI fluids (58). The degree of solubility by lipolysis mediated by bile components is dependent on the physical properties of the drug such as log P, molecular weight of the drugs, and intrinsic permeability properties (67). Further, this natural lipolysis process enables the uptake of highly hydrophobic drugs like halofantrine (log P > 6) via the lymphatic system of absorption (57).
However, the rate-limiting factor to oral bioavailability while using lipids as oral drug delivery excipients is the requirement of digestion prior to absorption (65). The lipid digestion of the triglycerides results in conversion to corresponding mixed glyceride components (mono- and diglycerides, free fatty acids) (18). However, as compared to triglycerides, the use of mixed glyceride systems has shown improved solvent capacity profiles and better bioavailability associated with better miscibility in GI fluid for low solubility drugs as evidenced in the case of cyclosporine (54,59,60). The substitution of corn oil (Sandimmune®) as lipid component with mono- and diglycerides of corn oil (Neoral®) resulted in better clinical efficacy of cyclosporine capsules with improved pharmacokinetic parameters (59).
The self-emulsifying characteristics in the LBDDS are commonly imparted by the usage of surfactants which tend to enhance the drug solubility along with minimization of the formulation dependence on the GI digestion processes (20,57). The toxicological concerns associated with ionic surfactants have led to the formulators having restricted use of surfactants to nonionic class in the LBDDS (68,69). The usage of this drug solubility enhancement parameter can be evidenced with the usage of the surfactants in marketed formulations of the antiretroviral compounds (68). While vitamin E and TPGS have been used as both solubilizer and permeation enhancers in amprenavir capsules and tipranavir oral solution, polyoxyl 35 castor oil has been used in ritonavir and ritonavir/lopinavir, while tipranavir capsules use polyoxyl 35 castor oil as the surfactant. This surfactant has shown the added potential of modulation of the bioavailability deregulator P-gp activity on the apical membrane of the intestine thereby improving absorption of lipophilic drugs (70). The usage of co-solvents like propylene glycol and ethanol allows improvement in solubilization profile and dispersion rate of lipid formulations (71). However, their usage is often associated with the preferential rapid portioning into the aqueous phase after exposure to GI fluids resulting in the chances of drug precipitation (72). Thus, the work of the formulator would include the choice of such appropriate excipients which shall help in maintaining the high drug loads in the solubilized form in the matrix, on exposure to the GI fluids besides aiding in the biological processes such as absorption and downregulation of the drug efflux pumps (73,74).
Hydrophilicity, Melting Point of Drugs, and Potential of Precipitation
The potential challenges in the development of the marketable LBDDS as against the conventional oral dosage forms include the choice of excipients, maintenance of drug solubility, in vivo stability and prevention of drug precipitation, along with their scalability to the production scale (71,74). The choice of formulation excipients for the desired characteristics would include a balance between the solvent capacity of the formulation to deliver the required dose and the resultant fate of these systems on exposure to the variable environmental conditions of the GI tract (18,51,65). Similar to formulation development approaches followed in conventional dosage forms, the physicochemical properties like log P and melting point of the active ingredients play an important role in formulating the most promising drug products (68). Reports suggest that log P values equivalent to 2 and those with high melting points are poor candidates for formulation into lipid oral carriers which may be preferentially formulated by size reduction and amorphous formulations (58). Such drug substances when formulated as lipid carriers may partition and permeate into the lipid bilayers freely but have higher chances of drug precipitation in the lumen (18,59,60). In such cases, the choice of the excipients plays a major role while deciding on the type of the formulation to be formulated to ensure that the required dose of the drug may be made bioavailable with lower dose of the drug (74).
Further, the solubility of the drug molecules measured as log P plays a major role while deciding the formulation type and the fate of the LBDDS in the GI tract. Poorly water-soluble drugs (log P > 2) are further subdivided in two classes: brick dust (nonlipophilic hydrophobic) and grease balls (lipophilic hydrophobic). LBDDS formulation strategies for “brick dust” compounds would include the use of the co-solvents and surfactants to counter lower solubility afforded by their tight crystal lattices and higher melting points. The compounds belonging to the second class need solubilization using oils (51,57).
While highly lipophilic drugs (log P > 5) are the most suitable agents for inclusion into the lipid systems, the incorporation of the drugs having moderate lipophilic properties (log P 2–5) needs further evaluation of other parameters like aqueous and lipid-based nonaqueous solubility, melting point, and chemical stability (52). The solubility profiles of the drugs in both environments play a major role in the development of formulations as they are correlated with the preferential partitioning characteristics of the drugs in the matrix, stability of the formulation in vivo in GI fluids, prevention of intermittent precipitation of the drug in the GI tract along with the potential of interactions with surface receptors associated with the absorption in the human system (18,67). Further reports suggest that drugs exhibiting physicochemical characters like low aqueous solubility, high hydrophobicity and high solubility in the lipids are to be used in the preparation of the formulations (75).
Depending on the potency of the drug, the drug load, and the solvent capacity, one needs to ascertain the optimal characteristics of the formulation like dissolution, and absorption characteristics are preserved for the efficacy of the drug product (40). Drugs with low potency are preferentially incorporated into oil-based systems only. Reports suggest that surfactant and co-solvent-based lipid systems undergo faster gastric emptying than the pure triglyceride systems. These emulsion-based systems exhibit rapid absorption rates and better bioavailability than the conventional forms (18).
Further, the use of the co-solvents for the solubilization of the hydrophobic drugs results in the loss of the solvent capacity of the formulation making them prone to precipitation on exposure to dilution in GI fluids (20,75). In such cases, the hydrophobicity of the drug becomes the critical factor determining the degree of reduction of the solvent capacity of the drug and its subsequent precipitation in the lumen (74).
Chemical Structure and Molecular Weight of the Drugs
The physicochemical properties like the chemical structure and molecular weight of the active ingredients play an important role for deciding upon the formulation strategies (56). One approach to find the suitability of the newer drug candidates for incorporation into the LBDDS would be to draw analogies between their chemical structure and the ones that have existed in the domain of these delivery systems (18). The multidimensional online tools like ChemMine among others may be used for such predictions (using various 2D similarity indices) to predict the probability of the molecule’s stability in the formulation and upon exposure to the external in vivo gastric environment (51,68). There are various 2D similarity and distance indices (Manhattan distance, Euclidean distance, cosine coefficient, Dice coefficient, Tanimoto coefficient, Soergel distance, substructure similarity, and superstructure similarity) which help in the quantitative molecular fingerprinting-based correlation of the newer molecular structures of new chemical entities with the molecules belonging to the classes having definite molecular patterns (76). Usage of ChemMine and other similar cheminformatics software enables the segregation of the molecules into distinct classes suitable for the development of the lipidic systems (63). This segregation of the molecules into the various classes of chemical structure based on their similarity indices would not only enable the formulation scientist to have an empirical idea about the similarity of the newer molecules with various classes of compounds previously encapsulated in lipid systems (which include vitamin D analogs, retinoids, and protease inhibitors) but also present them with suitable insights about the various LBDDS formulation strategies. The structural similarity coefficient (Tanimoto values) nearer to 1 with any of the classes of the drugs may be predictive of the better chances of the development of oral lipid formulation (20).
Molecular weight as a biopharmaceutical parameter plays an important role in deciding upon the choice of the components for the lipid-based systems (66). Although a wide molecular range of drugs (valproic acid 144 Da to cyclosporine 1202.64 Da) have been incorporated in the LBDDS marketed formulations, active ingredients with molecular weight higher than 500 Da are generally preferred for these formulations (67). These high molecular weight drugs would include protease inhibitors, cyclosporine A, dutasteride, nintedanib, and sirolimus among others (51,68).
Significance of the Particle Size of the Lipid-Based Systems
The emulsion droplet size plays a very important role in the future fate of the formulations. Reports suggest that the absorption rates of the drug cyclosporin A were higher when administered using emulsion with smaller droplet size (from the marketed formulations—Neoral and Sandimmune) (60). The impact of the finer particle size may be attributed to their higher susceptibility to lipolysis and solubilization by bile components to form the mixed micelles which holds true for the systems having only oil components (61). The incorporation of the co-solvents and the surfactants in the emulsion systems reduces their susceptibility to lipolysis (63). In such lipid-based forms, the particle size generated during the initial emulsification process becomes the limiting factor for the absorption and bioavailability of these formulations (64). Generally, it is assumed that the finer the particle size, the better shall be the lipolysis-mediated solubilization of the drug containing formulation and the greater the bioavailability (70).
FORMULATION ASPECTS
Lipophilic drugs in lipid-based oral solid dosage forms often invite the problem of low bioavailability. Many factors are accountable for this reason, but poor absorption due to slow and/or inadequate drug dissolution in the gastrointestinal tract lumen is the main culprit specifically. The enhancement in drug solubilization rate and/or extent into aqueous intestinal fluids may improve the bioavailability in this case. The lipophilic drug must dissolve in the liquid vehicle to accomplish a formulation, and the drug may get precipitated as fine suspension or form a liquid dispersion after dilution of the liquid vehicle surrounding the physiological fluid of the gastrointestinal tract. The precipitation risk of a drug may be minimized by the selection of optimum liquid vehicle composition. Different varieties of vehicles can be utilized to formulate a lipidic carrier and administered as oral solid dosage form like oral liquids, soft gelatin capsules, hard gelatin capsules, and tablets (77).
The use of formulations made up of natural lipids along with surfactants, co-surfactants, and co-solvents has increased in the past few years. The two main reasons as listed are the driving force to formulate more complex systems. First, water-insoluble drugs have very low solubility in triglyceride lipids, thus resulting into limited drug loading capacity of oral solutions. Second, bioavailability gets enhanced after self-emulsification in contact with gastrointestinal fluids to provide very fine particle size in case of highly dispersed formulations. It is not of much significance whether lipidic formulation is introduced in solid or liquid form; indeed, the aim should be to determine whether drug precipitation is prevented or not after formulation comes in contact with the gastrointestinal environment (78).
Selection of a Suitable Drug Candidate
BCS II drugs have less absorption due to poor aqueous solubility and reduced dissolution rate. However, these drugs present no issue in their permeability capacity across the intestinal membrane. Lipid-based oral formulations should be formulated when current formulation strategies like size reduction, salt form, surfactant addition, solid dispersion, etc. have failed. Many water-insoluble drugs are hydrophobic but not sufficiently lipophilic enough to be the ideal candidate for lipid-based oral formulation (79). There are mainly three criteria which need to be fulfilled for being selected a drug candidate in this system. First, the drug should be highly lipophilic, i.e., log P value greater than 5. Second, there should be a minimum solubility of 50 mg/g of long chain triglyceride lipids, and third, there should be significant differences in strong, positive food effect in case of co-administration of the drug with a fatty meal (20).
Excipient Selection and Classification
Lipids can be classified into categories according to Fig. 3 based upon their chemical composition. Table I depicts the percentage distribution of fatty acids in soya lecithin phospholipids (28,77). As drug absorption is influenced by lipids to a large extent, it is necessary to know the characteristics of lipids for lipid-based oral formulations (80). Solvent miscibility, solvent capacity, self-dispersibility of the formulation, digestibility and fate of digested products, physical form at room temperature, capsule shell compatibility, purity, chemical stability, irritancy, and toxicity are the main factors which govern the selection of lipid-based excipients in this type of formulation (17,80). The lists of excipients for these formulations are given in detail and different fatty acids are classified according to Table II. LCT, MCT, surfactants, and co-solvents are selected very often. Many lipids have hydrophobic and hydrophilic portion in their structure which ultimately decides the fate of the formulation in the gastrointestinal tract. Toxicity is an important issue which directly affects the selection of the surfactant. Biological membranes may be penetrated and fluidized by water-insoluble surfactants, and membrane components may be solubilized by water-soluble surfactants. Toxicity of cationic surfactant on biological membrane is more than anionic and nonionic surfactants. Several marketed products have been successfully formulated using nonionic surfactants (53). As nonionic surfactants have LD50 values of 50 and 5 g/kg for the oral and IV routes, respectively, levels up to 1 g surfactant can be easily tolerated in oral formulations (81,82). The Inactive Ingredients Guide (IIG) from the USFDA’s Center for Drug Evaluation and Research database comes handy for determining the maximum amount of excipient that can be given by a particular route of administration for a dosage form (61). Table III describes the excipients used in commercial lipid-based oral formulations (83).

Lipid classification
Regulatory Status of Lipid Excipients
Traditionally, excipients have been considered to be pharmacologically inert substances having no interactions with the active pharmaceutical ingredient (84). However, with the advent of novel pharmaceutical excipients, the probability of interactions with the drug substance (enhancement or decrement of activity), container closure system, and various biological systems have increased quite a few folds. Such novel excipients may be possessing certain toxicological and irritation potentials which may affect the safety of the human patients (84,85). The United States drug regulatory agency, Food and Drug Administration (US-FDA), has published a list of safe excipients in the Code of Federal Regulations (CFR) which has sufficient clinical evidence of being generally regarded as safe (GRAS) excipients (84). Further, as mentioned above, the agency maintains a guiding list of excipients under the name of IIG which has been previously used in approved marketed drug products and has substantial toxicological data (86). This list offers qualitative (pharmacopoeial and nonpharmacopoeial approved agents) and quantitative (maximal permissible dose) guidance to the usage of the various classes of inactive ingredients based on the route of administration or the dosage form for each excipient (76,86). The list provides a comprehensive account to aid in the development of the drug product intended to be administered through various routes (86,87). The review of the usage of the excipients in newer drug development process depends entirely on whether it has been previously used in any approved formulation through the specified route of administration. In such cases, usage of the inactive ingredient in the new drug formulations is permitted at a potency equivalent to that used in the approved safe formulation for use in humans, provided the excipient is used in the same physicochemical state (salt, polymorph, hydrophobic-hydrophilic nature, etc.) and given through the same route of administration (88). However, in case of newer ingredients like modified lipids, oils, etc., the sponsor needs to ascertain the preclinical and clinical safety of the usage of the same prior to usage in any drug product. The studies to be performed have been illustrated in great detail by the United States Pharmacopoeia under chapter <1074> Excipient Biological Safety Evaluation Guidelines (guidance on conducting a safety assessment of a novel excipient) and by Center for Drug Evaluation and Research, Center for Biologics Evaluation and Research (CDER, CBER-USFDA) under the Guidance for Industry: Nonclinical studies for the safety evaluation of pharmaceutical excipients in conjunction with the International Conference on Harmonization safety guideline 7A (ICH S7A) titled Safety Pharmacology studies for Human Pharmaceuticals (89). The USFDA guidance document gives a detailed overview of the different requirements of toxicological evaluation that may be necessary for deeming the inactive ingredient to be safe for usage in any human prescription medicines along with the various possible testing strategies that the inactive ingredient manufacturer needs to ascertain based on the duration of the usage of the drug product (short term, intermediate, and long term). This guidance details the need of scientific toxicological study-based risk assessment of usage of newer excipients in human drug formulations along with the establishment of maximal permissible safety limits (90,91). It is to be noted that in many cases the presence of clinical safety data of similar class of excipients may suffice for the preclinical studies. Further, usage of the excipient for drug delivery through a different route of administration than that present in the IIG database may not necessitate the need of carrying all the toxicological evaluations (84,85,88,89).
In case of the lipid-based dosage forms, the abovementioned quality and safety issues would be of primary concern prior to its approval for usage into drug formulations. It is to be noted that the USFDA does not necessitate the individual safety evaluation of the excipients (90). Scientifically, this is a more appropriate and regulatory approach as inactive ingredients as an integral part of formulations may behave in a different pattern with regard to safety and efficacy concerns as against the neat excipients (84,89). This approach is of particular importance in lipid- and oil-based systems due to their varied physicochemical properties and significant potential of interactions with other components of the drug formulation (85). The typical studies for acceptability of newer lipids/oils to the drug regulators would include drug stability studies, toxicological evaluations, immunological reaction studies, effects on the drug release, and their mechanism along with evaluation of the effect of the biopharmaceutical factors and safety assessment (85,88). In addition, the mechanistic molecular evaluation and assessment of the possible interactions of the newer lipids with the other excipients, drug substance, and the physiological environment becomes an important parameter when the oral route of administration is intended (84,91).
Further, lipids may be classified as follows:
-
Vegetable oils/triglycerides
Vegetable oils are digested and absorbed completely so they are safe to use. LCT, MCT, and short chain triglycerides (SCT) are the main triglycerides in which the ester group concentration decides solvent capacity. The solvent capacity of MCT is higher than that of LCT and is less prone to oxidation. Oils from different vegetable sources have different proportions of each fat. D-α-tocopheryl PEG 1000 succinate (TPGS) is derived from vegetable tocopherols which is water soluble and an absorption enhancer and is responsible for increment in bioavailability for many water-insoluble drugs (28). Table IV enlists the name of vegetable oils and animal oils/fats included in different pharmacopoeias, respectively.
-
Mixed glycerides and polar oils
When vegetable oil is partially hydrogenated, monoglycerides, diglycerides, and triglycerides like mixed species are produced. Span 85, oleic acid, etc. are used in commercial products (92). They are added in type II or III lipid formulation system. High lipid loads are necessary for drug solubilization in formulation containing MCT, and solubilization is reduced in response to reduction in mass of exogeneous lipids; moreover, surfactant digestion may lead to a decrease in solubilization capacity, whereas effective drug solubilization may take place even at low lipid concentrations. Due to luminal digestion of lipids, drug precipitation may occur in MCT type of lipid-based formulations (78).
-
Water-insoluble surfactants
Lipid excipients having intermediate hydrophilic-lipophilic balance (HLB of 8–12) are adsorbed at the oil-water interface. Their water solubility is dependent on the degree of ethoxylation. If shear is applied, then they produce emulsion and may be termed as dispersible. They do not self-emulsify due to lack of hydrophilicity. Oleate esters such as polyoxyethylene sorbitan trioleate (Tween 85) and polyoxyethylene glyceryl trioleate (Tagot-TO) are examples of this class of surfactants (93,94). These excipients are used in type III formulation.
-
Water-soluble surfactant
They are used in self-emulsifying drug delivery (SEDDS or SMEDDS) having an HLB value greater than 12 and form micelles in water above their critical micellar concentration (95,96). They are produced by mixing polyethylene glycols (PEG) with hydrolyzed vegetable oils. Cremophor RH 40 and RH 60 (ethoxylated hydrogenated castor oil) are produced from hydrogenation of vegetable oils. Cremophor inhibits the efflux pump thereby enhancing the absorption of the drug (97). It penetrates into the membrane, gets adsorbed, and interacts with intramolecular domains of efflux pump thereby imposing nonspecific conformational change (98).
-
Co-solvents
Most marketed products use co-solvents like propylene glycol, ethanol, glycerol, and PEG 400 in order to enhance the solubilization process (54). They help in the dispersion system and increase the solvent capacity of formulation (53). However, the incompatibility of solvents with capsule shells, immiscibility of some co-solvents with oils, and precipitation of the drug due to loss of solvent capacity are few disadvantages associated with them (99).
-
Additives
Varieties of lipid-soluble antioxidants such as α-tocopherol, propyl gallate, β-carotene, butylated hydroxyl toluene (BHT), or butylated hydroxyanisole (BHA) can be used to prevent oxidation of the drug (17) and are added as additives in the formulations.
Lipid Formulation Classification
The main aim of the lipid formulation classification system is to simplify the task of the formulator to select the most suitable type of formulation with respect to the physicochemical property of drug. Most of the marketed formulations belong to type III which is further classified into IIIA (predominantly oils) and IIIB (predominantly water soluble). Table V describes the classification of lipid-based oral formulations (78,79).
Key Formulation Parameters
There are few key principles which ultimately govern the choice of formulation component that affects the performance of lipid-based formulations (79).
Solvent Capacity
Dispersible, nondispersible, or self-emulsified molecular-dispersed formulations are preferred irrespective of solid or liquid filled into hard and soft gelatin capsules.
Lipid Polymorphism
Amphiphilic molecules can self-assemble themselves upon dispersion in water and exist in varieties of shapes and structures due to polymorphic phase behavior. Lipids are amphiphilic and play a functional role in biomembranes. They provide basic permeability barrier, structures for fusion and membrane permeation-related phenomenon, and shapes for bilayer organization (100). The lipid polymorphism is very well understood by molecular shape theory. Figure 4 describes the phenomenon of lipid polymorphism, which gives an idea of the geometry of the lipids that gives rise to different structures (101).

Lipid polymorphism
Impurity Profiling
The presence of minute quantities of residual fatty acid or residual solvent is important in lipid formulations. It is also important to determine which type of surfactants is used.
Solid-State Characteristics
Drug dissolution rate and crystallinity are highly influenced by interconversion of one physical form to another which leads to storage stability concerns.
Maintenance of Drug Solubilization on Dispersion and Digestion
After coming in contact with endogenous solubilizing components like bile salts and phospholipids, the formulation must retain the drug in a solubilized state. The risk of drug precipitation is higher in case the excipient is water insoluble or water immiscible to maintain drug solubilization capacity after dilution. The kinetic of drug precipitation is a key factor in all events to support drug absorption. Gastric lipase and co-lipase act at the lipid surface to digest glycerides and other excipients with lipid-based groups. In the presence of endogenous components, the capacity of exogeneous components to solubilize the drug may be increased or decreased. The long chain triglycerides are less prone to loss of solubilization capacity for highly lipophilic drugs. Lipid digestion in vitro models are key tools to determine the effect of digestion on drug solubilization (20).
Lymphatic Transport Stimulation
Long chain lipid-containing formulation may enhance the intrinsic capacity of lymphatic transport, i.e., the amount or dose of the drug that will preferentially go into systemic circulation via the lymph over hepatic portal vein thereby reducing first-pass hepatic metabolism.
Lipid-Based Oral Formulations
A lipid-based formulation triggers the production of chylomicrons, which help in the loading and solubilization of lipid and promote absorption into the lymphatic systems (102). Nanocarriers have distinct features like size and lipophilicity which can be used to target any specific tissue or organ. Here are some of the formulations which can be used for oral delivery of lipid-based formulations to target lymphatic systems. The lipid-based oral formulations may be selected according to formulation objectives. HLB values of excipients, lipophilicity, solvent capacity, dispersion-dissolution characteristics, stability, compatibility, in vitro lipid digestion model, and appropriate animal models are few important checkpoints to design and formulate these formulations and they are classified according to the sections that follow.
Solid Lipid Nanoparticles
Activation of chylomicron production is stimulated by the core of the lipid present in solid lipid nanoparticles. The increase in chylomicron production increases the absorption of the drug through the intestine to lymphatic systems via transcellular mechanism (103). Radioactive isotope containing SLN after administration shows transport by lymphatic systems rather than by systemic circulation. However, the uptake of SLN by lymphatic systems is mainly due to blockage of lymph to blood transport of SLN. So from this study, the lymphatic target of the drug might be possible with the help of SLN formulation which can bypass hepatic metabolism (104). Solid lipid nanoparticles loaded with methotrexate show increase in bioavailability up to 10 times due to lymphatic uptake of methotrexate, hence an increase in systemic circulation (105). Lipid is not the only affected parameter for bioavailability of the drug through SLN but the charge of the lipid also affect a lot as the SLN formulated from positively charged lipid gives improvement in bioavailability of nitrendipine from 3.21- to 5.35-fold compared to negatively charged lipid (106). The oral lymphatic system is a focal point for antiretroviral therapy nowadays. Efavirenz SLN containing long chain triglycerides gives promising results for increased bioavailability. The increase in bioavailability affirms that it bypasses portal circulation, and the spleen has the highest concentration of Efavirenz as part of lymphatic systems (107).
From these examples, we can say that solid lipid nanoparticles can be as useful compared to other systems for increasing bioavailability of the lipophilic drug to lymphatic targeting. Moreover, SLN shows a potential increase in stability of acid labile drug and volatile substance with increase in bioavailability and even used for controlled release of drugs. As the drug gets targeted to lymphatic systems rather than systemic circulation, the toxicity of drugs can be altered with the help of SLNs.
Nanostructured Lipid Carrier
An unsaturated mixture of solid lipid and liquid lipids as oil phase and surfactant containing aqueous phase are used for the preparation of nanostructured lipid carriers (NLCs). Long chain lipids like glyceryl monostearate and Labrafil WL were used to formulate NLCs of tamoxifen to target intestinal lymphatic systems for anticancer efficacy. When confirmed against the MCF-7 cell line for activity, a study suggests a relatively improved action of tamoxifen compared to free drug suspension (108). NLCs of etoposide formulated using soya bean oil and monostearin give promising results for lymphatic targeting by the oral route. A pharmacokinetic study demonstrated an increase in bioavailability up to 3.6 times when compared to drug solution (109). Likewise, NLCs of simvastatin and lovastatin also give prominent results for bioavailability enhancement and lymphatic system targeting. So NLC can be a potential approach to bypass first-pass metabolism and reduce the affinity of the P-gp efflux transporters along with lymphatic targeting.
Emulsions
Two or more immiscible phases mixed together with a third component like an emulsifier or surfactant to form a stable formulation is called emulsion. Types of emulsion can be defined by the nature of the continuous phase and disperse phase. It can be oil in water or water in oil. Penclomedine has a lymphatic transport of only 3% regardless of its higher P value (5.48) and high lipid solubility (175 mg/ml) (110). Due to stronger affinity of penclomedine to blood plasma proteins and red blood cells, it may compete with chylomicrons for lymphatic delivery. When emulsion of penclomedine with soya bean oil is prepared, it increases the partition coefficient of penclomedine; thus, it can prevent binding to plasma protein and increase lymphatic transport. For the effect of systemic blood levels of a drug emulsion, a formulation of ontazolast, a potent LTB4 inhibitor, was tested on rat. Emulsion prepared with 20% soya bean oil and other three lipids showed an increase in bioavailability and lymphatic transport compared to suspension almost 9 times (19). Prolong gastric emptying time and rapid absorption due to concurrent triglyceride transport favored directly the amount of ontazolast transport by lymph.
Self-Micro/Emulsifying Drug Delivery Systems
SEDDS monopolize peerless ability in improving oral bioavailability of highly lipophilic drugs. By definition, self-emulsifying drug delivery systems are isotropic mixtures of oils (natural or synthetic), surfactant (liquid or solid) with one or more hydrophilic solvents with co-solvent or liquid surfactant (111). Soft gelatin capsules or hard gelatin capsules can be used to deliver SEDDS orally and upon administration, due to dilution and gentle agitation of GI fluids, and it forms a very fine emulsion (o/w). SMEDDS of silymarin was formulated to increase its bioavailability and rate of release. SMEDDS were developed using GMO as oil phase due to the highest solubility of silymarin in GMO. The release rate of silymarin increased up to 2.5 times and bioavailability increased 36% higher than the reference capsule formulation (112). SEDDS of halofantrine is formulated using two different lipids 1,3-dioctanoyl-2-linoleyl-sn-glycerol (C8:0-C18:2-C8:0) (MLM) and 1,3-dilinoyl-2-octanoyl-sn-glycerol (C18:2-C8:0-C18:2) (LML) and Cremophor EL as surfactant. An optimized formulation of halofantrine promised a total bioavailability of 74.9% which was higher than any other formulations of halofantrine (113). Likewise, SMEDDS of raloxifene and vinpocetine gave prominent results in increasing bioavailability compared to oral suspension. The increase in bioavailability of raloxifene SMEDDS mainly was due to increased penetration ability through SMEDDS (114). Enhanced lymphatic transport, efficient release profile, and improved penetration are the key factors for increasing bioavailability of vinpocetine (115). These results suggest that S(M)EDDS play a key role for oral delivery of highly lipophilic drug and lymphatic targeting.
Liposomes
Liposomes are nanosized vesicles with an aqueous core surrounded by a biodegradable lipid bilayer structure (116). For poorly water-soluble drugs, liposomal systems were evaluated previously. Proliposomes with negatively charged lipid containing cefotaxime were evaluated for pharmacokinetic study, wherein the bioavailability of cefotaxime increased 2.7 and 2.3 times compared to the aqueous solution and the physical mixture of drug and lipid, respectively (117). This comparative study shows the role of liposomes in increasing bioavailability. Oral delivery of RNA and DNA is challenging even today. Liposomal formulation containing plasmid DNA pRc/CMV HBS encoding the S (small) region of hepatitis B surface antigen (HBsAg) might be helpful to solve this challenge. As liposomes composed of different lipids like phosphatidyl-choline (PC), 1,2-dioleoyl-3-(trimethylammonium propane (DOTAP), dioleoyl phosphatidylethanolamine (DOPE), and distearoyl phosphatidylcholine (DSPC) gave higher entrapment efficiency around 89–93% and showed resistance to displacement against sodium dodecyl sulfate which is anionic surfactant (118). Although liposomes might be used for lymphatic transport, they are not able to withstand bile salt digestion, and to overcome this problem concept of surface, modified liposomes with PEG are introduced. The surface PEG may produce a stearic barrier which will help to inhibit plasma-protein adsorption or opsonization. Steric coating also helps in electrophoretic mobility. Reduced affinity of sterically stabilized liposomes against mononuclear phagocyte systems can avoid its detection for uptake. Improved stability to GI environment was noted to a surface-modified PEG-liposomes containing recombinant human epidermal growth factor (rhEGF). These liposomes are useful carriers for intestinal lymphatic delivery of drugs and area under the curve for concentration increased up to 1.7- and 2.5-fold (119). These results show the effectiveness of liposomes to deliver therapeutic molecule to lymphatic systems.
Oily Liquids
Highly lipophilic drugs are soluble in oils only, e.g., steroids. The high quantity of oil required to dissolve the unit dose of the drug limits the use of this class of drug. Fractionated coconut oil and castor oil were used to formulate oily solution of bupivacaine-free base (120).
Mixed Micelles
They are formed above the critical micelle concentration (CMC) of lipids having more than one molecular species with a disc-like structure similar to a lipid bilayer. Paclitaxel was formulated with mixed micelles of PEG 2000—distearoyl phosphatidylethanolamine (DSPE) and vitamin E TPGS, and increased antitumor activity was obtained (121).
Conversion of Liquid-Lipid Components into Solids
Both liquids and solids can be administered through the oral route, but stability issues in liquids and semisolids are more than solids so liquid-lipid components may be converted into solids and filled into hard gelatin capsules or formulated as tablet. Spray congealing (122), spray drying (123), adsorption onto the solid carrier, melt granulation (124), and supercritical fluid-based method (125) may be utilized for conversion of liquid lipids into solids. Lyophilization is the most promising approach to enhance the stability of a lipid-based drug delivery system which in turn increases the shelf life of the delivery system. The water replacement model and the vitrification model are the two main hypothesis which explains the lyoprotective mechanism. The excess water contained in bilayers of liposomes represents loose state which remains in compacted state during freezing, and packing defects arise in the absence of lyoprotectant during the drying stage which is responsible for the leakiness of the lipid bilayer system. Lyoprotectant steadily substitutes the water molecules and also maintains the head group distance with phospholipids head groups. They also decrease the van der Waals interactions among phospholipid acyl chains in dry state which leads to reduction in Tm (gel to liquid crystalline phase transition temperature) so rehydration of freeze-dried cake takes place in an efficient manner. The gel-to-liquid crystalline phase transition temperature (Tm), encapsulated solute (drug) retention (ESR), and glass transition temperature (Tg) are the prime parameters to evaluate the performance of this process. Thus, a lyoprotective effect can be achieved more efficiently by optimizing the freeze drying protocol which decreases the freezing damage and enhances the morphology of freeze dried material (126).
The limited successful commercialization of lipid-based formulations like self-emulsifying formulations, emulsion, suspension, etc. leads to the development of a solid-state lipid-based drug delivery system in which liquid lipid formulation is absorbed onto an inert solid carrier material. The drug release modulation and control of lipase activity as well as lipid digestion are the key parameters to be optimized. The formulation should have good redispersibility, high lipid loading efficiency, and optimum flowability and mechanical strength for tablet compression. The solid-state lipid-based formulations are classified into SLH (silica-lipid hybrid nanoparticles), dry emulsions, and solid self (nano)-emulsifying drug delivery system. Silicon dioxide, magnesium stearate, polyvinyl alcohol (PVA), etc. are used traditionally as solid carriers. Mesoporous carbon, montmorillonite, layered double hydroxides, and porous starch are novel adsorbents used in this system (127).
CHARACTERIZATION OF LIPID SYSTEMS
Lipid-based oral formulations may be characterized by the tests in the sections that follow.
Physicochemical Study
Crystallization temperature, glass transition, melting point, and determination of solid fat content of the excipient may be examined using differential scanning calorimetry (DSC). Hot stage microscopy can be used to judge the organization of the lipid during heating or cooling, and X-ray diffraction (XRD) can be utilized to determine the crystallinity of a lipid excipient. The fatty acid distribution profiles may be identified by high performance-liquid chromatography (HPLC) and gas chromatography (GC). The saponification value tells the molecular weight of fatty acids. An iodine-based assay gives an indication of saturation of hydrocarbon chains (7).
Dissolution Testing and Dispersion Study
A conventional USP dissolution tester is recommended by the USFDA, but it does not match the in vivo resemblance of lipid-based formulations. Gastric and pancreatic lipases are present in the GIT which alter the emulsification, dispersion, and solubilization capacity in vivo; hence, digestibility of the formulation should be checked. Therefore, to evaluate such effects, dissolution testing in biorelevant media can be useful. Self-emulsifying formulation effectiveness can be judged by dispersion testing, i.e., emulsification capacity and particle size which may be done by visual observation and photon correlation spectroscopy (PCS), respectively (28).
In Vitro Study
In vitro studies can be designed and performed according to preliminary guidelines for formulation development and drug release. These studies also serve the purpose of piloting studies for vivo evaluation for proof of the concept. Lipid digestion models may be utilized to assess the performance of lipid-based oral formulation according to Fig. 5 (78). A model intestinal fluid containing a digestion buffer, phospholipids, and bile salts are added in temperature-controlled (37 °C) vessels in which lipid-based formulations are added with the addition of pancreatic lipase and co-lipase to initiate lipid digestion. Fatty acid (FA) is liberated by digestion of lipids which causes a drop in pH which is computed by a pH electrode thermocouple attached to a pH-stat meter controller and burette, which titrates the liberated FA by addition of NaOH. It quantifies digestion potential indirectly by measuring rate of addition of NaOH. The samples are processed for ultracentrifugation to isolate a poorly dispersed oil phase, well-dispersed aqueous phase, and a precipitated pellet phase. The aqueous phase is quantified for drug amount which does not precipitate thereby giving a prediction of tendency of formulation with respect to in vivo precipitation and its behavior. It gives a mechanistic approach to justify in vivo performance of lipid-based oral formulation. The wide range of physiological processes like delayed gastric emptying, increase in membrane fluidity, stimulation of bile flow and pancreatic juice secretion, promoting lymphatic drug transport, inhibition of efflux pump, etc. enhances the oral absorption of the drug in lipid-based formulation, and the assessment of these effects can be done with the help of in vitro models like in situ perfusion assays, intestinal microsomes, Caco-2 cells, and everted gut sac.

In vitro lipid digestion model
In Vivo Study
Bioavailability and pharmacokinetic profile of drugs can be altered by excipients and other parameters like size which can be estimated by proper in vivo models. Lipid-based oral systems have the capability to altered absorption through intestinal transport for lymphatic targeting so detailed study of absorption pathway is necessary. Insufficient clinical study data and inappropriate animal models are reasons for the weak prediction of in vivo lymphatic targeting.
Small Animal Models
For lymphatic transport estimation, there are numerous methods shown in different literature but preferably it is performed on anesthetized rats. In vivo efficiency of lymphatic transport may be depending on different animal models used for determination.
The Unconscious Rat Model
This model described by Porter et al. is used in the laboratory for research purpose. In this model, intravenous administration can be given by the jugular vein, blood samples can be collected from cannulated carotid artery, intestinal lymph fluid can be collected from mesenteric lymph duct, and for rehydration of fluids, the duodenum was used (128).
Intraduodenal dosing is generally performed for oral dosing of animals. Mesenteric lymph duct was used to collect all the fluid necessary for the calculation of lymphatic drug transport though drug gets absorbed through intestine and goes to portal vein circulation should also be calculated by collecting blood samples. For calculation, we can assume that systemic bioavailability of an orally administered drug directly relies on portal vein absorption (129). Therefore, area under the curve for portal blood concentration in lymph fistulized rats against a parallel group like intravenous administration can be compared for lymph concentration of the drugs. Bioavailability of saquinavir mesylate lipid-based formulation improved almost three times when compared with available marketed formulation of saquinavir hard gelatin capsule. A mesenteric lymph duct cannulated model was used to identify mechanisms related to the increase in bioavailability (130). The same approach was used to elucidate the lymphatic transport of vitamin D3 and chylomicrons’ flow was blocked for better elucidation of lymphatic flow. Results confirm that 75% of vitamin D3 was absorbed through the lymphatic systems (131).
Conscious Restrained Rat Model
The methodology for this model is similar to the unconscious model, but jugular and lymph cannulas are fixed under the skin at the back of the neck of the animal. Further, they are connected to swivel arrangements for sampling and infusion. Then, the animals are placed in a restrainer cage after recovering from anesthesia (132). Rats are orally dosed in this model for intraduodenal blood collection and cannulation can be neglected in this method. Lymph is collected in a harness containing changeable lymph bottles which can be used for drug estimation (133).
Conscious Unrestrained Rat Model
This model was developed for the simultaneous study of both oral and intravenous administration. For lymph fluid collection, mesenteric lymph duct fistulation is performed along with carotid artery cannulation and intraduodenal cannulation for systemic blood collection and overnight rehydration, respectively. A second group has only carotid artery cannulation for systemic blood collection for intravenous administration. For absolute bioavailability assessment, dose administration can be done from the right jugular vein and blood sampling can be done from the left carotid artery. Nonfasted animals are used for surgical procedures as they offer better visualization of mesenteric lymph duct. Lymph and plasma TG levels should return to basal level before dosing so animals are fasted for the recovery period also.
In Vitro-In Vivo Correlation
In vitro lipolysis and ex vivo intestinal permeability prediction model was utilized in the enhancement of the bioavailability of dexamethasone. It had good IVIVC but ex vivo permeation study failed to comply with in vitro lipolysis results. The reason was attributed to low solubility of griseofulvin (5 mg/ml) which was predisposed due to the presence of phospholipids, bile salts, and lipolytic products in the GIT. Therefore, sound knowledge of in vitro and in vivo techniques, implementation, and data treatments is prerequisite for successful IVIVC establishment (131).
Stability Testing
The stability testing of lipid-based oral formulation is in its infancy still. The formulator should check the stability of lipids and drugs separately because lipids are prone to oxidation and to other degradative changes easily. A slower dissolution rate may take place due to cross-linking of peroxides with the gelatin capsule shell thus lowering dissolution rate (134). Lipids are affected by temperature-dependent degradation easily and so accelerated stability testing results at elevated temperatures may not be extrapolated to ambient temperatures easily (27). It is very important in semisolid dosage form which may melt at working temperature. The formulator may utilize interfacial properties to correlate its stability. The Teflon Wilhelmy plate method is used to measure dynamic interfacial tension. Microelectrophoresis and photon correlation spectroscopy are used to measure emulsion droplet charge and droplet size, respectively, as an indicator of stability (135). The thermal stability of lipids is another important aspect to characterize this system. Melting and crystallization characteristics can be determined using DSC, and thermal decomposition profile can be measured by thermogravimetric analysis. Lipid extracted from Rhodococcus opacus PD630 exhibited four stages of thermal decomposition profile which represent decomposition of unsaponifiable matters, saturated and monounsaturated fatty acids, and other degradation products; thus, the thermal stability of lipids can be confirmed by this process (136).
CASE STUDIES OF MARKETED PRODUCTS
The current trends in potential drug candidates from drug discovery research platforms indicate toward an increasing number of hydrophobic drugs seeing the light of the day (52,54). The regulatory approval of these drug substances in the form of lipid-based oral drug delivery platforms has increased significantly over the past few years (more so for the last two decades) (18,51,68). At the current rate, oral lipid carriers account for more than 6% of the total commercially available therapeutic approvals (51,57). Literature review suggests the presence of 27 distinct molecular entities which have been formulated into 36 different lipid formulations (Table VI). While certain formulations have been discontinued from the market, none of discontinuations have been associated with patient safety and efficacy concerns (18,58). While the early lipid-based formulations were primarily composed of oils, the approval of Neoral® saw the advent of the usage of the surfactants and the co-solvents as formulation strategies (18,57,58,59). The soft gelatin capsules have been the choice of these lipid-based systems with more than 25 of the 36 approved formulations being based on the same technology (51,68).
These carrier systems have been used to orally deliver a wide range of therapeutic classes of molecules ranging from vitamin D analogs, retinoids, and antiretroviral drugs to immunosuppressant anticancer agents (60). Ergocalciferol (Drisdol) was the first approved lipidic system in 1941 by the United States Food and Drug Association (USFDA) (6,67). This soya bean oil-based liquid-filled capsule formulation of vitamin D analog is indicated for use in rickets associated with vitamin D resistance and in hypoparathyroidism. However, the use of this liquid formulation is restricted due to potential chances of the propylene glycol toxicity (137).
Another vitamin D analog calcitriol has therapeutic effectiveness in the treatment of osteoporosis, glucocorticoid-induced osteoporosis, secondary hyperparathyroidism, and psoriasis (6,18). However, the limited aqueous solubility of the drug coupled with the thermal, oxygen, and photodegradation properties of the drug has posed challenges to the development of suitable, stable, and therapeutically effective formulations (53,54,138). The use of triglyceride forms of coconut oil in formulating Rocaltrol® soft capsule and Rocaltrol® oral solution has helped to overcome these formulation challenges (138).
Paricalcitol (Zemplar®) is another example of vitamin D analog which has been formulated in the form of soft gelatin capsule containing medium chain triglycerides of coconut and palm oil along with alcohol as the co-solvent. The hydrophobic highly potent drug is indicated for the prevention and treatment of secondary hyperparathyroidism associated with chronic kidney disease. This formulation was developed with the aim of the improvement in the plasma concentrations of the drug after its administration via the oral route (18,51).
The immunosuppressant cyclosporin A was one of the compounds to have been extensively explored on this LBDDS platform (59,61). The drug is known to possess a very narrow therapeutic range while being associated with toxicological clinical implications even with the slightest changes in its dosing (40,59,60,61). Sandimmune® was initially formulated as an olive oil matrixed formulation which was further substituted with corn oil and linoleic acid glyceride. Novartis Pharmaceuticals Inc. further developed another product Neoral® based on corn oil derivatives with surfactants and co-solvents with the aim to improve the pharmacokinetic profile of the drug delivery and having better clinical efficacy (59,60). Certain generic equivalents were further approved by the USFDA (Restasis®, Gengraf®) utilizing the same platform with the aim to reduce the toxicity of the drug with improved pharmacokinetic profiles (139). The formulation of sirolimus (Rapamune®) and topotecan (Hycamtin®) using lipid-based systems offers the advantage of reduced systemic toxicity which is afforded by the usage of the corresponding parenteral formulations (40).
The antiretroviral drugs have been primarily formulated as oil-based systems with addition of surfactants and co-solvents (40,62,63,64). These drugs have relatively high polar surface areas and high molecular weights thereby affording their incorporation into the lipid-based systems (62). The utility of such formulation strategies is highlighted by the fact that the addition of these excipients would help in the solubilization of the drug to greater extent besides affording better permeation rates (69). The excipient vitamin E TPGS is one such excipient used in amprenavir capsules and tipranavir oral solution (63). The use of polyoxyl 35 castor oil as surfactant in the formulation not only improves the solubilization potential but also modulates the drug efflux activity of P-gp on the intestinal apical membrane thereby improving the absorption of the lipophilic drugs and drug combinations like ritonavir, ritonavir/lopinavir, saquinavir, and tipranavir (64). It is to be noted that the P-gp present on the intestinal villi has been reported to limit the extent of the absorption of these drugs through the oral route (71). This P-gp substrate polyoxyl 35 castor oil plays a dual role of improvement in the solubility and modulation of the permeability of these protease drugs thereby improving the bioavailability of these drugs (70).
However, certain formulations have been replaced by newer ones primarily on the basis of affordability of better formulations in place. A reduction in the daily pill burden in patients was highlighted for the discontinuation of saquinavir (Fortovase®—replaced by film-coated tablets) and ritonavir/lopinavir (Kaletra®—replaced by solid dispersion as it further afforded higher drug loading). Amprenavir (Agenerase®) was discontinued due to the availability of a better prodrug, fosamprenavir (18,51,57,63).
CHALLENGES AND FUTURE PERSPECTIVES
In order to optimize and identify the active lead compound, new molecule entity (NME) research very often results into highly lipophilic and water-insoluble compounds. Such NME may possess an excellent efficacy, but it suffers from poor solubilization and dissolution rate. Lipophilicity is central to the outcome of this type of lead, so efforts should be made that the compound should possess not only lipophilicity but also sufficient hydrophilicity and should also be a successful candidate for lipid-based oral formulations. The regulatory guidelines are still in its infancy for lipid-based oral formulations. Further research should be carried out to develop relevant in vitro lipid digestion models and other in vitro and in vivo models which might be able to predict perfect in vitro-in vivo correlation.
References
Prabhu S, Ortega M, Ma C. Novel lipid-based formulations enhancing the in vitro dissolution and permeability characteristics of a poorly water-soluble model drug, piroxicam. Int J Pharm. 2005;301(1–2):209–16.
Lipinski CA. Drug-like properties and the causes of poor solubility and poor permeability. J Pharmacol Toxicol Methods. 2000;44(1):235–49.
Porter CJ, Charman WN. In vitro assessment of oral lipid based formulations. Adv Drug Deliv Rev. 2001;50:S127–S47.
Tiwle R, Giri TK, Tripathi DK, Jain V, Alexander A. An exhaustive review on solubility enhancement for hydrophobic compounds by possible applications of novel techniques. Trends Appl Sci Res. 2012;7(8):596.
Chaudhary A, Nagaich U, Gulati N, Sharma V, Khosa R, Partapur M. Enhancement of solubilization and bioavailability of poorly soluble drugs by physical and chemical modifications: a recent review. J Adv Pharm Educ Res. 2012;2(1):32–67.
Pouton CW. Formulation of poorly water-soluble drugs for oral administration: physicochemical and physiological issues and the lipid formulation classification system. Eur J Pharm Sci. 2006;29(3–4):278–87.
Jannin V, Musakhanian J, Marchaud D. Approaches for the development of solid and semi-solid lipid-based formulations. Adv Drug Deliv Rev. 2008;60(6):734–46.
Charman WN, Porter CJ, Mithani S, Dressman JB. Physicochemical and physiological mechanisms for the effects of food on drug absorption: the role of lipids and pH. J Pharm Sci. 1997;86(3):269–82.
Fleisher D, Li C, Zhou Y, Pao L-H, Karim A. Drug, meal and formulation interactions influencing drug absorption after oral administration. Clin Pharmacokinet. 1999;36(3):233–54.
Sahbaz Y, Williams HD, Nguyen T-H, Saunders J, Ford L, Charman SA, et al. Transformation of poorly water-soluble drugs into lipophilic ionic liquids enhances oral drug exposure from lipid based formulations. Mol Pharm. 2015;12(6):1980–91.
Yin N, Brimble M, Harris P, Wen J. Enhancing the oral bioavailability of peptide drugs by using chemical modification and other approaches. Med Chem. 2014;4:763–9.
Patil S, Vhora I, Amrutiya J, Lalani R, Misra A. Role of nanotechnology in delivery of protein and peptide drugs. Curr Pharm Des. 2015;21(29):4155–73.
New RR, Kirby CJ. Solubilisation of hydrophilic drugs in oily formulations. Adv Drug Deliv Rev. 1997;25(1):59–69.
Shiau Y-F. In: Johnson LR, editor. Lipid digestion and absorption. 2nd ed. New York: Raven; 1986. p. 1527–56.
Wang C-S. Hydrolysis of dietary glycerides and phosphoglycerides: fatty acid and positional specificity of lipases and phospholipases. Fat absorption: CRC Press; 2018. p. 83–118.
Hunt J, Knox M. A relation between the chain length of fatty acids and the slowing of gastric emptying. J Physiol. 1968;194(2):327–36.
Gibson L. Lipid-based excipients for oral drug delivery. Drugs and the Pharmaceutical Sciences. 2007;170:33.
Pouton CW. Lipid formulations for oral administration of drugs: non-emulsifying, self-emulsifying and ‘self-microemulsifying’ drug delivery systems. Eur J Pharm Sci. 2000;11:S93–S8.
Hauss DJ, Fogal SE, Ficorilli JV, Price CA, Roy T, Jayaraj AA, et al. Lipid-based delivery systems for improving the bioavailability and lymphatic transport of a poorly water-soluble LTB4 inhibitor. J Pharm Sci. 1998;87(2):164–9.
Pouton CW, Porter CJ. Formulation of lipid-based delivery systems for oral administration: materials, methods and strategies. Adv Drug Deliv Rev. 2008;60(6):625–37.
Charman W, Stella V. Estimating the maximal potential for intestinal lymphatic transport of lipophilic drug molecules. Int J Pharm. 1986;34(1–2):175–8.
Erlanson-Albertsson C. Pancreatic colipase. Structural and physiological aspects. Biochim Biophys Acta. 1992;1125(1):1–7.
Bosch H, Postema N. Haas GHd, Van Deenen L. On the positional specificity of phospholipase A from pancreas. Biochim Biophys Acta. 1965;98(3):657–9.
Hoffman N. The relationship between uptake in vitro of oleic acid and micellar solubilization. Biochim Biophys Acta Biomembr. 1970;196(2):193–203.
Westergaard H, Dietschy JM. The mechanism whereby bile acid micelles increase the rate of fatty acid and cholesterol uptake into the intestinal mucosal cell. J Clin Invest. 1976;58(1):97–108.
Simmonds W. The role of micellar solubilization in lipid absorption. Aust J Exp Biol Med Sci. 1972;50(4):403–21.
Wasan KM. Formulation and physiological and biopharmaceutical issues in the development of oral lipid-based drug delivery systems. Drug Dev Ind Pharm. 2001;27(4):267–76.
Kalepu S, Manthina M, Padavala V. Oral lipid-based drug delivery systems—an overview. Acta Pharm Sin B. 2013;3(6):361–72.
Pouton CW, Charman WN. The potential of oily formulations for drug delivery to the gastro-intestinal tract. Elsevier; 1997.
Ananthakrishnan P, Mariani G, Moresco L, Giuliano AE. The anatomy and physiology of lymphatic circulation. Radioguided Surgery: Springer; 2008. p. 57–71.
Hiroshi Y, Shozo M, Chiharu K, Hitoshi S. Bifunctional delivery system for selective transfer of bleomycin into lymphatics via enteral route. Int J Pharm. 1981;8(4):291–302.
Beier R, Gebert A. Kinetics of particle uptake in the domes of Peyer’s patches. Am J Physiol Gastrointest Liver Physiol. 1998;275(1):G130–G7.
Eldridge JH, Hammond CJ, Meulbroek JA, Staas JK, Gilley RM, Tice TR. Controlled vaccine release in the gut-associated lymphoid tissues. I. Orally administered biodegradable microspheres target the Peyer’s patches. J Control Release. 1990;11(1–3):205–14.
Ahn H, Park J-H. Liposomal delivery systems for intestinal lymphatic drug transport. Biomater Res. 2016;20(1):36.
Iqbal J, Hussain MM. Intestinal lipid absorption. Am J Physiol Endocrinol Metab. 2009;296(6):E1183–E94.
Wilson FA, Dietschy JM. The intestinal unstirred layer: its surface area and effect on active transport kinetics. Biochim Biophys Acta Biomembr. 1974;363(1):112–26.
Thomson A, Schoeller C, Keelan M, Smith L, Clandinin M. Lipid absorptions passing through the unstirred layers, brush-border membrane, and beyond. Can J Physiol Pharmacol. 1993;71(8):531–55.
Humberstone AJ, Charman WN. Lipid-based vehicles for the oral delivery of poorly water soluble drugs. Adv Drug Deliv Rev. 1997;25(1):103–28.
Yáñez JA, Wang SW, Knemeyer IW, Wirth MA, Alton KB. Intestinal lymphatic transport for drug delivery. Adv Drug Deliv Rev. 2011;63(10–11):923–42.
Wacher VJ, Silverman JA, Zhang Y, Benet LZ. Role of P-glycoprotein and cytochrome P450 3A in limiting oral absorption of peptides and peptidomimetics. J Pharm Sci. 1998;87(11):1322–30.
Borst P, Schinkel A, Smit J, Wagenaar E, Van Deemter L, Smith A, et al. Classical and novel forms of multidrug resistance and the physiological functions of P-glycoproteins in mammals. Pharmacol Ther. 1993;60(2):289–99.
Thiebaut F, Tsuruo T, Hamada H, Gottesman MM, Pastan I, Willingham MC. Cellular localization of the multidrug-resistance gene product P-glycoprotein in normal human tissues. Proc Natl Acad Sci. 1987;84(21):7735–8.
Parkinson A. An overview of current cytochrome P450 technology for assessing the safety and efficacy of new materials. Toxicol Pathol. 1996;24(1):45–57.
Wacher VJ, Salphati L, Benet LZ. Active secretion and enterocytic drug metabolism barriers to drug absorption. Adv Drug Deliv Rev. 1996;20(1):99–112.
Zordan-Nudo T, Ling V, Liu Z, Georges E. Effects of nonionic detergents on P-glycoprotein drug binding and reversal of multidrug resistance. Cancer Res. 1993;53(24):5994–6000.
Batrakova EV, Li S, Miller DW, Kabanov AV. Pluronic P85 increases permeability of a broad spectrum of drugs in polarized BBMEC and Caco-2 cell monolayers. Pharm Res. 1999;16(9):1366–72.
Regev R, Assaraf YG, Eytan GD. Membrane fluidization by ether, other anesthetics, and certain agents abolishes P-glycoprotein ATPase activity and modulates efflux from multidrug-resistant cells. FEBS J. 1999;259(1–2):18–24.
Yoo J-SH, Smith TJ, Ning SM, Mao-Jung L, Thomas PE, Yang CS. Modulation of the levels of cytochromes P450 in rat liver and lung by dietary lipid. Biochem Pharmacol. 1992;43(12):2535–42.
Soldner A, Christians U, Susanto M, Wacher VJ, Silverman JA, Benet LZ. Grapefruit juice activates P-glycoprotein-mediated drug transport. Pharm Res. 1999;16(4):478–85.
Dahan A, Hoffman A. Rationalizing the selection of oral lipid based drug delivery systems by an in vitro dynamic lipolysis model for improved oral bioavailability of poorly water soluble drugs. J Control Release. 2008;129(1):1–10.
Savla R, Browne J, Plassat V, Wasan KM, Wasan EK. Review and analysis of FDA approved drugs using lipid-based formulations. Drug Dev Ind Pharm. 2017;43(11):1743–58.
Feeney OM, Crum MF, McEvoy CL, Trevaskis NL, Williams HD, Pouton CW, et al. 50 years of oral lipid-based formulations: provenance, progress and future perspectives. Adv Drug Deliv Rev. 2016;101:167–94.
Strickley RG. Currently marketed oral lipid-based dosage forms: drug products and excipients. Drugs and the Pharmaceutical Sciences. 2007;170:1.
Strickley RG. Solubilizing excipients in oral and injectable formulations. Pharm Res. 2004;21(2):201–30.
Alexander A. A review on novel therapeutic strategies for the enhancement of solubility for hydrophobic drugs through lipid and surfactant based self micro emulsifying drug delivery system: a novel approach. Am J Drug Disc Dev. 2012;2(4):143–83.
Charman WN, Porter CJ. Oral lipid-based formulations: using preclinical data to dictate formulation strategies for poorly water-soluble drugs. Oral lipid-based formulations: CRC Press; 2007. p. 207–28.
Griffin B. Advances in lipid-based formulations: overcoming the challenge of low bioavailability for poorly water soluble drug compounds. Am Pharm Rev http://www.americanpharmaceuticalreview.com/Featured-Articles/39299 [Consulted February 11, 2016]. 2012.
MacGregor KJ, Embleton JK, Lacy JE, Perry EA, Solomon LJ, Seager H, et al. Influence of lipolysis on drug absorption from the gastro-intestinal tract. Adv Drug Deliv Rev. 1997;25(1):33–46.
Kahan BD, Dunn J, Fitts C, Van DB, Wombolt D, Pollak R, et al. Reduced inter- and intrasubject variability in cyclosporine pharmacokinetics in renal transplant recipients treated with a microemulsion formulation in conjunction with fasting, low-fat meals, or high-fat meals. Transplantation. 1995;59(4):505–11.
Mendez R, Abboud H, Burdick J, Copley B, Freeman R, Batiuk TD, et al. Reduced intrapatient variability of cyclosporine pharmacokinetics in renal transplant recipients switched from oral Sandimmune to Neoral. Clin Ther. 1999;21(1):160–71.
Katneni K, Charman SA, Porter CJ. Impact of Cremophor-EL and Polysorbate-80 on digoxin permeability across rat jejunum: delineation of thermodynamic and transporter related events using the reciprocal permeability approach. J Pharm Sci. 2007;96(2):280–93.
Kim AE, Dintaman JM, Waddell DS, Silverman JA. Saquinavir, an HIV protease inhibitor, is transported by P-glycoprotein. J Pharmacol Exp Ther. 1998;286(3):1439–45.
Schmitt C, Kaeser B, Riek M, Bech N, Kreuzer C. Effect of saquinavir/ritonavir on P-glycoprotein activity in healthy volunteers using digoxin as a probe. Int J Clin Pharmacol Ther. 2010;48(3):192–9.
Perloff MD, Von Moltke LL, Marchand JE, Greenblatt DJ. Ritonavir induces P-glycoprotein expression, multidrug resistance-associated protein (MRP1) expression, and drug transporter-mediated activity in a human intestinal cell line. J Pharm Sci. 2001;90(11):1829–37.
Chen XQGO, Hageman MJ. Application of lipid-based formulations in drug discovery. J Med Chem. 2012;55:7945–56.
Gao P, Morozowich W. Case studies: rational development of self-emulsifying formulations for improving the oral bioavailability of poorly soluble, lipophilic drugs. Drugs and the Pharmaceutical Sciences. 2007;170:273.
Backman TW, Cao Y, Girke T. ChemMine tools: an online service for analyzing and clustering small molecules. Nucleic Acids Res. 2011;39(suppl_2):W486–W91.
Constantinides PP, Wasan KM. Lipid formulation strategies for enhancing intestinal transport and absorption of P-glycoprotein (P-gp) substrate drugs: in vitro/In vivo case studies. J Pharm Sci. 2007;96(2):235–48.
Aungst BJ. Absorption enhancers: applications and advances. AAPS J. 2012;14(1):10–8.
Akhtar N, Ahad A, Khar RK, Jaggi M, Aqil M, Iqbal Z, et al. The emerging role of P-glycoprotein inhibitors in drug delivery: a patent review. Expert Opin Ther Pat. 2011;21(4):561–76.
Kayser O, Olbrich C, Yardley V, Kiderlen A, Croft S. Formulation of amphotericin B as nanosuspension for oral administration. Int J Pharm. 2003;254(1):73–5.
Lindmark T, Kimura Y, Artursson P. Absorption enhancement through intracellular regulation of tight junction permeability by medium chain fatty acids in Caco-2 cells. J Pharmacol Exp Ther. 1998;284(1):362–9.
Veber DF, Johnson SR, Cheng H-Y, Smith BR, Ward KW, Kopple KD. Molecular properties that influence the oral bioavailability of drug candidates. J Med Chem. 2002;45(12):2615–23.
Flaten GE, Luthman K, Vasskog T, Brandl M. Drug permeability across a phospholipid vesicle-based barrier: 4. The effect of tensides, co-solvents and pH changes on barrier integrity and on drug permeability. Eur J Pharm Sci. 2008;34(2–3):173–80.
Müllertz A, Ogbonna A, Ren S, Rades T. New perspectives on lipid and surfactant based drug delivery systems for oral delivery of poorly soluble drugs. J Pharm Pharmacol. 2010;62(11):1622–36.
Bajusz D, Rácz A, Héberger K. Why is Tanimoto index an appropriate choice for fingerprint-based similarity calculations? J Chem Inf. 2015;7(1):20.
Stuchlík M, Zak S. Lipid-based vehicle for oral drug delivery. Biomed Pap. 2001;145(2):17–26.
Porter CJ, Trevaskis NL, Charman WN. Lipids and lipid-based formulations: optimizing the oral delivery of lipophilic drugs. Nat Rev Drug Discov. 2007;6(3):231.
Hauss DJ. Oral lipid-based formulations. Adv Drug Deliv Rev. 2007;59(7):667–76.
Hauss DJ. Oral lipid-based formulations: enhancing the bioavailability of poorly water-soluble drugs. CRC Press; 2007.
van Oss CJ. Nonionic surfactants: physical chemistry (surfactant science series Vol. 23). MJ Schick (ed) Marcel Dekker, Inc., New York and Basel, 1987, pp. xv + 1136, $225.00. J Dispers Sci Technol. 1990;11(4):437–8.
Attwood D, Florence A. Surfactant systems—their chemistry, pharmacy and biology. New York: Chapman and Hall; 1983.
Padley FB, Gunstone FD, Harwood JL. Occurrence and characteristics of oils and fats. The lipid handbook. Berlin: Springer; 1986. p. 49–170.
Chen M-L. Lipid excipients and delivery systems for pharmaceutical development: a regulatory perspective. Adv Drug Deliv Rev. 2008;60(6):768–77.
Maincent P. The regulatory environment: the challenges for lipid-based formulation. Bulletin Technique Gattefossé. 2007;100:47–9.
U.S. Food and Drug Administration, Center for Drug Evaluation and Research, Inactive ingredient guide. Division of Drug Information Resources. [Internet]. Office of Management, Center for Drug Evaluation and Research, Food and Drug Administration. 1996. Available from: http://www.fda.gov/cder/drug/iig/default.htm.
U.S. Food and Drug Administration, Title 21, Code of Federal Regulations, Part 182, 184, 186. Office of the Federal Register [Internet]. National Archives and Records Administration. 2007.
Ghosh S, Roy T. Nanoparticulate drug-delivery systems: lymphatic uptake and its gastrointestinal applications. 2014 [cited 4 06]. 123–30].
Guidance for Industry: Nonclinical studies for the safety evaluation of pharmaceutical excipients. Office of Training and Communication, Division of Drug Information, HFD-240, Center for Drug Evaluation and Research, Food and Drug Administration, or Office of Communication, Training, and Manufacturers Assistance, HFM-40, Center for Biologics Evaluation and Research, Food and Drug Administration; 2005.
Mukherjee B, Maji R, Roychowdhury S, Ghosh S. Toxicological concerns of engineered nanosize drug delivery systems. Am J Ther. 2016;23(1):e139–e50.
Szebeni J, Muggia F, Gabizon A, Barenholz Y. Activation of complement by therapeutic liposomes and other lipid excipient-based therapeutic products: prediction and prevention. Adv Drug Deliv Rev. 2011;63(12):1020–30.
Kaukonen AM, Boyd BJ, Porter CJ, Charman WN. Drug solubilization behavior during in vitro digestion of simple triglyceride lipid solution formulations. Pharm Res. 2004;21(2):245–53.
Pouton CW. Formulation of self-emulsifying drug delivery systems. Adv Drug Deliv Rev. 1997;25(1):47–58.
Pouton CW. Self-emulsifying drug delivery systems: assessment of the efficiency of emulsification. Int J Pharm. 1985;27(2–3):335–48.
Rowe RC, Sheskey PJ, Owen SC. Handbook of pharmaceutical excipients. London: Pharmaceutical Press; 2006.
Schick MJ. Nonionic surfactants: physical chemistry. Boca Raton: CRC Press; 1987.
Shono Y, Nishihara H, Matsuda Y, Furukawa S, Okada N, Fujita T, et al. Modulation of intestinal P-glycoprotein function by cremophor EL and other surfactants by an in vitro diffusion chamber method using the isolated rat intestinal membranes. J Pharm Sci. 2004;93(4):877–85.
Hugger ED, Novak BL, Burton PS, Audus KL, Borchardt RT. A comparison of commonly used polyethoxylated pharmaceutical excipients on their ability to inhibit P-glycoprotein activity in vitro. J Pharm Sci. 2002;91(9):1991–2002.
Cole ET, Cadé D, Benameur H. Challenges and opportunities in the encapsulation of liquid and semi-solid formulations into capsules for oral administration. Adv Drug Deliv Rev. 2008;60(6):747–56.
Cullis PR, Hope MJ, Tilcock CP. Lipid polymorphism and the roles of lipids in membranes. Chem Phys Lipids. 1986;40(2–4):127–44.
Hafez IM, Cullis PR. Roles of lipid polymorphism in intracellular delivery. Adv Drug Deliv Rev. 2001;47(2–3):139–48.
Holm R, Porter CJ, Edwards GA, Müllertz A, Kristensen HG, Charman WN. Examination of oral absorption and lymphatic transport of halofantrine in a triple-cannulated canine model after administration in self-microemulsifying drug delivery systems (SMEDDS) containing structured triglycerides. Eur J Pharm Sci. 2003;20(1):91–7.
Cavalli R, Zara GP, Caputo O, Bargoni A, Fundarò A, Gasco MR. Transmucosal transport of tobramycin incorporated in SLN after duodenal administration to rats. Part I—a pharmacokinetic study. Pharmacol Res. 2000;42(6):541–5.
Bargoni A, Cavalli R, Caputo O, Fundarò A, Gasco MR, Zara GP. Solid lipid nanoparticles in lymph and plasma after duodenal administration to rats. Pharm Res. 1998;15(5):745–50.
Battaglia L, Serpe L, Muntoni E, Zara G, Trotta M, Gallarate M. Methotrexate-loaded SLNs prepared by coacervation technique: in vitro cytotoxicity and in vivo pharmacokinetics and biodistribution. Nanomedicine. 2011;6(9):1561–73.
Manjunath K, Venkateswarlu V. Pharmacokinetics, tissue distribution and bioavailability of nitrendipine solid lipid nanoparticles after intravenous and intraduodenal administration. J Drug Target. 2006;14(9):632–45.
Makwana V, Jain R, Patel K, Nivsarkar M, Joshi A. Solid lipid nanoparticles (SLN) of Efavirenz as lymph targeting drug delivery system: elucidation of mechanism of uptake using chylomicron flow blocking approach. Int J Pharm. 2015;495(1):439–46.
Shete H, Chatterjee S, De A, Patravale V. Long chain lipid based tamoxifen NLC. Part II: pharmacokinetic, biodistribution and in vitro anticancer efficacy studies. Int J Pharm. 2013;454(1):584–92.
Zhang T, Chen J, Zhang Y, Shen Q, Pan W. Characterization and evaluation of nanostructured lipid carrier as a vehicle for oral delivery of etoposide. Eur J Pharm Sci. 2011;43(3):174–9.
Myers R, Stella V. Factors affecting the lymphatic transport of penclomedine (NSC-338720), a lipophilic cytotoxic drug: comparison to DDT and hexachlorobenzene. Int J Pharm. 1992;80(1–3):51–62.
Gursoy RN, Benita S. Self-emulsifying drug delivery systems (SEDDS) for improved oral delivery of lipophilic drugs. Biomed Pharmacother. 2004;58(3):173–82.
Woo JS, Kim T-S, Park J-H, Chi S-C. Formulation and biopharmaceutical evaluation of silymarin using SMEDDS. Arch Pharm Res. 2007;30(1):82–9.
Khumpirapang N, Pikulkaew S, Müllertz A, Rades T, Okonogi S. Self-microemulsifying drug delivery system and nanoemulsion for enhancing aqueous miscibility of Alpinia galanga oil. PloS One. 2017;12(11):e0188848.
Thakkar H, Nangesh J, Parmar M, Patel D. Formulation and characterization of lipid-based drug delivery system of raloxifene-microemulsion and self-microemulsifying drug delivery system. J Pharm Bioallied Sci. 2011;3(3):442.
Chen Y, Li G, Wu X, Chen Z, Hang J, Qin B, et al. Self-microemulsifying drug delivery system (SMEDDS) of vinpocetine: formulation development and in vivo assessment. Biol Pharm Bull. 2008;31(1):118–25.
Nishioka Y, Yoshino H. Lymphatic targeting with nanoparticulate system. Adv Drug Deliv Rev. 2001;47(1):55–64.
Sheue Nee Ling S, Magosso E, Abdul Karim Khan N, Hay Yuen K, Anne Barker S. Enhanced oral bioavailability and intestinal lymphatic transport of a hydrophilic drug using liposomes. Drug Dev Ind Pharm. 2006;32(3):335–45.
Perrie Y, Obrenovic M, McCarthy D, Gregoriadis G. Liposome (Lipodine™)-mediated DNA vaccination by the oral route. Journal of Liposome Research. 2002;12(1–2):185–97.
Li H, Song J-H, Park J-S, Han K. Polyethylene glycol-coated liposomes for oral delivery of recombinant human epidermal growth factor. Int J Pharm. 2003;258(1–2):11–9.
Larsen DB, Joergensen S, Olsen NV, Hansen SH, Larsen C. In vivo release of bupivacaine from subcutaneously administered oily solution. Comparison with in vitro release. J Control Release. 2002;81(1–2):145–54.
Gill KK, Kaddoumi A, Nazzal S. Mixed micelles of PEG2000-DSPE and vitamin-E TPGS for concurrent delivery of paclitaxel and parthenolide: enhanced chemosenstization and antitumor efficacy against non-small cell lung cancer (NSCLC) cell lines. Eur J Pharm Sci. 2012;46(1–2):64–71.
Passerini N, Albertini B, Perissutti B, Rodriguez L. Evaluation of melt granulation and ultrasonic spray congealing as techniques to enhance the dissolution of praziquantel. Int J Pharm. 2006;318(1–2):92–102.
Chauhan B, Shimpi S, Paradkar A. Preparation and evaluation of glibenclamide-polyglycolized glycerides solid dispersions with silicon dioxide by spray drying technique. Eur J Pharm Sci. 2005;26(2):219–30.
Chauhan B, Shimpi S, Paradkar A. Preparation and characterization of etoricoxib solid dispersions using lipid carriers by spray drying technique. AAPS PharmSciTech. 2005;6(3):E405–E9.
Sethia S, Squillante E. Solid dispersion of carbamazepine in PVP K30 by conventional solvent evaporation and supercritical methods. Int J Pharm. 2004;272(1–2):1–10.
Chen C, Han D, Cai C, Tang X. An overview of liposome lyophilization and its future potential. J Control Release. 2010;142(3):299–311.
Dening TJ, Rao S, Thomas N, Prestidge CA. Novel nanostructured solid materials for modulating oral drug delivery from solid-state lipid-based drug delivery systems. AAPS J. 2016;18(1):23–40.
Porter CJ, Charman WN. Model systems for intestinal lymphatic transport studies. Models for assessing drug absorption and metabolism. Springer; 1996. p. 85–102.
Griffin BT, O’Driscoll CM. A comparison of intestinal lymphatic transport and systemic bioavailability of saquinavir from three lipid-based formulations in the anaesthetised rat model. J Pharm Pharmacol. 2006;58(7):917–25.
Perry CM, Noble S. Saquinavir soft-gel capsule formulation. Drugs. 1998;55(3):461–86.
Dahan A, Hoffman A. The effect of different lipid based formulations on the oral absorption of lipophilic drugs: the ability of in vitro lipolysis and consecutive ex vivo intestinal permeability data to predict in vivo bioavailability in rats. Eur J Pharm Biopharm. 2007;67(1):96–105.
Hauss DJ, Fogal SE, Ficorilli Ficorilli JV. Chronic collection of mesenteric lymph from conscious, tethered rats. J Am Assoc Lab Anim Sci. 1998;37(3):56–8.
Trevaskis NL, Hu L, Caliph SM, Han S, Porter CJ. The mesenteric lymph duct cannulated rat model: application to the assessment of intestinal lymphatic drug transport. J Vis Exp. 2015;97.
Chen G-L, Hao W-H. Factors affecting zero-order release kinetics of porous gelatin capsules. Drug Dev Ind Pharm. 1998;24(6):557–62.
Kim Y-H, Koczo K, Wasan DT. Dynamic film and interfacial tensions in emulsion and foam systems. J Colloid Interface Sci. 1997;187(1):29–44.
Yang S, Simmonds RS, Birch EJ. Physicochemical characterization and thermal properties of lipids from R. opacus PD630. Food and Public Health. 2014;4(3):87–92.
Shah BR, Finberg L. Single-day therapy for nutritional vitamin D-deficiency rickets: a preferred method. J Pediatr. 1994;125(3):487–90.
Yuan T, Qin L, Wang Z, Nie J, Guo Z, Li G, et al. Solid lipid dispersion of calcitriol with enhanced dissolution and stability. AJPS. 2013;8(1):39–47.
Singh A, Narsipur S. Cyclosporine: a commentary on brand versus generic formulation exchange. J Trans. 2011;2011.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Additional information
Guest Editor: Sanyog Jain
Rights and permissions
About this article

Cite this article
Patel, V., Lalani, R., Bardoliwala, D. et al. Lipid-Based Oral Formulation Strategies for Lipophilic Drugs. AAPS PharmSciTech 19, 3609–3630 (2018). https://doi.org/10.1208/s12249-018-1188-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1208/s12249-018-1188-8