
Abstract
Severe viral pneumonia is a significant cause of morbidity and mortality globally, whether due to outbreaks of endemic viruses, periodic viral epidemics, or the rarer but devastating global viral pandemics. While limited anti-viral therapies exist, there is a paucity of direct therapies to directly attenuate viral pneumonia-induced lung injury, and management therefore remains largely supportive. Mesenchymal stromal/stem cells (MSCs) are receiving considerable attention as a cytotherapeutic for viral pneumonia. Several properties of MSCs position them as a promising therapeutic strategy for viral pneumonia-induced lung injury as demonstrated in pre-clinical studies in relevant models. More recently, early phase clinical studies have demonstrated a reassuring safety profile of these cells. These investigations have taken on an added importance and urgency during the COVID-19 pandemic, with multiple trials in progress across the globe. In parallel with clinical translation, strategies are being investigated to enhance the therapeutic potential of these cells in vivo, with different MSC tissue sources, specific cellular products including cell-free options, and strategies to ‘licence’ or ‘pre-activate’ these cells, all being explored. This review will assess the therapeutic potential of MSC-based therapies for severe viral pneumonia. It will describe the aetiology and epidemiology of severe viral pneumonia, describe current therapeutic approaches, and examine the data suggesting therapeutic potential of MSCs for severe viral pneumonia in pre-clinical and clinical studies. The challenges and opportunities for MSC-based therapies will then be considered.
Similar content being viewed by others

Introduction
Severe viral pneumonia is a significant cause of morbidity and mortality globally – one need look no further than the current COVID-19 pandemic to appreciate this. Outside of pandemic settings, viral-induced pneumonias remain common, both due to periodic epidemics, and from seasonal outbreaks of endemic disease. While anti-viral therapies exist, there is a paucity of direct therapies to directly attenuate viral pneumonia-induced lung injury. Management therefore remains largely supportive, including assistance with gas exchange and management of complications and super-infections.
Mesenchymal stromal/stem cells (MSCs) can be isolated from several tissues, such as bone marrow, adipose tissue, and umbilical cord[1, 2], and are receiving considerable attention as a cytotherapeutic for viral pneumonia. Several properties of MSCs position them as a promising therapeutic strategy for viral pneumonia-induced lung injury. Early phase clinical studies have demonstrated a reassuring safety profile of these cells when administered to healthy and ill patients. In addition, strategies are being investigated to enhance the therapeutic potential of these cells in vivo, with different tissue sources, cellular products (cell-free options), and methods of ‘licencing’ or ‘pre-activation’ of these cells, all being explored.
The aim of this review is to assess the therapeutic potential of MSC-based therapies for severe viral pneumonia of different aetiologies. It will describe the aetiology and epidemiology of severe viral pneumonia, describe current therapeutic approaches, and examine the data suggesting therapeutic potential of MSCs for severe viral pneumonia in pre-clinical and clinical studies. The challenges and opportunities for MSC-based therapies will then be considered.
Aetiology and epidemiology of severe viral pneumonia
Viruses are an increasingly frequent cause of pneumonia [3] in the general population. Before the COVID-19 pandemic, rhinovirus (23.6%), parainfluenza (20.8%), metapneumovirus (18.1%), influenza (16.7%) and respiratory syncytial virus (13.9%) were the main viral causes of severe pneumonia [4] (Table 1).
Influenza Virus A and B are common causes of severe pneumonia mainly in the fall or winter seasons. In the USA, it is estimated that influenza caused 38,000,000 cases in 2019–2020 season and 22,000 deaths [5]. Worldwide it is estimated that influenza causes 4–9 deaths per 100,000 individuals annually [6]. Over 10% of influenza patients require ICU admission. Severe disease develops mainly in patients with increased age and underlying conditions [7,8,9], due to isolated influenza infection or by a secondary bacterial co-infection, frequently with Staphylococcus aureus (often methicillin-resistant) or Streptococcus pneumoniae [10].
Influenza can cause pandemic disease. In 1918 Spanish flu caused 20–100 million deaths worldwide [11]. In 2009, the Swine-Origin Influenza A (H1N1) virus pandemic started in Mexico [12] and rapidly spread worldwide. Influenza A H1N1 caused severe disease with pneumonia and acute respiratory distress syndrome (ARDS) and extrapulmonary disease. Younger patients, female, immunosuppression, obesity and pregnancy were risk factors for severe disease [13]. ICU mortality in H1N1-infected patients was approximately 40% [12]. Avian-origin Influenza has also been reported to cause human disease in isolated cases, but to date widespread human dissemination has not occurred [14].
Since 2000, several novel coronaviruses (CoVs) causing severe pneumonia have emerged. In 2003, severe acute respiratory syndrome (SARS), caused by the novel SARS-CoV emerged in the southeast of China, spreading throughout southeast Asia. More than 8000 patients were diagnosed, with 774 deaths reported in 26 countries during 2003 [15]. Spread was rapid, with nosocomial outbreaks affecting health care workers a feature [16]. In 2012, Middle-Eastern respiratory syndrome (MERS), caused by MERS-CoV, was first isolated in Saudi Arabia [17]. Since 2012 MERS outbreaks have been reported in 27 countries and 35% of patients died [18].
The SARS-CoV2 virus, which emerged in Wuhan China in late 2019, and was declared a pandemic by the WHO in March 2020, has infected over 250 million people, with over 4.5 million deaths reported (likely underestimated) to date [19]. Older age, immunosuppression, obesity, diabetes, hypertension, COPD, higher renal and cardiovascular SOFA score components, lower PaO2/FiO2 ratio, neutrophilia, higher LDH, D-dimer and a shorter time between first symptoms and ICU admission are associated with mortality [20,21,22]. Patients requiring invasive mechanical ventilation have a 45% mortality [23], with up to 80% of patients over 80 years of age dying [24]. Long-term morbidity after COVID-19 (‘Long COVID’), with severe alteration on chest-CT, decreased pulmonary functions and quality of life, is widely reported [25, 26].
Mechanisms of lung injury in viral pneumonia
The alveolar–capillary unit is disrupted during severe viral pneumonia [27], with upregulation of pro-inflammatory mediators, infiltration of neutrophils and monocyte–macrophages into the vascular and alveolar compartments increasing capillary endothelial permeability, resulting in ARDS [27]. Notable differences exist in the biologic profile of COVID-19 relative to classical ARDS, with lower expression of interferons (IFNs) and an increase in thrombotic mediators [28]. An understanding of the viral replication cycle, and the associated host response, highlights the unique pathophysiology of viral pneumonia.
Influenza viruses may infect a variety of lung cells, including ciliated epithelial cells, type I and II alveolar cells, and immune cells [29]. Virus tropism is due to the ability of influenza viruses to bind different isoforms of sialic acid present on host cells via hyaluronidase. The higher virulence of some influenza subtypes (e.g. the “avian” H5N1) may also be related to their greater affinity for the sialyl-galactosyl residues present in the distal respiratory tract, thus leading to more severe lung involvement [29].
The coronaviruses (SARS-CoV, MERS-CoV, and SARS-CoV-2) exploit distinct receptors to enter host cells. MERS-CoV binds to the dipeptidyl peptidase-4 (DPP4), a surface protein mainly expressed on alveolar macrophages and, to a lesser extent, on alveolar epithelial cells and T cells [30, 31]. This marked tropism for immune rather than lung cells is the basis of MERS-CoV immunopathogenesis. SARS-CoV-2 has tropism for ciliated airway epithelial cells and type II alveolar epithelial cells, which is conferred by its dependence on both the human angiotensin converting enzyme-2 (ACE2) receptor, as well as the host transmembrane serine protease-2 (TMPRSS2) for S-protein cleavage and subsequent activation [32]. An important, receptor-mediated pathogenic process derives from the dysregulation of the renin–angiotensin system (RAS) induced by SARS-CoV and SARS-CoV-2. By binding to ACE2, they induce both its internalization and shedding through ADAM17 activation; a reduced ACE2 activity results in increased vascular permeability, enhanced lung oedema, and worsening lung damage and the pro-inflammatory response [33, 34].
Upon viral sensing, pattern recognition receptors (PRRs), such as retinoic acid-inducible gene (RIG-I)-like receptors (RLRs) and nucleotide-binding oligomerization domain (NOD)-like receptors (NLR), such as NLRP3, activate signalling pathways that trigger the release of type I and III IFNs, as well as pro-inflammatory mediators, including cytokines, chemokines, and antimicrobial peptides, that assist in the prevention and clearance of respiratory viral infections [35]. The lung injury in the case of primary viral pneumonia is caused, in part, from the overproduction of inflammatory cytokines resulting from virus replication in lung cells [36]. Early work performed to characterize the host immune response of COVID-19 suggested an immune signature consisting of elevated serum cytokines [particularly Interleukin (IL)-1β, IL-6 and tumour necrosis factor (TNF)-α], impaired interferon responses, and peripheral lymphopenia as markers of severe disease; other associated inflammatory serum markers include elevated levels of ferritin, lactate dehydrogenase, d-dimer, C-reactive protein, and coagulation factors [36, 37]. The pro-inflammatory immune signature of SARS-CoV-2 has been likened to macrophage-activation syndrome (MAS), a life-threatening clinical entity observed in autoimmune diseases and mimicked in many viral infections, including influenza [38, 39]. However, reported plasma IL-6 levels in COVID-19 patients appear to be significantly lower on average (10- to 40-fold) when compared with those reported in other non-COVID-19 ARDS cohorts that display signs of a cytokine storm [40]. It is important to note that IL-6 has several important anti-inflammatory as well as anti-viral functions [41]. Recent findings also suggest that in addition to an increase in pro-inflammatory mediator production there may also be disruptions in specific resolution pathways in patients with COVID-19 [42].
Coronavirus replication is generally associated with a delayed and dramatically reduced IFN induction in most cell types, and COVID-19 severity correlates with the degree of impairment [43, 44]. The ability to evade the innate immune response seems to be the highest for SARS-CoV-2, followed by SARS-CoV and MERS-CoV and, generally, human endemic CoVs are worse inhibitors than epidemic and pandemic viruses [45]. In the case of SARS-CoV, this leads to the dysregulated activation of the inflammatory monocyte–macrophage response, in turn causing vascular leakage and impaired B- and T-cell activation [46].
Data from patients who died from COVID-19 suggest that SARS-CoV-2 infects endothelial cells to cause inflammation (endothelialitis) [47]. This observed endothelialitis supports the hypothesis that SARS-CoV-2 has tropism for vascular endothelial cells, which express the ACE receptor [48]. Indeed, viral cytotoxicity is likely contributing to the pathogenesis of severe COVID-19, since post-mortem detection of replicating virus is common [49]. Direct endothelial damage could also explain the multi-system organ failure and hypercoagulable state associated with severe COVID-19, since local pulmonary endothelialitis could result in activation of the coagulation cascade and exuberant production of endothelium-derived pro-inflammatory cytokines [50].
Current therapies for viral pneumonia
Approved anti-viral medications with activity against influenza viruses [51] are used as prophylactic adjuncts to the influenza vaccine to control the infection and/or as treatment to reduce symptoms from influenza (Table 2). Antiviral treatment is recommended as early as possible for any patient with confirmed or suspected influenza who are hospitalized, have severe, complicated, or progressive illness; or are at higher risk for influenza complications [51]. Antivirals may also be considered for healthy, symptomatic outpatients not at high risk for influenza complications, who are diagnosed with confirmed or suspected influenza, based on clinical judgment, if treatment can be initiated within 48 h of illness onset.
Neuraminidase inhibitors (oseltamivir, zanamivir, and peramivir) block enzymes that cleave sialic acid groups from glycoproteins, effectively preventing release of viral particles—virions—from the host cell [52]. These have activity against both Influenza A and B viruses, and are approved by the U.S. Food and Drug Administration (FDA) and by the European Center for Disease Prevention and Control [53]. Baloxavir is a cap-dependent endonuclease inhibitor that interferes with viral RNA transcription and blocks virus replication [54] and has shown significant promise in a multicenter randomized clinical trial of post-exposure prophylaxis [55,56,57]. The adamantanes (amantadine, rimantadine) are antivirals that target the Influenza A virus M2 ion channel protein [58]. However, high levels of resistance (> 99%) to adamantanes among circulating Influenza A (H3N2) and influenza A(H1N1)pdm09 (“2009 H1N1”) viruses preclude their recommendation for anti-viral treatment or chemoprophylaxis of currently circulating Influenza A viruses (for detailed discussion see [59]).
Multiple compounds are currently at various stages of investigation for influenza treatment [60,61,62]. The Influenza Therapeutics Program [63] is a major focus of BARDA (The Biomedical Advanced Research and Development Authority) [64]. Prior to the pandemic, the International Society for Influenza and other Respiratory Virus Diseases held its 6th Antiviral Group (isirvAVG) conference to review emerging therapeutics towards seasonal and pandemic influenza, respiratory syncytial virus, coronaviruses including MERS-CoV and SARS-CoV, human rhinovirus, and other respiratory viruses [65]. Multiple additional compounds are under investigation for the treatment of non-coronavirus, lower respiratory tract infections (Additional file 1: Table S1). In addition to potential toxicity and the rapid development of resistance, the limitation of anti-viral-directed therapy is that it obviates management of the response of the host to infection—a fundamental determinant of outcome in the development of ARDS [66,67,68].
In summary, while specific anti-viral therapies exist, they must be used early in the disease course to be effective. Their limited efficacy later in the infection process means that the cornerstone of management remains supportive care with respiratory support (oxygen, ventilatory assistance), rest, antipyretics, analgesics, nutrition, and close observation [65]. New therapies to directly attenuate viral-induced pneumonia and lung injury are a priority.
MSCs—therapeutic potential for viral pneumonia
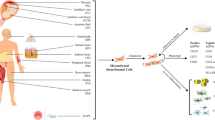
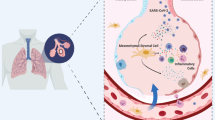
MSCs have been proposed as a promising therapeutic strategy in viral pneumonia because they possess immunomodulatory, anti-microbial, and pro-resolution properties (Fig. 1) [2]. MSCs can be easily sourced from various tissue types, and while the MSC optimal tissue source remains unclear, bone marrow (BM) and umbilical cord (UC) derived MSCs may be more effective than adipose tissue derived MSCs in pre-clinical acute lung injury models [69].

Mechanisms of action of MSCs which can counteract viral infection. (1) Viral infection leads to tissue damage at the delicate blood-air barrier in the lung. The release of inflammatory cytokines initiates further tissue damage with (2) inflammatory T-cell proliferation and differentiation to Th-1 and Th-17s, (3) inflammatory white cell recruitment from the blood and tissues leading to further inflammation, creation of neutrophil NETS, fibroblast differentiation, oedema fluid accumulation and significant barrier disruption. MSCs have been demonstrated to act on several of the injurious processes that occur in infection such as (4) Release of cytokines and chemokines which promote anti-inflammatory innate and adaptive cell phenotypes, (5) release of factors which prevent the formation of NETS, reduce barrier disruption, and (6) prevent fibroblast differentiation and promote PMN apoptosis. (7) MSC IL-10 production and production from anti-inflammatory monocytes induces regulatory B and T cells and promotes tissue protection and repair, and MSC IDO production regulates inflammatory T-cell proliferation
MSC administration kinetics
Following systemic administration, MSCs are initially trapped in the lungs and subsequently ‘home’ and are retained in injured or inflamed areas [70]. Tracking studies utilizing radiolabelled or fluorescently labelled MSCs demonstrate MSCs lodge in the pulmonary vascular bed where they remain detectable for a few days with the majority of cells being cleared within 24–48 h although this may be prolonged in injured lungs [71, 72]. While in the pulmonary capillary bed, MSCs appear to ‘sense’ the surrounding inflammatory environment through cell surface damage and pathogen molecular pattern receptors [73,74,75] releasing a range of soluble elements of its ‘secretome’ in response.
MSC homing to sites of injury is mediated via chemokine receptors, adhesion proteins, and matrix metalloproteinase (MMPs) [76]. At injury sites, MSCs interact with target cells through cell-to-cell contact and paracrine/endocrine effects, the latter by secreting soluble mediators (including anti-inflammatory cytokines, antimicrobial peptides, and angiogenic growth factors) and extracellular vesicles (EVs), and/or by transferring organelles such as mitochondria to target immune cells (leukocytes [monocytes, macrophages, lymphocytes] and structural cells [including endothelial, epithelial, and smooth-muscle cells]) [77]. MSC-derived EVs contain proteins, lipids, mRNA, microRNAs, DNAs [78]. Mitochondrial transfer from MSCs can occur through tunnelling nanotubes (TNTs), gap junctions, or via EVs [1, 78,79,80].
Systemically administered MSCs may also undergo rapid tissue factor-mediated apoptosis in a phenomenon designated the ‘instant blood-mediated inflammatory response’ (IBMIR) [81]. The response of the host immune system to these dead or dying MSCs driving potential beneficial responses for the underlying lung injury [82]. This may be particularly relevant in both non-viral and viral-induced ARDS in the context of increasing recognition of different inflammatory ARDS phenotypes [83].
Microenvironmental responsiveness and MSC pre-activation
MSCs respond to microenvironmental cues as demonstrated by their release of anti-inflammatory mediators when placed in an inflammatory lung environment [84]. This is mediated by MSC response to damage and pathogen-associated molecular patterns (DAMPs and PAMPs, respectively) [85]. MSC toll-like receptors (TLRs) 3 and 9 are activated by viral RNA and viral unmethylated CpG-DNA, respectively, leading to activation of downstream signalling pathways [2]. Recent reports have characterized MSCs responsiveness to the in vivo inflammatory lung environment by exposing them to either clinical bronchoalveolar lavage fluid (BALF) or serum samples from patients with ARDS [85,86,87,88,89]. Exposure of MSCs to a healthy lung environment (i.e., BALF obtained from healthy volunteers) induced expression of genes encoding for recognition as foreign to the host immune system and for inflammation [85]. In contrast, MSCs exposed to BALF samples from patients with cystic fibrosis or ARDS demonstrated disease-specific responses in gene and protein expression and in downstream effects on immune effector cells such as alveolar macrophages [85, 87, 89].
Pre-activation or ‘licencing’ of MSCs may enhance or direct their therapeutic potential by pre-exposure to conditions that mimic specific microenvironmental conditions [74, 85], or via approaches such as genetic modification [90].
Direct versus indirect MSC effects
Direct MSC effects may be mediated via cell–cell contact or via its secretome [91]. MSC soluble and insoluble extracellular products can be utilized in place of a whole cell therapy [92], facilitating delivery directly into the lung via nebulization [93, 94]. MSC-secreted angiopoietin-1 (Ang-1) and keratinocyte growth factor (KGF) enhance restoration of disrupted alveolar–capillary barrier, while specific regulatory mRNAs in EVs mediate the protective effects of MSCs in pre-clinical models of bacterial or non-infectious acute lung injuries [95, 96].
MSCs—insights from pre-clinical studies
There is a large body of literature demonstrating efficacy of either systemic or direct intratracheal (IT) MSC administration in pre-clinical models of acute pneumonia/pneumonitis and acute lung injury. Most of these studies involve acute lung injury induced by bacteria or bacterial products (endotoxin) or other means [97]. The models include both rodents, as well as large animals (pig, sheep) and explanted human lungs [98]. A range of approaches have been utilized for dose size, dosing route, and MSC source. In contrast, there are a relatively small number of pre-clinical studies investigating effects of MSC administration in pre-clinical models of respiratory virus infections.
MSC effects in viral pneumonia
Systemic MSC administration may reduce the chemokines responsible for lung leukocyte infiltration, such as granulocyte–macrophage colony-stimulating factor (GM-CSF), monocyte chemoattractant protein-1 (MCP-1), and macrophage inflammatory protein-1 alpha (MIP-1α). Antiviral immune responses include increased levels of IFN-γ, which alone or together with pro-inflammatory cytokines activate MSCs to release anti-inflammatory mediators [99]. In vitro, MSCs suppress lymphocyte proliferation in response to the activation of influenza-specific T cells and inhibit the cytotoxicity of specific T cells against H1N1 influenza virus [100]. MSCs may also promote lung epithelial and endothelial cell repair, which may be associated with the anti-inflammatory, anti-apoptotic, and anti-oxidative effects of MSCs, thus promoting endothelial–epithelial barrier integrity, which helps the surfactant to recover followed by a decrease in alveolar oedema and atelectasis [101].
Strain-dependent efficacy in influenza models
MSC administration improved dysregulated alveolar fluid clearance and protein permeability induced by H5N1 and H7N9 influenza viruses in in vitro models, in part by releasing soluble mediators including Ang-1 and KGF that up-regulated sodium and chloride transporters [102]. In parallel studies, systemic administration of human bone marrow-derived MSCs 5 days after induction of H5N1 infection in aged, immunocompetent mice reduced virus-induced mortality, weight loss, lung oedema, BALF CD4+ T cells and natural killer (NK) cells, lung histopathological lesions, pro-inflammatory cytokines and chemokines in the absence of reducing lung virus titres. Notably, no effects were observed in mortality and body weight loss in young mice. These observations suggest that systemic MSC administration may provide benefit in older patients who are at higher risk for severe pulmonary illness caused by H5N1 influenza and also possibly SARS-CoV-2 infection.
Avian influenza virus infection can trigger a very intense pro-inflammatory response compared to other influenza viruses. Avian Influenza virus (H9N2) infection increases serum and lung levels of GM-CSF, MCP-1, MIP-1α and inflammatory leukocyte chemoattractants. In a pre-clinical model of H9N2-induced lung injury using young immunocompetent mice, a single systemic MSC administration (105 MSCs) 3 days after injury resulted in reduction in mortality, lung oedema, histologic injury, BALF and serum chemokines and cytokines (including MCP-1 and MIP-1a), and improved gas-exchange and increased anti-inflammatory mediator concentrations [103]. In another study, systemic administration of UC-MSCs were more effective than BM-MSCs when administered 5 days after induction of Influenza A (H5N1) infection in young female immunocompetent mice with respect to decreasing body weight loss, lung oedema, and inflammation [104]. However, neither type of MSC improved mortality or decreased virus titre. Nonetheless, this has provided one platform for use of UC-MSCs in large number of clinical investigations in COVID-19-induced ARDS.
Despite the potential for MSCs to be effective in the resolution of viral pneumonia, two studies found MSCs not to be protective against influenza respiratory infections in mice. In one, the effects of a single systemic administration in immunocompetent mice of either murine or xenogeneic human bone marrow-derived MSCs were assessed in lung injury induced by mouse-adapted H1N1 or swine-origin pandemic H1N1 [105]. Neither MSC administration, either alone or as an adjuvant therapy with oseltamivir, was effective either when administered prior to virus inoculation, or when therapeutically administered. In another study, neither systemic nor IT administration of 2 doses of MSCs (5 × 105 cells) improved H1N1-mediated lung injury [106].
MSCs in pre-clinical COVID-19
There are no pre-clinical data investigating effects of MSC administration in models of coronavirus respiratory infection, mostly due to the lack of an established animal model. SARS-CoV-2 replication was observed in several non-human primates and in inbred strains of mice following intranasal infection, but these models failed to show clinical signs of pulmonary disease as seen in humans [107]. Human ACE2-overexpressing transgenic mice infected with SARS-CoV-2 demonstrated interstitial pneumonia with lymphocyte and monocyte infiltration into the alveolar interstitium and accumulation of macrophages in alveolar spaces [108]. While these models require further evaluation, they may facilitate the testing of therapeutics including cell-based therapies for COVID-19. We can appreciate the promise of MSCs for the treatment of COVID-19 infection by taking what we know from pre-clinical studies using infectious reagents of bacterial and viral origin.
Cell-free therapies
MSCs-derived EVs have been demonstrated to have comparable and in some cases more effective than MSCs themselves in ameliorating inflammation and injury in a range of pre-clinical lung injury models [109, 110]. Systemic administration of porcine MSC-EVs was found to be safe and reduced virus shedding in nasal swabs, influenza replication in the lungs, BALF pro-inflammatory cytokines and chemokines, histopathological changes when administered 12 h after viral inoculation in a mixed swine (H3N2, H1N1) and avian (H9N5, H7N2) influenza-induced pig lung injury model [111]. These findings suggest systemic EV administration has therapeutic potential for respiratory virus-induced lung injuries.
Summary
There is a substantial pre-clinical literature examining the potential for MSCs to reduce lung injury, via effects that address injury mechanisms directly relevant to viral pneumonia. However, the data from pre-clinical studies of respiratory virus infections are limited to influenza viruses and have produced conflicting results. MSC-derived cell products, particularly EVs, demonstrate significant therapeutic promise.
MSCs—insights from clinical studies
MSC safety profile in clinical trials
A recent systematic review and meta-analysis of MSC therapy included 55 randomized control trials with 2696 patients [112] across a wide variety of clinical conditions. Other than an increased risk of pyrexia, no safety signals (including in relation to infection, thromboembolic events or malignancy) were reported. Furthermore, the risk of death was significantly lower in the MSC-treated group compared to controls. Further follow-up data are needed to determine the safety of MSCs in the longer term.
Clinical trials in ARDS and sepsis
Several ongoing and completed trials of MSCs for ARDS and sepsis support the investigation of MSCs in patients with viral pneumonia. These studies demonstrate that MSC therapy in critically ill ARDS and sepsis patients is feasible and safe, albeit the numbers treated to date is small (see detailed review [113]). Two phase 2 studies in early ARDS demonstrate that MSCs were well tolerated [87, 114]. Post hoc analyses suggested patients with more severe impairment of oxygenation had a better treatment response [114] and that higher MSC viability was associated with greater improvements in Ang-2 and oxygenation [122]. Intravenous MSC therapy reduced lung permeability injury and lung injury severity versus placebo, decreasing mediators of lung injury, such as angiopoietin-2 [115].
MSCs in viral pneumonia
An observational study investigating the use of menstrual blood-derived MSCs in 17 patients with H7N9 viral-induced moderate-to-severe ARDS reported no safety issues [116]. Patients received a dose of 1 × 106 cells/kg MSCs on multiple (up to 4) occasions in early versus later phase ARDS. MSC therapy in COVID-19 patients has been reported in multiple small uncontrolled studies. There was significant heterogeneity in the population receiving MSCs as well as the regimens used in terms of dose and frequency of administration. In general, these studies reported no safety issues and promising efficacy data, however findings must be considered exploratory due to the many methodological limitations. Randomized controlled clinical trials of MSC therapy for COVID-19 are now being reported (Table 3). The dose used was variable and several of these trials tested multiple doses. Together these trials indicate that MSCs were well tolerated and showed promising efficacy in COVID-19 infection ([117,118,119] and NCT04371393). Other trials have recently completed recruitment and results are awaited [120]. Further larger trials are required to adequately assess the question of efficacy of MSCs in viral pneumonia.
Future directions: opportunities and challenges
While allogeneic MSCs demonstrate considerable therapeutic promise for severe viral pneumonia significant knowledge gaps and challenges remain (Table 4). Translation of MSCs to effective therapy in patients with severe viral pneumonia will require additional studies to determine: (1) the optimal MSCs therapeutic; (2) the optimal dose regimen; (3) the optimal patient population, and (5) the potential interaction of concomitant therapies.
Optimal MSC therapeutic
Allogeneic MSCs can be harvested from bone marrow, umbilical cord, and adipose tissue, and each of these MSC sources have been tested in small ARDS trials that reported no safety issues [121, 122]. MSCs derived from older donors may have impaired function [123]. Recently, MSCs that were produced with induced pluripotent stem cell (iPSC) technology were tested in a sheep model of severe pneumonia and sepsis over 48 h; based on a preliminary report, i-MSCs markedly improved oxygenation and they reduced protein rich lung lymph flow indicating reduced lung vascular injury compared to placebo controls [124]. There is also pre-clinical work indicating that EVs derived from MSCs can deliver most of the biological cargo from MSCs and might be adapted for clinical treatment, currently being developed for therapy of premature infants with neonatal respiratory distress syndrome [125]. It is not possible at this time to determine the relative efficacy of the various sources of MSCs, although there is considerable interest in iPSC-derived MSCs as they might accelerate and simplify production providing there are no safety issues that emerge in pre-clinical studies, especially regarding oncogenic risk.
Optimal MSC dosing regimen
The optimal dosing regimen of MSCs for viral pneumonia is uncertain, and pre-clinical studies in small animals provide limited insights regarding dosing or timing of MSC therapy in the clinical setting. In pre-clinical studies, a dose-dependent effect, with greater efficacy seen with higher doses, has been frequently reported [126]. However, in a human LPS model, dose-dependent adverse effects were demonstrated at the highest dose investigated with enhanced febrile response and coagulation activation reported [127]. Furthermore, in a human study of diabetic nephropathy, 150 but not 300 million MSCs improved renal function [128]. Neither lower nor higher doses may be optimal [129]. The dose used in phase 1 and 2a trials [121, 122], and an ongoing 2b clinical trial in ARDS (https://stattrial.com; password—StemCells4All NCT03818854) has been based on a study in a sheep bacterial pneumonia and sepsis model indicating that 10 × 106 MSC/kg was superior to 5 × 106 MSC/kg for reducing pulmonary oedema [130]. Other trials have used lower doses ranging from 2–4-10 × 106 MSCs/kg [114]. More studies will be needed to establish optimal dosing which may also depend on the source of the MSCs and their viability and potency. It is also possible that a higher dose may be needed for ARDS if the patient’s clinical course includes evidence of systemic injury with shock.
Frequency of MSC administration is a further consideration. Although studies in ARDS and sepsis have used a single intravenous infusion of MSCs, data emerging from studies in COVID-19 support efficacy with multiple doses. There is also some pre-clinical evidence to support multiple dosing regiments. A preliminary report in bacterial pneumonia and sepsis in sheep that showed efficacy of two intravenous doses of 10 × 106 i-MSCs/kg spaced 24 h apart [124]. The new evidence that MSCs can reduce biologic evidence of lung injury in BALF 48 h after intravenous administration of 10 × 106 MSCs/kg suggests that perhaps a second dose could be administered between 48 and 72 h to maximize efficacy, especially if the patient was not clinically improving by oxygenation and other pulmonary and systemic criteria.
A further challenge related to dosing regimens is that differing MSC products may not have consistent efficacy. A potency assay to standardize therapeutic efficacy of a dose would be an important development to allow direct comparison of MSC products, however at present this is not available.
Optimal patient population
The immunomodulatory effect of MSCs raises the potential that they may be more likely to exert therapeutic effects in the subgroup of patients with a dysfunctional pro-inflammatory response to viral infection. Proof-of-concept for this approach comes from research into patients with ARDS, where hypo- and hyper-inflammatory endotypes have been identified [131, 132] that respond differently to therapeutic interventions [133]. Calfee and colleagues reported that a parsimonious set of three biologic markers identified patients with higher mortality versus lower mortality for classical ARDS prior to COVID-19 (interleukin-8, Protein C, and bicarbonate), and this approach worked very well even in a re-analysis of the START-2 trial 60 patient trial of MSCs versus placebo [134]. Other approaches could include focusing on viral pneumonia patients with the highest mortality risk, which would include patients in shock requiring vasopressors. Another approach would be to use a radiographic severity score showing more pulmonary oedema with a RALE score greater than 20 for example, since higher RALE scores is an independent marker of higher mortality [135].
Impairment of MSCs by viral infection
A potential concern is that viruses may directly infect and impair MSC function due to their expression of receptors which allow the entry of several types of viruses. However, because MSCs are negative for the aACE2 and TMPRSS2 proteins, they are not susceptible to SARS-CoV-2 [136]. Therefore, MSCs would be safe and effective for treating patients with COVID-19 pneumonia. CD147 is another entry receptor for SARS-CoV-2, expressed by tissue-specific stem cells [137] and certain pulmonary cells. Viral infection by either route and intracellular replication results in both loss of airway epithelial cells and regenerating stem cells, thus diminishing cellular and lung regeneration.
MSCs use with concomitant therapy
One final challenge relates to the potential interaction of concomitant therapies, particularly in the setting of COVID-19 pneumonia. Treatment with steroids is now standard of care for hospitalized patients with COVID-19 requiring oxygen therapy. However, there are data to suggest that steroids may attenuate the beneficial effects of MSCs which will have implications for the impact of MSCs in this setting [138].
Summary and conclusions
Mesenchymal stem cells display considerable promise for the treatment of more severe viral pneumonia, display potentially relevant mechanisms of action, and have a demonstrated safety profile in early phase studies, MSCS are currently being evaluated in multiple larger phase 2 clinical trials for COVID-19 pneumonia. Important insights will emerge from these studies in the coming months. Nevertheless, important knowledge gaps remain to be elucidated, including the optimal cell type, dose regimen and dose timing, as well as the optimal patient subpopulations for these therapies. There are also challenges to the scale-up of MSC production to conduct large phase 3 studies and for their eventual clinical use should they prove effective for the treatment of viral pneumonia. Further research aimed at optimizing the therapeutic potential of these MSCs for viral pneumonia will continue to be an important priority if we are to realise their therapeutic potential for this devastating condition.
Availability of data and materials
Not applicable.
References
Abreu SC, Lopes-Pacheco M, Weiss DJ, Rocco PRM (2021) Mesenchymal Stromal Cell-Derived Extracellular Vesicles in Lung Diseases: Current Status and Perspectives. Front Cell Dev Biol 9:600711
Khoury M, Cuenca J, Cruz FF, Figueroa FE, Rocco PRM, Weiss DJ (2020) Current status of cell-based therapies for respiratory virus infections: applicability to COVID-19. Eur Respir J 55:8
Jain S, Self WH, Wunderink RG, Team CES (2015) Community-Acquired Pneumonia Requiring Hospitalization. N Engl J Med 373:2382
Choi SH, Hong SB, Ko GB, Lee Y, Park HJ, Park SY, Moon SM, Cho OH, Park KH, Chong YP, Kim SH, Huh JW, Sung H, Do KH, Lee SO, Kim MN, Jeong JY, Lim CM, Kim YS, Woo JH, Koh Y (2012) Viral infection in patients with severe pneumonia requiring intensive care unit admission. Am J Respir Crit Care Med 186:325–332
Centers for Disease Control and Prevention NCfIaRDN (2021) Burden of Influenza. Book Burden of Influenza, New York
Iuliano AD, Roguski KM, Chang HH, Muscatello DJ, Palekar R, Tempia S, Cohen C, Gran JM, Schanzer D, Cowling BJ, Wu P, Kyncl J, Ang LW, Park M, Redlberger-Fritz M, Yu H, Espenhain L, Krishnan A, Emukule G, van Asten L, Pereira da Silva S, Aungkulanon S, Buchholz U, Widdowson MA, Bresee JS, Collaborator G-A, N, (2018) Estimates of global seasonal influenza-associated respiratory mortality: a modelling study. Lancet 391:1285–1300
Babcock HM, Merz LR, Fraser VJ (2006) Is influenza an influenza-like illness? Clinical presentation of influenza in hospitalized patients. Infect Control Hosp Epidemiol 27:266–270
Chow EJ, Doyle JD, Uyeki TM (2019) Influenza virus-related critical illness: prevention, diagnosis, treatment. Crit Care 23:214
Ramsey CD, Kumar A (2013) Influenza and endemic viral pneumonia. Crit Care Clin 29:1069–1086
Dandachi D, Rodriguez-Barradas MC (2018) Viral pneumonia: etiologies and treatment. J Investig Med 66:957–965
Murray CJ, Lopez AD, Chin B, Feehan D, Hill KH (2006) Estimation of potential global pandemic influenza mortality on the basis of vital registry data from the 1918–20 pandemic: a quantitative analysis. Lancet 368:2211–2218
Perez-Padilla R, de la Rosa-Zamboni D, Ponce de Leon S, Hernandez M, Quinones-Falconi F, Bautista E, Ramirez-Venegas A, Rojas-Serrano J, Ormsby CE, Corrales A, Higuera A, Mondragon E, Cordova-Villalobos JA, Influenza IWGo, (2009) Pneumonia and respiratory failure from swine-origin influenza A (H1N1) in Mexico. N Engl J Med 361:680–689
Writing Committee of the WHOCoCAoPI, Bautista E, Chotpitayasunondh T, Gao Z, Harper SA, Shaw M, Uyeki TM, Zaki SR, Hayden FG, Hui DS, Kettner JD, Kumar A, Lim M, Shindo N, Penn C, Nicholson KG (2010) Clinical aspects of pandemic 2009 influenza A (H1N1) virus infection. N Engl J Med 362:1708–1719
To KK, Chan JF, Yuen KY (2014) Viral lung infections: epidemiology, virology, clinical features, and management of avian influenza A(H7N9). Curr Opin Pulm Med 20:225–232
Peiris JS, Yuen KY, Osterhaus AD, Stohr K (2003) The severe acute respiratory syndrome. N Engl J Med 349:2431–2441
Lee N, Qureshi ST (2013) Other viral pneumonias: coronavirus, respiratory syncytial virus, adenovirus, hantavirus. Crit Care Clin 29:1045–1068
Zaki AM, van Boheemen S, Bestebroer TM, Osterhaus AD, Fouchier RA (2012) Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N Engl J Med 367:1814–1820
WHO (2021) Middle East respiratory syndrome coronavirus (MERS-CoV). In: Book Middle East respiratory syndrome coronavirus (MERS-CoV)
Medicine JHU (2021) Coronavirus Resource Center - Global Map. In: Book Coronavirus Resource Center - Global Map
Network C-IGobotR, the C-ICUI (2021) Clinical characteristics and day-90 outcomes of 4244 critically ill adults with COVID-19: a prospective cohort study. Intensive Care Med 47:60–73
Grasselli G, Greco M, Zanella A, Albano G, Antonelli M, Bellani G, Bonanomi E, Cabrini L, Carlesso E, Castelli G, Cattaneo S, Cereda D, Colombo S, Coluccello A, Crescini G, Forastieri Molinari A, Foti G, Fumagalli R, Iotti GA, Langer T, Latronico N, Lorini FL, Mojoli F, Natalini G, Pessina CM, Ranieri VM, Rech R, Scudeller L, Rosano A, Storti E, Thompson BT, Tirani M, Villani PG, Pesenti A, Cecconi M, Network C-LI (2020) Risk Factors Associated With Mortality Among Patients With COVID-19 in Intensive Care Units in Lombardy, Italy. JAMA Intern Med 180:1345–1355
Wu C, Chen X, Cai Y, Xia J, Zhou X, Xu S, Huang H, Zhang L, Zhou X, Du C, Zhang Y, Song J, Wang S, Chao Y, Yang Z, Xu J, Zhou X, Chen D, Xiong W, Xu L, Zhou F, Jiang J, Bai C, Zheng J, Song Y (2020) Risk factors associated with acute respiratory distress syndrome and death in patients with coronavirus disease 2019 pneumonia in Wuhan, China. JAMA Intern Med 180:934–943
Lim ZJ, Subramaniam A, Ponnapa Reddy M, Blecher G, Kadam U, Afroz A, Billah B, Ashwin S, Kubicki M, Bilotta F, Curtis JR, Rubulotta F (2021) Case fatality rates for patients with COVID-19 requiring invasive mechanical ventilation. a meta-analysis. Am J Respir Crit Care Med 203:54–66
Jung C, Flaatten H, Fjolner J, Bruno RR, Wernly B, Artigas A, Bollen Pinto B, Schefold JC, Wolff G, Kelm M, Beil M, Sviri S, van Heerden PV, Szczeklik W, Czuczwar M, Elhadi M, Joannidis M, Oeyen S, Zafeiridis T, Marsh B, Andersen FH, Moreno R, Cecconi M, Leaver S, Boumendil A, De Lange DW, Guidet B (2021) The impact of frailty on survival in elderly intensive care patients with COVID-19: the COVIP study. Crit Care 25:149
Gonzalez J, Benitez ID, Carmona P, Santisteve S, Monge A, Moncusi-Moix A, Gort-Paniello C, Pinilla L, Carratala A, Zuil M, Ferrer R, Ceccato A, Fernandez L, Motos A, Riera J, Menendez R, Garcia-Gasulla D, Penuelas O, Bermejo-Martin JF, Labarca G, Caballero J, Torres G, de Gonzalo-Calvo D, Torres A, Barbe F, Project C (2021) Pulmonary Function and Radiologic Features in Survivors of Critical COVID-19: A 3-Month Prospective Cohort. Chest 160:187–198
Barbeta E, Benegas M, Sanchez M, Motos A, Ferrer M, Ceccato A, Lopez R, Bueno L, Artigas RM, Ferrando C, Fernandez-Barat L, Albacar N, Badia JR, Lopez T, Sandoval E, Toapanta D, Castro P, Soriano A, Torres A, Care CCC, G, (2021) Risk Factors and Clinical Impact of Fibrotic-Like Changes and the Organizing Pneumonia Pattern in Patients with COVID-19- and Non-COVID-19-induced Acute Respiratory Distress Syndrome. Arch Bronconeumol 23:9
Matthay MA, Zemans RL, Zimmerman GA, Arabi YM, Beitler JR, Mercat A, Herridge M, Randolph AG, Calfee CS (2019) Acute respiratory distress syndrome. Nat Rev Dis Primers 5:18
Hue S, Beldi-Ferchiou A, Bendib I, Surenaud M, Fourati S, Frapard T, Rivoal S, Razazi K, Carteaux G, Delfau-Larue MH, Mekontso-Dessap A, Audureau E, de Prost N (2020) Uncontrolled Innate and Impaired Adaptive Immune Responses in Patients with COVID-19 Acute Respiratory Distress Syndrome. Am J Respir Crit Care Med 202:1509–1519
Matrosovich MN, Matrosovich TY, Gray T, Roberts NA, Klenk HD (2004) Human and avian influenza viruses target different cell types in cultures of human airway epithelium. Proc Natl Acad Sci U S A 101:4620–4624
Wang N, Shi X, Jiang L, Zhang S, Wang D, Tong P, Guo D, Fu L, Cui Y, Liu X, Arledge KC, Chen YH, Zhang L, Wang X (2013) Structure of MERS-CoV spike receptor-binding domain complexed with human receptor DPP4. Cell Res 23:986–993
Meyerholz DK, Lambertz AM, McCray PB Jr (2016) Dipeptidyl Peptidase 4 Distribution in the Human Respiratory Tract: Implications for the Middle East Respiratory Syndrome. Am J Pathol 186:78–86
Hoffmann M, Kleine-Weber H, Schroeder S, Kruger N, Herrler T, Erichsen S, Schiergens TS, Herrler G, Wu NH, Nitsche A, Muller MA, Drosten C, Pohlmann S, (2020) SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 181: 271–280
Lambert DW, Yarski M, Warner FJ, Thornhill P, Parkin ET, Smith AI, Hooper NM, Turner AJ (2005) Tumor necrosis factor-alpha convertase (ADAM17) mediates regulated ectodomain shedding of the severe-acute respiratory syndrome-coronavirus (SARS-CoV) receptor, angiotensin-converting enzyme-2 (ACE2). J Biol Chem 280:30113–30119
Heurich A, Hofmann-Winkler H, Gierer S, Liepold T, Jahn O, Pohlmann S (2014) TMPRSS2 and ADAM17 cleave ACE2 differentially and only proteolysis by TMPRSS2 augments entry driven by the severe acute respiratory syndrome coronavirus spike protein. J Virol 88:1293–1307
Clementi N, Ghosh S, De Santis M, Castelli M, Criscuolo E, Zanoni I, Clementi M, Mancini N (2021) Viral Respiratory Pathogens and Lung Injury. Clin Microbiol Rev 34:87
McElvaney OJ, McEvoy NL, McElvaney OF, Carroll TP, Murphy MP, Dunlea DM, Ni Choileain O, Clarke J, O’Connor E, Hogan G, Ryan D, Sulaiman I, Gunaratnam C, Branagan P, O’Brien ME, Morgan RK, Costello RW, Hurley K, Walsh S, de Barra E, McNally C, McConkey S, Boland F, Galvin S, Kiernan F, O’Rourke J, Dwyer R, Power M, Geoghegan P, Larkin C, O’Leary RA, Freeman J, Gaffney A, Marsh B, Curley GF, McElvaney NG (2020) Characterization of the Inflammatory Response to Severe COVID-19 Illness. Am J Respir Crit Care Med 202:812–821
Chen G, Wu D, Guo W, Cao Y, Huang D, Wang H, Wang T, Zhang X, Chen H, Yu H, Zhang X, Zhang M, Wu S, Song J, Chen T, Han M, Li S, Luo X, Zhao J, Ning Q (2020) Clinical and immunological features of severe and moderate coronavirus disease 2019. J Clin Invest 130:2620–2629
Schulert GS, Zhang M, Fall N, Husami A, Kissell D, Hanosh A, Zhang K, Davis K, Jentzen JM, Napolitano L, Siddiqui J, Smith LB, Harms PW, Grom AA, Cron RQ (2016) Whole-Exome Sequencing Reveals Mutations in Genes Linked to Hemophagocytic Lymphohistiocytosis and Macrophage Activation Syndrome in Fatal Cases of H1N1 Influenza. J Infect Dis 213:1180–1188
Mehta P, McAuley DF, Brown M, Sanchez E, Tattersall RS, Manson JJ, Collaboration HAS, UK, (2020) COVID-19: consider cytokine storm syndromes and immunosuppression. Lancet 395:1033–1034
Sinha P, Matthay MA, Calfee CS (2020) Is a “Cytokine Storm” Relevant to COVID-19? JAMA Intern Med 180:1152–1154
McElvaney OJ, Curley GF, Rose-John S, McElvaney NG (2021) Interleukin-6: obstacles to targeting a complex cytokine in critical illness. Lancet Respir Med 9:643–654
Palmas F, Clarke J, Colas RA, Gomez EA, Keogh A, Boylan M, McEvoy N, McElvaney OJ, McElvaney O, Alalqam R, McElvaney NG, Curley GF, Dalli J (2021) Dysregulated plasma lipid mediator profiles in critically ill COVID-19 patients. PLoS ONE 16:e0256226
Blanco-Melo D, Nilsson-Payant BE, Liu WC, Uhl S, Hoagland D, Moller R, Jordan TX, Oishi K, Panis M, Sachs D, Wang TT, Schwartz RE, Lim JK, Albrecht RA, tenOever BR (2020) Imbalanced Host Response to SARS-CoV-2 Drives Development of COVID-19. Cell 181:1036–1045
Zhou Z, Ren L, Zhang L, Zhong J, Xiao Y, Jia Z, Guo L, Yang J, Wang C, Jiang S, Yang D, Zhang G, Li H, Chen F, Xu Y, Chen M, Gao Z, Yang J, Dong J, Liu B, Zhang X, Wang W, He K, Jin Q, Li M, Wang J (2020) Heightened Innate Immune Responses in the Respiratory Tract of COVID-19 Patients. Cell Host Microbe 27:883–890
Shuai H, Chu H, Hou Y, Yang D, Wang Y, Hu B, Huang X, Zhang X, Chai Y, Cai JP, Chan JF, Yuen KY (2020) Differential immune activation profile of SARS-CoV-2 and SARS-CoV infection in human lung and intestinal cells: Implications for treatment with IFN-beta and IFN inducer. J Infect 81:e1–e10
Channappanavar R, Fehr AR, Vijay R, Mack M, Zhao J, Meyerholz DK, Perlman S (2016) Dysregulated Type I Interferon and Inflammatory Monocyte-Macrophage Responses Cause Lethal Pneumonia in SARS-CoV-Infected Mice. Cell Host Microbe 19:181–193
Ackermann M, Verleden SE, Kuehnel M, Haverich A, Welte T, Laenger F, Vanstapel A, Werlein C, Stark H, Tzankov A, Li WW, Li VW, Mentzer SJ, Jonigk D (2020) Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in Covid-19. N Engl J Med 383:120–128
Hamming I, Timens W, Bulthuis ML, Lely AT, Navis G, van Goor H (2004) Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J Pathol 203:631–637
Varga Z, Flammer AJ, Steiger P, Haberecker M, Andermatt R, Zinkernagel AS, Mehra MR, Schuepbach RA, Ruschitzka F, Moch H (2020) Endothelial cell infection and endotheliitis in COVID-19. Lancet 395:1417–1418
Ward SE, Fogarty H, Karampini E, Lavin M, Schneppenheim S, Dittmer R, Morrin H, Glavey S, Ni Cheallaigh C, Bergin C, Martin-Loeches I, Mallon PW, Curley GF, Baker RI, Budde U, O’Sullivan JM, Krish JS (2021) ADAMTS13 regulation of VWF multimer distribution in severe COVID-19. J Thromb Haemost 19:1914–1921
Prevention CCfdca (2021) Influenza (Flu) For Clinicians: Antiviral Medication. In: Book Influenza (Flu) For Clinicians: Antiviral Medication
Bai Y, Jones JC, Wong SS, Zanin M (2021) Antivirals Targeting the Surface Glycoproteins of Influenza Virus: Mechanisms of Action and Resistance. Viruses 13:3
Control EECfDPa (2017) Expert opinion on neuraminidase inhibitors for the prevention and treatment of influenza. In: Book Expert opinion on neuraminidase inhibitors for the preventiona and treatment of influenza. ECDC
Hayden FG, Shindo N (2019) Influenza virus polymerase inhibitors in clinical development. Curr Opin Infect Dis 32:176–186
Ikematsu H, Kawai N, Iwaki N, Kashiwagi S (2017) Duration of fever and other symptoms after the inhalation of laninamivir octanoate hydrate; comparison of the 2011/12 to 2015/16 Japanese influenza seasons. J Infect Chemother 23:627–633
Hayden F (2009) Developing new antiviral agents for influenza treatment: what does the future hold? Clin Infect Dis 48(Suppl 1):S3-13
Ison MG, Portsmouth S, Yoshida Y, Shishido T, Mitchener M, Tsuchiya K, Uehara T, Hayden FG (2020) Early treatment with baloxavir marboxil in high-risk adolescent and adult outpatients with uncomplicated influenza (CAPSTONE-2): a randomised, placebo-controlled, phase 3 trial. Lancet Infect Dis 20:1204–1214
Mtambo SE, Amoako DG, Somboro AM, Agoni C, Lawal MM, Gumede NS, Khan RB, Kumalo HM (2021) Influenza Viruses: Harnessing the Crucial Role of the M2 Ion-Channel and Neuraminidase toward Inhibitor Design. Molecules 26:67
Han J, Perez J, Schafer A, Cheng H, Peet N, Rong L, Manicassamy B (2018) Influenza Virus: Small Molecule Therapeutics and Mechanisms of Antiviral Resistance. Curr Med Chem 25:5115–5127
Herold S, Hoegner K, Vadasz I, Gessler T, Wilhelm J, Mayer K, Morty RE, Walmrath HD, Seeger W, Lohmeyer J (2014) Inhaled granulocyte/macrophage colony-stimulating factor as treatment of pneumonia-associated acute respiratory distress syndrome. Am J Respir Crit Care Med 189:609–611
Paine R 3rd, Standiford TJ, Dechert RE, Moss M, Martin GS, Rosenberg AL, Thannickal VJ, Burnham EL, Brown MB, Hyzy RC (2012) A randomized trial of recombinant human granulocyte-macrophage colony stimulating factor for patients with acute lung injury. Crit Care Med 40:90–97
Halstead ES, Umstead TM, Davies ML, Kawasawa YI, Silveyra P, Howyrlak J, Yang L, Guo W, Hu S, Hewage EK, Chroneos ZC (2018) GM-CSF overexpression after influenza a virus infection prevents mortality and moderates M1-like airway monocyte/macrophage polarization. Respir Res 19:3
Services. USDoHH (2019) Influenza Therapeutics Program. In: Book Influenza Therapeutics Program
Services. USDoHH (2019) Influenza & emerging infectious diseases. In: Book influenza & emerging infectious diseases
Beigel JH, Nam HH, Adams PL, Krafft A, Ince WL, El-Kamary SS, Sims AC (2019) Advances in respiratory virus therapeutics - A meeting report from the 6th isirv Antiviral Group conference. Antiviral Res 167:45–67
Weis S, TeVelthuis AJW (2021) Influenza Virus RNA Synthesis and the Innate Immune Response. Viruses 13:8
Bahadoran A, Bezavada L, Smallwood HS (2020) Fueling influenza and the immune response: Implications for metabolic reprogramming during influenza infection and immunometabolism. Immunol Rev 295:140–166
Land WG (2021) Role of DAMPs in respiratory virus-induced acute respiratory distress syndrome-with a preliminary reference to SARS-CoV-2 pneumonia. Genes Immun 22:141–160
McIntyre LA, Moher D, Fergusson DA, Sullivan KJ, Mei SH, Lalu M, Marshall J, McLeod M, Griffin G, Grimshaw J, Turgeon A, Avey MT, Rudnicki MA, Jazi M, Fishman J, Stewart DJ, Biology CCCT, G, (2016) Efficacy of Mesenchymal Stromal Cell Therapy for Acute Lung Injury in Preclinical Animal Models: A Systematic Review. PLoS ONE 11:e0147170
Masterson CH, Curley GF, Laffey JG (2019) Modulating the distribution and fate of exogenously delivered MSCs to enhance therapeutic potential: knowns and unknowns. Intensive Care Med Exp 7:41
Masterson CH, Tabuchi A, Hogan G, Fitzpatrick G, Kerrigan SW, Jerkic M, Kuebler WM, Laffey JG, Curley GF (2021) Intra-vital imaging of mesenchymal stromal cell kinetics in the pulmonary vasculature during infection. Sci Rep 11:5265
Armitage J, Tan DBA, Troedson R, Young P, Lam KV, Shaw K, Sturm M, Weiss DJ, Moodley YP (2018) Mesenchymal stromal cell infusion modulates systemic immunological responses in stable COPD patients: a phase I pilot study. Eur Respir J 51:8
Waterman RS, Tomchuck SL, Henkle SL, Betancourt AM (2010) A new mesenchymal stem cell (MSC) paradigm: polarization into a pro-inflammatory MSC1 or an Immunosuppressive MSC2 phenotype. PLoS ONE 5:e10088
Kusuma GD, Carthew J, Lim R, Frith JE (2017) Effect of the Microenvironment on Mesenchymal Stem Cell Paracrine Signaling: Opportunities to Engineer the Therapeutic Effect. Stem Cells Dev 26:617–631
Islam D, Huang Y, Fanelli V, Delsedime L, Wu S, Khang J, Han B, Grassi A, Li M, Xu Y, Luo A, Wu J, Liu X, McKillop M, Medin J, Qiu H, Zhong N, Liu M, Laffey J, Li Y, Zhang H (2019) Identification and modulation of microenvironment is crucial for effective mesenchymal stromal cell therapy in acute lung injury. Am J Respir Crit Care Med 199:1214–1224
Ullah M, Liu DD, Thakor AS (2019) Mesenchymal Stromal Cell Homing: Mechanisms and Strategies for Improvement. Science 15:421–438
Murray LMA, Krasnodembskaya AD (2019) Concise Review: Intercellular Communication Via Organelle Transfer in the Biology and Therapeutic Applications of Stem Cells. Stem Cells 37:14–25
Maron-Gutierrez T, Rocco PRM (2020) Cell-Free Therapies: Novel Approaches for COVID-19. Front Immunol 11:583017
Islam MN, Das SR, Emin MT, Wei M, Sun L, Westphalen K, Rowlands DJ, Quadri SK, Bhattacharya S, Bhattacharya J (2012) Mitochondrial transfer from bone-marrow-derived stromal cells to pulmonary alveoli protects against acute lung injury. Nat Med 18:759–765
de Carvalho LRP, Abreu SC, de Castro LL, Andrade da Silva LH, Silva PM, Vieira JB, Santos RT, Cabral MR, Khoury M, Weiss DJ, Lopes-Pacheco M, Silva PL, Cruz FF, Rocco PRM (2021) Mitochondria-Rich Fraction Isolated From Mesenchymal Stromal Cells Reduces Lung and Distal Organ Injury in Experimental Sepsis. Crit Care Med 49:e880
Moll G, Drzeniek N, Kamhieh-Milz J, Geissler S, Volk HD, Reinke P (2020) MSC Therapies for COVID-19: Importance of Patient Coagulopathy, Thromboprophylaxis, Cell Product Quality and Mode of Delivery for Treatment Safety and Efficacy. Front Immunol 11:1091
Weiss DJ, English K, Krasnodembskaya A, Isaza-Correa JM, Hawthorne IJ, Mahon BP (2019) The Necrobiology of Mesenchymal Stromal Cells Affects Therapeutic Efficacy. Front Immunol 10:1228
Reddy K, Calfee CS, McAuley DF (2021) Acute Respiratory Distress Syndrome Subphenotypes beyond the Syndrome: A Step toward Treatable Traits? Am J Respir Crit Care Med 203:1449–1451
Horie S, Gaynard S, Murphy M, Barry F, Scully M, O’Toole D, Laffey JG (2020) Cytokine pre-activation of cryopreserved xenogeneic-free human mesenchymal stromal cells enhances resolution and repair following ventilator-induced lung injury potentially via a KGF-dependent mechanism. Intensive Care Med Exp 8:8
Rolandsson Enes S, Hampton TH, Barua J, McKenna DH, Dos Santos CC, Amiel E, Ashare A, Liu KD, Krasnodembskaya AD, English K, Stanton BA, Rocco PRM, Matthay MA, Weiss DJ (2021) Healthy versus inflamed lung environments differentially effect MSCs. Eur Respir J 7:89
Bustos ML, Huleihel L, Meyer EM, Donnenberg AD, Donnenberg VS, Sciurba JD, Mroz L, McVerry BJ, Ellis BM, Kaminski N, Rojas M (2013) Activation of human mesenchymal stem cells impacts their therapeutic abilities in lung injury by increasing interleukin (IL)-10 and IL-1RN levels. Stem Cells Transl Med 2:884–895
Abreu SC, Rolandsson Enes S, Dearborn J, Goodwin M, Coffey A, Borg ZD, Dos Santos CC, Wargo MJ, Cruz FF, Loi R, DeSarno M, Ashikaga T, Antunes MA, Rocco PRM, Liu KD, Lee JW, Matthay MA, McKenna DH, Weiss DJ (2019) Lung inflammatory environments differentially alter mesenchymal stromal cell behavior. Am J Physiol Lung Cell Mol Physiol 317:L823–L831
Abreu SC, Xisto DG, de Oliveira TB, Blanco NG, de Castro LL, Kitoko JZ, Olsen PC, Lopes-Pacheco M, Morales MM, Weiss DJ, Rocco PRM (2019) Serum from Asthmatic Mice Potentiates the Therapeutic Effects of Mesenchymal Stromal Cells in Experimental Allergic Asthma. Stem Cells Transl Med 8:301–312
Abreu SC, Hampton TH, Hoffman E, Dearborn J, Ashare A, Singh Sidhu K, Matthews DE, McKenna DH, Amiel E, Barua J, Krasnodembskaya A, English K, Mahon B, Dos Santos C, Cruz FF, Chambers DC, Liu KD, Matthay MA, Cramer RA, Stanton BA, Rocco PRM, Wargo MJ, Weiss DJ, Rolandsson Enes S (2020) Differential effects of the cystic fibrosis lung inflammatory environment on mesenchymal stromal cells. Am J Physiol Lung Cell Mol Physiol 319:L908–L925
Noronha NC, Mizukami A, Caliari-Oliveira C, Cominal JG, Rocha JLM, Covas DT, Swiech K, Malmegrim KCR (2019) Priming approaches to improve the efficacy of mesenchymal stromal cell-based therapies. Stem Cell Res Ther 10:131
Hayes M, Curley GF, Masterson C, Devaney J, O’Toole D, Laffey JG (2015) Mesenchymal stromal cells are more effective than the MSC secretome in diminishing injury and enhancing recovery following ventilator-induced lung injury. Intensive Care Med Exp 3:29
Eleuteri S, Fierabracci A (2019) Insights into the Secretome of Mesenchymal Stem Cells and Its Potential Applications. Int J Mol Sci 20:7
McCarthy SD, Horgan E, Ali A, Masterson C, Laffey JG, MacLoughlin R, O’Toole D (2020) Nebulized Mesenchymal Stem Cell Derived Conditioned Medium Retains Antibacterial Properties Against Clinical Pathogen Isolates. J Aerosol Med Pulm Drug Deliv 33:140–152
Shi M-M, Yang Q-Y, Monsel A, Yan J-Y, Dai C-X, Zhao J-Y, Shi G-C, Zhou M, Zhu X-M, Li S-K, Li P, Wang J, Li M, Lei J-G, Xu D, Zhu Y-G, Qu J-M (2021) Preclinical efficacy and clinical safety of clinical-grade nebulized allogenic adipose mesenchymal stromal cells-derived extracellular vesicles. J Extracellular Vesicles 10:e12134
Lee JW, Krasnodembskaya A, McKenna DH, Song Y, Abbott J, Matthay MA (2013) Therapeutic effects of human mesenchymal stem cells in ex vivo human lungs injured with live bacteria. Am J Respir Crit Care Med 187:751–760
Monsel A, Zhu YG, Gennai S, Hao Q, Hu S, Rouby JJ, Rosenzwajg M, Matthay MA, Lee JW (2015) Therapeutic Effects of Human Mesenchymal Stem Cell-derived Microvesicles in Severe Pneumonia in Mice. Am J Respir Crit Care Med 192:324–336
Cruz FF, Weiss DJ, Rocco PR (2016) Prospects and progress in cell therapy for acute respiratory distress syndrome. Expert Opin Biol Ther 16:1353–1360
Laffey JG, Matthay MA (2017) Fifty Years of Research in ARDS. Cell-based Therapy for Acute Respiratory Distress Syndrome. Biology and Potential Therapeutic Value. Am J Respir Crit Care Med 196:266–273
Polchert D, Sobinsky J, Douglas G, Kidd M, Moadsiri A, Reina E, Genrich K, Mehrotra S, Setty S, Smith B, Bartholomew A (2008) IFN-gamma activation of mesenchymal stem cells for treatment and prevention of graft versus host disease. Eur J Immunol 38:1745–1755
Liu X, Feng T, Gong T, Shen C, Zhu T, Wu Q, Li Q, Li H (2015) Human Umbilical Cord Mesenchymal Stem Cells Inhibit the Function of Allogeneic Activated Vgamma9Vdelta2 T Lymphocytes In Vitro. Biomed Res Int 2015:317801
Monguio-Tortajada M, Bayes-Genis A, Rosell A, Roura S (2021) Are mesenchymal stem cells and derived extracellular vesicles valuable to halt the COVID-19 inflammatory cascade? Current evidence and future perspectives. Thorax 76:196–200
Chan MC, Kuok DI, Leung CY, Hui KP, Valkenburg SA, Lau EH, Nicholls JM, Fang X, Guan Y, Lee JW, Chan RW, Webster RG, Matthay MA, Peiris JS (2016) Human mesenchymal stromal cells reduce influenza A H5N1-associated acute lung injury in vitro and in vivo. Proc Natl Acad Sci U S A 113:3621–3626
Li Y, Xu J, Shi W, Chen C, Shao Y, Zhu L, Lu W, Han X (2016) Mesenchymal stromal cell treatment prevents H9N2 avian influenza virus-induced acute lung injury in mice. Stem Cell Res Ther 7:159
Loy H, Kuok DIT, Hui KPY, Choi MHL, Yuen W, Nicholls JM, Peiris JSM, Chan MCW (2019) Therapeutic Implications of Human Umbilical Cord Mesenchymal Stromal Cells in Attenuating Influenza A(H5N1) Virus-Associated Acute Lung Injury. J Infect Dis 219:186–196
Darwish I, Banner D, Mubareka S, Kim H, Besla R, Kelvin DJ, Kain KC, Liles WC (2013) Mesenchymal stromal (stem) cell therapy fails to improve outcomes in experimental severe influenza. PLoS ONE 8:e71761
Gotts JE, Abbott J, Matthay MA (2014) Influenza causes prolonged disruption of the alveolar-capillary barrier in mice unresponsive to mesenchymal stem cell therapy. Am J Physiol Lung Cell Mol Physiol 307:L395-406
Deng W, Bao L, Liu J, Xiao C, Liu J, Xue J, Lv Q, Qi F, Gao H, Yu P, Xu Y, Qu Y, Li F, Xiang Z, Yu H, Gong S, Liu M, Wang G, Wang S, Song Z, Liu Y, Zhao W, Han Y, Zhao L, Liu X, Wei Q, Qin C (2020) Primary exposure to SARS-CoV-2 protects against reinfection in rhesus macaques. Science 369:818–823
Bao L, Deng W, Huang B, Gao H, Liu J, Ren L, Wei Q, Yu P, Xu Y, Qi F, Qu Y, Li F, Lv Q, Wang W, Xue J, Gong S, Liu M, Wang G, Wang S, Song Z, Zhao L, Liu P, Zhao L, Ye F, Wang H, Zhou W, Zhu N, Zhen W, Yu H, Zhang X, Guo L, Chen L, Wang C, Wang Y, Wang X, Xiao Y, Sun Q, Liu H, Zhu F, Ma C, Yan L, Yang M, Han J, Xu W, Tan W, Peng X, Jin Q, Wu G, Qin C (2020) The pathogenicity of SARS-CoV-2 in hACE2 transgenic mice. Nature 583:830–833
Abreu SC, Weiss DJ, Rocco PR (2016) Extracellular vesicles derived from mesenchymal stromal cells: a therapeutic option in respiratory diseases? Stem Cell Res Ther 7:53
Phelps J, Sanati-Nezhad A, Ungrin M, Duncan NA, Sen A (2018) Bioprocessing of Mesenchymal Stem Cells and Their Derivatives: Toward Cell-Free Therapeutics. Stem Cells Int 2018:9415367
Khatri M, Richardson LA, Meulia T (2018) Mesenchymal stem cell-derived extracellular vesicles attenuate influenza virus-induced acute lung injury in a pig model. Stem Cell Res Ther 9:17
Thompson M, Mei SHJ, Wolfe D, Champagne J, Fergusson D, Stewart DJ, Sullivan KJ, Doxtator E, Lalu M, English SW, Granton J, Hutton B, Marshall J, Maybee A, Walley KR, Santos CD, Winston B, McIntyre L (2020) Cell therapy with intravascular administration of mesenchymal stromal cells continues to appear safe: An updated systematic review and meta-analysis. E Clin Med 19:100249
Gorman E, Millar J, McAuley D, O’Kane C (2021) Mesenchymal stromal cells for acute respiratory distress syndrome (ARDS), sepsis, and COVID-19 infection: optimizing the therapeutic potential. Expert Rev Respir Med 15:301–324
Bellingan G, Jacono F, Bannard-Smith J, Brealey D, Meyer N, Thickett D, Young D, Bentley A, McVerry BJ, Wunderink RG, Doerschug KC, Summers C, Rojas M, Ting A, Jenkins ED (2021) Safety and efficacy of multipotent adult progenitor cells in acute respiratory distress syndrome (MUST-ARDS): a multicentre, randomised, double-blind, placebo-controlled phase 1/2 trial. Intensive Care Med 23:1–9. https://doi.org/10.1007/s00134-021-06570-4
Wick KD, Leligdowicz A, Zhuo H, Ware LB, Matthay MA (2021) Mesenchymal stromal cells reduce evidence of lung injury in patients with ARDS. JCI Insight 6:45
Chen J, Hu C, Chen L, Tang L, Zhu Y, Xu X, Chen L, Gao H, Lu X, Yu L, Dai X, Xiang C, Li L (2020) Clinical Study of Mesenchymal Stem Cell Treatment for Acute Respiratory Distress Syndrome Induced by Epidemic Influenza A (H7N9) Infection: A Hint for COVID-19 Treatment. Engineering (Beijing) 6:1153–1161
Shi L, Huang H, Lu X, Yan X, Jiang X, Xu R, Wang S, Zhang C, Yuan X, Xu Z, Huang L, Fu JL, Li Y, Zhang Y, Yao WQ, Liu T, Song J, Sun L, Yang F, Zhang X, Zhang B, Shi M, Meng F, Song Y, Yu Y, Wen J, Li Q, Mao Q, Maeurer M, Zumla A, Yao C, Xie WF, Wang FS (2021) Effect of human umbilical cord-derived mesenchymal stem cells on lung damage in severe COVID-19 patients: a randomized, double-blind, placebo-controlled phase 2 trial. Signal Transduct Target Ther 6:58
Lanzoni G, Linetsky E, Correa D, Messinger Cayetano S, Alvarez RA, Kouroupis D, Alvarez Gil A, Poggioli R, Ruiz P, Marttos AC, Hirani K, Bell CA, Kusack H, Rafkin L, Baidal D, Pastewski A, Gawri K, Lenero C, Mantero AMA, Metalonis SW, Wang X, Roque L, Masters B, Kenyon NS, Ginzburg E, Xu X, Tan J, Caplan AI, Glassberg MK, Alejandro R, Ricordi C (2021) Umbilical cord mesenchymal stem cells for COVID-19 acute respiratory distress syndrome: A double-blind, phase 1/2a, randomized controlled trial. Stem Cells Transl Med 10:660–673
Dilogo IH, Aditianingsih D, Sugiarto A, Burhan E, Damayanti T, Sitompul PA, Mariana N, Antarianto RD, Liem IK, Kispa T, Mujadid F, Novialdi N, Luviah E, Kurniawati T, Lubis AMT, Rahmatika D (2021) Umbilical cord mesenchymal stromal cells as critical COVID-19 adjuvant therapy: A randomized controlled trial. Stem Cells Transl Med 8:563
Gorman E, Shankar-Hari M, Hopkins P, Tunnicliffe WS, Perkins GD, Silversides J, McGuigan P, Jackson C, Boyle R, McFerran J, McDowell C, Campbell C, McFarland M, Smythe J, Thompson J, Williams B, Curley G, Laffey JG, Clarke M, O’Kane C, McAuley DF (2020) Repair of Acute Respiratory Distress Syndrome by Stromal Cell Administration in COVID-19 (REALIST-COVID-19): A structured summary of a study protocol for a randomised, controlled trial. Trials 21:462
Wilson JG, Liu KD, Zhuo H, Caballero L, McMillan M, Fang X, Cosgrove K, Vojnik R, Calfee CS, Lee JW, Rogers AJ, Levitt J, Wiener-Kronish J, Bajwa EK, Leavitt A, McKenna D, Thompson BT, Matthay MA (2015) Mesenchymal stem (stromal) cells for treatment of ARDS: a phase 1 clinical trial. Lancet Respir Med 3:24–32
Matthay MA, Calfee CS, Zhuo H, Thompson BT, Wilson JG, Levitt JE, Rogers AJ, Gotts JE, Wiener-Kronish JP, Bajwa EK, Donahoe MP, McVerry BJ, Ortiz LA, Exline M, Christman JW, Abbott J, Delucchi KL, Caballero L, McMillan M, McKenna DH, Liu KD (2019) Treatment with allogeneic mesenchymal stromal cells for moderate to severe acute respiratory distress syndrome (START study): a randomised phase 2a safety trial. Lancet Respir Med 7:154–162
Bustos ML, Huleihel L, Kapetanaki MG, Lino-Cardenas CL, Mroz L, Ellis BM, McVerry BJ, Richards TJ, Kaminski N, Cerdenes N, Mora AL, Rojas M (2014) Aging mesenchymal stem cells fail to protect because of impaired migration and antiinflammatory response. Am J Respir Crit Care Med 189:787–798
Hashimoto KBN, Enkhbaatar P, Angel M, Lader A, Czuczman M, Matthay MA (2021) Novel Induced-Mesenchymal Stem Cells (i-MSCs) Attenuate Severity of ARDS in Septic Sheep. Cytotherapy 23:5
Lee JW, Matthay MA (2019) Is a part better than the whole for cell-based therapy for acute respiratory distress syndrome? Anesthesiology 130:683–685
Devaney J, Horie S, Masterson C, Elliman S, Barry F, O’Brien T, Curley GF, O’Toole D, Laffey JG (2015) Human mesenchymal stromal cells decrease the severity of acute lung injury induced by E. coli in the rat. Thorax 70:625–635
Perlee D, van Vught LA, Scicluna BP, Maag A, Lutter R, Kemper EM, van ’t Veer C, Punchard MA, Gonzalez J, Richard MP, Dalemans W, Lombardo E, de Vos AF, van der Poll T, (2018) Intravenous Infusion of Human Adipose Mesenchymal Stem Cells Modifies the Host Response to Lipopolysaccharide in Humans: A Randomized, Single-Blind, Parallel Group, Placebo Controlled Trial. Stem Cells 36:1778–1788
Packham DK, Fraser IR, Kerr PG, Segal KR (2016) Allogeneic Mesenchymal Precursor Cells (MPC) in Diabetic Nephropathy: A Randomized, Placebo-controlled, Dose Escalation Study. EBioMedicine 12:263–269
Kabat M, Bobkov I, Kumar S, Grumet M (2020) Trends in mesenchymal stem cell clinical trials 2004–2018: Is efficacy optimal in a narrow dose range? Stem Cells Transl Med 9:17–27
Asmussen S, Ito H, Traber DL, Lee JW, Cox RA, Hawkins HK, McAuley DF, McKenna DH, Traber LD, Zhuo H, Wilson J, Herndon DN, Prough DS, Liu KD, Matthay MA, Enkhbaatar P (2014) Human mesenchymal stem cells reduce the severity of acute lung injury in a sheep model of bacterial pneumonia. Thorax 69:819–825
Calfee CS, Delucchi K, Parsons PE, Thompson BT, Ware LB, Matthay MA, Network NA (2014) Subphenotypes in acute respiratory distress syndrome: latent class analysis of data from two randomised controlled trials. Lancet Respir Med 2:611–620
Calfee CS, Delucchi KL, Sinha P, Matthay MA, Hackett J, Shankar-Hari M, McDowell C, Laffey JG, O’Kane CM, McAuley DF, Trials ICC, G, (2018) Acute respiratory distress syndrome subphenotypes and differential response to simvastatin: secondary analysis of a randomised controlled trial. Lancet Respir Med 6:691–698
Matthay MA, Arabi YM, Siegel ER, Ware LB, Bos LDJ, Sinha P, Beitler JR, Wick KD, Curley MAQ, Constantin JM, Levitt JE, Calfee CS (2020) Phenotypes and personalized medicine in the acute respiratory distress syndrome. Intensive Care Med 46:2136–2152
Sinha P, Delucchi KL, McAuley DF, O’Kane CM, Matthay MA, Calfee CS (2020) Development and validation of parsimonious algorithms to classify acute respiratory distress syndrome phenotypes: a secondary analysis of randomised controlled trials. Lancet Respir Med 8:247–257
Warren MA, Zhao Z, Koyama T, Bastarache JA, Shaver CM, Semler MW, Rice TW, Matthay MA, Calfee CS, Ware LB (2018) Severity scoring of lung oedema on the chest radiograph is associated with clinical outcomes in ARDS. Thorax 73:840–846
Leng Z, Zhu R, Hou W, Feng Y, Yang Y, Han Q, Shan G, Meng F, Du D, Wang S, Fan J, Wang W, Deng L, Shi H, Li H, Hu Z, Zhang F, Gao J, Liu H, Li X, Zhao Y, Yin K, He X, Gao Z, Wang Y, Yang B, Jin R, Stambler I, Lim LW, Su H, Moskalev A, Cano A, Chakrabarti S, Min KJ, Ellison-Hughes G, Caruso C, Jin K, Zhao RC (2020) Transplantation of ACE2(-) Mesenchymal Stem Cells Improves the Outcome of Patients with COVID-19 Pneumonia. Aging Dis 11:216–228
Ulrich H, Pillat MM (2020) CD147 as a Target for COVID-19 Treatment: Suggested Effects of Azithromycin and Stem Cell Engagement. Stem Cell Rev Rep 16:434–440
Chen X, Gan Y, Li W, Su J, Zhang Y, Huang Y, Roberts AI, Han Y, Li J, Wang Y, Shi Y (2014) The interaction between mesenchymal stem cells and steroids during inflammation. Cell Death Dis 5:e1009
Dobson J, Whitley RJ, Pocock S, Monto AS (2015) Oseltamivir treatment for influenza in adults: a meta-analysis of randomised controlled trials. Lancet 385:1729–1737
Butler D (2014) Tamiflu report comes under fire. Nature 508:439–440
Michiels B, Van Puyenbroeck K, Verhoeven V, Vermeire E, Coenen S (2013) The value of neuraminidase inhibitors for the prevention and treatment of seasonal influenza: a systematic review of systematic reviews. PLoS ONE 8:e60348
Ebell MH, Call M, Shinholser J (2013) Effectiveness of oseltamivir in adults: a meta-analysis of published and unpublished clinical trials. Fam Pract 30:125–133
Heneghan CJ, Onakpoya I, Thompson M, Spencer EA, Jones M, Jefferson T (2014) Zanamivir for influenza in adults and children: systematic review of clinical study reports and summary of regulatory comments. BMJ 348:g2547
Bradley JS, Blumer JL, Romero JR, Michaels MG, Munoz FM, Kimberlin DW, Pahud B, DeBiasi RL, Yamamoto G, Roberts G, Hossain M, Shortino D, Yates PJ, Adams B, Peppercorn A (2017) Intravenous Zanamivir in Hospitalized Patients With Influenza. Pediatrics 140:8
Marty FM, Vidal-Puigserver J, Clark C, Gupta SK, Merino E, Garot D, Chapman MJ, Jacobs F, Rodriguez-Noriega E, Husa P, Shortino D, Watson HA, Yates PJ, Peppercorn AF (2017) Intravenous zanamivir or oral oseltamivir for hospitalised patients with influenza: an international, randomised, double-blind, double-dummy, phase 3 trial. Lancet Respir Med 5:135–146
Kohno S, Yen MY, Cheong HJ, Hirotsu N, Ishida T, Kadota J, Mizuguchi M, Kida H, Shimada J, Group SCS (2011) Phase III randomized, double-blind study comparing single-dose intravenous peramivir with oral oseltamivir in patients with seasonal influenza virus infection. Antimicrob Agents Chemother 55:5267–5276
Thorlund K, Awad T, Boivin G, Thabane L (2011) Systematic review of influenza resistance to the neuraminidase inhibitors. BMC Infect Dis 11:134
Hayden FG, Sugaya N, Hirotsu N, Lee N, de Jong MD, Hurt AC, Ishida T, Sekino H, Yamada K, Portsmouth S, Kawaguchi K, Shishido T, Arai M, Tsuchiya K, Uehara T, Watanabe A, Baloxavir Marboxil Investigators G (2018) Baloxavir Marboxil for Uncomplicated Influenza in Adults and Adolescents. N Engl J Med 379:913–923
Baker J, Block SL, Matharu B, Burleigh Macutkiewicz L, Wildum S, Dimonaco S, Collinson N, Clinch B, Piedra PA (2020) Baloxavir Marboxil Single-dose Treatment in Influenza-infected Children: A Randomized, Double-blind, Active Controlled Phase 3 Safety and Efficacy Trial (miniSTONE-2). Pediatr Infect Dis J 39:700–705
Yokoyama T, Sakaguchi H, Ishibashi T, Shishido T, Piedra PA, Sato C, Tsuchiya K, Uehara T (2020) Baloxavir Marboxil 2% Granules in Japanese Children With Influenza: An Open-label Phase 3 Study. Pediatr Infect Dis J 39:706–712
Ikematsu H, Hayden FG, Kawaguchi K, Kinoshita M, de Jong MD, Lee N, Takashima S, Noshi T, Tsuchiya K, Uehara T (2020) Baloxavir Marboxil for Prophylaxis against Influenza in Household Contacts. N Engl J Med 383:309–320
Koshimichi H, Ishibashi T, Kawaguchi N, Sato C, Kawasaki A, Wajima T (2018) Safety, Tolerability, and Pharmacokinetics of the Novel Anti-influenza Agent Baloxavir Marboxil in Healthy Adults: Phase I Study Findings. Clin Drug Investig 38:1189–1196
Nakano T, Yamaguchi H, Chiba T, Shiosakai K, Chikada S, Matsuoka Y (2021) The safety and efficacy of the long-acting neuraminidase inhibitor laninamivir octanoate hydrate for Inhalation Suspension Set in children under the age of 5 in a post-marketing surveillance. J Infect Chemother 27:1436–1446
Watanabe A, Chang SC, Kim MJ, Chu DW, Ohashi Y, Group MS (2010) Long-acting neuraminidase inhibitor laninamivir octanoate versus oseltamivir for treatment of influenza: A double-blind, randomized, noninferiority clinical trial. Clin Infect Dis 51:1167–1175
Yamashita M, Tomozawa T, Kakuta M, Tokumitsu A, Nasu H, Kubo S (2009) CS-8958, a prodrug of the new neuraminidase inhibitor R-125489, shows long-acting anti-influenza virus activity. Antimicrob Agents Chemother 53:186–192
Hirotsu N, Saisho Y, Hasegawa T, Shishido T (2018) Clinical and virologic effects of four neuraminidase inhibitors in influenza A virus-infected children (aged 4–12 years): an open-label, randomized study in Japan. Expert Rev Anti Infect Ther 16:173–182
Acknowledgements
Not applicable.
Funding
Science Foundation Ireland Future Research Leaders Award (16/FRL-3845) awarded to J. Laffey.
Author information
Authors and Affiliations
Contributions
All authors contributed to the writing of the original draft and to the critical revision of the manuscript for important intellectual content.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
See COI forms for each author.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
Supplementary Table 1. Antiviral Medications Under investigation for treatment of Influenza.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Masterson, C.H., Ceccato, A., Artigas, A. et al. Mesenchymal stem/stromal cell-based therapies for severe viral pneumonia: therapeutic potential and challenges. ICMx 9, 61 (2021). https://doi.org/10.1186/s40635-021-00424-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40635-021-00424-5